

Abstract
Expression of the sodium iodide symporter (NIS) is required for efficient iodide uptake in thyroid and lactating breast. Since most differentiated thyroid cancer expresses NIS, β-emitting radioactive iodide is routinely utilized to target remnant thyroid cancer and metastasis after total thyroidectomy. Stimulation of NIS expression by high levels of thyroid-stimulating hormone is necessary to achieve radioiodide uptake into thyroid cancer that is sufficient for therapy. The majority of breast cancer also expresses NIS, but at a low level insufficient for radioiodine therapy. Retinoic acid is a potent NIS inducer in some breast cancer cells. NIS is also modestly expressed in some non-thyroidal tissues, including salivary glands, lacrimal glands and stomach. Selective induction of iodide uptake is required to target tumors with radioiodide. Iodide uptake in mammalian cells is dependent on the level of NIS gene expression, but also successful translocation of NIS to the cell membrane and correct insertion. The regulatory mechanisms of NIS expression and membrane insertion are regulated by signal transduction pathways that differ by tissue. Differential regulation of NIS confers selective induction of functional NIS in thyroid cancer cells, as well as some breast cancer cells, leading to more efficient radioiodide therapy for thyroid cancer and a new strategy for breast cancer therapy. The potential for systemic radioiodide treatment of a range of other cancers, that do not express endogenous NIS, has been demonstrated in models with tumor-selective introduction of exogenous NIS.
Keywords: Sodium iodide symporter, thyroid cancer, breast cancer, Transcriptional regulation, Posttranslational regulation
1. Introduction
Sodium iodide symporter (NIS, or SLC5A5, solute carrier family 5, member 5)(Dai et al., 1996; Smanik et al., 1997) is expressed at the highest level in the thyroid and lactating breast (Dohan et al., 2003). Since NIS confers highly efficient iodide accumulation in cells, its expression in cancer cells allows for the diagnostic and therapeutic application of radioactive substrates of NIS, such as iodide (123I, 124I, and 131I) and pertechnetate (99mTcO4−). A majority (68–86%) of thyroid cancer retains functional NIS expression (Castro et al., 2001; Wapnir et al., 2003). β-emitting radioiodide-131 (131I) is, therefore, routinely used for ablation of remnant tumors after total thyroidectomy. In thyroid cancer, the native NIS expression and radioiodide uptake is reduced. Stimulation of NIS expression by increasing the serum levels of thyroid-stimulating hormone (TSH), is required, prior to 131I administration. Most differentiated thyroid cancer responds to these high levels of serum TSH with an increase in NIS expression and iodide uptake (Schlumberger, 1998). The elevation of serum TSH can be achieved either by withdrawal of thyroid hormone supplement after thyroidectomy or administration of recombinant TSH (thyrogen) (Ladenson et al., 1997).
The majority of breast cancer (70–80%) also expresses NIS (Tazebay et al., 2000; Wapnir et al., 2003), although iodide uptake is usually reduced or absent (Moon et al., 2001; Wapnir et al., 2004). Enhancement of the endogenous NIS expression in breast cancer has been proposed as an approach that would allow 131I therapy (Boelaert & Franklyn, 2003). NIS, however, is expressed in the thyroid gland and other sites, such as stomach and salivary glands (Dohan et al., 2003), so selective induction of NIS in the target cancer is required.
The efficacy of 131I to destroy target tumors is dependent on the tissue-selective NIS gene induction, but also the effective translocation of NIS protein to the cell membrane and correct membrane insertion. 131I retention in the target tumors, and the biological half-life of 131I in the body, also influence treatment efficacy. Normal thyroid tissue incorporates the trapped iodide into thyroglobulin (Tg), referred to as organification, resulting in longer iodide retention. Iodide in most thyroid cancer, as well as breast cancer, however, is not efficiently incorporated into proteins and hence more easily discharged from cancer tissues (Schlumberger et al., 2007).
In this review, we will describe recent findings of pathways and agents that stimulate endogenous NIS gene expression, as well as intracellular NIS translocation, in thyroid cells and breast cancer cells. Dissection of signal transduction pathways for NIS regulation confers novel potential targets to increase the efficacy of radioiodide therapy and expand its application to radioiodide-refractory thyroid cancer, as well as breast cancer and other NIS-expressing tumors.
2. Physiology of iodide metabolism and NIS
The thyroid must trap ~60 μg iodide/day from the bloodstream to produce adequate thyroid hormone. The thyroid contains 70–90% of the iodide in the body (9–10 mg) (Riggs, 1952), and this iodide accumulation is dependent on NIS (Dai et al., 1996), expressed on the basolateral membrane of thyroid follicular cells (Fig. 1). NIS is a glycosylated protein with 13 trans-membrane domains, transporting 2 Na+ and one I−, dependent on the Na+ gradient maintained by Na+/K+ ATPase (Dohan et al., 2003). NIS activity produces the iodide concentration gradient from blood to NIS-expressing cells, up to 30-fold. Iodide taken up into the thyroid follicular cell by NIS, is released to the lumen via pendrin, oxidized by thyroid peroxidase (TPO) with hydrogen peroxide (H2O2) produced mainly by dual oxidase-2 (DUOX2), and binds to tyrosine residues of Tg accumulated in the lumen (Fig. 1). The process of iodide incorporation into Tg is termed “organification”. The iodized tyrosine residues are then used for thyroid hormone synthesis. The transport of iodide into and through the thyroid gland is tightly regulated by TSH from the pituitary gland (Dohan et al., 2003; Kogai et al., 2006; Pesce et al., 2012). TSH stimulates NIS transcription (Kogai et al., 2000a; Kogai et al., 1997; Saito et al., 1997), prolongs NIS protein half-life, and stimulates translocation of NIS into the cell membrane (Riedel et al., 2001), maximizing iodide uptake in thyroid cells.
Fig. 1.
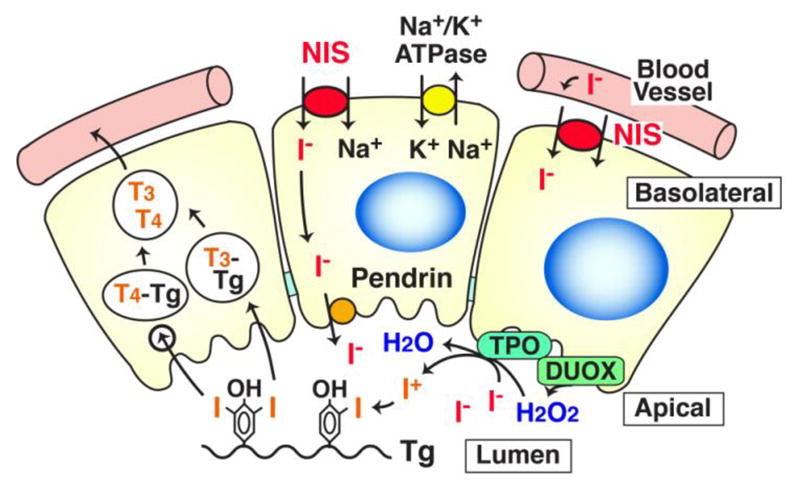
Schematic representation of iodide transport in the thyroid gland. The thyroid gland consist of follicles with one layer of epithelial cells surrounding the lumen. Iodide (I−) in circulation is transported into the lumen via basolateral NIS and apical pendrin. The activity of NIS requires the Na+-gradient maintained by Na+-K+ ATPase. Iodide in the lumen is organified with Tg by TPO in the presence of H2O2 produced mainly by DUOX2. The iodinated tyrosine residues are used for synthesis of thyroid hormones, triiodothyronine (T3) or thyroxine (T4).
Infants need ~90 μg/day of iodide to produce thyroid hormone, essential for normal brain development. Lactating mammary glands efficiently accumulates iodide so that breast milk contains 150–180 μg/L iodide (Semba & Delange, 2001). NIS is expressed on the basolateral membrane of lactating mammary alveolar cells (Cho et al., 2000), and accumulates iodide from the bloodstream into milk. Expression of breast NIS is induced by oxytocin secreted from the posterior pituitary, and this action is enhanced by the elevated levels of serum prolactin and estrogen present in the postnatal period (Cho et al., 2000; Tazebay et al., 2000).
Several other extra-thyroidal tissues express NIS, including salivary glands, stomach, intestine, and lacrimal glands (Dohan et al., 2003). In the gastrointestinal system, salivary ductal cells, as well as gastric mucosa, express NIS on the basolateral membrane (Altorjay et al., 2007; Josefsson et al., 2002), while epithelium of the small intestine expresses NIS on the brush border membrane (apical side)(Nicola et al., 2009a). The iodide in food and water taken orally is absorbed in the intestines through the apical NIS (Nicola et al., 2009a), and transferred into circulation (Fig. 2). In contrast, the salivary glands (mainly parotid glands) and stomach take iodide from the bloodstream and release it into gastrointestinal tract (Brown-Grant, 1961). The kidneys excrete more than 90% of ingested iodide (Cavalieri, 1997). Renal clearance of iodide is mainly dependent on glomerular filtration rate and not re-absorption by renal tubules (Bricker & Hlad, 1955). The iodide secretion by salivary glands and stomach into the gastrointestinal tract, followed by re-absorption through intestine, is likely a mechanism to conserve iodide (Fig. 2), as demonstrated in the cow (Miller et al., 1975). The factors that regulate NIS expression and function in the gastrointestinal system, however, have not been identified (Josefsson et al., 2006).
Fig. 2.
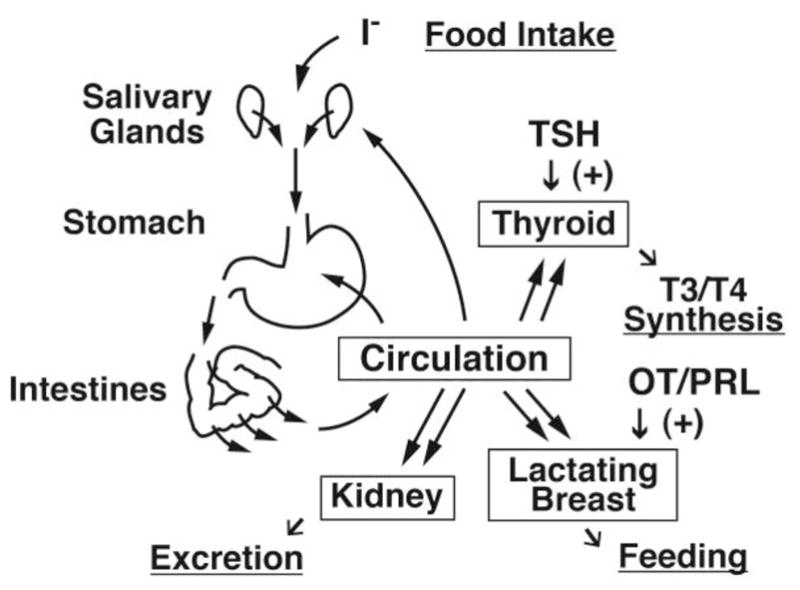
A simplified model of the free iodide cycle in the human body. Most iodine is ingested as iodide (I−) or iodate (IO3−), which is rapidly reduced to iodide (Burgi et al., 2001). Iodide is absorbed by small intestine via the apical NIS, transferred into the circulation, and then taken up in the thyroid gland, as well as lactating breast, although ~90% of ingested iodide will be excreted by the kidneys. A fraction of circulating iodide is released again to the gastrointestinal tract through the salivary glands and stomach that express basolateral NIS. The sodium-dependent multivitamin transporter (SLC5A6) has also been proposed to mediate sodium-coupled iodide transport in the intestines (de Carvalho & Quick, 2011). OT, oxytocin; PRL, prolactin.
NIS-expressing extra-thyroidal tissues, such as lacrimal glands, salivary glands, stomach, and lactating breast tissues, also express the lactoperoxidase system, a natural antimicrobial system (Bosch et al., 2000). Its bactericidal activities are dependent on generation of H2O2, hypoiodite (IO−), and/or thiocyanate (SCN−). A fraction of iodide in those tissues is oxidized to the antibacterial compound IO− by endogenous lactoperoxidase, a possible function of iodide in these tissues (Majerus & Courtois, 1992).
3. Radioiodide therapy in thyroid cancer treatment
131I is widely used in patients with differentiated thyroid cancer for ablation of the remnant of normal thyroid tissue after a total thyroidectomy and for residual or metastatic thyroid cancer. Ablation of the thyroid remnant, following the removal of the primary tumor, may decrease recurrence of differentiated thyroid cancer (Sawka et al., 2004). If the 131I uptake is observed in distant metastases, 131I treatment is highly effective and markedly increases the survival rate, especially in younger patients with small metastases (Durante et al., 2006).
More than 70% of differentiated thyroid cancer, including papillary cancer and follicular cancer, expresses NIS and actively take up 131I. The de-differentiation of thyroid cancer, however, influences the regulation of NIS and reduces functional NIS expression (Kogai et al., 2006). As a result, the tumor is visualized on a radioiodide imaging study as a relatively “cold” nodule with reduced tracer uptake, compared to the surrounding normal tissue. Differentiated thyroid cancer usually retains expression of the TSH receptor (TSHR), although less differentiated thyroid cancer has reduced expression of TSHR (Mizukami et al., 1994; Ohta et al., 1991). The majority of well-differentiated thyroid cancers respond to TSH stimulation with an increase in endogenous NIS expression and 131I accumulation.
The increase in serum TSH level to stimulate NIS after total thyroidectomy, is achieved by the withdrawal of thyroid hormone treatment, which increases secretion of endogenous TSH from the pituitary due to reduced feedback of circulating thyroid hormone. The resulting hypothyroidism reduces the renal clearance of 131I (Maruca et al., 1984; Meier et al., 1994; Riggs, 1952), and may increase efficacy by prolonging the retention of 131I in target cancer. The hypothyroidism, however, is associated with fatigue, weakness, cognitive impairment, and mood disorders. In addition, the thyroid hormone withdrawal is not well tolerated in patients with advanced cancer, heart failure, as well as renal failure. Administration of recombinant human TSH is utilized as an alternative and has similar efficacy to thyroxine withdrawal, but without significant side effects (Haugen et al., 1999; Ladenson et al., 1997).
To achieve sufficient effective dose of 131I (>80 gray (Gy)) in target tumor(s), a high dose (>95 mCi) of 131I is frequently ingested, resulting in 0.1 to 27% of administrated 131I taken up by tumor tissues (Maxon et al., 1983). Iodide uptake in the stomach and salivary glands is often observed in whole body scans with radioiodide, but absorbed radiation dose in 131I therapy is significantly smaller than thyroid (less than 0.1%)(MIRD, 1975). This is likely due to modest NIS expression and rapid release of 131I into the gastrointestinal tract. Moderate side effects in salivary glands and lacrimal glands, however, are still relatively common (10 to 60%) after 131I treatment (Van Nostrand, 2009), including sialoadenitis, dry mouth, dry eyes, and conjunctivitis. These are usually temporary, but become permanent with increasing lifetime cumulative dose. Agents to promote saliva flow, such as lemon candy, have been recommended, but are not clearly shown to reduce salivary gland damage. Pilocarpin, a M3 muscarinic acetylcholine receptor agonist, was also utilized to stimulate salivation but was not effective (Alexander et al., 1998).
In the normal thyroid, the retention time of organified iodine in follicles is significantly longer than that of free iodide, which is readily discharged from thyroid glands, likely by simple diffusion. Iodine organification, however, is reduced in thyroid cancer (Field et al., 1973; Valenta, 1966; Wolff et al., 1959), due to reduced activity of the TPO enzyme and/or DUOXs (Gerard et al., 2003; Ohye & Sugawara, 2010; Takamatsu et al., 1992). As a result, the effective half-life of 131I in tumors (0.5–3 days) is significantly reduced compared to that in normal thyroid tissue (3–7 days)(Menzel et al., 2003; Schlumberger et al., 2007). Radioiodine therapy, however, remains very effective in patients with differentiated thyroid cancer, even without extensive organification.
A significant fraction of metastatic thyroid cancer, in the range of 30–40%, does not respond to 131I therapy, even in the presence of an elevated TSH (Maxon & Smith, 1990). Greater NIS expression in thyroid cancer is associated with greater uptake of radioiodide (Castro et al., 2001), as well as a better prognosis (Ward et al., 2003). Increased NIS expression is desired to improve the efficacy of 131I. The regulation of NIS in thyroid follicular cells and thyroid cancer cells, therefore, has been intensively studied, and is summarized in Table 1.
Table 1.
NIS stimulators in thyroid cells in vitro.
| Agent | Mechanism of actiona | Cell lineb | NIS mRNA | NIS protein | I− uptakec | References |
|---|---|---|---|---|---|---|
| TSH | TSHR agonist | FRTL5 | up | up | 10~15 | (Kogai et al., 1997) |
| TSH | TSHR agonist | PCCL3 | up | ~30 | (Trapasso et al., 1999) | |
| TSH | TSHR agonist | Primary human thyroid | up | up | ~13 | (Kogai et al., 2000a; Saito et al., 1997) |
| Adenosine | ADRA1 agonist | FRTL5 | up | up | ~7.5 | (Harii et al., 1999) |
| TSH/LPSd | TLR4 agonist | FRTL5, PCCL3 | up | ~2.0 | (Nicola et al., 2009b) | |
| TSH/L-NAMEd | NOS inhibitor | FRTL5 | up | ~1.3 | (Fozzatti et al., 2007) | |
| TSH/KT5823d | cGK inhibitor | FRTL5 | ~2.2 | (Fozzatti et al., 2007) | ||
| TSH/LY294002d | PI3K inhibitor | FRTL5, PCCL3 | up | up | ~3.0 | (Kogai et al., 2008b) |
| TSH/Rapamycind | mTOR inhibitor | PCCL3 | up | ~2.5 | (de Souza et al., 2010) | |
| TSH/Resveratrold | Sirtuin activator | FRTL5 | up | ~3.0 | (Sebai et al., 2010) | |
| LY294002 | PI3K inhibitor | NIS-BHP2–7 (TPC1) | up | ~3.5 | (Kogai et al., 2008b) | |
| Akti-1/2 | AKT inhibitor | NIS-BHP2–7 (TPC1) | up | (Kogai et al., 2008b) | ||
| Depsipeptide | HDACi | FTC133, SW1736 | up | ~10 | (Kitazono et al., 2001) | |
| Trichostatin A | HDACi | BHP18–21v (TPC1) K1, FTC133, C643 |
up | up | ~3.0 | (Furuya et al., 2004) |
| SAHA | HDACi | KAT18, OCUT1 | up | (Hou et al., 2010) | ||
| SAHA/perifosinee | HDACi/AKTi | K1, C643 | up | (Hou et al., 2010) | ||
| SAHA/RDEA119e | HDACi/MEKi | K1, C643 | up | (Hou et al., 2010) | ||
| SAHA/RDEA119/perifosinef | HDACi/MEKi/AKTi | C643, FTC133 | up | (Hou et al., 2010) | ||
| SAHA/RDEA119/perifosine/TSH | HDACi/MEKi/AKTi/TSHR | K1, C643, KAT18 | up | 4~8 | (Hou et al., 2010) | |
| Sunitinib/forskolin | RTKi/AC agonist | TPC1 (BHP) | up | (Fenton et al., 2010) | ||
| Troglitazone | PPAR agonist | FTC133, TPC1 | up | (Park et al., 2005) | ||
| tRA | RAR agonist | FTC133 | up | (Schmutzler et al., 1997) | ||
| Lovastatin | HMGR inhibitor | CGTH | up | ~3 | (Frohlich et al., 2009) |
Abbreviations: ADRA1, adrenergic receptor α 1; NOS, nitric oxide synthase; cGK, cGMP-dependent protein kinase; mTOR, mammalian target of rapamycin; HMGR, HMG-CoA reductase; HDACi, HDAC inhibitor; AKTi, AKT inhibitor; MEKi, MEK inhibitor; RTKi, RTK inhibitor.
