

Abstract
Human NSCLCs with activating mutations in EGFR frequently respond to treatment with EGFR tyrosine kinase inhibitors (TKIs) such as erlotinib but responses are not durable as tumors acquire resistance. Secondary mutations in EGFR (T790M) or upregulation of the MET kinase are found in over 50% of resistant tumors. Here, we report increased activation of AXL and evidence of epithelial-to-mesenchymal transition (EMT) in multiple in vitro and in vivo EGFR-mutant lung cancer models with erlotinib acquired resistance in the absence of EGFR T790M or MET activation. Genetic or pharmacologic inhibition of AXL restored sensitivity to erlotinib in these tumor models. Increased expression of AXL, and in some cases its ligand GAS6, was found in EGFR-mutant lung cancers obtained from patients with EGFR TKI acquired resistance. These data identify AXL as a promising therapeutic target whose inhibition could prevent or overcome EGFR TKI acquired resistance in EGFR-mutant lung cancer patients.
Introduction
Non-small cell lung cancer (NSCLC) has served as a model for genotype-directed targeted cancer therapy. NSCLC patients whose tumors harbor activating kinase domain mutations in the epidermal growth factor receptor (EGFR) often initially respond to treatment with an EGFR tyrosine kinase inhibitor (TKI) such as erlotinib.1–4However, acquired resistance to EGFR TKI treatment invariably develops.5,6 There is no effective therapy for patients who develop such resistance. Work by our group and others has shown that resistance to EGFR TKI treatment can occur through a secondary resistance mutation in EGFR (T790M), activation of the MET kinase, and activation of the NF-kB pathway.7–11. 12The mechanisms underlying acquired resistance to EGFR TKI treatment are unknown in over 40% of EGFR-mutant NSCLC patients.
Recent studies indicate that multiple resistance mechanisms may operate within an individual tumor to promote EGFR TKI acquired resistance in NSCLC patients. For example, the EGFR T790M resistance mutation and activation of MET can co-occur in some EGFR-mutant NSCLCs with acquired resistance to EGFR TKI treatment.13,12 Furthermore, recent evidence suggests that the acquisition of EGFR TKI resistance in EGFR-mutant NSCLCs may be associated not only with genotypic alterations but also histological changes that occur during adaptation to therapy.14
By studying human EGFR-mutant NSCLC tumor models and clinical specimens we aimed to: 1) identify novel mechanisms of acquired resistance to EGFR TKI treatment, and 2) further clarify the extent to which distinct and co-existent genotypic and histological changes promote the acquisition of EGFR TKI treatment resistance in NSCLC patients.
Results
AXL promotes resistance in vivo and in vitro
To identify novel mechanisms of acquired resistance to EGFR TKI treatment, our 3 groups independently established new in vivo and in vitro (n=5) models of acquired resistance to the EGFR TKI erlotinib using EGFR exon 19 deletion mutant (delE746-A750) HCC827 human NSCLC cells. HCC827 cells are initially sensitive to erlotinib treatment (in vitro IC50 ~5nM) and we and others have used them to develop in vitro models of EGFR TKI acquired resistance in studies that have led to the identification of clinically relevant mechanisms of EGFR TKI resistance.10,11 To establish the in vivo model, cohorts of 5 mice (2 tumors/mouse) with established HCC827 tumors were treated with vehicle or 4 escalating doses of erlotinib(from 6.25 mg/kg/day to 50 mg/kg/day) over ~ 5 months to derive erlotinib-resistant tumors. Erlotinib treatment of HCC827 xenograft tumors (10 tumors/dose, daily treatment) resulted in an initial dose-dependent decrease in tumor volume and the subsequent development of acquired resistance (>25% re-growth from max reduction) after 6–10 weeks of treatment in each tumor (Figure 1a, Table 1).Sequencing of EGFR in each erlotinib resistant tumor showed that none harbored the EGFR T790M mutation nor other secondary mutations in EGFR associated with erlotinib resistance (D761Y, L474S, T854A) (data not shown). To examine whether the erlotinib resistant tumors harbored increased expression of either known or potential novel drivers of resistance, we conducted microarray expression profiling of 17 xenograft tumors across each treatment group as well as 2 vehicle treated control tumors. We asked which genes were differentially regulated in the erlotinib resistant tumors compared to the control tumors (unpaired T-test, P<0.05). The analysis showed that 21 genes were increased (≥1 log2 fold change) specifically in the erlotinib resistant tumors (Supplementary Table 1). Unexpectedly, we found that the receptor tyrosine kinase AXL was the most highly overexpressed gene in the tumors with acquired erlotinib resistance (Supplementary Table 1). Consistent with prior studies9,13, we also observed that MET was among the genes upregulated, although to a much lesser extent that AXL. We found increased mRNA expression of MET (≥1 log2 fold change) in 5/17 (29%) of the tumors with erlotinib acquired resistance (Figure 1b, Supplementary Table 2). The analysis did not identify overexpression of IGF-1R, Ras, or EGFR in the erlotinib-resistant tumors (Supplementary Tables 1, 2). Compared to control tumors, expression of AXL was increased (≥1 log2 fold change) in 15/17 (88%) of the tumors with erlotinib resistance (Figure 1c, Supplementary Table 2). Moreover, we also found increased expression (≥ 1 log2 fold change) of GAS6, the ligand for AXL, in 8/17 (47%) of the tumors with erlotinib resistance (Figure 1d, Supplementary Table 2). AXL was overexpressed in each tumor that had increased MET or GAS6 levels. In 10 of the 15 tumors (66.6%) with AXL upregulation, MET overexpression was not observed. In each tumor in which AXL and MET were both increased AXL was overexpressed to a higher degree. AXL and GAS6 overexpression was unique to treatment resistant tumors and was not the result of acute effects of erlotinib treatment as their levels were not increased in erlotinib-sensitive tumors harvested after 48h of erlotinib treatment (Supplementary Figure 1). aCGH analysis showed that neither AXL or GAS6 was amplified and sequencing of AXL revealed that it was not mutated in the erlotinib resistant tumors (data not shown). Based on these findings and recent data demonstrating that AXL can promote cell growth in several cancer cell lines15,16, we hypothesized that AXL overexpression and activation may promote acquired resistance to erlotinib in EGFR-mutant NSCLCs.
Figure 1. AXL is overexpressed in EGFR-mutant NSCLC tumor xenografts with acquired resistance to erlotinib.