Origins of FRTL5 and PCCL3 are rat thyroid glands; BHP2–7, BHP18–21, K1, TPC1, papillary thyroid cancer; CGTH, FTC133, follicular thyroid cancer; C643, KAT18, OCUT1, undifferentiated (anaplastic) thyroid cancer.
Approximate fold-induction over the group without treatment is shown. Since specific activity of radioiodide in each study varies, values may not be compared among studies.
Enhances the TSH-induced NIS expression.
Enhances the SAHA-induced NIS expression.
Enhances the effects of double combination treatments.
4. Transcriptional regulation of NIS in thyroid
TSH is the primary regulator of NIS expression in thyroid glands. Stimulation of TSHR activates adenylyl cyclase through the Gs-protein, resulting in cyclic AMP (cAMP) accumulation in thyroid cells. The elevation of endogenous cAMP induces NIS transcription by stimulating several signal pathways of cis-regulatory elements in a NIS locus (reviewed in (Kogai et al., 2006), including the NIS upstream enhancer (NUE), the most potent TSH-responsive enhancer contained in the NIS promoter (Ohno et al., 1999; Taki et al., 2002).
4.1 NIS gene regulation via NUE
The NUE in the human genome is located 9242 to 9300 base-pairs upstream of the coding region of NIS, overlapping with the RPL18A gene (Fig. 3A), due to a high density of coding sequences in NIS-encoding chromosome 19 (Grimwood et al., 2004). The human NUE consists of one Pax8 (thyroid-specific transcription factor) binding site and one cAMP-response element (CRE)-like site (Fig. 3B), both of which are required for the full activity of NUE (Taki et al., 2002). The NUE sequence is conserved among several species, although the surrounding sequences are quite different (Kogai et al., 2006).
Fig. 3.
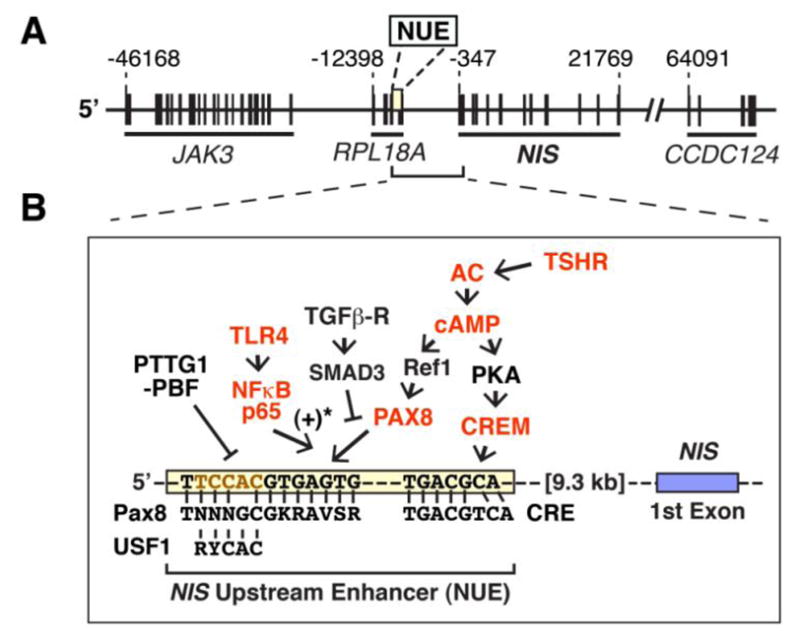
Regulation of the NUE in thyroid cells. A. Map of the human chromosome 19p around the NIS gene locus. The “A” in the translation start site (ATG) of NIS is referred to as +1. B. TSHR signaling pathways to NUE. NIS expression in thyroid cells is predominantly regulated by the TSHR signaling to NUE. Gain-of-function studies of the molecules, indicated by red color, have demonstrated stimulation of the NUE activity. The consensus sequences of cis-elements of PAX8, CRE, and USF1 are indicated along with the sequence of human NUE. *, Stimulatory effects have been reported with rat NUE, which contains an additional Pax8 element and an NFκB element (Nicola et al., 2010). AC, adenylyl cyclase; Ref-1, apurinic apyrimidinic endonuclease redox effector factor-1.
cAMP stimulates the NUE through both protein kinase-A (PKA)-dependent and -independent pathways in thyroid cells (Fig. 3B) (Chun et al., 2004; Ohno et al., 1999; Taki et al., 2002). PKA phosphorylates the cAMP-responsive element binding protein (CREB) and other basic-leucine zipper (B-ZIP) proteins, such as activating transcription factor-1 (ATF-1) and CRE-modulator (CREM), leading to recruitment of these B-ZIP proteins by the CRE-like element in NUE (Chun et al., 2004; Taki et al., 2002). Over-expression of a CREM activator, τ2α, enhances the NUE activity in FRTL-5 rat thyroid cells when treated with forskolin (Fenton et al., 2008), indicating an important role of the CREM activator in the PKA-dependent activation of NUE.
Pax8 is a key transcription factor for thyroid development and differentiation (Mansouri et al., 1998). Transcription of thyroid specific genes, including TSHR, Tg, TPO, and NIS, is dependent on PAX8 activity. Binding of PAX8 to the NUE, in response to TSH stimulation (Costamagna et al., 2004), is the primary requirement for significant activation of NUE (Ohno et al., 1999; Taki et al., 2002). The TSH signaling facilitates the reduction of PAX8 (Kambe et al., 1996) through redox effector factor-1 (Ref-1), which stimulates PAX8 binding to its cis-elements (Fig. 3B)(Tell et al., 1998).
4.2 NIS expression and NUE regulation in thyroid cancer cells
The RET proto-oncogene encodes a receptor tyrosine kinase (RTK) which mediates extracellular neurotrophin signaling to intracellular signal transduction pathways, including the MAPK (mitogen-activated protein kinase)/ERK (extracellular signal-regulated kinase) pathway. The activation of the RET-RAS -BRAF-MEK (MAP/ERK kinase)-ERK pathway is critical for tumor initiation and/or promotion in papillary thyroid cancer (Fagin, 2004). Constitutively active mutants of RET, RET/PTC rearrangement, and BRAF V600E are hallmarks of papillary thyroid cancer. Activating mutations in BRAF are most common in sporadic papillary thyroid cancer in adults, while RET/PTC rearrangement is expressed more frequently in pediatric and radiation-induced cancers. The RET/PTC rearrangement is a characteristic finding in well-differentiated papillary thyroid cancer without aggressive behavior (Ricarte-Filho et al., 2009). In contrast, BRAF activating mutations are often observed in radioiodide-refractory thyroid cancer, especially clinically aggressive papillary thyroid cancer with metastasis (present in more than 95%)(Ricarte-Filho et al., 2009). Other genetic modifications in thyroid cancer, such as mutations of N-RAS (Volante et al., 2009), a catalytic subunit of phosphatidylinositol 3-kinase (PI3KCA), and AKT (Ricarte-Filho et al., 2009), are also associated with a poor prognosis.
An experimental model with constitutive expression of RET/PTC in PCCL3 rat thyroid cells has been utilized for several studies. The exogenous RET/PTC significantly suppresses the expression of Pax8 (De Vita et al., 1998) and the activity of PKA (Venkateswaran et al., 2004), leading to reduced NIS expression (Trapasso et al., 1999; Venkateswaran et al., 2004). The reduced PKA activity is associated with down-regulation of B-ZIP proteins. Indeed, expression of B-ZIP proteins that bind to NUE was significantly decreased in BHP 2–7 cells, variants of RET/PTC-positive TPC1 papillary thyroid cancer cells (Schweppe et al., 2008), resulting in reduced NUE activity (Taki et al., 2002), as well as low NIS expression (Kogai et al., 2001; Ohta et al., 1997).
BRAF mediates the inhibitory effects of RET/PTC on NIS expression through the MEK-ERK pathway (Mitsutake et al., 2006). The activating mutation in BRAF induces transforming growth factor (TGF)-β secretion from thyroid cancer cells, resulting in its paracrine action in tumor tissues (Riesco-Eizaguirre et al., 2009). Increased TGFβ is associated with tumor invasion, stimulation of cell mobility, as well as suppression of NIS expression through SMA- and MAD-related protein (SMAD)-3 (Costamagna et al., 2004) (Figure 3). The BRAF mutation, therefore, contributes to the down-regulation of NIS via both the MEK-ERK pathway and the TGFβ-SMAD3 pathway (Riesco-Eizaguirre et al., 2009), which negatively affect the PAX8 action on NUE activation. The expression of PAX8 is significantly decreased in ~70% of thyroid cancers, along with reduced NIS expression, especially in poorly differentiated thyroid cancers (Fabbro et al., 1994; Puglisi et al., 2000). These observations indicate that the constitutive activation of RET-BRAF signaling reduced NIS expression in papillary thyroid cancer cells, at least in part by suppressing the two major regulators of NUE, PAX8 and B-ZIP proteins.
4.3 Regulation of NUE by TSH-independent signaling pathways
Recent studies have demonstrated regulation of the NUE in thyroid cells by TSH-independent mechanisms that affect the PAX8 binding to the NUE (Fig. 3B).
The NUE is negatively regulated by the pituitary tumor-transforming gene-1 product (PTTG1)(Boelaert et al., 2007). PTTG1 was originally identified as a proto-oncogene product expressed in pituitary tumors (Pei & Melmed, 1997), functioning as a transcription factor for cell cycle-regulating genes, and some differentiation-related genes (Tong & Eigler, 2009). A selective cofactor is required for PTTG1 to function in regulation of its respective target genes. The cofactor for the suppression of NUE is the PTTG1-binding factor (PBF or PTTG1-interacting protein, PTTG1-IP) (Boelaert et al., 2007). The PAX8 element, as well as an overlapping element of upstream transcription factor (USF)-1 (Fig. 3B), in the NUE is important for negative regulation by the PTTG1/PBF complex. Abundant expression of PTTG1 (Saez et al., 2006), as well as PBF (Stratford et al., 2005), has been observed in most thyroid cancer samples, suggesting contribution of those factors to the reduced NIS expression in thyroid cancer. In addition, high PTTG1 expression is associated with the reduced efficacy of radioiodide therapy in thyroid cancer (Saez et al., 2006). A recent in vivo study with a transgenic mouse model of PBF has demonstrated that the thyroid-selective over-expression of PBF reduces functional NIS expression in thyroid glands, and induces thyroid enlargement with macrofollicular lesions (Read et al., 2011).
Several in vitro studies have demonstrated that TGFβ suppresses the differentiated function of thyroid cells, including iodide uptake (Pang et al., 1992) and iodide organification (Pisarev et al., 2009). TGFβ significantly decreased NIS mRNA expression in FRTL-5 rat thyroid cells (Kawaguchi et al., 1997; Pekary & Hershman, 1998). The suppressive effects are partially due to interaction between PAX8 and SMAD3, a downstream modulator of TGFβ signaling, negatively affecting NUE activity (Costamagna et al., 2004).
Bacterial and viral infection, followed by activation of innate immune response through Toll-like receptor (TLR) signaling, is a proposed link between infection and autoimmune thyroid diseases (Harii et al., 2005; Yamazaki et al., 2007). The Gram-negative bacterial endotoxin, lipopolysaccharide (LPS), a ligand of TLR-4, significantly enhanced the TSH-stimulated NIS mRNA expression and iodide uptake (~2.0 fold) in rat thyroid cells, by activating the nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB)(Nicola et al., 2009b). A member of the class II NF-κB, p65, directly interacts with Pax8, and activates the NIS transcription via the rat NUE (Nicola et al., 2010).
4.4 PI3K inhibition stimulates NIS expression in thyroid cells
Insulin, as well as insulin-like growth factor (IGF)-1, significantly decreases iodide uptake in rat thyroid cells in vitro (Kogai et al., 2008b; Saji & Kohn, 1991). Signaling pathways of RTK, including insulin receptors, are frequently mediated by PI3K. Inhibition of PI3K by LY294002, as well as Wortmannin, has been shown to significantly induce NIS mRNA expression and iodide uptake (~3.0-fold) in rat thyroid cells (Garcia & Santisteban, 2002; Kogai et al., 2008b). The NIS induction by LY294002 is dependent on newly synthesized protein(s), including Pax8, in PCCL3 rat thyroid cells (Fig. 4). LY294002 also increased iodide uptake in exogenous NIS-expressing BHP 2–7 cells, likely due to stabilization of NIS mRNA (Kogai et al., 2008b).
Fig. 4.
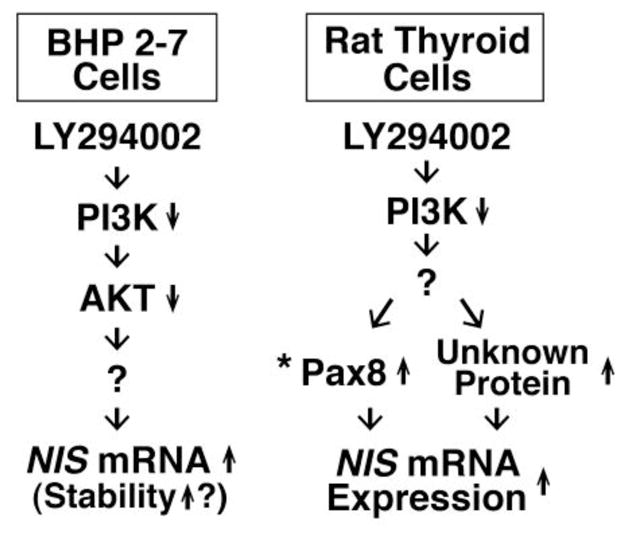
Differential mechanisms of NIS up-regulation by PI3K inhibition with LY294002 in rat thyroid cells and BHP 2–7 papillary thyroid cancer cells. *, LY294002 induces Pax8 in PCCL3 cells, but not in FRTL-5 cells, resulting in a more robust induction of NIS in PCCL3 cells (Kogai et al., 2008b).
The mechanisms of enhancement of iodide uptake by PI3K are distinct in rat thyroid cells and thyroid cancer cells (Fig. 4). The effect of PI3K in NIS-expressing BHP human papillary thyroid cancer cells was mimicked by an inhibitor of AKT, a major effector of PI3K, indicating a contribution of the canonical PI3K-AKT pathway (Kogai et al., 2008b). In contrast, the FRTL-5 rat thyroid cells did not respond to the AKT inhibitor (Kogai et al., 2008b). In addition, modulation of IGF-PI3K signaling affects porcine NIS expression opposite to the effect on rat thyroid NIS. IGF-1 significantly increases NIS mRNA expression in porcine primary thyroid cells (Norden et al., 2007), while LY294002 reduces iodide uptake (Kogai et al., 2008b). Additional studies with human thyroid cells, therefore, are necessary to evaluate the impact of inhibitors of the PI3K pathway on NIS expression in thyroid cancer.
Most RTK inhibitors, clinically used for treatment of non-thyroid cancers, induce hypothyroidism in 20–50% of patients, by several mechanisms. These mechanisms include attenuating thyroid blood flow and increased metabolism of thyroid hormone by type 3 deiodinase (Hamnvik et al., 2011). A multi-targeted RTK inhibitor, sunitinib, transiently induces hypothyroidism, in part due to reduced iodide uptake in the thyroid (Mannavola et al., 2007). The effect, however, does not likely require the suppression of NIS expression, but other mechanisms, such as impairment of iodide organification (Salem et al., 2008). In contrast, an in vitro study with BHP 2–7 papillary thyroid cancer cells demonstrated stimulatory effects of sunitinib on NIS mRNA expression in the presence of an adenylyl cyclase activator, forskolin (Fenton et al., 2010). Since sunitinib down-regulates the PI3K-AKT pathway (Keefe et al., 2010), it likely mimics the effects of PI3K inhibition on NIS expression, at least partially through PAX8 induction (Fenton et al., 2010).
4.5 Effects of HDAC inhibitors and combination treatments with signal transduction inhibitors in less-differentiated thyroid cancer cells
Epigenetic modifications of chromatin, including histone deacetylation and hypermethylation, are associated with poorly differentiated cancer cells. Histone deacetylase (HDAC) inhibitors induce differentiation and expression of thyroid-selective genes in poorly differentiated thyroid cancer cells (Furuya et al., 2004; Kitazono et al., 2001). A member of the bicyclic peptide class of HDAC inhibitor, FR901228 (or depsipeptide), significantly induces NIS mRNA expression and iodide uptake in thyroid cancer cell lines, including BHP 18–21v papillary thyroid cancer cells (Furuya et al., 2004), FTC-133 follicular thyroid cancer cells, and SW-1736 undifferentiated thyroid cancer cells (Kitazono et al., 2001). Depsipeptide significantly induced expression of Tg and TPO, resulting in recovery of iodide organification in BHP 18–21v cells (Furuya et al., 2004), favorable for increasing radioiodide retention.
Most papillary thyroid cancer expresses RET/PTC or BRAF mutants, which activate the MAPK pathway of MEK-ERK. The PI3K-AKT signaling also plays a role in tumor progression in thyroid cancer (Shinohara et al., 2007). Modulation of these signaling pathways may reconstitute the thyroid-specific functions in poorly differentiated thyroid cancer cells. Recently, combination treatments with an HDAC inhibitor, a MEK inhibitor, and/or an AKT inhibitor have been tested in several thyroid cancer cell lines with successful recovery of NIS expression (Hou et al., 2010), although the combination required for NIS induction varied among the cell lines (Table 1). An HDAC inhibitor, SAHA, was required for significant NIS induction in the tested cell lines, including K1 papillary cancer cells, FTC-133 follicular cancer cells, and OCUT1 and C643 undifferentiated cancer cell lines. Since TSHR was also induced by SAHA, treatment with TSH further enhanced the SAHA-induced NIS expression in some cell lines. The addition of a MEK inhibitor RDEA119 and/or an AKT inhibitor perifosine, variably affected the SAHA-induced NIS expression among the tested cell lines. The triple combination of SAHA, RDEA119, and perifosine significantly (4 to 8-fold) induced the iodide uptake in K1 cells, as well as two undifferentiated cancer cell lines, C643 and KAT18.