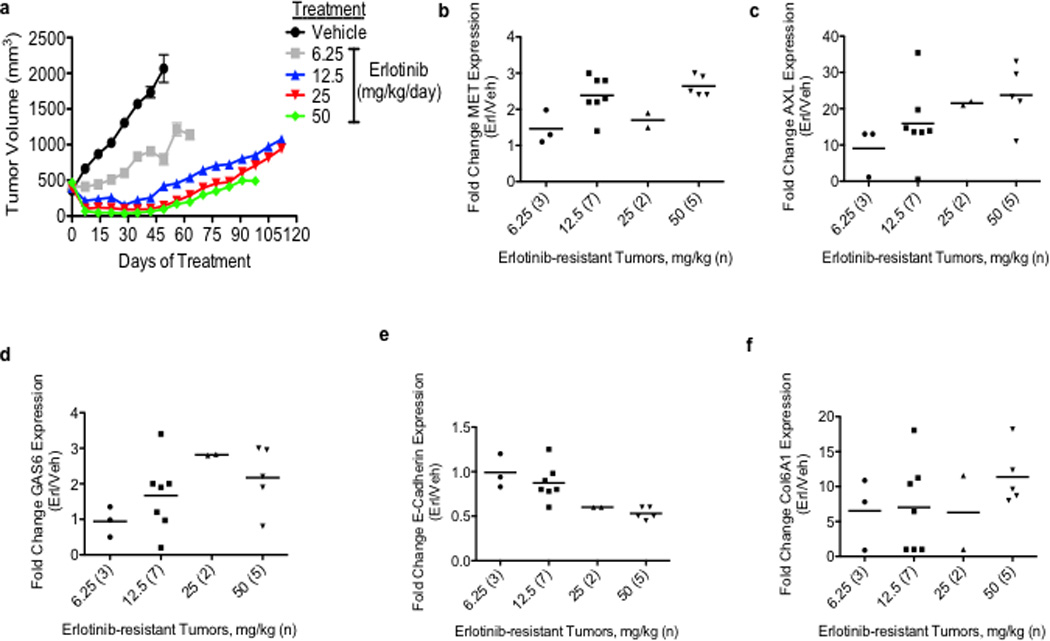
a, Effects of a dose-response of erlotinib in HCC827 xenograft tumors in immunocompromised mice (n=10 tumors/treatment group). b–f, mRNA expression of indicated genes in HCC827 erlotinib resistant tumor xenografts (treated at the indicated erlotinib doses) relative to vehicle-treated control tumors. The number of tumors analyzed from each treatment cohort is indicated in parentheses. Data are expressed as the mean ± SEM of the fold change relative to the mean expression of the genes in 2 vehicle treated control xenograft tumors.
Table 1. Erlotinib treatment response in HCC827 tumor xenografts.
Data from 10 tumors per treatment group are expressed as average tumor volume reduction, median time to maximum reduction, and median time to erlotinib resistance ± SEM.
| Erlotinib Dose (mg/kg) |
Maximum Volume Reduction (% change from baseline) |
Time to Maximum Volume Reduction (weeks) |
Median Time to Resistance (weeks) |
|---|---|---|---|
| 6.25 | N/A | N/A | N/A |
| 12.5 | −56 | 4 ± 0.3 | 6 ± 0.5 |
| 25 | −69 | 5 ± 0.2 | 8 ± 0.8 |
| 50 | −84 | 4 ± 0.4 | 10 ± 1 |
AXL has been previously associated with EMT and recent studies suggest that therapeutic resistance may, in some cases, be associated with histological changes including EMT in NSCLCs.14,17,18 We noted that the tumor xenografts with acquired erlotinib resistance harbored alterations in the expression level of several genes that are established biomarkers of EMT. For example, we found increased levels of COL6A1 (a type IV collagen), HMGA1 and HMGA2 and decreased levels of keratin genes (including KRT6A, KRT14, KRT5) (Supplementary Table 1) in the tumors with acquired erlotinib resistance compared with the control tumors. We further examined whether the erlotinib resistant xenograft tumors exhibit molecular changes known to occur during EMT. Indeed, we found downregulation of E-cadherin (≥0.5 log2 fold change) in 8/15 (53%) (Figure 1e, Supplementary Table 2), upregulation of COL6A1 (≥1 log2 fold change) in 14/15 (93%) (Figure 1f, Supplementary Table 2) and increased vimentin (≥1 log2 fold change) in 10/15 (66%) (Supplementary Figure 2) of the erlotinib-resistant tumors with AXL overexpression compared to the vehicle treated control tumors.
Consistent with the microarray data, we found increased levels of AXL and phosphorylated (p) AXL proteins in representative resistant HCC827 tumor xenografts at each dose of erlotinib compared with vehicle-treated tumors (Figure 2a). We did not find alterations in the levels of IGF-1R, pIGF-1R, or Ras in the erlotinib resistant tumors (Figure 2a). In contrast, we observed decreased levels of pEGFR, pErbB3, and pMET in response to erlotinib treatment both in sensitive tumors harvested at 48h of treatment (Figure 2b) and in resistant tumors at each dose of erlotinib compared to vehicle treated tumors (Figure 2a). Erlotinib treatment for 48h decreased pAKT, pERK, and pRelA and increased Parp cleavage in sensitive tumors (a marker of apoptosis) (Figure 2b) but pAKT, pERK, pRelA levels were maintained in each tumor with acquired erlotinib resistance (Figure 2a). Furthermore, we observed increased expression of the EMT marker vimentin in several erlotinib-resistant tumors (Figure 2a). These data suggest that AXL upregulation may activate AKT, MAPK or NF-kB signaling to promote resistance to erlotinib treatment in EGFR-mutant NSCLCs perhaps in association with an EMT.
Figure 2. AXL overexpression is necessary for acquired resistance to erlotinib treatment in EGFR-mutant NSCLC tumors in vivo.