These findings have raised the possibility of radioiodide therapy in some aggressive radioiodide-refractory thyroid cancer after treatment with an HDAC inhibitor. The inhibition of MAPK pathway and/or AKT possibly enhances the effects of HDAC inhibitor on the NIS expression. K1 cells, harboring the BRAF mutation but not RET/PTC (Schweppe et al., 2008), responded well to the MEK inhibitor RDEA119 to enhance the SAHA-induced NIS expression (Hou et al., 2010). In contrast, another MEK inhibitor, PD98059, significantly decreased the iodide uptake in the exogenous RET/PTC-expressing PCCL3 cells (Vadysirisack et al., 2007). The difference of genetic background may confer the differential responses to signal transduction inhibitors.
5. Potential application of radioiodide therapy to non-thyroidal cancers
5.1 NIS gene therapy for non-thyroidal cancers
Due to the success of radioiodide therapy for thyroid cancer, the NIS gene has been introduced into other cancers to achieve sufficient 131I accumulation for tumor shrinkage. Early studies of the antitumor effects of 131I after NIS gene therapy, however, did not show a consistent response of increased uptake (reviewed in (Riesco-Eizaguirre & Santisteban, 2006). This may have been the result of insufficient delivery or expression of NIS. Recent improvements in NIS delivery systems, as well as the addition of radiation-sensitizing agents and oncolytic treatments, has resulted in greater tumor shrinkage and significant tumor growth inhibition in models of many types of cancer (summarized in Tables 2 and 3).
Table 2.
Recent experimental NIS gene therapy with xenografts of cancer cells constitutively expressing NISa.
In these studies, cancer cells were stably transfected with NIS driven by the indicated promoter, and subcutaneously implanted in rodents to develop xenografts. The animals were then systemically treated with 131I.
| Cancer type | Cell line | Vector | NIS-driving promoter | Additive agent | Tumor shrinkageb | Reference |
|---|---|---|---|---|---|---|
| anaplastic thyroid cancer | ARO | lentivirus | PGK | HKII shRNA | (+) | (J. E. Kim et al., 2011) |
| colon cancer | CT26 | lentivirus | ubiquitin C | (±) | (H. J. Kim et al., 2007) | |
| CT26 (MUC-introduced) | lentivirus | ubiquitin C | MUC1 DNA vaccine | (±) | (Jeon et al., 2007) | |
| HCT-15 | plasmid | CMV | MDR1 shRNA/doxorubicin | (++) | (Jeon et al., 2010) | |
| glioma | F98 | retrovirus | LTR | (±) | (Shen et al., 2004) | |
| hepatoma | HepG2 | plasmid | AFP | (±) | (Willhauck et al., 2008a) | |
| Hep3B | retrovirus | 5xMyc-TERT | (±) | (S. H. Kim et al., 2008) | ||
| Hep3B | retrovirus | TERT | TERT siRNA | (+) | (S. Kim et al., 2012) | |
| uterine cervical cancer | TC1 | retrovirus | CMV | DC-E7 vaccine | (++) | (Jeon et al., 2011) |
Abbreviations: AFP, alpha-fetoprotein; CMV, cytomegalovirus; DC-E7, E7 antigen-presenting dendritic cells; HKII, hexokinase II; LTR, long terminal repeat from moloney murine leukemia virus; MUC1, mucin-1; MDR1, multidrug resistance-1; PGK, phosphoglycerate kinase; shRNA, short hairpin RNA; TERT, telomerase reverse transcriptase.
(++), complete tumor eradication; (+), significant tumor shrinkage; (±), significant inhibition of tumor growth.
Table 3.
Recent studies of experimental NIS gene therapy with xenografts of cancer cells without endogenous NIS expression.
In these studies, animals with subcutaneous xenografts of cancer cells expressing no endogenous NIS were administered with the indicated NIS-expressing vector and/or the additive agent, followed by systemic 131I treatment.
| Cancer type | Cell line | Vectorb | NIS-driving promoterg | Administrationh | Additive agent/effecti | Tumor shrinkagej | Reference |
|---|---|---|---|---|---|---|---|
| breast cancer | ZR 75-1 | Ad5ΔE1ΔE3 | ERE-SV40 | it. | (±) | (Montiel-Equihua et al., 2008) | |
| colon cancer | HCT15 | Ad5ΔE1ΔE3 | CMV | it. | MDR1 shRNA & DOX | (±) | (Ahn et al., 2010) |
| HT-29 | Ad5ΔE1ΔE3 | hTERT or hTR | iv. | (±) | (Riesco-Eizaguirre et al., 2011) | ||
| colorectal cancer | HCT116 | Ad5ΔE1ΔE3 | hTR | it. | EBRT/DNA-PKi | (+) | (Hingorani et al., 2010b) |
| HCT116 | Ad5-vKH1c | Wnt-responsive TCF4 | it. | oncolytic virus | (+) | (Peerlinck et al., 2009) | |
| hepatoma | HepG2 | Ad5ΔE1ΔE3 | HIP | it. | (++) | (Herve et al., 2008) | |
| Huh7 | plasmid/polyplexd | CMV | iv. | (±) | (Klutz et al., 2011) | ||
| Huh7 | NIS-MSCe | CMV in MSC | iv of NIS-MSC | (±) | (Knoop et al., 2011) | ||
| medullary thyroid cancer | TT | Ad5ΔE1 | CEA | it. | (±) | (Spitzweg et al., 2007) | |
| melanoma | M14 | Ad5ΔE1ΔE3 | hTERT or hTR | iv. | (±) | (Riesco-Eizaguirre et al., 2011) | |
| mesothelioma | H513 | MV-Edmf | it. | IFNβ, oncolytic virus | (±) | (Li et al., 2010) | |
| multiple myeloma | 5TGM1 | VSVΔ51f | it. or iv. | oncolytic virus | (±) | (Goel et al., 2007) | |
| MM1, KAS6/1 | MV-Edmf | iv. | oncolytic virus | (++) | (Dingli et al., 2004) | ||
| neuroblastoma | Neuro2A | plasmid/polyplexd | CMV | iv. | (±) | (Klutz et al., 2009) | |
| pancreatic cancer | Capan-2 | Ad5ΔE1 | mucin-1 (MUC1) | it. | (+) | (Dwyer et al., 2006) | |
| PC3 | Ad5ΔE1ΔE3 | survivin | it. | (±) | (Huang et al., 2011) | ||
| PC3-MM2a | Ad5/3-Δ24 | E3 | it. | oncolytic virus | (+) | (Hakkarainen et al., 2009) | |
| BxPC-3 | MV-Edmf | it. | oncolytic virus | (±) | (Carlson et al., 2009) | ||
| prostate cancer | LNCaP | Ad5ΔE3 | Provasin/RSV | it. | (±) | (Trujillo et al., 2010) |
a hormone-refractory metastatic subline of the PC-3 cell line.
abbreviations: Ad, adenovirus; MV-Edm, Edmonston lineage of measles virus; VSV, vesicular stomatitis virus.
self-replicates in Wnt-overexpressing cells by using the exogenous TCF4 Wnt-responsive element.
an expression vector of NIS was condensed with EGF receptor-targeting polyplex.
NIS-constitutively expressing mesenchymal stem cells (MSCs) are used as a vihecle of NIS.
negative-sense single-stranded RNA virus.
abbreviations: CEA, carcinoembryonic antigen; CMV, cytomegalovirus; ERE, estrogen responsive element; HIP, hepatocarcinoma-intestine-pancreas gene; hTERT, human telomerase reverse transcriptase; hTR, human telomerase RNA; RSV, rous sarcoma virus; SV40, simian virus-40; TCF, T-cell factor.
it., intratumoral injection; iv., intravenous injection
abbreviations: DNA-PKi, DNA-dependent protein kinase inhibitor; DOX, doxorubicin; EBRT, external beam radiotherapy; IFNβ, tumor necrosis factor-β; MDR1, multidrug resistance gene-1.
(++), complete tumor eradication; (+), significant tumor shrinkage; (±), significant inhibition of tumor growth.
Previous studies of NIS gene therapy, without adjuvant therapies, has shown that the antitumor efficacy of 131I is dependent on the magnitude of NIS gene expression. To achieve complete tumor destruction, more than 20% of the injected radioiodide dose, per gram of tumor (%ID/g), needs to accumulate in a tumor. A successful prostate cancer xenograft model has been described that accumulates 25 to 30%ID/g in the tumors (Spitzweg et al., 2000). For comparison, poorly differentiated thyroid cancer xenografts accumulated only 4.9–9.3%ID/g and were not effectively treated with radioiodine (Shimura et al., 1997). A NIS gene delivered with an adenovirus vector and a tissue specific gene promoter, the prostate-specific antigen gene (PSA) promoter, confered efficient functional NIS expression in prostate cancer xenografts (Dwyer et al., 2005; Spitzweg et al., 2001), and phase 2 trials are currently being conducted in prostate cancer patients. Other tumor specific promoters, such as the human telomerase reverse transcriptase (hTERT) promoter, the carcinoembryonic antigen (CEA) promoter, and the alpha-fetoprotein (AFP) promoter, have also directed robust and selective NIS expression in the tumor, leading to remarkable inhibition of tumor growth by 131I (Table 2).
To achieve synergistic or additive cytotoxic effects, combined treatments with NIS gene therapy and a tumor targeting strategy, such as utilization of an oncolytic vector (Goel et al., 2007; Hakkarainen et al., 2009; Li et al., 2010; Peerlinck et al., 2009), vaccination against a tumor-specific antigen (Jeon et al., 2011), or inhibition of intracellular glucose metabolism by knockdown of hexokinase II (J. E. Kim et al., 2011), have been studied, resulting in significant growth inhibition or eradication of tumor (Tables 2 and 3). Inhibition of DNA repair by the DNA-dependent protein kinase inhibitor (DNA-PKi) enhanced the antitumor effects by combination treatment with 131I and external beam radiotherapy in colorectal cancer cells, as well as head and neck cancer cells (Hingorani et al., 2010b).
Conventionally, NIS gene therapy has been performed with virus vectors. Results of initial clinical studies of gene therapy for X-linked severe combined immunodeficiency (X-SCID), however, have indicated a high incidence of leukemia due to unexpected integration of viral DNA to the host genomes, raising safety concerns about the gene delivery by virus vectors. The majority of recent experimental NIS gene therapies have been performed with replication-defective adenoviruses (Table 3) preventing unfavorable genomic integration. Some oncolytic viruses used for NIS gene therapy (Carlson et al., 2009; Dingli et al., 2004; Goel et al., 2007; Hakkarainen et al., 2009) are negative-sense single-stranded RNA viruses, not generally integrated into the host genomes. A plasmid vector conjugated with polyplex targeted to EGF receptor (Klutz et al., 2009) is also a promising strategy for safe and highly selective delivery of NIS into the targeted tumor.
5.2 Induction of endogenous NIS in breast cancer
The majority (70–80%) of breast cancers express NIS (Tazebay et al., 2000; Wapnir et al., 2003), while only 20–30% take-up radioiodide, due to low functional NIS expression (Moon et al., 2001; Wapnir et al., 2004). Induction of endogenous NIS may allow us to utilize radioiodide therapy in breast cancer (Boelaert & Franklyn, 2003; Welcsh & Mankoff, 2000), therefore, NIS inducible agents and their regulatory mechanisms have been well investigated in breast cancer cells (summarized in Table 4). Among those agents, retinoic acid (RA) is the most potent single-agent NIS inducer in breast cancer cells (Table 4). Treatment with RA significantly increases the cytotoxicity of 131I in MCF-7 breast cancer cells (Kogai et al., 2000b). An in vivo study demonstrated that systemic RA treatment achieves approximately 20–40%ID/g of iodide uptake in MCF-7 xenograft tumors (Kogai et al., 2004), which is in the range of iodide uptake providing successful tumor shrinkage in prostate cancer xenografts with exogenous NIS expression (Spitzweg et al., 2001). RA does not induce, but reduces NIS in FRTL-5 rat thyroid cells (Schmutzler et al., 1997). The differential regulation of NIS in thyroid glands and breast cancer confers selective NIS induction by RA in breast cancer in mouse models (Kogai et al., 2004).
Table 4.
Stimulator of endogenous NIS expression in breast cancer cells in vitro.
| Agenta | Mechanism of actionb | Cell line | NIS mRNA | NIS protein | I− uptakec | References |
|---|---|---|---|---|---|---|
| tRA, 9-cis RA | RAR/RXR agonist | MCF7 | up | up | 10~13 | (Kogai et al., 2000b) |
| tRA, 9-cis RA | RAR/RXR agonist | BT474, T47D | up | (Sponziello et al., 2010; Tanosaki et al., 2003) | ||
| AGN190168 | RARβ/γagonist | MCF7 | up | 10~13 | (Kogai et al., 2005) | |
| Am80 | RARα/βagonist | MCF7 | up | up | ~7.0 | (Ohashi et al., 2009) |
| Dex+tRAd | GR+RAR agonists | MCF7 | up | ~3.5d | (Kogai et al., 2005; Unterholzner et al., 2006) | |
| HC+tRAe | GR+RAR agonists | MCF7 | up | ~1.7e | (Dohan et al., 2006) | |
| IBMX+tRAe | P2Y2+RAR agonists | MCF7 | up | ~1.4e | (Dohan et al., 2006) | |
| IBMX+HC+tRAe | P2Y2+GR+RAR agonists | MCF7 | up | ~2.5e | (Dohan et al., 2006) | |
| CBZ+tRAe | PXR+RAR agonists | MCF7 | up | up | ~1.8e | (Willhauck et al., 2011) |
| CBZ+Dex+tRAe | PXR+GR+RAR agonists | MCF7 | up | up | ~4.4e | (Willhauck et al., 2011) |
| Troglitazone+9cisRAf | PPARγ+RAR agonists | MCF7 | up | ~1.8f | (Tanosaki et al., 2003) | |
| Theophylline | PDE antagonist/P2R inhibitorg | MCF7 | up | ~4.7 | (Yoon et al., 2009) | |
| LBH589 | HDAC inhibitor | MCF7 | up | up | ~2.3 | (Fortunati et al., 2010) |
| LBH589 | HDAC inhibitor | T47D | up | up | ~4.8 | (Fortunati et al., 2010) |
| LBH589 | HDAC inhibitor | MDA-MB231 | up | up | ~2.7 | (Fortunati et al., 2010) |
| NaB, TSA | HDAC inhibitor | MCF7, HCC-1937 | up | (Sponziello et al., 2010) |
Abbreviations: IBMX, 3-isobutyl-1-methyl xanthine; HC, hydrocortisone; NaB, sodium butyrate; TSA, Trichostatin A.
Abbreviations: P2Y2, P2Y purinergic receptor-2; PXR, pregnane X receptor; PDE, phosphodiesterase; P2R, P2 purinergic receptor.
Values are approximate fold-induction over the group without RA, unless otherwise noted. Since specific activity of radioiodide in each study varies, values may not be compared.
Additive/synergistic effects with 10−7M tRA. Values are approximate fold-increase over the group with 10−7M tRA.
Additive/synergistic effects with 10−6M tRA. Values are approximate fold-increase over the group with 10−6M tRA.
Additive/synergistic effects with 10−6M 9-cis RA. Values are approximate fold-increase over the group with 10−7M 9-cis RA.
Theophylline increase cAMP accumulation by inhibiting PDE and P2R, however, cAMP does not significantly induce NIS (Dohan et al., 2006; Kogai et al., 2000b).
5.3 Retention of radioiodide in non-thyroidal cancer
In non-thyroidal tumors, trapped radioiodide is not organified, resulting in shorter iodide retention, compared to thyroid glands. The biological half-life of radioiodide in NIS-expressing non-thyroidal tumor, 5 to 6 hours in rodent models (Kogai et al., 2004; Shimura et al., 1997; Spitzweg et al., 2001), is correlated to the half-life in serum and the whole body (Shimura et al., 1997). Radioiodide retention in serum in human (~20 hours) (Maruca et al., 1984) is much longer than that in rodents (~6 hours)(Shimura et al., 1997). Higher radiation dose of 131I, thus, would be expected in humans with NIS-expressing tumors.
6. NIS regulation by retinoic acid in breast cancer cells
RA significantly induces NIS in several breast cancer cell lines, including MCF-7, T47D, and BT474 (Kogai et al., 2000b; Sponziello et al., 2010; Tanosaki et al., 2003), as well as mouse models, including MCF-7 xenografts (Cheong et al., 2011; Kogai et al., 2004) and the murine mammary tumor virus-polyoma virus middle T antigen (MMTV-PyVT) transgenic breast cancer mouse model (Kogai et al., 2004). Since MCF-7 cells are most responsive to RA treatment, this cell line has been primarily utilized for studies of endogenous NIS induction in breast cancer (Kogai et al., 2006). All-trans RA (tRA) (Tretinoin), 9-cis RA (Alitretinoin), 13-cis RA (Isotretinoin), as well as several synthetic ligands of retinoic acid receptor (RAR), significantly induce NIS and iodide uptake (Fig. 5) in MCF7 cells (Kogai et al., 2005; Kogai et al., 2000b; Tanosaki et al., 2003). The three isomers of RA, tRA, 9-cis RA, and 13-cis RA, are enzymatically converted to tRA, a potent agonist of RAR, as well as 9-cis RA, a potent agonist of retinoid-X receptor (RXR), by endogenous isomerases (Fig. 6). Among the three RAR isoforms, α, β, and γ, RAR α and γ are predominantly expressed in MCF-7 cells (Kogai et al., 2004; Titcomb et al., 1994). RARβ is rapidly induced by RA treatment, providing a positive feedback mechanism for RA stimulation (de The et al., 1990). Among several synthetic RAR-selective agonists and RXR agonists, AGN 190168 (Tazarotene), a RARβ/γ agonist, is the most potent inducer of NIS in MCF-7 cells (Fig. 5)(Kogai et al., 2005; Ohashi et al., 2009). A loss-of-function study has validated a critical role of RARβ for NIS induction (Ohashi et al., 2009), although the expression level of the RARβ isoform is relatively low (Kogai et al., 2004; Titcomb et al., 1994).
Fig. 5.
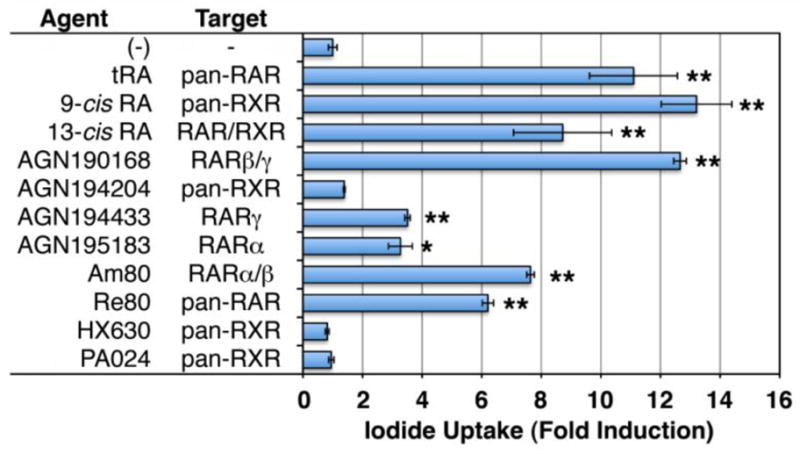
Effects of retinoid receptor agonists on iodide uptake in MCF-7 cells in vitro. Cells were treated with 10−6 M of each agonist for 48 hours, and iodide uptake assay was performed with 20 mCi/mmol of Na125I, as described (Kogai et al., 2008b; Weiss et al., 1984). The uptake was normalized by cellular protein amount or cell number. Fold-induction over the group without retinoid treatment is presented. *, P < 0.02; ** P < 0.01, when compared to the negative control (n = 3 or 4).
Fig. 6.
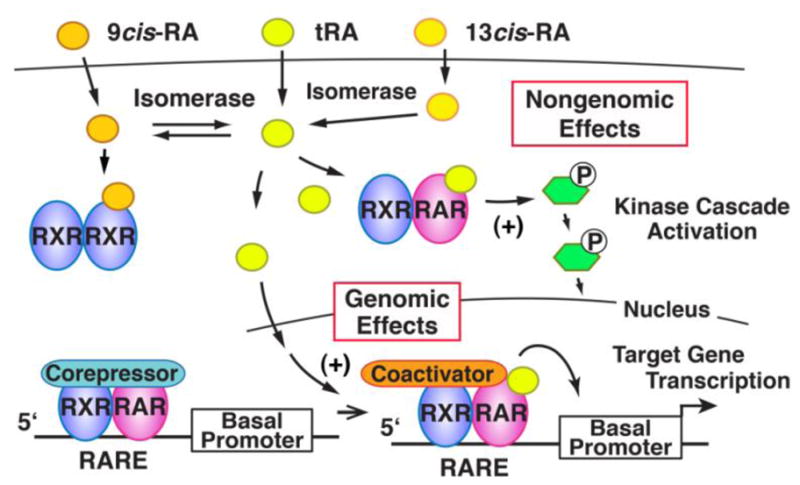
Comparison of genomic and non-genomic effects of RA. Conversion of isomers of RA is also indicated. The RAR/RXR heterodimer, not bound to chromatin contributes to kinase cascade activation, whereas the RAR/RXR bound to an RARE (retinoic acid response element) regulates expression of the target gene. Retinoic acids are hydrophobic compounds and associate with soluble retinoid-binding proteins (not shown in this schema) in the intracellular as well as extracellular compartments.
RAR is a type II nuclear hormone receptor, localized mainly in the nucleus, does not bind heat shock protein and makes a heterodimer with RXR (Mangelsdorf et al., 1995). The RAR/RXR heterodimer binds to a cis-element on its target genes and stimulates transcription (‘genomic effects’, see Fig. 6). RAR/RXR not bound to chromatin directly activates some signal transduction pathways, including PI3K (‘non-genomic effects’, Fig. 6). In the case of NIS induction, both genomic and nongenomic actions have been proposed to stimulate NIS gene expression by tRA (Alotaibi et al., 2010; Kogai et al., 2012; Kogai et al., 2008a; Ohashi et al., 2009). RAR agonists, but not RXR agonists, induce NIS (Kogai et al., 2005; Ohashi et al., 2009), while RXR selective antagonists, as well as knockdown of RXRα, the predominant isoform of RXR in MCF-7 cells (Kogai et al., 2004; Titcomb et al., 1994), block NIS induction by tRA (Ohashi et al., 2009). These findings demonstrate the requirement of RAR/RXR hetero-dimers, particularly RARβ/RXRα, for NIS induction.
6.1 Non-genomic effects of RA through PI3K participate in NIS induction in breast cancer cells
Although most of type II nuclear receptors are distributed in the nucleus, catalytic activity of PI3K, predominantly localized in cytoplasm, is directly modulated by those receptors, including thyroid hormone receptor (Cao et al., 2005; Furuya et al., 2006), RAR (Day et al., 2006; del Rincon et al., 2003; Masia et al., 2007; Ohashi et al., 2009), and peroxisome proliferator-activated receptors (PPARs)(Han et al., 2005; Lin et al., 2005). RA temporally activates a major PI3K effector, AKT, within the first 10 min of RA treatment in MCF-7 cells (Ohashi et al., 2009), as well as SH-SY5Y neuroblastoma cells (Masia et al., 2007). A regulatory subunit of PI3K, p85, directly interacts with RAR isoforms, including RARα (Day et al., 2006; Masia et al., 2007) and RARβ (Ohashi et al., 2009). Co-immunoprecipitation studies have demonstrated the association between p85 and the RARβ/RXRα heterodimer (Ohashi et al., 2009). Since loss-of-function analysis demonstrates the requirement of both RARβ and p85, the crosstalk between RARβ signaling and PI3K signaling may mediate NIS induction by RA (Ohashi et al., 2009).
6.2 Rac1/p38β contributes RA-induced NIS expression in breast cancer cells
The p38 kinase is a MAPK, regulating cell proliferation, differentiation, and migration. Four p38 isoforms, α, β, γ, and δ, are found in mammalian cells with variable tissue distribution and substrate specificity, producing differential activation of downstream effector pathways (Jiang et al., 1996; Pramanik et al., 2003). tRA stimulates phosphorylation of p38 isoforms, α and β, in MCF7 breast cancer cells through a small GTPase Rac1 (Alsayed et al., 2001; Kogai et al., 2012). The NIS expression in MCF-7 cells requires one of the p38 pathways, MKK3B-p38β (Fig. 7)(Kogai et al., 2012). Over-expression of p38β, as well as Rac1, significantly enhances the tRA-induced NIS expression and iodide uptake (Kogai et al., 2012). The p38α is considered to be an important mediator of stress signaling, cell proliferation and differentiation in cancer cells (Wagner & Nebreda, 2009), whereas the p38β isoform is thought to be a minor pathway in rodent development and physiology, based on the findings in p38β-deficient mice (Beardmore et al., 2005). The requirement of p38β for the NIS expression in breast cancer cells, therefore, may provide a strategy for relatively specific induction of NIS in some breast cancer cells.
Fig. 7.
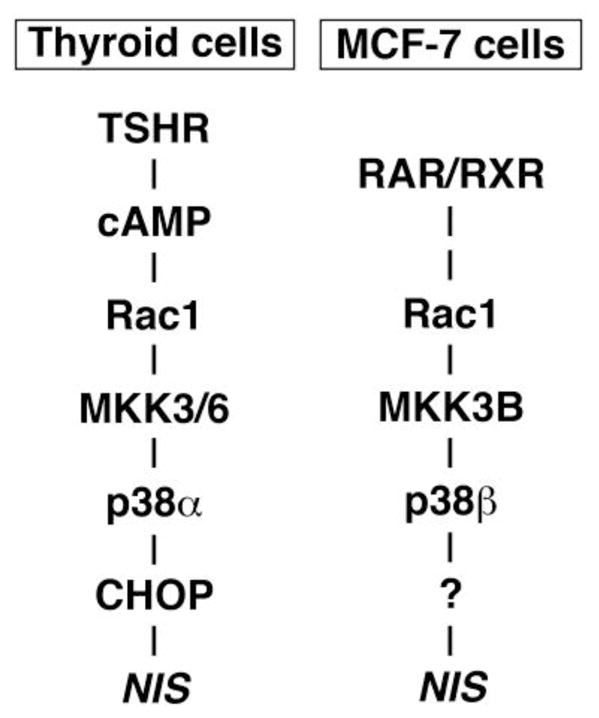
Distinct p38 pathways regulate NIS expression in FRTL-5 rat thyroid cells and MCF-7 breast cancer cells. CHOP, CCAAT/enhancer-binding protein-homologous protein. This figure is reproduced from (Kogai et al., 2012).
6.3 Genomic-effects of RAR and NIS expression
The genomic effects of RAR is mediated by its cis-elements, retinoic acid response elements, with diverse orientations of half sites, 5′-PuG(G/T)(T/A)CA-3′, often a direct repeat with spacing of 2 or 5 bases (DR-2 or DR-5). Among these consensus sequences, several DR-2 sequences are located in the NIS intron sequences (Kogai et al., 2008a). Binding of RARα, as well as RNA polymerase II, to the intronic DR-2 elements have been shown within 30 min of initiation of tRA stimulation in MCF-7 cells (Alotaibi et al., 2010), indicating a potential role of the intronic DR-2 elements in the initiation of human NIS transcription.
DR-2 elements are often located in Alu elements, one of the most abundant repeated elements in the human genome (Laperriere et al., 2007). Alu elements are retrotransposons, proposed to contribute to primate evolution. The NIS-carrying chromosome 19 has higher density (25.8%) of the Alu repeats compared to other chromosomes (Grimwood et al., 2004). All DR-2 elements in the human NIS introns are located in such repeated retrotransposons (Table 5), while the mouse NIS gene sequence does not contain any DR-2 element. tRA, however, significantly induces mouse NIS expression in breast tumors of the transgenic mouse model of MMTV-PyVT (Kogai et al., 2004). The full induction of NIS by tRA, therefore, must include non-genomic effects of RA, as discussed above.
Table 5.
Putative retinoic acid response elements in human NIS intron sequences and retrotransposon.
| Position | Sequence | RARE | Strandb | Repeated elementc |
|---|---|---|---|---|
| 1st intron | AGGTCAggAGTTCA | DR-2a | (+) | AluSx1 |
| AGGTCAggAGTTCA | DR-2a | (+) | AluSg | |
| 5th intron | TCACCTgAGGTCAcAGTTCA | DR-1, ER-8d | (−) | AluSx1 |
| 7th intron | AGGTCAatGGGCAA | DR-2 | (+) | MIRc |
| 8th intron | AGGTCAggAGTTCA | DR-2a | (−) | AluSx |
| 12th intron | AGGTCAggAGTTCA | DR-2a | (+) | AluSg |
| AGGTCAggAGTTCA | DR-2a | (−) | AluSx | |
| AGGTCAggAGTTCA | DR-2a | (−) | AluSx | |
| 13th intron | AGGTCAggAGTTCA | DR-2a | (+) | AluSx1 |
| 14th intron | GGTTCAatTGTTCA | DR-2 | (−) | AluSx2 |
The sequence is equivalent.
(+), top strand; (−), bottom strand
According to RepeatMasker (http://www.repeatmasker.org).
ER-8, everted repeat with 8-base pair separation.
6.4 Alternatives to tRA treatment for the NIS induction
tRA is commonly used for treatment of acute promyelocytic leukemia (APL). The remission of APL, however, is often for a limited duration of time (Frankel et al., 1994), partially due to a short biological half-life of tRA (Warrell, 1993). In addition, tRA treatment frequently causes a cardio-respiratory distress syndrome, called “retinoic acid syndrome”, in patients with APL. New retinoid preparations, therefore, have been sought that are more biologically stable and selective for RA signaling, with less toxicity (Kagechika, 2002; Nagpal & Chandraratna, 2000). The majority of synthetic retinoids, however, have only been used for topical skin treatment.
The 50% effective concentration (EC50) of tRA in vitro for NIS induction is ~10−7 M (Kogai et al., 2005), consistent with that for transcriptional regulation of other tRA-regulated genes (Idres et al., 2002). The systemic dose of tRA required for maximum NIS induction in rodent models, however, is likely higher than would be tolerated for routine treatment in humans (Kogai et al., 2004). In addition, several in vivo studies have demonstrated variable effects of systemic tRA treatment on radioiodide uptake (~1.2 to ~15-fold) in MCF-7 xenografts (Cheong et al., 2011; Kogai et al., 2004; Willhauck et al., 2008b), possibly due to clonal variation of MCF-7 cells (Kogai et al., 2004; Lacroix & Leclercq, 2004; Seibert et al., 1983). To search for more efficient agents, several other retinoids have been tested for the ability to induce NIS in MCF-7 cells in vitro (Fig. 5) (Kogai et al., 2005; Ohashi et al., 2009).
Among the retinoids that markedly induce NIS, 13-cis RA is the only retinoid, other than tRA, commonly used for systemic administration. 13-cis RA is widely used for treatment of cystic acne, as well as neuroblastoma and other cancers. Endogenous isomerases, such as glutathione S-transferases (Chen & Juchau, 1998), convert 13-cis RA to tRA in target cells so 13-cis RA works as a prodrug of tRA (Fig. 6) with less side effects and a longer biological half-life. The efficacy of enzymatic conversion is distinct in each tissue, and isomerase activity is relatively low in tumor tissues. After systemic administration of 13-cis RA in MCF-7 xenograft mice, the fraction converted to tRA in the xenograft tumors (~20%) is significantly smaller than that in liver (~68%) (Conley et al., 1999). The magnitude of NIS mRNA induction by 13-cis RA is actually lower (~65%) than that by tRA in MCF-7 cells in vitro (Kogai et al., 2005).
An in vitro study has demonstrated that AGN190168 (Tazarotene) is the most effective synthetic retinoid for the NIS induction in MCF-7 cells (Fig. 5)(Kogai et al., 2005). AGN190168 is clinically used for acne and psoriasis, but limited to topical application, due to a very short half-life (< 1 hour) of its active metabolite AGN 190299 in the serum (Chien et al., 1992; Hsyu et al., 1994). Selective RARβ agonists with longer half-lives would establish more effective and less toxic treatment for the NIS induction.
6.5 Enhancement of tRA-induced NIS expression by other nuclear receptor ligands
A nuclear receptor dimmer recognizes a specific response element, typically containing two common half-sites, 5′-AGGTCA-3′, or its variants. The selectivity of cis-element to each receptor dimmer is dependent on spacing and orientation of the two half-sites. The consensus half-site, therefore, is occasionally shared by different receptors. Co-activators and co-repressors are also often shared among various nuclear receptors. In addition, RXR is shared among type II nuclear receptors to form hetero-dimers. These mechanisms result in crosstalk among nuclear receptor signaling pathways (Yen, 2001). To potentially enhance the effects of RAR agonists on NIS expression, a number of nuclear receptor ligands have been tested in breast cancer cells (Table 4).
Agonists of glucocorticoid receptor (GR), such as dexamethasone (Dex) and hydrocortisone, synergistically increase expression of genes induced by tRA (Medh & Schmidt, 1997; Tsai et al., 2000), including NIS (Dohan et al., 2006; Kogai et al., 2005; Unterholzner et al., 2006). Dex significantly increases iodide uptake (>3-fold with 10−7 M tRA) in MCF-7 cells both in the presence and absence of tRA (Kogai et al., 2005). Significant reduction of the EC50 of tRA (from ~10−7 M to 6.8×10−9 M) for iodide uptake by Dex (10−7 M), shown in an in vitro study (Kogai et al., 2005), provides an approach to decrease the in vivo dose of tRA for NIS induction.
The combination of Dex and AGN190168 is effective for NIS induction in MCF-7 breast cancer cells. Sustained treatment with tRA is associated with significant attenuation of iodide uptake after the peak of induction at 48 hours (Kogai et al., 2005). The addition of Dex to AGN190168 prolonged the peak period of iodide-uptake for up to 4 days, while the addition of Dex to tRA did not extend the peak period (Kogai et al., 2005). It is likely, therefore, that the combination of AGN190168 and Dex could confer an increased cumulative radiation dose of 131I, although in vivo use of AGN190168 is not feasible because of its rapid metabolism (Chien et al., 1992; Hsyu et al., 1994).
Addition of carbamazepine (CBZ), an agonist of pregnane X receptor (PXR), has been described to significantly (~1.8-fold) enhance tRA-induced iodide uptake both in the presence and absence of Dex (Willhauck et al., 2011), although a relatively high concentration (100 μM) of CBZ is required for the maximum stimulatory effect. The addition of Dex, as well as CBZ, significantly enhances the cytotoxic effects of 131I induced by tRA in MCF-7 cells (Kogai et al., 2005; Willhauck et al., 2011; Willhauck et al., 2008b).
Troglitazone, a PPARγ agonist, has also been reported to significantly (~1.8-fold) enhance 9-cis RA-induced NIS expression in MCF-7 cells (Tanosaki et al., 2003). Other PPARγ agonists, pioglitazone and rosiglitazone, however, did not significantly enhance iodide uptake or NIS mRNA expression in MCF-7 cells (Kogai et al., 2005; Tanosaki et al., 2003). The enhancement of NIS expression by troglitazone is likely due to PPARγ-independent off target effects, as is the case in its effects on cell growth and apoptosis (Wei et al., 2009).
A GR agonist is the most effective enhancer of tRA-induced NIS expression in MCF-7 cells. The combination of AGN190168 and Dex (Kogai et al., 2005), as well as the triple combination of tRA, Dex and CBZ (Willhauck et al., 2011), are the most effective for iodide uptake in vitro. An in vivo study with MCF-7 xenografts has demonstrated significant enhancement of tRA-stimulated tumor radioiodide uptake by systemic Dex treatment (Willhauck et al., 2008b). The magnitude of induction, however, is modest (~3.5-fold), achieving iodide accumulation with only 25% or less activity of the radioiodide required for tumor shrinkage (Willhauck et al., 2008b). In contrast, another in vivo study with only tRA demonstrated robust induction of iodide uptake (up to 15-fold) (Kogai et al., 2004). The discrepancy could be due to differential responses of MCF7 cells to systemic tRA treatment in NIS mRNA induction, almost no significant induction (Willhauck et al., 2008b) vs. ~40-fold induction (Kogai et al., 2004). The difference in findings may be due to the heterogeneity of MCF-7 cells (Lacroix & Leclercq, 2004).
6.6 Effects of RA and/or Dex on the NIS expression in normal breast cells
To establish a potential therapeutic and diagnostic application of NIS induction by RA in breast cancer, the effects of RA on NIS expression in normal breast tissues is important. In vitro studies with human normal breast-derived cells have demonstrated no significant effects of tRA on the NIS mRNA and iodide uptake in MCF12A cells (Kogai et al., 2000b), as well as in HB-2 cells (Willhauck et al., 2008b). Although systemic tRA treatment does not promote significant iodide uptake in breast tissues in severe combined immunodeficient (SCID)/beige mice (Kogai et al., 2004), another study with sensitive imaging has demonstrated radioiodide uptake in normal mammary glands in ~75% of tRA/Dex-treated CD1 mice (Willhauck et al., 2008b). The effects of tRA/Dex combination treatment on normal breast tissues will need to be investigated in other animal models, as well as human primary cell models.
7. Differential regulation of NIS expression in thyroid cells and breast cancer cells
Since iodide accumulation is critical for thyroid hormone synthesis, NIS expression is persistently maintained in thyroid glands by TSH stimulation. In contrast, NIS expression in breast tissue is not dependent on TSH and is transient, just during lactation due to stimulation by oxytocin, prolactin, and estradiol (Dohan et al., 2003). RA significantly induces NIS in some breast cancer cells (Kogai et al., 2006) and thyroid follicular cancer cells (Schmutzler & Kohrle, 2000), but not other normal tissues, including thyroid (Kogai et al., 2004; Schmutzler et al., 1997). Such differential regulation of NIS (summarized in Table 6) allows for selective induction or reduction of endogenous NIS expression in target tissue(s). An example is the thyroid-specific regulation of NIS by the TSHR signaling pathway brings about the selective induction of NIS in thyroid cancer required for 131I therapy.
Table 6.