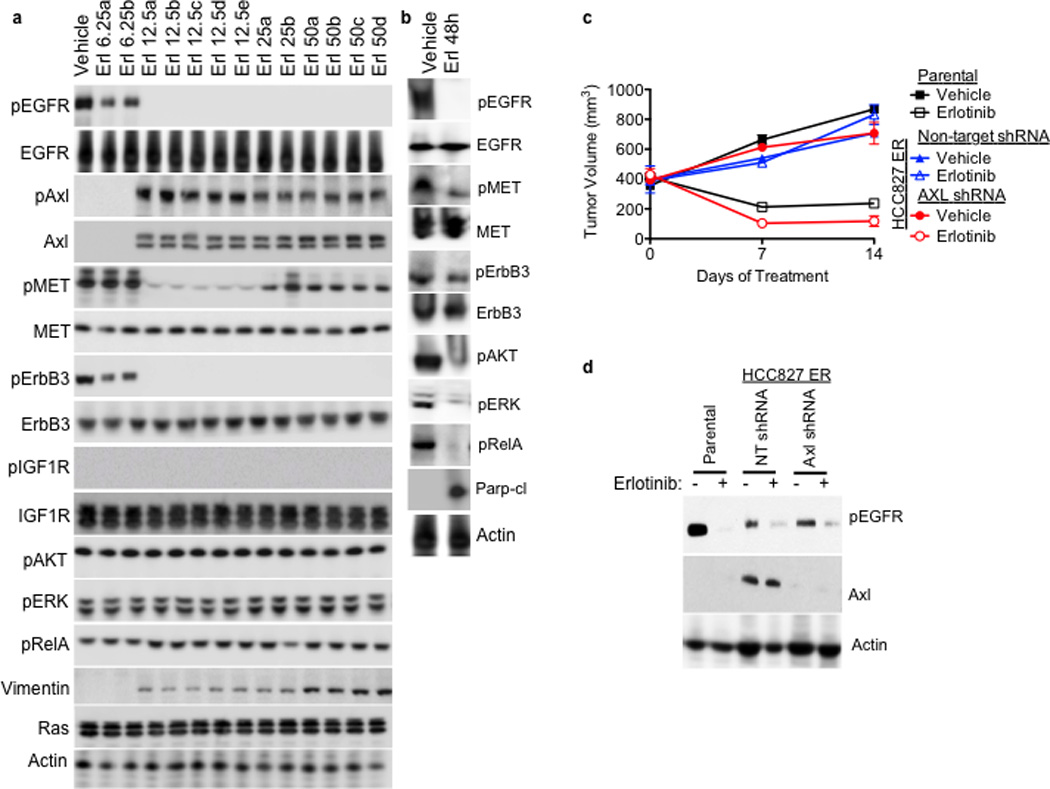
a, AXL and pAXL protein levels are increased in the absence of pEGFR and increased pMET in HCC827 tumors from each erlotinib treatment cohort compared with vehicle treated tumors. Tumors were harvested for analysis at the completion of the study for each treatment group (Vehicle: Day 45; 6.25 Erl: Day 60; 12.5 Erl: Day 110; 25 Erl: Day 110; 50 Erl: Day 100) b, Acute and transient treatment of HCC827 tumors with erlotinib (12.5 mg/kg/day) decreases pAKT, pERK, pRELa and increases the levels of cleaved Parp. c, Response of parental HCC827 xenograft tumors or HCC827 ER xenograft tumors (n= 10 tumors/group) transduced with a non-target shRNA or an shRNA targeting AXL to treatment with either vehicle or erlotinib (12.5 mg/kg/day). Tumor volumes are expressed as mean ± SEM. d, Validation of AXL knockdown and the effects of erlotinib treatment on pEGFR in representative tumor xenografts (c) by western blot analysis.
AXL inhibition restores erlotinib sensitivity
To determine whether inhibition of AXL may restore erlotinib sensitivity in vivo, we took a genetic approach because the pharmacokinetics and specificity of currently available AXL inhibitors in vivo are suboptimal. We generated xenograft tumors in immunocompromised mice using either parental HCC827 cells or an erlotinib resistant subclone of HCC827 cells that we established, which we found to express increased levels of AXL (HCC827 subline “ER1”, discussed further below). We transduced the AXL-overexpressing HCC827 ER1 cells with either a non-targeting shRNA or an shRNA targeting AXL prior to engraftment into the mice and then treated them with either vehicle or erlotinib (12.5 mg/kg/day). As expected, the parental HCC827 tumors were sensitive to erlotinib treatment whereas the AXL-overexpressing HCC827ER1 tumors transduced with the non-target shRNA were erlotinib-resistant (Figure 2c). In contrast, knockdown of AXL restored sensitivity to erlotinib in the HCC827 ER1 tumors (Figure 2c–d). The data show that AXL was required for erlotinib resistance in this in vivo model.
To further explore the role of AXL in EGFR TKI acquired resistance we next focused on the novel in vitro models of erlotinib acquired resistance that we generated in conjunction with the in vivo tumor xenograft models. Five erlotinib resistant HCC827 clonal sublines were established (“ER” 1–5) independently in 3 laboratories, each with an erlotinib IC50> 10 µM, through prolonged (>5 months) and continuous exposure to erlotinib (Figure 3a). Each of these cell lines was also resistant to the irreversible EGFR kinase inhibitor BIBW2992 (afatinib) (Supplementary Figure 3). Thus resistance in these cellular models is unlikely a consequence of the secondary drug resistance mutation (T790M) in EGFR and is generalizable to distinct classes of EGFR TKIs. Indeed, the EGFR T790M resistance mutation was not detected by sequencing in any ER subline (data not shown). We sought to determine whether AXL or other genes previously implicated in EGFR TKI acquired resistance were upregulated in the HCC827 ER1-5 sublines compared to parental cells. To identify genes that were differentially regulated (threshold 3-fold change, FDR<0.1) in the context of erlotinib acquired resistance, we profiled the ER1-3 sublines as compared to parental HCC827 cells by genome-wide microarrays (Supplementary Table 3) and validated our findings by Q RT PCR and western blots in all 5 sublines. First, we noted that expression of EGFR, MET, IGF-1R, pIGF-1R and Ras was unaltered in each ER subline (Figure 3b). We did not detect activating mutations in K-Ras or BRAF by sequencing in the ER sublines (data not shown). Consistent with our findings in the erlotinib resistant tumor xenografts, we found significant upregulation of AXL and its ligand GAS6 in each HCC827 ER subline compared to HCC827 parental cells (Figure 3b, Supplementary Table 3, Supplementary Figure 4). We also found increased expression of vimentin and decreased expression of E-cadherin in each ER subline (Figure 3b,Supplementary Figure 4). Neither somatic mutations in nor genomic amplification of AXL were found by sequencing and aCGH (or FISH), respectively, in the HCC827 ER sublines (data not shown).