Differential regulation of NIS expression in MCF7 cells and FRTL5 cells.
| Agent | Mechanism of actiona | MCF7 cells | FRTL5 cells | References |
|---|---|---|---|---|
| FSK | AC stimulator | down | up | (Dohan et al., 2006; Kogai et al., 2000b) |
| tRA | RAR agonist | up | down | (Kogai et al., 2000b; Schmutzler et al., 1997) |
| Insulin | IR agonist | up | down | (Arturi et al., 2005; Kogai et al., 2008b; Saji & Kohn, 1991) |
| Dex | GR agonist | up | down | (Kogai et al., 2005; Saji & Kohn, 1990) |
| Estradiol | ER agonist | down | down | (Furlanetto et al., 1999; Kogai et al., 2005) |
| LY294002 | PI3K inhibitor | down | up | (Kogai et al., 2008a; Kogai et al., 2008b) |
| SB203580 | p38α/β inhibitor | down (p38β) | down (p38α) | (Kogai et al., 2012; Pomerance et al., 2000) |
Abbreviations: AC, adenylyl cyclase; IR, insulin receptor; ER, estrogen receptor.
In the 131I therapy proposed for non-thyroidal cancer treatment, 131I accumulation by thyroid glands should be minimized to avoid thyroid damage and to maximize 131I content available to target the tumor. Elevated serum thyroid hormones suppress secretion of TSH from the pituitary gland, followed by suppression of thyroid NIS expression. Thyroxine does not significantly affect the iodide uptake in tRA-stimulated MCF-7 breast cancer xenografts (Kogai et al., 2004), or NIS-introduced xenograft tumors (Boland et al., 2000; Shimura et al., 1997). An inhibitor of iodide organification, methimazole (MMI), reduces retention of 131I in thyroid glands. A pilot clinical trial (Wapnir et al., 2004) demonstrated that the combination treatment with triiodothyronine and MMI markedly reduced the estimated radiation dose of 131I in thyroid glands after ingestion of 100 mCi from ~270 Gy to a cumulative dose of ~3 Gy in thyroid (Wapnir et al., 2004).
The RA signaling for NIS induction in breast cancer cells is mediated by the PI3K and p38β MAPK pathways (Kogai et al., 2012; Ohashi et al., 2009). The PI3K pathway inhibits expression of NIS in thyroid cancer, opposite to the effect in breast cancer (Kogai et al., 2008a; Kogai et al., 2008b), as shown in Table 6. Thyroid cells, as well as breast cancer cells, require the Rac1-p38 MAPK pathway for the full induction of NIS (Kogai et al., 2012; Pomerance et al., 2000). Distinct isoforms of p38 and MKK, as well as downstream effectors, however, mediate the signaling toward NIS expression in those cell types (Fig. 7)(Kogai et al., 2012). These differential regulatory mechanisms could allow stimulation of a selective NIS-inducing pathway in target tumors expectedly with less side effects.
8. Posttranslational regulation of NIS
NIS functions as a transporter, only when it is properly distributed to the cell surface membrane. Posttranslational regulatory mechanisms, especially translocation of NIS, have been proposed as an important factor determining the functionality of NIS, and of interest as a target to augment iodide uptake in NIS-expressing cancer cells.
8.1 Regulation of NIS translocation in thyroid cells by TSH
When FRTL-5 rat thyroid cells were stimulated by TSH, iodide uptake, as well as NIS protein production, was significantly induced in 24 hours (Kogai et al., 1997). NIS protein induction reached ~80% of the maximum at 36 hours, while iodide uptake at 36 hours was still 30–40% of the maximum reached in 72 hours (Kogai et al., 1997). The time lag between iodide uptake and NIS protein induction has suggested the posttranslational regulation of NIS by TSH (Kogai et al., 1997). In the presence of TSH, NIS in FRTL-5 cells is mainly distributed to the cell surface membrane, while when TSH is removed NIS is mainly localized in the intracellular compartments (Riedel et al., 2001). A thyroid-specific NIS translocation mechanism, therefore, has been proposed, which is responsive to TSH stimulation (Kogai et al., 1997; Riedel et al., 2001). In Graves’ disease thyroid tissues, NIS is predominantly expressed on the basolateral membrane (Dohan et al., 2001), likely due to the activation of TSHR signaling by circulating stimulating antibody associated with Graves’ disease.
8.2 Impairment of NIS translocation in cancer cells
NIS mRNA expression is decreased in some differentiated thyroid cancer tissues, likely due to failure of transcriptional regulation of NIS (Kogai et al., 2001; Puppin et al., 2004; Taki et al., 2002). Several studies, however, have reported abundant expression of NIS in differentiated thyroid cancer (Dohan et al., 2001; Saito et al., 1998; Wapnir et al., 2003), demonstrating abundant NIS expression in the cytoplasm, but little on the cell surface membrane. Similar observations have been described in breast tissues. Lactating breast alveolar cells express intense membrane NIS (Cho et al., 2000; Tazebay et al., 2000), while the majority of breast cancer NIS is localized in the cytoplasm (Kogai et al., 2004; Wapnir et al., 2003). The failure of NIS translocation to the cell surface membrane, therefore, has been proposed to contribute to reduced radioiodide accumulation in those cancers.
8.3 PBF as a NIS translocation regulator
PBF, one of the NUE regulators (Fig. 3B), also has been characterized as a protein interacting and co-localizing with NIS protein in the cytoplasm (Smith et al., 2009). Exogenous PBF in Cos-7 cells is predominantly expressed in CD63-positive late endosome with NIS, co-localizing with NIS in clathrin-coated vesicles (Smith et al., 2009). In fact, NIS has a dileucine motif, which is able to directly interact with the clathrin-coated machinery (Bonifacino & Traub, 2003), at the intracellular C-terminal portion (Dohan et al., 2003). In NIS-introduced Cos-7 cells, exogenous PBF expression significantly reduced iodide uptake and cell surface NIS expression (Smith et al., 2009). Expression of PBF is significantly increased in thyroid cancer, compared to normal thyroid (Stratford et al., 2005). Most breast cancers express abundant PBF, while expression of PBF in normal breast tissues is only modest (Watkins et al., 2010). The abundant expression of PBF, therefore, is likely associated with the reduced cell surface NIS expression in those cancers.
8.4 Signal transduction pathways and NIS translocation
Cultures of the rat thyroid cell lines, FRTL-5 and PCCL3, require both TSH and insulin to maintain cell differentiation and proliferation. The stimulatory effects of PI3K inhibitor LY294002 on iodide uptake in FRTL-5 rat thyroid cells are, at least partially, due to the up-regulation of NIS mRNA expression (Kogai et al., 2008b). Removal of insulin from culture media completely abolished the augmentation of NIS mRNA and protein expression by LY294002. Meanwhile, iodide uptake was increased by LY294002 even without insulin (Kogai et al., 2008b). The discrepancy between the effects on NIS protein expression and iodide uptake indicates some posttranslational mechanism(s), including NIS translocation to the cell surface membrane, in the regulation by PI3K inhibition. Our preliminary study has indicated stimulation of NIS translocation by a PI3K-AKT-mTOR signaling inhibitor, PP242, in BHP 2–7 thyroid cancer cells (unpublished observation).
Regulation of NIS translocation by PI3K has also been reported in breast cancer cells. A constitutively active mutant of PI3K, p110αCAAX, suppressed the expression of cell surface NIS, as well as iodide uptake, in MCF7 cells (Knostman et al., 2007). The over-expression of PI3K increased expression of unglycosylated forms of NIS (~50 kDa) in the NIS-induced MCF-7 cells (Knostman et al., 2007). Consistently, PI3K inhibition abolished the expression of unglycosylated NIS in FRTL-5 rat thyroid cells (Kogai et al., 2008b). Mutation of NIS at the glycosylation sites reduced the iodide uptake up to ~50%, likely due to reduced NIS expression on the cell surface membrane (Levy et al., 1998). PI3K may regulate the NIS translocation by modulating the glycosylation status of NIS.
The NIS translocation to the cell surface membrane is enhanced by EGF (epidermal growth factor) receptor stimulation in NIS-introduced non-thyroidal cancer cells (Jung et al., 2008). Treatment with epidermal growth factor increased iodide uptake in NIS-transfected T47D human breast cancer cells, as well as PC12 rat pheochromocytoma cells. This effect was abolished by PD98059 (Jung et al., 2008), a MEK-1 inhibitor, indicating a role of the MEK-ERK signaling cascade in the NIS translocation.
9. Conclusion
Over 60 years of experience validates significant efficacy of 131I therapy in most differentiated thyroid cancer. TSH stimulation in thyroid cancer maximizes the effect of 131I, likely by enhancing NIS gene expression and facilitating the translocation of NIS to the cell surface membrane. Despite these actions, more than 90 mCi of 131I, however, is still typically required to achieve the sufficient effective radiation dose in the target tumors. High doses of radioiodine are associated with adverse effects, including dysfunction of salivary and lacrimal glands, and a small increased risk of secondary cancers and leukemia (Alexander et al., 1998). Recent progress in the study of NIS regulation has brought about possibilities of new therapeutic approaches, which may decrease the ingested dose in 131I therapy, and expand application of 131I therapy to some radioiodide-refractory thyroid cancers.
PI3K inhibitors induce NIS in rat thyroid cells as well as RET/PTC-positive papillary thyroid cancer cells. To enhance the NIS expression in well-differentiated thyroid cancer, modulation of PI3K-AKT pathway is a promising strategy (de Souza et al., 2010; Kogai et al., 2008b), especially in cancer that retains TSH-responsiveness. HDAC inhibitors restore NIS expression in poorly differentiated thyroid cancer cells (Furuya et al., 2004; Kitazono et al., 2001). Since over-activation of the MAPK pathway and the PI3K-AKT pathway is critical for development and progression of aggressive thyroid cancers (Fagin, 2004; Shinohara et al., 2007), inhibition of these pathways may also induce re-differentiation and restore NIS expression. A very recent clinical pilot study has actually demonstrated increased radioiodide uptake with a MEK inhibitor Selumetinib in 11 of 17 cases with metastatic thyroid cancer (Ho et al., 2011). The triple combination treatment with inhibitors of HDAC, MAPK, and AKT is a new approach to restore NIS expression and radioiodide accumulation in the poorly differentiated thyroid cancer (Hou et al., 2010). Previous observations, however, have shown variable effects in different cell lines (Hou et al., 2010), possibly due to different genetic backgrounds and culture conditions. Elucidation of detailed mechanisms of NIS induction, including isoform specificity of targeted kinases, as well as gene expression profiles in those cells, will be required to establish the efficient NIS induction in various types of thyroid cancer.
A number of animal studies of exogenous NIS introduction into non-thyroidal cancer have demonstrated efficient tumor shrinkage with 131I (Hingorani et al., 2010a). Breast cancer expresses endogenous NIS, which can be markedly induced by RA in some experimental models (Table 4). To achieve sufficient radioiodide uptake for tumor shrinkage, however, a high dose of RA is required that would not likely be tolerated in human (Kogai et al., 2004). Addition of some other nuclear hormone receptor ligands, such as Dex and CBZ, significantly enhances the tRA-induced NIS expression (Kogai et al., 2005; Unterholzner et al., 2006; Willhauck et al., 2011), and prolongs NIS induction by some RAR isoform-specific agonists (Kogai et al., 2005). The magnitude of NIS induction, however, has varied among experimental systems, especially in animal studies (Cheong et al., 2011; Kogai et al., 2004; Willhauck et al., 2008b). Genetic and epigenetic differences could influence the responsiveness to RA.
Recent studies elucidating RA signaling to NIS transcription have demonstrated significant roles for what were considered minor signal transduction mediators, such as RARβ, and p38β, in the NIS induction by RA (Kogai et al., 2012; Ohashi et al., 2009). Comprehensive studies of signal transduction, such as phospho-proteomics, may provide more detailed information of NIS-inducing pathways. Targeted modulation of such signaling pathways to NIS transcription would provide more selective, and hence more efficient and less toxic, treatment for the induction of iodide uptake in some breast cancer cells.
Stimulation of NIS translocation to the cell surface membrane is a novel approach to achieve higher iodide uptake in NIS-expressing cells. Targeting NIS-interacting protein(s), such as PBF (Smith et al., 2009), in the intracellular compartment would stimulate cell surface NIS expression. Signal transduction inhibitors, including PI3K/AKT inhibitors and MEK/ERK inhibitors, also have the potential to enhance the functional NIS expression in some thyroid cancer, as well as non-thyroid cancer cells (Jung et al., 2008; Kogai et al., 2008b).
Functional NIS expression can be augmented by up-regulation of both the transcriptional and post-translational pathways. Some isoform-specific signal transduction pathways play critical roles in the tissue-specific NIS regulation. Dissection of such signaling pathways should lead to methods to further enhance the functional NIS expression in thyroid and breast cancer, expanding the application of radioiodide therapy to radioiodide-refractory thyroid cancer and NIS-expressing breast cancer.
Acknowledgments
We would like to thank Drs. Jerome Hershman, Masahiro Sugawara, and Yan-Yun Liu for helpful discussions. This study was supported by NIH RO1 CA089364 and VA merit review funds.
Abbreviations
- APL
acute promyelocytic leukemia
- B-ZIP
basic-leucine zipper
- cAMP
cyclic AMP
- CBZ
carbamazepine
- CRE
cAMP-response element
- CREM
CRE-modulator
- Dex
dexamethasone
- DR
direct repeat
- DUOX
dual oxidase
- EC50
50% effective concentration
- ER
estrogen receptor
- ERK
extracellular signal-regulated kinase
- GR
glucocorticoid receptor
- Gy
gray
- HDAC
histone deacetylase
- IGF
insulin-like growth factor
- MAPK
mitogen-activated protein kinase
- MEK
MAP/ERK kinase
- MKK
MAPK kinase
- MMI
methimazole
- MMTV-PyVT
murine mammary tumor virus-polyoma virus middle T antigen
- NIS
sodium iodide symporter
- NUE
NIS upstream enhancer
- PBF
PTTG1-binding factor
- %ID/g
% of injected dose per gram of tumor
- PI3K
phosphatidylinositol 3-kinase
- PKA
protein kinase-A
- PPAR
peroxisome proliferator-activated receptor
- PTTG1
pituitary tumor-transforming gene-1
- RA
retinoic acid
- RAR
retinoic acid receptor
- RTK
receptor tyrosine kinase
- RXR
retinoid-X receptor
- Tg
thyroglobulin
- TGF
transforming growth factor
- TLR
Toll-like receptor
- TPO
thyroid peroxidase
- tRA
All-trans RA
- TSH
thyroid-stimulating hormone
- TSHR
TSH Receptor
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn SJ, Jeon YH, Lee YJ, Lee YL, Lee SW, Ahn BC, et al. Enhanced anti-tumor effects of combined MDR1 RNA interference and human sodium/iodide symporter (NIS) radioiodine gene therapy using an adenoviral system in a colon cancer model. Cancer Gene Ther. 2010;17:492–500. doi: 10.1038/cgt.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander C, Bader JB, Schaefer A, Finke C, Kirsch CM. Intermediate and long-term side effects of high-dose radioiodine therapy for thyroid carcinoma. J Nucl Med. 1998;39:1551–1554. [PubMed] [Google Scholar]
- Alotaibi H, Yaman E, Salvatore D, Di Dato V, Telkoparan P, Di Lauro R, et al. Intronic elements in the Na+/I− symporter gene (NIS) interact with retinoic acid receptors and mediate initiation of transcription. Nucleic Acids Res. 2010;38:3172–3185. doi: 10.1093/nar/gkq023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsayed Y, Uddin S, Mahmud N, Lekmine F, Kalvakolanu DV, Minucci S, et al. Activation of Rac1 and the p38 mitogen-activated protein kinase pathway in response to all-trans-retinoic acid. J Biol Chem. 2001;276:4012–4019. doi: 10.1074/jbc.M007431200. [DOI] [PubMed] [Google Scholar]
- Altorjay A, Dohan O, Szilagyi A, Paroder M, Wapnir IL, Carrasco N. Expression of the Na+/I− symporter (NIS) is markedly decreased or absent in gastric cancer and intestinal metaplastic mucosa of Barrett esophagus. BMC Cancer. 2007;7:5. doi: 10.1186/1471-2407-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arturi F, Ferretti E, Presta I, Mattei T, Scipioni A, Scarpelli D, et al. Regulation of iodide uptake and sodium/iodide symporter expression in the MCF-7 human breast cancer cell line. Journal of Clinical Endocrinology and Metabolism. 2005;90:2321–2326. doi: 10.1210/jc.2004-1562. [DOI] [PubMed] [Google Scholar]
- Beardmore VA, Hinton HJ, Eftychi C, Apostolaki M, Armaka M, Darragh J, et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25:10454–10464. doi: 10.1128/MCB.25.23.10454-10464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelaert K, Franklyn JA. Sodium iodide symporter: a novel strategy to target breast, prostate, and other cancers? Lancet. 2003;361:796–797. doi: 10.1016/s0140-6736(03)12720-1. [DOI] [PubMed] [Google Scholar]
- Boelaert K, Smith VE, Stratford AL, Kogai T, Tannahill LA, Watkinson JC, et al. PTTG and PBF repress the human sodium iodide symporter. Oncogene. 2007;26:4344–4356. doi: 10.1038/sj.onc.1210221. [DOI] [PubMed] [Google Scholar]
- Boland A, Ricard M, Opolon P, Bidart JM, Yeh P, Filetti S, et al. Adenovirus-mediated transfer of the thyroid sodium/iodide symporter gene into tumors for a targeted radiotherapy. Cancer Res. 2000;60:3484–3492. [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Bosch EH, van Doorne H, de Vries S. The lactoperoxidase system: the influence of iodide and the chemical and antimicrobial stability over the period of about 18 months. J Appl Microbiol. 2000;89:215–224. doi: 10.1046/j.1365-2672.2000.01098.x. [DOI] [PubMed] [Google Scholar]
- Bricker NS, Hlad CJ., Jr Observations on the mechanism of the renal clearance of I131. J Clin Invest. 1955;34:1057–1072. doi: 10.1172/JCI103155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Grant K. Extrathyroidal iodide concentrating mechanisms. Physiol Rev. 1961;41:189–213. [Google Scholar]
- Burgi H, Schaffner TH, Seiler JP. The toxicology of iodate: a review of the literature. Thyroid. 2001;11:449–456. doi: 10.1089/105072501300176408. [DOI] [PubMed] [Google Scholar]
- Cao X, Kambe F, Moeller LC, Refetoff S, Seo H. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol Endocrinol. 2005;19:102–112. doi: 10.1210/me.2004-0093. [DOI] [PubMed] [Google Scholar]
- Carlson SK, Classic KL, Hadac EM, Dingli D, Bender CE, Kemp BJ, et al. Quantitative molecular imaging of viral therapy for pancreatic cancer using an engineered measles virus expressing the sodium-iodide symporter reporter gene. AJR Am J Roentgenol. 2009;192:279–287. doi: 10.2214/AJR.08.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro MR, Bergert ER, Goellner JR, Hay ID, Morris JC. Immunohistochemical analysis of sodium iodide symporter expression in metastatic differentiated thyroid cancer: correlation with radioiodine uptake. J Clin Endocrinol Metab. 2001;86:5627–5632. doi: 10.1210/jcem.86.11.8048. [DOI] [PubMed] [Google Scholar]
- Cavalieri RR. Iodine metabolism and thyroid physiology: current concepts. Thyroid. 1997;7:177–181. doi: 10.1089/thy.1997.7.177. [DOI] [PubMed] [Google Scholar]
- Chen H, Juchau MR. Recombinant human glutathione S-transferases catalyse enzymic isomerization of 13-cis-retinoic acid to all-trans-retinoic acid in vitro. Biochem J. 1998;336(Pt 1):223–226. doi: 10.1042/bj3360223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong SJ, Jang D, Jeong HJ, Lim ST, Sohn MH, Katzenellenbogen JA, et al. Reduction of stimulated sodium iodide symporter expression by estrogen receptor ligands in breast cancer cells. Nucl Med Biol. 2011;38:287–294. doi: 10.1016/j.nucmedbio.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Chien DS, Sandri RB, Tang-Liu DS. Systemic pharmacokinetics of acitretin, etretinate, isotretinoin, and acetylenic retinoids in guinea pigs and obese rats. Drug Metab Dispos. 1992;20:211–217. [PubMed] [Google Scholar]
- Cho JY, Leveille R, Kao R, Rousset B, Parlow AF, Burak WE, Jr, et al. Hormonal regulation of radioiodide uptake activity and Na+/I− symporter expression in mammary glands. J Clin Endocrinol Metab. 2000;85:2936–2943. doi: 10.1210/jcem.85.8.6727. [DOI] [PubMed] [Google Scholar]
- Chun JT, Di Dato V, D’Andrea B, Zannini M, Di Lauro R. The CRE-like element inside the 5′-upstream region of the rat sodium/iodide symporter gene interacts with diverse classes of b-Zip molecules that regulate transcriptional activities through strong synergy with Pax-8. Mol Endocrinol. 2004;18:2817–2829. doi: 10.1210/me.2004-0020. [DOI] [PubMed] [Google Scholar]
- Conley BA, Ramsland TS, Sentz DL, Wu S, Rosen DM, Wollman M, et al. Antitumor activity, distribution, and metabolism of 13-cis-retinoic acid as a single agent or in combination with tamoxifen in established human MCF-7 xenografts in mice. Cancer Chemother Pharmacol. 1999;43:183–197. doi: 10.1007/s002800050883. [DOI] [PubMed] [Google Scholar]
- Costamagna E, Garcia B, Santisteban P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J Biol Chem. 2004;279:3439–3446. doi: 10.1074/jbc.M307138200. [DOI] [PubMed] [Google Scholar]
- Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature. 1996;379:458–460. doi: 10.1038/379458a0. [DOI] [PubMed] [Google Scholar]
- Day RM, Lee YH, Park AM, Suzuki YJ. Retinoic acid inhibits airway smooth muscle cell migration. Am J Respir Cell Mol Biol. 2006;34:695–703. doi: 10.1165/rcmb.2005-0306OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho FD, Quick M. Surprising substrate versatility in SLC5A6: Na+-coupled I− transport by the human Na+/multivitamin transporter (hSMVT) J Biol Chem. 2011;286:131–137. doi: 10.1074/jbc.M110.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza EC, Padron AS, Braga WM, de Andrade BM, Vaisman M, Nasciutti LE, et al. MTOR downregulates iodide uptake in thyrocytes. J Endocrinol. 2010;206:113–120. doi: 10.1677/JOE-09-0436. [DOI] [PubMed] [Google Scholar]
- de The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H, Dejean A. Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature. 1990;343:177–180. doi: 10.1038/343177a0. [DOI] [PubMed] [Google Scholar]
- De Vita G, Zannini M, Cirafici AM, Melillo RM, Di Lauro R, Fusco A, et al. Expression of the RET/PTC1 oncogene impairs the activity of TTF-1 and Pax-8 thyroid transcription factors. Cell Growth Differ. 1998;9:97–103. [PubMed] [Google Scholar]
- del Rincon SV, Rousseau C, Samanta R, Miller WH., Jr Retinoic acid-induced growth arrest of MCF-7 cells involves the selective regulation of the IRS-1/PI 3-kinase/AKT pathway. Oncogene. 2003;22:3353–3360. doi: 10.1038/sj.onc.1206485. [DOI] [PubMed] [Google Scholar]
- Dingli D, Peng KW, Harvey ME, Greipp PR, O’Connor MK, Cattaneo R, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103:1641–1646. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- Dohan O, Baloch Z, Banrevi Z, Livolsi V, Carrasco N. Rapid communication: predominant intracellular overexpression of the Na(+)/I(−) symporter (NIS) in a large sampling of thyroid cancer cases. J Clin Endocrinol Metab. 2001;86:2697–2700. doi: 10.1210/jcem.86.6.7746. [DOI] [PubMed] [Google Scholar]
- Dohan O, De la Vieja A, Carrasco N. Hydrocortisone and purinergic signaling stimulate sodium/iodide symporter (NIS)-mediated iodide transport in breast cancer cells. Mol Endocrinol. 2006;20:1121–1137. doi: 10.1210/me.2005-0376. [DOI] [PubMed] [Google Scholar]
- Dohan O, De la Vieja A, Paroder V, Riedel C, Artani M, Reed M, et al. The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocr Rev. 2003;24:48–77. doi: 10.1210/er.2001-0029. [DOI] [PubMed] [Google Scholar]
- Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP, et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91:2892–2899. doi: 10.1210/jc.2005-2838. [DOI] [PubMed] [Google Scholar]
- Dwyer RM, Bergert ER, O’Connor MK, Gendler SJ, Morris JC. Adenovirus-mediated and targeted expression of the sodium-iodide symporter permits in vivo radioiodide imaging and therapy of pancreatic tumors. Hum Gene Ther. 2006;17:661–668. doi: 10.1089/hum.2006.17.661. [DOI] [PubMed] [Google Scholar]
- Dwyer RM, Schatz SM, Bergert ER, Myers RM, Harvey ME, Classic KL, et al. A preclinical large animal model of adenovirus-mediated expression of the sodium-iodide symporter for radioiodide imaging and therapy of locally recurrent prostate cancer. Mol Ther. 2005;12:835–841. doi: 10.1016/j.ymthe.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Fabbro D, Di Loreto C, Beltrami CA, Belfiore A, Di Lauro R, Damante G. Expression of thyroid-specific transcription factors TTF-1 and PAX-8 in human thyroid neoplasms. Cancer Res. 1994;54:4744–4749. [PubMed] [Google Scholar]
- Fagin JA. Challenging dogma in thyroid cancer molecular genetics--role of RET/PTC and BRAF in tumor initiation. J Clin Endocrinol Metab. 2004;89:4264–4266. doi: 10.1210/jc.2004-1426. [DOI] [PubMed] [Google Scholar]
- Fenton MS, Marion KM, Hershman JM. Identification of cyclic adenosine 3′,5′-monophosphate response element modulator as an activator of the human sodium/iodide symporter upstream enhancer. Endocrinology. 2008;149:2592–2606. doi: 10.1210/en.2007-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton MS, Marion KM, Salem AK, Hogen R, Naeim F, Hershman JM. Sunitinib inhibits MEK/ERK and SAPK/JNK pathways and increases sodium/iodide symporter expression in papillary thyroid cancer. Thyroid. 2010;20:965–974. doi: 10.1089/thy.2010.0008. [DOI] [PubMed] [Google Scholar]
- Field JB, Larsen PR, Yamashita K, Mashiter K, Dekker A. Demonstration of iodide transport defect but normal iodide organification in nonfunctioning nodules of human thyroid glands. J Clin Invest. 1973;52:2404–2410. doi: 10.1172/JCI107430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunati N, Catalano MG, Marano F, Mugoni V, Pugliese M, Bosco O, et al. The pan-DAC inhibitor LBH589 is a multi-functional agent in breast cancer cells: cytotoxic drug and inducer of sodium-iodide symporter (NIS) Breast Cancer Res Treat. 2010;124:667–675. doi: 10.1007/s10549-010-0789-z. [DOI] [PubMed] [Google Scholar]
- Fozzatti L, Velez ML, Lucero AM, Nicola JP, Mascanfroni ID, Maccio DR, et al. Endogenous thyrocyte-produced nitric oxide inhibits iodide uptake and thyroid-specific gene expression in FRTL-5 thyroid cells. J Endocrinol. 2007;192:627–637. doi: 10.1677/joe.1.06967. [DOI] [PubMed] [Google Scholar]
- Frankel SR, Eardley A, Heller G, Berman E, Miller WH, Jr, Dmitrovsky E, et al. All-trans retinoic acid for acute promyelocytic leukemia. Results of the New York Study. Ann Intern Med. 1994;120:278–286. doi: 10.7326/0003-4819-120-4-199402150-00004. [DOI] [PubMed] [Google Scholar]
- Frohlich E, Brossart P, Wahl R. Induction of iodide uptake in transformed thyrocytes: a compound screening in cell lines. Eur J Nucl Med Mol Imaging. 2009;36:780–790. doi: 10.1007/s00259-008-1024-6. [DOI] [PubMed] [Google Scholar]
- Furlanetto TW, Nguyen LQ, Jameson JL. Estradiol increases proliferation and down-regulates the sodium/iodide symporter gene in FRTL-5 cells. Endocrinology. 1999;140:5705–5711. doi: 10.1210/endo.140.12.7197. [DOI] [PubMed] [Google Scholar]
- Furuya F, Hanover JA, Cheng SY. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc Natl Acad Sci U S A. 2006;103:1780–1785. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya F, Shimura H, Suzuki H, Taki K, Ohta K, Haraguchi K, et al. Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology. 2004;145:2865–2875. doi: 10.1210/en.2003-1258. [DOI] [PubMed] [Google Scholar]
- Garcia B, Santisteban P. PI3K is involved in the IGF-I inhibition of TSH-induced sodium/iodide symporter gene expression. Mol Endocrinol. 2002;16:342–352. doi: 10.1210/mend.16.2.0774. [DOI] [PubMed] [Google Scholar]
- Gerard AC, Daumerie C, Mestdagh C, Gohy S, De Burbure C, Costagliola S, et al. Correlation between the loss of thyroglobulin iodination and the expression of thyroid-specific proteins involved in iodine metabolism in thyroid carcinomas. J Clin Endocrinol Metab. 2003;88:4977–4983. doi: 10.1210/jc.2003-030586. [DOI] [PubMed] [Google Scholar]
- Goel A, Carlson SK, Classic KL, Greiner S, Naik S, Power AT, et al. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood. 2007;110:2342–2350. doi: 10.1182/blood-2007-01-065573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwood J, Gordon LA, Olsen A, Terry A, Schmutz J, Lamerdin J, et al. The DNA sequence and biology of human chromosome 19. Nature. 2004;428:529–535. doi: 10.1038/nature02399. [DOI] [PubMed] [Google Scholar]
- Hakkarainen T, Rajecki M, Sarparanta M, Tenhunen M, Airaksinen AJ, Desmond RA, et al. Targeted radiotherapy for prostate cancer with an oncolytic adenovirus coding for human sodium iodide symporter. Clin Cancer Res. 2009;15:5396–5403. doi: 10.1158/1078-0432.CCR-08-2571. [DOI] [PubMed] [Google Scholar]
- Hamnvik OP, Larsen PR, Marqusee E. Thyroid dysfunction from antineoplastic agents. J Natl Cancer Inst. 2011;103:1572–1587. doi: 10.1093/jnci/djr373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Ritzenthaler JD, Wingerd B, Roman J. Activation of peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) increases the expression of prostaglandin E2 receptor subtype EP4. The roles of phosphatidylinositol 3-kinase and CCAAT/enhancer-binding protein beta. J Biol Chem. 2005;280:33240–33249. doi: 10.1074/jbc.M507617200. [DOI] [PubMed] [Google Scholar]
- Harii N, Endo T, Ohmori M, Onaya T. Extracellular adenosine increases Na+/I− symporter gene expression in rat thyroid FRTL-5 cells. Mol Cell Endocrinol. 1999;157:31–39. doi: 10.1016/s0303-7207(99)00166-5. [DOI] [PubMed] [Google Scholar]
- Harii N, Lewis CJ, Vasko V, McCall K, Benavides-Peralta U, Sun X, et al. Thyrocytes express a functional toll-like receptor 3: overexpression can be induced by viral infection and reversed by phenylmethimazole and is associated with Hashimoto’s autoimmune thyroiditis. Mol Endocrinol. 2005;19:1231–1250. doi: 10.1210/me.2004-0100. [DOI] [PubMed] [Google Scholar]
- Haugen BR, Pacini F, Reiners C, Schlumberger M, Ladenson PW, Sherman SI, et al. A comparison of recombinant human thyrotropin and thyroid hormone withdrawal for the detection of thyroid remnant or cancer. J Clin Endocrinol Metab. 1999;84:3877–3885. doi: 10.1210/jcem.84.11.6094. [DOI] [PubMed] [Google Scholar]
- Herve J, Cunha AS, Liu B, Valogne Y, Longuet M, Boisgard R, et al. Internal radiotherapy of liver cancer with rat hepatocarcinoma-intestine-pancreas gene as a liver tumor-specific promoter. Hum Gene Ther. 2008;19:915–926. doi: 10.1089/hum.2007.153. [DOI] [PubMed] [Google Scholar]
- Hingorani M, Spitzweg C, Vassaux G, Newbold K, Melcher A, Pandha H, et al. The biology of the sodium iodide symporter and its potential for targeted gene delivery. Curr Cancer Drug Targets. 2010a;10:242–267. doi: 10.2174/156800910791054194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani M, White CL, Zaidi S, Pandha HS, Melcher AA, Bhide SA, et al. Therapeutic effect of sodium iodide symporter gene therapy combined with external beam radiotherapy and targeted drugs that inhibit DNA repair. Mol Ther. 2010b;18:1599–1605. doi: 10.1038/mt.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AL, Grewal R, Leboeuf R, Sherman E, Fury M, Pfister D, et al. Reacquisition of RAI uptake in RAI-refractory metastatic thyroid cancers by pretreatment with the selective MEK inhibitor selumetinib. Thyroid. 2011;21(S1):A-111. [Google Scholar]
- Hou P, Bojdani E, Xing M. Induction of thyroid gene expression and radioiodine uptake in thyroid cancer cells by targeting major signaling pathways. J Clin Endocrinol Metab. 2010;95:820–828. doi: 10.1210/jc.2009-1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsyu PH, Bowen B, Tang-Liu D. Pharmacokinetics of a novel retinoid AGN 190168 and its metabolite AGN 190299 after intravenous administration of AGN 190168 to rats. Biopharm Drug Dispos. 1994;15:347–357. doi: 10.1002/bdd.2510150502. [DOI] [PubMed] [Google Scholar]
- Huang R, Zhao Z, Ma X, Li S, Gong R, Kuang A. Targeting of tumor radioiodine therapy by expression of the sodium iodide symporter under control of the survivin promoter. Cancer Gene Ther. 2011;18:144–152. doi: 10.1038/cgt.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idres N, Marill J, Flexor MA, Chabot GG. Activation of retinoic acid receptor-dependent transcription by all-trans-retinoic acid metabolites and isomers. J Biol Chem. 2002;277:31491–31498. doi: 10.1074/jbc.M205016200. [DOI] [PubMed] [Google Scholar]
- Jeon YH, Ahn SJ, Lee YJ, Lee YL, Lee SW, Park SY, et al. Human sodium iodide symporter added to multidrug resistance 1 small hairpin RNA in a single gene construct enhances the therapeutic effects of radioiodine in a nude mouse model of multidrug resistant colon cancer. Cancer Biother Radiopharm. 2010;25:671–679. doi: 10.1089/cbr.2010.0837. [DOI] [PubMed] [Google Scholar]
- Jeon YH, Choi Y, Kim HJ, Kim CW, Jeong JM, Lee DS, et al. Human sodium iodide symporter gene adjunctive radiotherapy to enhance the preventive effect of hMUC1 DNA vaccine. Int J Cancer. 2007;121:1593–1599. doi: 10.1002/ijc.22837. [DOI] [PubMed] [Google Scholar]
- Jeon YH, Lee HW, Lee YL, Kim JE, Hwang MH, Jeong SY, et al. Combined E7-dendritic cell-based immunotherapy and human sodium/iodide symporter radioiodine gene therapy with monitoring of antitumor effects by bioluminescent imaging in a mouse model of uterine cervical cancer. Cancer Biother Radiopharm. 2011;26:671–679. doi: 10.1089/cbr.2011.1081. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, et al. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta) J Biol Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- Josefsson M, Evilevitch L, Westrom B, Grunditz T, Ekblad E. Sodium-iodide symporter mediates iodide secretion in rat gastric mucosa in vitro. Exp Biol Med (Maywood) 2006;231:277–281. doi: 10.1177/153537020623100306. [DOI] [PubMed] [Google Scholar]