Figure 3. AXL upregulation is necessary and sufficient for erlotinib acquired resistance in EGFR mutant NSCLC cellular models.
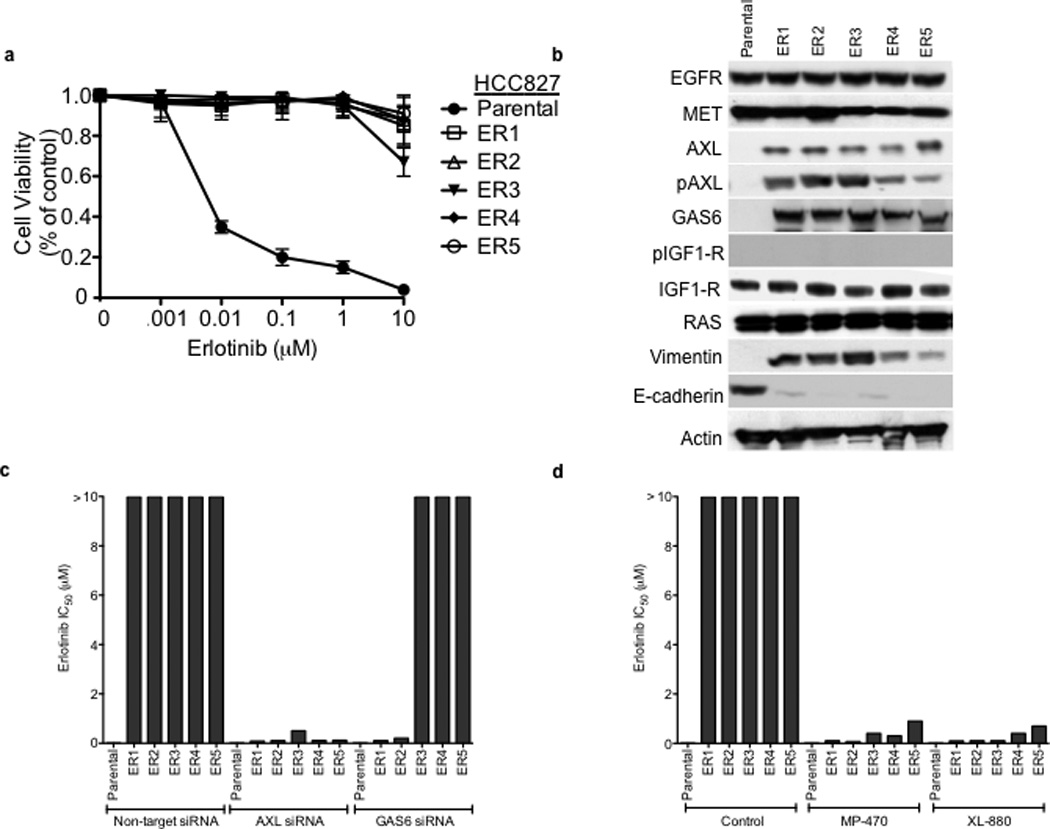
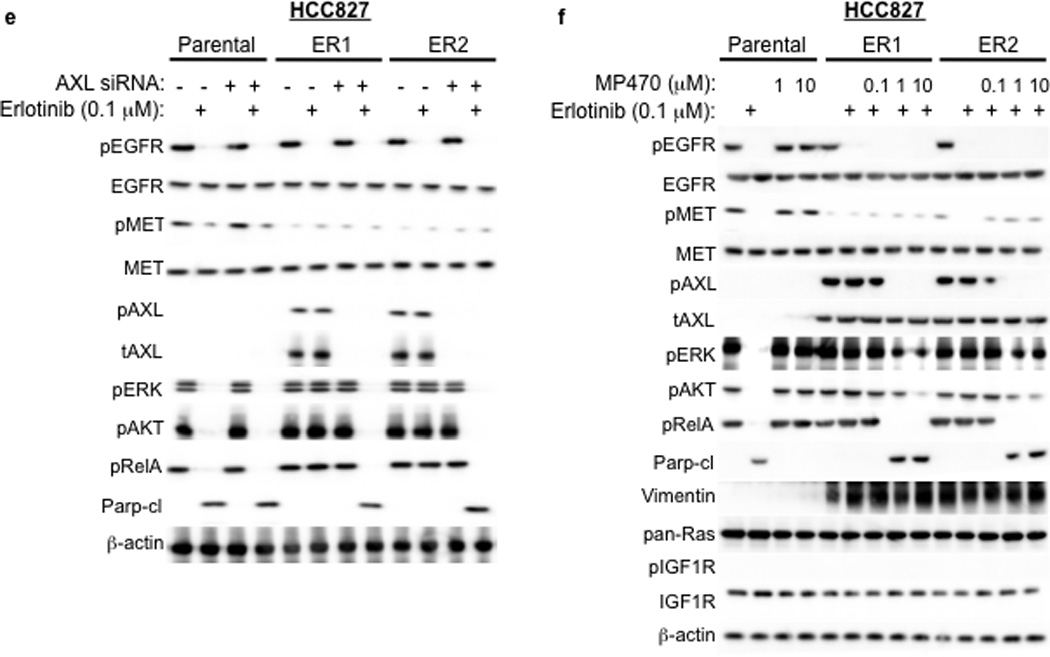
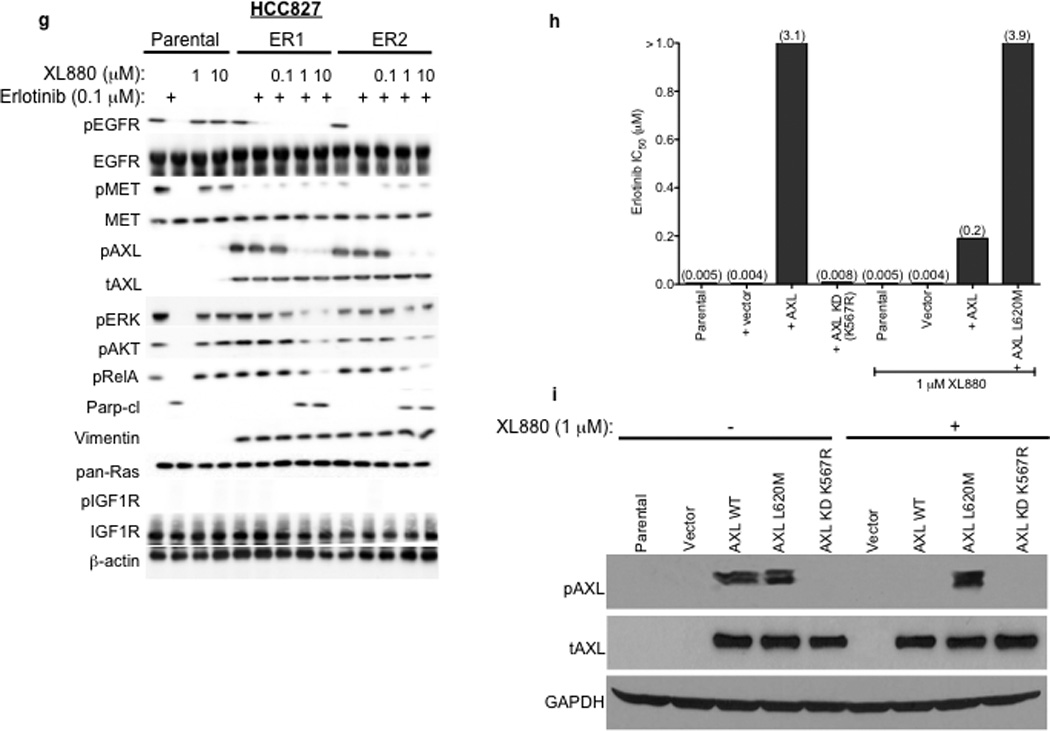
a, HCC827 ER1-ER5 sublines are resistant to erlotinib treatment as measured by CellTiterGLO cell viability assay. Data are from 3 independent experiments and are expressed as percent of vehicle treated cells and mean ± SEM. b, Expression of AXL and GAS6 in the ER sublines compared with parental HCC827 cells (data are from Western blot analysis). c, Erlotinib IC50 in HCC827 cell lines (as indicated) measured 48h after treatment with a non-targeting or AXL or GAS6 siRNA. Erlotinib IC50 is shown in parentheses. Data are representative of 3 independent experiments. d, Erlotinib IC50 in HCC827 cell lines (as indicated) measured 48h after treatment with vehicle (control) or with MP-470 (1 µM) or XL-880 (1 µM) and erlotinib. Erlotinib IC50 is shown in parentheses. Data are representative of 3 independent experiments. e, Effects of treatment for 48h with a non-targeting (−) or AXL siRNA in parental or ER1 and ER2 cell lines in the absence and presence of erlotinib on the indicated biomarkers. Data represent 3 independent experiments. f–g, Effects of treatment for 48h with a vehicle or the indicated doses of (f) MP-470 or (g) XL-880 in parental or ER1 and ER2 cell lines in the absence and presence of erlotinib on the indicated biomarkers. Data represent 3 independent experiments. h,Erlotinib IC50 in HCC827 cells measured 5 days after transfection the cDNA constructs encoding the indicated proteins and treated with either vehicle (left) or with XL-880 (1 µM) and erlotinib. Erlotinib IC50 is shown in parentheses. Data are representative of 3 independent experiments. i, Western blot for the indicated proteins in lysates from cells transfected with the indicated cDNA constructs and treated with XL-880 (1 µM) for 3 hours prior to cell lysis (h).
We next asked whether AXL overexpression and activation was necessary for erlotinib acquired resistance in vitro by genetically or pharmacologically inhibiting AXL in the HCC827 ER sublines. First we found that knockdown of AXL using an AXL siRNA had no effect on erlotinib sensitivity in parental HCC827 cells but restored erlotinib sensitivity in each ER subline (Figure 3c). Interestingly, we found that knockdown of GAS6 restored erlotinib sensitivity in the ER1 and ER2 sublines but not in the ER3, ER4 or ER5 sublines (Figure 3c). This observation is reminiscent of recent data showing that upregulation of the MET ligand HGF can promote resistance to gefitinib treatment in some EGFR mutant NSCLC cells.10 Because we observed residual, albeit diminished, levels of pMET in some of the ER sublines, we asked whether MET knockdown by siRNA could restore erlotinib sensitivity in this system. We found that knockdown of MET did not significantly affect erlotinib sensitivity either in parental or HCC827 ER1 cells or further enhance the effects of AXL knockdown on erlotinib sensitivity in ER1 cells (Supplementary Figure 5).