- Josefsson M, Grunditz T, Ohlsson T, Ekblad E. Sodium/iodide-symporter: distribution in different mammals and role in entero-thyroid circulation of iodide. Acta Physiol Scand. 2002;175:129–137. doi: 10.1046/j.1365-201X.2002.00968.x. [DOI] [PubMed] [Google Scholar]
- Jung KH, Paik JY, Ko BH, Lee KH. Mitogen-activated protein kinase signaling enhances sodium iodide symporter function and efficacy of radioiodide therapy in nonthyroidal cancer cells. J Nucl Med. 2008;49:1966–1972. doi: 10.2967/jnumed.108.055764. [DOI] [PubMed] [Google Scholar]
- Kagechika H. Novel synthetic retinoids and separation of the pleiotropic retinoidal activities. Curr Med Chem. 2002;9:591–608. doi: 10.2174/0929867024606975. [DOI] [PubMed] [Google Scholar]
- Kambe F, Nomura Y, Okamoto T, Seo H. Redox regulation of thyroid-transcription factors, Pax-8 and TTF-1, is involved in their increased DNA-binding activities by thyrotropin in rat thyroid FRTL-5 cells. Mol Endocrinol. 1996;10:801–812. doi: 10.1210/mend.10.7.8813721. [DOI] [PubMed] [Google Scholar]
- Kawaguchi A, Ikeda M, Endo T, Kogai T, Miyazaki A, Onaya T. Transforming growth factor-beta1 suppresses thyrotropin-induced Na+/I− symporter messenger RNA and protein levels in FRTL-5 rat thyroid cells. Thyroid. 1997;7:789–794. doi: 10.1089/thy.1997.7.789. [DOI] [PubMed] [Google Scholar]
- Keefe SM, Cohen MA, Brose MS. Targeting vascular endothelial growth factor receptor in thyroid cancer: the intracellular and extracellular implications. Clin Cancer Res. 2010;16:778–783. doi: 10.1158/1078-0432.CCR-08-2743. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Jeon YH, Kang JH, Lee YJ, Kim KI, Chung HK, et al. In vivo long-term imaging and radioiodine therapy by sodium-iodide symporter gene expression using a lentiviral system containing ubiquitin C promoter. Cancer Biol Ther. 2007;6:1130–1135. doi: 10.4161/cbt.6.7.4342. [DOI] [PubMed] [Google Scholar]
- Kim JE, Ahn BC, Hwang MH, Jeon YH, Jeong SY, Lee SW, et al. Combined RNA interference of hexokinase II and (131)I-sodium iodide symporter gene therapy for anaplastic thyroid carcinoma. J Nucl Med. 2011;52:1756–1763. doi: 10.2967/jnumed.111.090266. [DOI] [PubMed] [Google Scholar]
- Kim S, Youn H, Song MG, Kang JH, Chung HK, Lee DS, et al. Complementary treatment of siTERT for improving the antitumor effect of TERT-specific I-131 therapy. Cancer Gene Ther. 2012;19:263–270. doi: 10.1038/cgt.2011.88. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung HK, Kang JH, Kim KI, Jeon YH, Jin YN, et al. Tumor-targeted radionuclide imaging and therapy based on human sodium iodide symporter gene driven by a modified telomerase reverse transcriptase promoter. Hum Gene Ther. 2008;19:951–957. doi: 10.1089/hum.2008.030. [DOI] [PubMed] [Google Scholar]
- Kitazono M, Robey R, Zhan Z, Sarlis NJ, Skarulis MC, Aikou T, et al. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na(+)/I(−) symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J Clin Endocrinol Metab. 2001;86:3430–3435. doi: 10.1210/jcem.86.7.7621. [DOI] [PubMed] [Google Scholar]
- Klutz K, Russ V, Willhauck MJ, Wunderlich N, Zach C, Gildehaus FJ, et al. Targeted radioiodine therapy of neuroblastoma tumors following systemic nonviral delivery of the sodium iodide symporter gene. Clin Cancer Res. 2009;15:6079–6086. doi: 10.1158/1078-0432.CCR-09-0851. [DOI] [PubMed] [Google Scholar]
- Klutz K, Willhauck MJ, Dohmen C, Wunderlich N, Knoop K, Zach C, et al. Image-guided tumor-selective radioiodine therapy of liver cancer after systemic nonviral delivery of the sodium iodide symporter gene. Hum Gene Ther. 2011;22:1563–1574. doi: 10.1089/hum.2011.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoop K, Kolokythas M, Klutz K, Willhauck MJ, Wunderlich N, Draganovici D, et al. Image-guided, tumor stroma-targeted 131I therapy of hepatocellular cancer after systemic mesenchymal stem cell-mediated NIS gene delivery. Mol Ther. 2011;19:1704–1713. doi: 10.1038/mt.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knostman KA, McCubrey JA, Morrison CD, Zhang Z, Capen CC, Jhiang SM. PI3K activation is associated with intracellular sodium/iodide symporter protein expression in breast cancer. BMC Cancer. 2007;7:137. doi: 10.1186/1471-2407-7-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogai T, Curcio F, Hyman S, Cornford EM, Brent GA, Hershman JM. Induction of follicle formation in long-term cultured normal human thyroid cells treated with thyrotropin stimulates iodide uptake but not sodium/iodide symporter messenger RNA and protein expression. J Endocrinol. 2000a;167:125–135. doi: 10.1677/joe.0.1670125. [DOI] [PubMed] [Google Scholar]
- Kogai T, Endo T, Saito T, Miyazaki A, Kawaguchi A, Onaya T. Regulation by thyroid-stimulating hormone of sodium/iodide symporter gene expression and protein levels in FRTL-5 cells. Endocrinology. 1997;138:2227–2232. doi: 10.1210/endo.138.6.5189. [DOI] [PubMed] [Google Scholar]
- Kogai T, Hershman JM, Motomura K, Endo T, Onaya T, Brent GA. Differential regulation of the human sodium/iodide symporter gene promoter in papillary thyroid carcinoma cell lines and normal thyroid cells. Endocrinology. 2001;142:3369–3379. doi: 10.1210/endo.142.8.8344. [DOI] [PubMed] [Google Scholar]
- Kogai T, Kanamoto Y, Che LH, Taki K, Moatamed F, Schultz JJ, et al. Systemic retinoic acid treatment induces sodium/iodide symporter expression and radioiodide uptake in mouse breast cancer models. Cancer Res. 2004;64:415–422. doi: 10.1158/0008-5472.can-03-2285. [DOI] [PubMed] [Google Scholar]
- Kogai T, Kanamoto Y, Li AI, Che LH, Ohashi E, Taki K, et al. Differential regulation of sodium/iodide symporter gene expression by nuclear receptor ligands in MCF-7 breast cancer cells. Endocrinology. 2005;146:3059–3069. doi: 10.1210/en.2004-1334. [DOI] [PubMed] [Google Scholar]
- Kogai T, Liu YY, Mody K, Shamsian DV, Brent GA. Regulation of sodium iodide symporter gene expression by the Rac1/p38beta MAP kinase signaling pathway in MCF-7 breast cancer cells. J Biol Chem. 2012;287:3292–3300. doi: 10.1074/jbc.M111.315523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogai T, Ohashi E, Jacobs MS, Sajid-Crockett S, Fisher ML, Kanamoto Y, et al. Retinoic acid stimulation of the sodium/iodide symporter in MCF-7 breast cancer cells is mediated by the insulin growth factor-I/phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase signaling pathways. J Clin Endocrinol Metab. 2008a;93:1884–1892. doi: 10.1210/jc.2007-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogai T, Sajid-Crockett S, Newmarch LS, Liu YY, Brent GA. Phosphoinositide-3-kinase inhibition induces sodium/iodide symporter expression in rat thyroid cells and human papillary thyroid cancer cells. J Endocrinol. 2008b;199:243–252. doi: 10.1677/JOE-08-0333. [DOI] [PubMed] [Google Scholar]
- Kogai T, Schultz JJ, Johnson LS, Huang M, Brent GA. Retinoic acid induces sodium/iodide symporter gene expression and radioiodide uptake in the MCF-7 breast cancer cell line. Proc Natl Acad Sci U S A. 2000b;97:8519–8524. doi: 10.1073/pnas.140217197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogai T, Taki K, Brent GA. Enhancement of sodium/iodide symporter expression in thyroid and breast cancer. Endocr Relat Cancer. 2006;13:797–826. doi: 10.1677/erc.1.01143. [DOI] [PubMed] [Google Scholar]
- Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat. 2004;83:249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- Ladenson PW, Braverman LE, Mazzaferri EL, Brucker-Davis F, Cooper DS, Garber JR, et al. Comparison of administration of recombinant human thyrotropin with withdrawal of thyroid hormone for radioactive iodine scanning in patients with thyroid carcinoma. N Engl J Med. 1997;337:888–896. doi: 10.1056/NEJM199709253371304. [DOI] [PubMed] [Google Scholar]
- Laperriere D, Wang TT, White JH, Mader S. Widespread Alu repeat-driven expansion of consensus DR2 retinoic acid response elements during primate evolution. BMC Genomics. 2007;8:23. doi: 10.1186/1471-2164-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy O, De la Vieja A, Ginter CS, Riedel C, Dai G, Carrasco N. N-linked glycosylation of the thyroid Na+/I− symporter (NIS). Implications for its secondary structure model. J Biol Chem. 1998;273:22657–22663. doi: 10.1074/jbc.273.35.22657. [DOI] [PubMed] [Google Scholar]
- Li H, Peng KW, Dingli D, Kratzke RA, Russell SJ. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010;17:550–558. doi: 10.1038/cgt.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Gurlo T, Haataja L, Hsueh WA, Butler PC. Activation of peroxisome proliferator-activated receptor-gamma by rosiglitazone protects human islet cells against human islet amyloid polypeptide toxicity by a phosphatidylinositol 3′-kinase-dependent pathway. J Clin Endocrinol Metab. 2005;90:6678–6686. doi: 10.1210/jc.2005-0079. [DOI] [PubMed] [Google Scholar]
- Majerus PM, Courtois PA. Susceptibility of Candida albicans to peroxidase-catalyzed oxidation products of thiocyanate, iodide and bromide. J Biol Buccale. 1992;20:241–245. [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannavola D, Coco P, Vannucchi G, Bertuelli R, Carletto M, Casali PG, et al. A novel tyrosine-kinase selective inhibitor, sunitinib, induces transient hypothyroidism by blocking iodine uptake. J Clin Endocrinol Metab. 2007;92:3531–3534. doi: 10.1210/jc.2007-0586. [DOI] [PubMed] [Google Scholar]
- Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet. 1998;19:87–90. doi: 10.1038/ng0598-87. [DOI] [PubMed] [Google Scholar]
- Maruca J, Santner S, Miller K, Santen RJ. Prolonged iodine clearance with a depletion regimen for thyroid carcinoma: concise communication. J Nucl Med. 1984;25:1089–1093. [PubMed] [Google Scholar]
- Masia S, Alvarez S, de Lera AR, Barettino D. Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol Endocrinol. 2007;21:2391–2402. doi: 10.1210/me.2007-0062. [DOI] [PubMed] [Google Scholar]
- Maxon HR, Smith HS. Radioiodine-131 in the diagnosis and treatment of metastatic well differentiated thyroid cancer. Endocrinol Metab Clin North Am. 1990;19:685–718. [PubMed] [Google Scholar]
- Maxon HR, Thomas SR, Hertzberg VS, Kereiakes JG, Chen IW, Sperling MI, et al. Relation between effective radiation dose and outcome of radioiodine therapy for thyroid cancer. N Engl J Med. 1983;309:937–941. doi: 10.1056/NEJM198310203091601. [DOI] [PubMed] [Google Scholar]
- Medh RD, Schmidt TJ. Trans-retinoic acid and glucocorticoids synergistically induce transcription from the mouse mammary tumor virus promoter in human embryonic kidney cells. J Steroid Biochem Mol Biol. 1997;62:129–142. doi: 10.1016/s0960-0760(97)00033-2. [DOI] [PubMed] [Google Scholar]
- Meier CA, Braverman LE, Ebner SA, Veronikis I, Daniels GH, Ross DS, et al. Diagnostic use of recombinant human thyrotropin in patients with thyroid carcinoma (phase I/II study) J Clin Endocrinol Metab. 1994;78:188–196. doi: 10.1210/jcem.78.1.8288703. [DOI] [PubMed] [Google Scholar]
- Menzel C, Kranert WT, Dobert N, Diehl M, Fietz T, Hamscho N, et al. rhTSH stimulation before radioiodine therapy in thyroid cancer reduces the effective half-life of (131)I. J Nucl Med. 2003;44:1065–1068. [PubMed] [Google Scholar]
- Miller JK, Swanson EW, Spalding GE. Iodine absorption, excretion, recycling, and tissue distribution in the dairy cow. J Dairy Sci. 1975;58:1578–1593. doi: 10.3168/jds.S0022-0302(75)84753-9. [DOI] [PubMed] [Google Scholar]
- MIRD. Summary of current radiation dose estimates to humans from 123I, 124I, 125I, 126I, 130I, 131I, and 132I as sodium iodide. J Nucl Med. 1975;16:857–860. [PubMed] [Google Scholar]
- Mitsutake N, Miyagishi M, Mitsutake S, Akeno N, Mesa C, Jr, Knauf JA, et al. BRAF mediates RET/PTC-induced mitogen-activated protein kinase activation in thyroid cells: functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology. 2006;147:1014–1019. doi: 10.1210/en.2005-0280. [DOI] [PubMed] [Google Scholar]
- Mizukami Y, Hashimoto T, Nonomura A, Michigishi T, Nakamura S, Noguchi M, et al. Immunohistochemical demonstration of thyrotropin (TSH)-receptor in normal and diseased human thyroid tissues using monoclonal antibody against recombinant human TSH-receptor protein. J Clin Endocrinol Metab. 1994;79:616–619. doi: 10.1210/jcem.79.2.8045985. [DOI] [PubMed] [Google Scholar]
- Montiel-Equihua CA, Martin-Duque P, de la Vieja A, Quintanilla M, Burnet J, Vassaux G, et al. Targeting sodium/iodide symporter gene expression for estrogen-regulated imaging and therapy in breast cancer. Cancer Gene Ther. 2008;15:465–473. doi: 10.1038/cgt.2008.6. [DOI] [PubMed] [Google Scholar]
- Moon DH, Lee SJ, Park KY, Park KK, Ahn SH, Pai MS, et al. Correlation between 99mTc-pertechnetate uptakes and expressions of human sodium iodide symporter gene in breast tumor tissues. Nucl Med Biol. 2001;28:829–834. doi: 10.1016/s0969-8051(01)00243-8. [DOI] [PubMed] [Google Scholar]
- Nagpal S, Chandraratna RA. Recent developments in receptor-selective retinoids. Curr Pharm Des. 2000;6:919–931. doi: 10.2174/1381612003400146. [DOI] [PubMed] [Google Scholar]
- Nicola JP, Basquin C, Portulano C, Reyna-Neyra A, Paroder M, Carrasco N. The Na+/I− symporter mediates active iodide uptake in the intestine. Am J Physiol Cell Physiol. 2009a;296:C654–662. doi: 10.1152/ajpcell.00509.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola JP, Nazar M, Mascanfroni ID, Pellizas CG, Masini-Repiso AM. NF-kappaB p65 subunit mediates lipopolysaccharide-induced Na(+)/I(−) symporter gene expression by involving functional interaction with the paired domain transcription factor Pax8. Mol Endocrinol. 2010;24:1846–1862. doi: 10.1210/me.2010-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola JP, Velez ML, Lucero AM, Fozzatti L, Pellizas CG, Masini-Repiso AM. Functional toll-like receptor 4 conferring lipopolysaccharide responsiveness is expressed in thyroid cells. Endocrinology. 2009b;150:500–508. doi: 10.1210/en.2008-0345. [DOI] [PubMed] [Google Scholar]
- Norden MM, Larsson F, Tedelind S, Carlsson T, Lundh C, Forssell-Aronsson E, et al. Down-regulation of the sodium/iodide symporter explains 131I-induced thyroid stunning. Cancer Res. 2007;67:7512–7517. doi: 10.1158/0008-5472.CAN-07-0823. [DOI] [PubMed] [Google Scholar]
- Ohashi E, Kogai T, Kagechika H, Brent GA. Activation of the PI3 kinase pathway by retinoic acid mediates sodium/iodide symporter induction and iodide transport in MCF-7 breast cancer cells. Cancer Res. 2009;69:3443–3450. doi: 10.1158/0008-5472.CAN-08-3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Zannini M, Levy O, Carrasco N, di Lauro R. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol. 1999;19:2051–2060. doi: 10.1128/mcb.19.3.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K, Endo T, Onaya T. The mRNA levels of thyrotropin receptor, thyroglobulin and thyroid peroxidase in neoplastic human thyroid tissues. Biochem Biophys Res Commun. 1991;174:1148–1153. doi: 10.1016/0006-291x(91)91540-s. [DOI] [PubMed] [Google Scholar]
- Ohta K, Pang XP, Berg L, Hershman JM. Growth inhibition of new human thyroid carcinoma cell lines by activation of adenylate cyclase through the beta-adrenergic receptor. J Clin Endocrinol Metab. 1997;82:2633–2638. doi: 10.1210/jcem.82.8.4136. [DOI] [PubMed] [Google Scholar]
- Ohye H, Sugawara M. Dual oxidase, hydrogen peroxide and thyroid diseases. Exp Biol Med (Maywood) 2010;235:424–433. doi: 10.1258/ebm.2009.009241. [DOI] [PubMed] [Google Scholar]
- Pang XP, Park M, Hershman JM. Transforming growth factor-beta blocks protein kinase-A-mediated iodide transport and protein kinase-C-mediated DNA synthesis in FRTL-5 rat thyroid cells. Endocrinology. 1992;131:45–50. doi: 10.1210/endo.131.1.1612026. [DOI] [PubMed] [Google Scholar]
- Park JW, Zarnegar R, Kanauchi H, Wong MG, Hyun WC, Ginzinger DG, et al. Troglitazone, the peroxisome proliferator-activated receptor-gamma agonist, induces antiproliferation and redifferentiation in human thyroid cancer cell lines. Thyroid. 2005;15:222–231. doi: 10.1089/thy.2005.15.222. [DOI] [PubMed] [Google Scholar]
- Peerlinck I, Merron A, Baril P, Conchon S, Martin-Duque P, Hindorf C, et al. Targeted radionuclide therapy using a Wnt-targeted replicating adenovirus encoding the Na/I symporter. Clin Cancer Res. 2009;15:6595–6601. doi: 10.1158/1078-0432.CCR-09-0262. [DOI] [PubMed] [Google Scholar]
- Pei L, Melmed S. Isolation and characterization of a pituitary tumor-transforming gene (PTTG) Mol Endocrinol. 1997;11:433–441. doi: 10.1210/mend.11.4.9911. [DOI] [PubMed] [Google Scholar]
- Pekary AE, Hershman JM. Tumor necrosis factor, ceramide, transforming growth factor-beta1, and aging reduce Na+/I- symporter messenger ribonucleic acid levels in FRTL-5 cells. Endocrinology. 1998;139:703–712. doi: 10.1210/endo.139.2.5760. [DOI] [PubMed] [Google Scholar]
- Pesce L, Bizhanova A, Caraballo JC, Westphal W, Butti ML, Comellas A, et al. TSH regulates pendrin membrane abundance and enhances iodide efflux in thyroid cells. Endocrinology. 2012;153:512–521. doi: 10.1210/en.2011-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev MA, Thomasz L, Juvenal GJ. Role of transforming growth factor beta in the regulation of thyroid function and growth. Thyroid. 2009;19:881–892. doi: 10.1089/thy.2007.0303. [DOI] [PubMed] [Google Scholar]
- Pomerance M, Abdullah HB, Kamerji S, Correze C, Blondeau JP. Thyroid-stimulating hormone and cyclic AMP activate p38 mitogen-activated protein kinase cascade. Involvement of protein kinase A, rac1, and reactive oxygen species. J Biol Chem. 2000;275:40539–40546. doi: 10.1074/jbc.M002097200. [DOI] [PubMed] [Google Scholar]
- Pramanik R, Qi X, Borowicz S, Choubey D, Schultz RM, Han J, et al. p38 isoforms have opposite effects on AP-1-dependent transcription through regulation of c-Jun. The determinant roles of the isoforms in the p38 MAPK signal specificity. J Biol Chem. 2003;278:4831–4839. doi: 10.1074/jbc.M207732200. [DOI] [PubMed] [Google Scholar]
- Puglisi F, Cesselli D, Damante G, Pellizzari L, Beltrami CA, Di Loreto C. Expression of Pax-8, p53 and bcl-2 in human benign and malignant thyroid diseases. Anticancer Res. 2000;20:311–316. [PubMed] [Google Scholar]
- Puppin C, Arturi F, Ferretti E, Russo D, Sacco R, Tell G, et al. Transcriptional regulation of human sodium/iodide symporter gene: a role for redox factor-1. Endocrinology. 2004;145:1290–1293. doi: 10.1210/en.2003-1250. [DOI] [PubMed] [Google Scholar]
- Read ML, Lewy GD, Fong JC, Sharma N, Seed RI, Smith VE, et al. Proto-oncogene PBF/PTTG1IP regulates thyroid cell growth and represses radioiodide treatment. Cancer Res. 2011;71:6153–6164. doi: 10.1158/0008-5472.CAN-11-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69:4885–4893. doi: 10.1158/0008-5472.CAN-09-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel C, Levy O, Carrasco N. Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J Biol Chem. 2001;276:21458–21463. doi: 10.1074/jbc.M100561200. [DOI] [PubMed] [Google Scholar]
- Riesco-Eizaguirre G, De la Vieja A, Rodriguez I, Miranda S, Martin-Duque P, Vassaux G, et al. Telomerase-driven expression of the sodium iodide symporter (NIS) for in vivo radioiodide treatment of cancer: a new broad-spectrum NIS-mediated antitumor approach. J Clin Endocrinol Metab. 2011;96:E1435–1443. doi: 10.1210/jc.2010-2373. [DOI] [PubMed] [Google Scholar]
- Riesco-Eizaguirre G, Rodriguez I, De la Vieja A, Costamagna E, Carrasco N, Nistal M, et al. The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res. 2009;69:8317–8325. doi: 10.1158/0008-5472.CAN-09-1248. [DOI] [PubMed] [Google Scholar]
- Riesco-Eizaguirre G, Santisteban P. A perspective view of sodium iodide symporter research and its clinical implications. Eur J Endocrinol. 2006;155:495–512. doi: 10.1530/eje.1.02257. [DOI] [PubMed] [Google Scholar]
- Riggs DS. Quantitative aspects of iodine metabolism in man. Pharmacol Rev. 1952;4:284–370. [PubMed] [Google Scholar]
- Saez C, Martinez-Brocca MA, Castilla C, Soto A, Navarro E, Tortolero M, et al. Prognostic significance of human pituitary tumor-transforming gene immunohistochemical expression in differentiated thyroid cancer. J Clin Endocrinol Metab. 2006;91:1404–1409. doi: 10.1210/jc.2005-2532. [DOI] [PubMed] [Google Scholar]
- Saito T, Endo T, Kawaguchi A, Ikeda M, Katoh R, Kawaoi A, et al. Increased expression of the sodium/iodide symporter in papillary thyroid carcinomas. J Clin Invest. 1998;101:1296–1300. doi: 10.1172/JCI1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Endo T, Kawaguchi A, Ikeda M, Nakazato M, Kogai T, et al. Increased expression of the Na+/I- symporter in cultured human thyroid cells exposed to thyrotropin and in Graves’ thyroid tissue. J Clin Endocrinol Metab. 1997;82:3331–3336. doi: 10.1210/jcem.82.10.4269. [DOI] [PubMed] [Google Scholar]
- Saji M, Kohn LD. Effect of hydrocortisone on the ability of thyrotropin to increase deoxyribonucleic acid synthesis and iodide uptake in FRTL-5 rat thyroid cells: opposite regulation of adenosine 3′,5′-monophosphate signal action. Endocrinology. 1990;127:1867–1876. doi: 10.1210/endo-127-4-1867. [DOI] [PubMed] [Google Scholar]
- Saji M, Kohn LD. Insulin and insulin-like growth factor-I inhibit thyrotropin-increased iodide transport in serum-depleted FRTL-5 rat thyroid cells: modulation of adenosine 3′,5′-monophosphate signal action. Endocrinology. 1991;128:1136–1143. doi: 10.1210/endo-128-2-1136. [DOI] [PubMed] [Google Scholar]
- Salem AK, Fenton MS, Marion KM, Hershman JM. Effect of sunitinib on growth and function of FRTL-5 thyroid cells. Thyroid. 2008;18:631–635. doi: 10.1089/thy.2007.0336. [DOI] [PubMed] [Google Scholar]
- Sawka AM, Thephamongkhol K, Brouwers M, Thabane L, Browman G, Gerstein HC. Clinical review 170: A systematic review and metaanalysis of the effectiveness of radioactive iodine remnant ablation for well-differentiated thyroid cancer. J Clin Endocrinol Metab. 2004;89:3668–3676. doi: 10.1210/jc.2003-031167. [DOI] [PubMed] [Google Scholar]
- Schlumberger M. Papillary and follicular thyroid carcinoma. N Engl J Med. 1998;338:297–306. doi: 10.1056/NEJM199801293380506. [DOI] [PubMed] [Google Scholar]
- Schlumberger M, Lacroix L, Russo D, Filetti S, Bidart JM. Defects in iodide metabolism in thyroid cancer and implications for the follow-up and treatment of patients. Nat Clin Pract Endocrinol Metab. 2007;3:260–269. doi: 10.1038/ncpendmet0449. [DOI] [PubMed] [Google Scholar]
- Schmutzler C, Kohrle J. Retinoic acid redifferentiation therapy for thyroid cancer. Thyroid. 2000;10:393–406. doi: 10.1089/thy.2000.10.393. [DOI] [PubMed] [Google Scholar]
- Schmutzler C, Winzer R, Meissner-Weigl J, Kohrle J. Retinoic acid increases sodium/iodide symporter mRNA levels in human thyroid cancer cell lines and suppresses expression of functional symporter in nontransformed FRTL-5 rat thyroid cells. Biochem Biophys Res Commun. 1997;240:832–838. doi: 10.1006/bbrc.1997.7715. [DOI] [PubMed] [Google Scholar]
- Schweppe RE, Klopper JP, Korch C, Pugazhenthi U, Benezra M, Knauf JA, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab. 2008;93:4331–4341. doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebai H, Hovsepian S, Ristorcelli E, Aouani E, Lombardo D, Fayet G. Resveratrol increases iodide trapping in the rat thyroid cell line FRTL-5. Thyroid. 2010;20:195–203. doi: 10.1089/thy.2009.0171. [DOI] [PubMed] [Google Scholar]
- Seibert K, Shafie SM, Triche TJ, Whang-Peng JJ, O’Brien SJ, Toney JH, et al. Clonal variation of MCF-7 breast cancer cells in vitro and in athymic nude mice. Cancer Res. 1983;43:2223–2239. [PubMed] [Google Scholar]
- Semba RD, Delange F. Iodine in human milk: perspectives for infant health. Nutr Rev. 2001;59:269–278. doi: 10.1111/j.1753-4887.2001.tb05512.x. [DOI] [PubMed] [Google Scholar]
- Shen DH, Marsee DK, Schaap J, Yang W, Cho JY, Hinkle G, et al. Effects of dose, intervention time, and radionuclide on sodium iodide symporter (NIS)-targeted radionuclide therapy. Gene Ther. 2004;11:161–169. doi: 10.1038/sj.gt.3302147. [DOI] [PubMed] [Google Scholar]
- Shimura H, Haraguchi K, Miyazaki A, Endo T, Onaya T. Iodide uptake and experimental 131I therapy in transplanted undifferentiated thyroid cancer cells expressing the Na+/I− symporter gene. Endocrinology. 1997;138:4493–4496. doi: 10.1210/endo.138.10.5571. [DOI] [PubMed] [Google Scholar]
- Shinohara M, Chung YJ, Saji M, Ringel MD. AKT in thyroid tumorigenesis and progression. Endocrinology. 2007;148:942–947. doi: 10.1210/en.2006-0937. [DOI] [PubMed] [Google Scholar]
- Smanik PA, Ryu KY, Theil KS, Mazzaferri EL, Jhiang SM. Expression, exon-intron organization, and chromosome mapping of the human sodium iodide symporter. Endocrinology. 1997;138:3555–3558. doi: 10.1210/endo.138.8.5262. [DOI] [PubMed] [Google Scholar]
- Smith VE, Read ML, Turnell AS, Watkins RJ, Watkinson JC, Lewy GD, et al. A novel mechanism of sodium iodide symporter repression in differentiated thyroid cancer. J Cell Sci. 2009;122:3393–3402. doi: 10.1242/jcs.045427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzweg C, Baker CH, Bergert ER, O’Connor MK, Morris JC. Image-guided radioiodide therapy of medullary thyroid cancer after carcinoembryonic antigen promoter-targeted sodium iodide symporter gene expression. Hum Gene Ther. 2007;18:916–924. doi: 10.1089/hum.2007.081. [DOI] [PubMed] [Google Scholar]
- Spitzweg C, Dietz AB, O’Connor MK, Bergert ER, Tindall DJ, Young CY, et al. In vivo sodium iodide symporter gene therapy of prostate cancer. Gene Ther. 2001;8:1524–1531. doi: 10.1038/sj.gt.3301558. [DOI] [PubMed] [Google Scholar]
- Spitzweg C, O’Connor MK, Bergert ER, Tindall DJ, Young CY, Morris JC. Treatment of prostate cancer by radioiodine therapy after tissue-specific expression of the sodium iodide symporter. Cancer Res. 2000;60:6526–6530. [PubMed] [Google Scholar]
- Sponziello M, Scipioni A, Durante C, Verrienti A, Maranghi M, Giacomelli L, et al. Regulation of sodium/iodide symporter and lactoperoxidase expression in four human breast cancer cell lines. J Endocrinol Invest. 2010;33:2–6. doi: 10.1007/BF03346542. [DOI] [PubMed] [Google Scholar]
- Stratford AL, Boelaert K, Tannahill LA, Kim DS, Warfield A, Eggo MC, et al. Pituitary tumor transforming gene binding factor: a novel transforming gene in thyroid tumorigenesis. J Clin Endocrinol Metab. 2005;90:4341–4349. doi: 10.1210/jc.2005-0523. [DOI] [PubMed] [Google Scholar]
- Takamatsu J, Hosoya T, Tsuji M, Yamada M, Murakami Y, Sakane S, et al. Peroxidase and coupling activities of thyroid peroxidase in benign and malignant thyroid tumor tissues. Thyroid. 1992;2:193–196. doi: 10.1089/thy.1992.2.193. [DOI] [PubMed] [Google Scholar]
- Taki K, Kogai T, Kanamoto Y, Hershman JM, Brent GA. A thyroid-specific far-upstream enhancer in the human sodium/iodide symporter gene requires Pax-8 binding and cyclic adenosine 3′,5′-monophosphate response element-like sequence binding proteins for full activity and is differentially regulated in normal and thyroid cancer cells. Mol Endocrinol. 2002;16:2266–2282. doi: 10.1210/me.2002-0109. [DOI] [PubMed] [Google Scholar]
- Tanosaki S, Ikezoe T, Heaney A, Said JW, Dan K, Akashi M, et al. Effect of ligands of nuclear hormone receptors on sodium/iodide symporter expression and activity in breast cancer cells. Breast Cancer Res Treat. 2003;79:335–345. doi: 10.1023/a:1024064424855. [DOI] [PubMed] [Google Scholar]
- Tazebay UH, Wapnir IL, Levy O, Dohan O, Zuckier LS, Zhao QH, et al. The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat Med. 2000;6:871–878. doi: 10.1038/78630. [DOI] [PubMed] [Google Scholar]
- Tell G, Pellizzari L, Cimarosti D, Pucillo C, Damante G. Ref-1 controls pax-8 DNA-binding activity. Biochem Biophys Res Commun. 1998;252:178–183. doi: 10.1006/bbrc.1998.9548. [DOI] [PubMed] [Google Scholar]
- Titcomb MW, Gottardis MM, Pike JW, Allegretto EA. Sensitive and specific detection of retinoid receptor subtype proteins in cultured cell and tumor extracts. Mol Endocrinol. 1994;8:870–877. doi: 10.1210/mend.8.7.7984149. [DOI] [PubMed] [Google Scholar]
- Tong Y, Eigler T. Transcriptional targets for pituitary tumor-transforming gene-1. J Mol Endocrinol. 2009;43:179–185. doi: 10.1677/JME-08-0176. [DOI] [PubMed] [Google Scholar]
- Trapasso F, Iuliano R, Chiefari E, Arturi F, Stella A, Filetti S, et al. Iodide symporter gene expression in normal and transformed rat thyroid cells. Eur J Endocrinol. 1999;140:447–451. doi: 10.1530/eje.0.1400447. [DOI] [PubMed] [Google Scholar]
- Trujillo MA, Oneal MJ, McDonough S, Qin R, Morris JC. A probasin promoter, conditionally replicating adenovirus that expresses the sodium iodide symporter (NIS) for radiovirotherapy of prostate cancer. Gene Ther. 2010;17:1325–1332. doi: 10.1038/gt.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LC, Hung MW, Chen YH, Su WC, Chang GG, Chang TC. Expression and regulation of alkaline phosphatases in human breast cancer MCF-7 cells. Eur J Biochem. 2000;267:1330–1339. doi: 10.1046/j.1432-1327.2000.01100.x. [DOI] [PubMed] [Google Scholar]
- Unterholzner S, Willhauck MJ, Cengic N, Schutz M, Goke B, Morris JC, et al. Dexamethasone stimulation of retinoic Acid-induced sodium iodide symporter expression and cytotoxicity of 131-I in breast cancer cells. J Clin Endocrinol Metab. 2006;91:69–78. doi: 10.1210/jc.2005-0779. [DOI] [PubMed] [Google Scholar]
- Vadysirisack DD, Venkateswaran A, Zhang Z, Jhiang SM. MEK signaling modulates sodium iodide symporter at multiple levels and in a paradoxical manner. Endocr Relat Cancer. 2007;14:421–432. doi: 10.1677/erc.1.01263. [DOI] [PubMed] [Google Scholar]
- Valenta L. Metastatic thyroid carcinoma in man concentrating iodine without organification. J Clin Endocrinol Metab. 1966;26:1317–1324. doi: 10.1210/jcem-26-12-1317. [DOI] [PubMed] [Google Scholar]
- Van Nostrand D. The benefits and risks of I-131 therapy in patients with well-differentiated thyroid cancer. Thyroid. 2009;19:1381–1391. doi: 10.1089/thy.2009.1611. [DOI] [PubMed] [Google Scholar]
- Venkateswaran A, Marsee DK, Green SH, Jhiang SM. Forskolin, 8-Br-3′,5′-cyclic adenosine 5′-monophosphate, and catalytic protein kinase A expression in the nucleus increase radioiodide uptake and sodium/iodide symporter protein levels in RET/PTC1-expressing cells. J Clin Endocrinol Metab. 2004;89:6168–6172. doi: 10.1210/jc.2004-1414. [DOI] [PubMed] [Google Scholar]
- Volante M, Rapa I, Gandhi M, Bussolati G, Giachino D, Papotti M, et al. RAS mutations are the predominant molecular alteration in poorly differentiated thyroid carcinomas and bear prognostic impact. J Clin Endocrinol Metab. 2009;94:4735–4741. doi: 10.1210/jc.2009-1233. [DOI] [PubMed] [Google Scholar]
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Wapnir IL, Goris M, Yudd A, Dohan O, Adelman D, Nowels K, et al. The Na+/I− symporter mediates iodide uptake in breast cancer metastases and can be selectively down-regulated in the thyroid. Clin Cancer Res. 2004;10:4294–4302. doi: 10.1158/1078-0432.CCR-04-0074. [DOI] [PubMed] [Google Scholar]
- Wapnir IL, van de Rijn M, Nowels K, Amenta PS, Walton K, Montgomery K, et al. Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J Clin Endocrinol Metab. 2003;88:1880–1888. doi: 10.1210/jc.2002-021544. [DOI] [PubMed] [Google Scholar]
- Ward LS, Santarosa PL, Granja F, da Assumpcao LV, Savoldi M, Goldman GH. Low expression of sodium iodide symporter identifies aggressive thyroid tumors. Cancer Lett. 2003;200:85–91. doi: 10.1016/s0304-3835(03)00392-6. [DOI] [PubMed] [Google Scholar]
- Warrell RP., Jr Retinoid resistance in acute promyelocytic leukemia: new mechanisms, strategies, and implications. Blood. 1993;82:1949–1953. [PubMed] [Google Scholar]
- Watkins RJ, Read ML, Smith VE, Sharma N, Reynolds GM, Buckley L, et al. Pituitary tumor transforming gene binding factor: a new gene in breast cancer. Cancer Res. 2010;70:3739–3749. doi: 10.1158/0008-5472.CAN-09-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Yang J, Lee SL, Kulp SK, Chen CS. PPARgamma-independent antitumor effects of thiazolidinediones. Cancer Lett. 2009;276:119–124. doi: 10.1016/j.canlet.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss SJ, Philp NJ, Grollman EF. Iodide transport in a continuous line of cultured cells from rat thyroid. Endocrinology. 1984;114:1090–1098. doi: 10.1210/endo-114-4-1090. [DOI] [PubMed] [Google Scholar]
- Welcsh PL, Mankoff DA. Taking up iodide in breast tissue. Nature. 2000;406:688–689. doi: 10.1038/35021173. [DOI] [PubMed] [Google Scholar]
- Willhauck MJ, DJOK, Wunderlich N, Goke B, Spitzweg C. Stimulation of retinoic acid-induced functional sodium iodide symporter (NIS) expression and cytotoxicity of (1)(3)(1)I by carbamazepine in breast cancer cells. Breast Cancer Res Treat. 2011;125:377–386. doi: 10.1007/s10549-010-0835-x. [DOI] [PubMed] [Google Scholar]
- Willhauck MJ, Sharif Samani BR, Klutz K, Cengic N, Wolf I, Mohr L, et al. Alpha-fetoprotein promoter-targeted sodium iodide symporter gene therapy of hepatocellular carcinoma. Gene Ther. 2008a;15:214–223. doi: 10.1038/sj.gt.3303057. [DOI] [PubMed] [Google Scholar]
- Willhauck MJ, Sharif-Samani B, Senekowitsch-Schmidtke R, Wunderlich N, Goke B, Morris JC, et al. Functional sodium iodide symporter expression in breast cancer xenografts in vivo after systemic treatment with retinoic acid and dexamethasone. Breast Cancer Res Treat. 2008b;109:263–272. doi: 10.1007/s10549-007-9646-0. [DOI] [PubMed] [Google Scholar]
- Wolff J, Robbins J, Rall JE. Iodide trapping without organification in a transplantable rat thyroid tumor. Endocrinology. 1959;64:1–11. doi: 10.1210/endo-64-1-1. [DOI] [PubMed] [Google Scholar]
- Yamazaki K, Suzuki K, Yamada E, Yamada T, Takeshita F, Matsumoto M, et al. Suppression of iodide uptake and thyroid hormone synthesis with stimulation of the type I interferon system by double-stranded ribonucleic acid in cultured human thyroid follicles. Endocrinology. 2007;148:3226–3235. doi: 10.1210/en.2006-1638. [DOI] [PubMed] [Google Scholar]
- Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. 2001;81:1097–1142. doi: 10.1152/physrev.2001.81.3.1097. [DOI] [PubMed] [Google Scholar]
- Yoon JK, Park BN, Paik JY, Jung KH, Ko BH, Lee KH. Effects of theophylline on radioiodide uptake in MCF-7 breast cancer and NIS gene-transduced SNU-C5 colon cancer cells. Cancer Biother Radiopharm. 2009;24:201–208. doi: 10.1089/cbr.2008.0555. [DOI] [PubMed] [Google Scholar]