Treatment with a monoclonal antibody against AXL was recently shown to sensitize NSCLC cell lines that express wild type (WT) EGFR to erlotinib.19 Thus we asked whether knockdown of AXL by siRNA enhances erlotinib sensitivity in 5 NSCLC cell lines with WT EGFR (A549, H460, H1573, H2009, Calu-1). We found that AXL knockdown had no effect on erlotinib sensitivity in these cell lines (Supplementary Figure 6). Our data suggest that inhibition of AXL enhances erlotinib sensitivity specifically in the context of NSCLCs with EGFR kinase domain activating mutations in cells with acquired EGFR TKI resistance.
Next we sought to validate our genetic findings using 2 commercially available small molecule inhibitors of AXL, MP-470 and XL-880.15 As expected, treatment with MP-470 (1 µM) or XL-880 (1 µM) alone did not significantly affect the viability of HCC827 parental or ER cells (Supplementary Figure 7). In contrast, we found that treatment with MP-470 (1 µM) or XL-880 (1 µM), restored sensitivity to concurrent erlotinib treatment in each ER subline (Figure 3d). To determine whether treatment with an AXL inhibitor is synergistic with concurrent erlotinib treatment we conducted combination index (CI) analysis in 2 of the ER sublines (ER1 and ER4). We found that treatment with either MP470 or XL880, but not the MET inhibitor PHA665752, was synergistic with concurrent erlotinib treatment (Supplementary Figure 8). Together, the genetic and pharmacologic data show that AXL is required for acquired erlotinib resistance in this system.
We aimed to determine which signaling events downstream of AXL might promote acquired erlotinib resistance in EGFR mutant NSCLCs. Because AXL has been shown to activate the MAPK, AKT and NF-kB pathways and regulate apoptosis in some cancer cell lines15,16, we examined activation of these pathways in HCC827 parental or ER cell lines treated with a non-targeting or AXL siRNA and either vehicle or erlotinib (100 nM). As expected, we found that erlotinib treatment decreased pERK, pAKT and pRelA (a marker of NF-kB signaling) and increased Parp cleavage (a marker of apoptosis) in parental HCC827 cells (Figure 3e). AXL siRNA treatment had no effect on these pathways in parental HCC827 cells (Figure 3e). Erlotinib treatment decreased pEGFR both in parental HCC827 cells and in the ER1 and ER2 sublines (Figure 3e). This observation is consistent with our finding that the ER sublines do not harbor a secondary resistance mutation in EGFR that would abrogate the ability of erlotinib to inhibit EGFR. In contrast, erlotinib treatment decreased pERK, pAKT and pRelA and increased the levels of cleaved Parp only upon AXL knockdown in the ER1 and ER2 cell lines (Figure 3e). Similar results were observed in HCC827 ER3, ER4 and ER5 sublines (Supplementary Figure 9).
We next sought to validate our genetic findings using pharmacologic inhibitors of AXL. As expected, erlotinib decreased pEGFR, pERK, pAKT, pRelA and increased the levels of cleaved Parp in parental HCC827 cells irrespective of concurrent treatment with MP-470 or XL-880 (Figure 3f–g). In contrast, these effects of erlotinib treatment were observed only upon concurrent treatment with MP-470 (Figure 3f) or XL-880 (Figure 3g) in the HCC827 ER1 and ER2 cells. Similar results were observed in HCC827 ER3, ER4 and ER5 sublines treated with erlotinib and XL-880 either alone or in combination (Supplementary Figure 9). Together, the data suggest that AXL activation is necessary for acquired erlotinib resistance and that the MAPK, AKT and NF-kB pathways may, in part, mediate erlotinib resistance downstream of AXL in this system.
To determine whether AXL is upregulated in the setting of erlotinib acquired resistance in cells other than HCC827, we used the same erlotinib treatment protocol to establish 2 isogenic, erlotinib-resistant sublines (IC50> 1 µM) derived from H3255 cells that express the EGFR L858R mutant commonly found in lung cancer patients and that are otherwise sensitive to erlotinib (IC50 ~15nM). Similar to HCC827 cells, we found (1) overexpression of AXL in the absence of EGFR T790M, pEGFR and increased pMET, (2) overexpression of the EMT marker vimentin, and (3) that inhibition of AXL by siRNA, MP-470 or XL880 restored sensitivity to concurrent erlotinib treatment in the H3255 ER sublines (Supplementary Figure 10). Together, these data show that AXL upregulation promotes erlotinib acquired resistance in the setting of EMT in EGFR-mutant NSCLC tumor models and that combined inhibition of AXL and EGFR overcomes acquired resistance to erlotinib in these models.
We next asked whether (1) forced expression of AXL is sufficient to induce erlotinib resistance and (2) whether AXL kinase activity is necessary for the induction of erlotinib resistance by AXL. To address these questions, we generated cDNA constructs that encode either wild type (WT) AXL or a kinase-impaired mutant of AXL in which a key conserved lysine within the kinase domain was changed to arginine (K567R). Transient overexpression of WT AXL, but not AXL KD (K567R), in HCC827 cells increased the levels of pAXL and the IC50 for XL880 (Supplementary Figure 11, Figure 3h–i), suggesting that the K567R impairs AXL kinase function. Overexpression of WT AXL, but not kinase impaired AXL KD (K567R), induced resistance to erlotinib in HCC827 cells that was reversed upon treatment with XL-880 (1 µM) (Figure 3h–i). In addition, we found that introduction of WT AXL was sufficient to induce partial resistance to erlotinib in PC9 cells that express an EGFR exon 19-deletion mutant and are otherwise erlotinib-sensitive (IC50~25 nM) (Supplementary Figure 12). Together, the data indicate that AXL kinase activation is necessary and sufficient to promote erlotinib resistance in these EGFR mutant NSCLC models.
To confirm that treatment with an AXL inhibitor restores erlotinib sensitivity via inhibition of AXL, and is not a consequence of an “off-target” effect of drug treatment, we identified the “gatekeeper” residue within the AXL kinase domain based by structural modeling of the co-crystal of MET bound to XL880 and of the location of the T790M gatekeeper residue in EGFR 20,21 (Supplementary Figure 13). The analysis indicated that an L620M mutation in AXL would be expected to result in steric clash with XL-880 and would block the ability of XL-880 to inhibit AXL. Thus we generated a cDNA encoding this gatekeeper mutant of AXL (L620M) and introduced it into HCC827 cells to determine if expression of it blocks the ability of XL-880 to restore erlotinib sensitivity in the setting of AXL overexpression. Expression of AXL L620M significantly increased the IC50 for XL-880 (Supplementary Figure 11) and blocked the ability of XL880 to decrease pAXL in HCC827 cells (Figure 3i). In contrast to overexpression of WT AXL, we found that expression of AXL L620M abrogated the ability of XL-880 (1 µM) to restore erlotinib sensitivity in HCC827 cells (Figure 3h–i). The data indicate that treatment with the AXL inhibitor XL880 restores erlotinib sensitivity by inhibiting AXL kinase activation in these EGFR mutant NSCLC models.
AXL-mediated resistance may occur with an EMT
We observed an association between AXL overexpression and markers of EMT, including increased expression of vimentin, in several models of acquired erlotinib resistance. Recent data indicate that vimentin can promote EMT via epigenetic regulation of genes that are critical for EMT in breast cancer cells.17 Thus we sought to determine whether vimentin overexpression was necessary for AXL overexpression and erlotinib resistance in HCC827 cells. We found that knockdown of vimentin by both pooled and 4 individual siRNAs decreased, but did not completely abolish, AXL expression (Figure 4a). Vimentin knockdown and concomitant downregulation of AXL enhanced erlotinib sensitivity in HCC827 ER3 cells (Figure 4b). Because vimentin has been shown to promote migration and adhesion in cells that have undergone an EMT17,22, we examined whether HCC827 ER cells exhibited increased migration and adhesion. We found that HCC827 cells with acquired erlotinib resistance (ER3) exhibit increased migration and adhesion compared to parental HCC827 cells (Figure 4c–d). We also found that knockdown of vimentin, AXL, or treatment with XL880 inhibited migration and adhesion in HCC827 ER3 cells but had no effect in parental HCC827 cells (Figure 4c–d). Together, the data support a possible role for an EMT that is marked by vimentin overexpression in the development of acquired EGFR TKI resistance that is driven by AXL in this model of human EGFR mutant lung cancer.
Figure 4. AXL-mediated erlotinib resistance occurs in association with EMT in EGFR-mutant NSCLC cellular models.
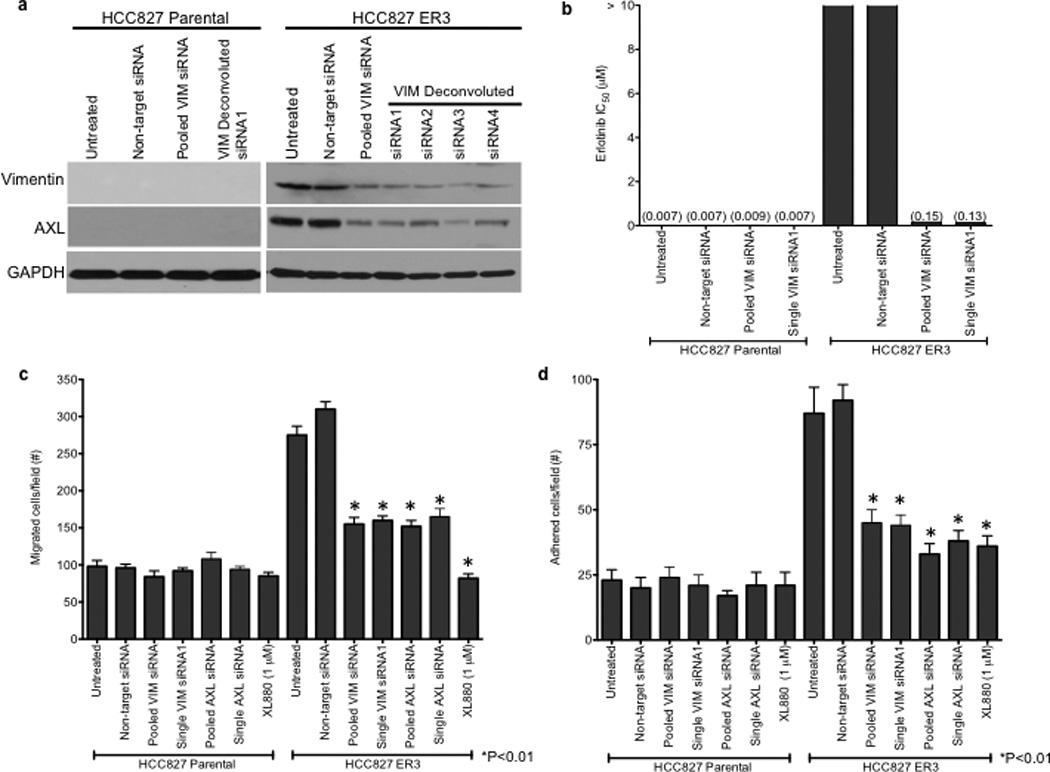
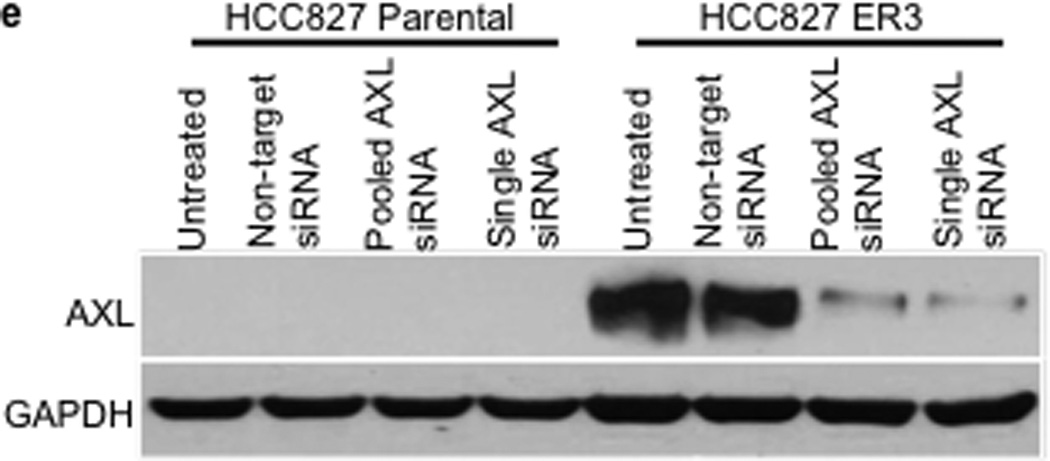
a, Effects of treatment with a non-targeting or the indicated vimentin siRNAs on AXL expression in HCC827 ER3 cells. b, Erlotinb IC50 in HCC827 parental or ER3 cells upon treatment with a non-targeting or the indicated vimentin siRNAs. Erlotinib IC50 is shown in parentheses. Data represent 3 independent experiments. c, Increased migration through a transwell chamber of ER3 compared to parental HCC827 cells in cells treated with a non-targeting or the indicated vimentin (VIM) or AXL siRNAs or XL-880 (1 µM); n=3, data expressed as mean ± SEM. d, Increased adherence to plastic of HCC827 ER3 cells compared to HCC827 parental cells in cells treated with a non-targeting or the indicated vimentin (VIM) or AXL siRNAs or XL-880 (1 µM). 5 100× microscopic fields per cell line were counted. n=3, data expressed as mean ± SEM. e, Western blot for the indicated proteins in lysates from cells transfected with the indicated siRNAs in (c–d).
AXL is upregulated in patients with acquired resistance
Based on the preclinical data, we hypothesized that upregulation of AXL may promote acquired resistance to EGFR TKI treatment in EGFR-mutant NSCLC patients. To test this hypothesis and to clinically validate our preclinical findings, we measured the expression of AXL by IHC (immunohistochemistry) in 35 matched EGFR-mutant NSCLC specimens obtained from patients both prior to treatment with the EGFR TKIs erlotinib or gefitinib and upon the development of EGFR TKI acquired resistance. In cases where enough material was available for additional studies, we also examined the specimens for GAS6 and vimentin (as a marker for EMT) expression by IHC (scoring system shown in Supplementary Figure 14), EGFR T790M by sequencing, and MET amplification by FISH. All patients in the cohort we analyzed had either an exon 19 deletion mutation or an exon 21 point mutation (L858R) in EGFR, were treated with either erlotinib or gefitinib and MET the established clinical definition of EGFR TKI acquired resistance (Table 2).23 The clinical sample set that we analyzed harbored the full spectrum of major alterations known to occur in EGFR mutant lung cancer patients with acquired EGFR TKI resistance.9,12,13,14 Indeed, we detected the EGFR T790M mutation in 8/28 (29%) and MET amplification in 6/31 (19%) of the resistant specimens examined (Tables 2–3 and Figure 5a, b). As compared to the pre-treatment specimen, we detected increased expression (an increase by 2+ or greater) of AXL in 7/35 (20%) and GAS6 in 7/28 (25%), and vimentin in 2/10 (20%) of the EGFR TKI resistant specimens evaluated (Tables 2–3 and Figure 5a–b). In 3 cases (#13,14,15) in which AXL was expressed at the same level in the baseline and matched resistance specimen we detected increased GAS6 specifically in the resistant specimen (Tables 2–3). This finding is consistent with the hypothesis that GAS6 overexpression may promote AXL activation in the setting of EGFR TKI acquired resistance in some cases. This hypothesis is supported by our preclinical data showing that knockdown of GAS6 can restore erlotinib sensitivity in some ER sublines that also overexpress AXL (Figure 3c). In order to provide independent validation of our IHC findings of increased expression of AXL and GAS6, we performed Q-RT-PCR on a small cohort of EGFR T790M mutation negative EGFR mutant lung cancer specimens with acquired EGFR TKI resistance compared to matched baseline specimens (n=5). Using stringent cut-offs (threshold >3 fold change in mRNA levels by Q RT PCR), we found increased AXL and GAS6 mRNA levels upon resistance in 20% and 60%, respectively, of these specimens (data not shown). We did not find increased AXL or GAS6 in the single specimen in which we detected increased MET expression (>3 fold change in mRNA by Q RT PCR) in this small cohort (data not shown).
Table 2. AXL is upregulated in human EGFR mutant NSCLC specimens from patients with acquired EGFR TKI resistance.
Clinical characteristics and expression of the indicated biomarkers in the 35 paired EGFR-mutant NSCLC specimens obtained from patients both prior to treatment and upon acquired resistance to treatment with either erlotinib or gefitinib (Median PFS= 18 months, range 7–59). NA= not enough tissue available for analysis. ACC= adenocarcinoma. NSC= non-small cell carcinoma. SqCC= squamous cell carcinoma. VIM = vimentin.
| ID | Age | Sex | Tumor type |
EGFR mutation |
TKI | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AXL | GAS6 |
EGFR T790M |
MET amp |
VIM | ||||||||
| Baseline | Resistance | Baseline | Resistance | |||||||||
| 1 | 49 | M | ACC | L858R | 0 | 3 | NA | NA | − | N | NA | gefitinib |
| 2 | 50 | M | ACC | Del19 | 0 | 3 | 0 | 3 | − | N | N | gefitinib |
| 3 | 80 | M | ACC | Del19 | 0 | 3 | 0 | 3 | + | N | N | gefitinib |
| 4 | 64 | F | ACC | L858R | 1 | 3 | NA | NA | − | N | N | gefitinib |
| 5 | 47 | F | NSC | Del19 | 0 | 2 | NA | NA | − | NA | N | gefitinib |
| 6 | 48 | F | ACC | L858R | 2 | 3 | 2 | 2 | − | N | N | gefitinib |
| 7 | 59 | F | NSC | Del19 | 0 | 3 | 1 | 0 | + | N | NA | erlotinib |
| 8 | 67 | F | NSC | Del19 | 0 | 1 | 0 | 0 | + | N | NA | erlotinib |
| 9 | 74 | M | ACC | Del19 | 1 | 3 | 0 | 3 | − | N | Y | erlotinib |
| 10 | 49 | F | ACC | L858R | 2 | 3 | 2 | 3 | − | N | N | gefinitib |
| 11 | 43 | M | NSC | Del19 | 2 | 3 | 0 | 3 | − | N | N | gefinitib |
| 12 | 56 | F | NSC | Del19 | 1 | 1 | 0 | 2 | − | N | N | erlotinib |
| 13 | 54 | F | ACC | Del19 | 3 | 3 | 0 | 3 | − | N | NA | gefitinib |
| 14 | 67 | M | ACC | Del19 | 3 | 3 | 0 | 2 | − | N | NA | gefitinib |
| 15 | 59 | M | ACC | Del19 | 3 | 3 | 0 | 1 | − | NA | Y | gefitinib |
| 16 | 50 | F | ACC | Del19 | 0 | 0 | NA | NA | NA | Y | NA | gefitinib |
| 17 | 76 | F | ACC | Del19 | 0 | 0 | 3 | 3 | NA | N | NA | gefitinib |
| 18 | 64 | F | SqCC | Del19 | 0 | 0 | 0 | 0 | + | Y | NA | gefitinib |
| 19 | 66 | F | ACC | L858R | 0 | 0 | 1 | 0 | − | N | NA | gefitinib |
| 20 | 54 | F | ACC | Del19 | 0 | 0 | 0 | 0 | − | N | NA | gefitinib |
| 21 | 67 | F | ACC | L858R | 0 | 0 | NA | NA | NA | NA | NA | gefitinib |
| 22 | 73 | F | ACC | L858R | 0 | 0 | 3 | 3 | − | N | NA | gefitinib |
| 23 | 58 | M | ACC | Del19 | 0 | 0 | NA | NA | + | Y | NA | gefitinib |
| 24 | 73 | F | ACC | L858R | 0 | 0 | 2 | 2 | + | Y | NA | gefitinib |
| 25 | 62 | F | ACC | Del19 | 0 | 0 | 3 | 3 | NA | Y | NA | gefitinib |
| 26 | 77 | M | ACC | L858R | 0 | 0 | NA | NA | NA | NA | NA | gefitinib |
| 27 | 72 | F | ACC | Del19 | 0 | 0 | 2 | 3 | − | N | NA | gefitinib |
| 28 | 51 | F | ACC | L858R | 0 | 0 | 0 | 0 | − | N | NA | gefitinib |
| 29 | 67 | M | ACC | L858R | 0 | 0 | 3 | 3 | + | N | NA | gefitinib |
| 30 | 63 | M | ACC | L858R | 0 | 0 | 3 | 0 | − | N | NA | gefitinib |
| 31 | 57 | F | ACC | Del19 | 0 | 0 | 0 | 0 | NA | N | NA | gefitinib |
| 32 | 71 | F | ACC | Del19 | 0 | 0 | 0 | 0 | NA | N | NA | gefitinib |
| 33 | 54 | F | ACC | Del19 | 0 | 0 | 0 | 0 | − | N | NA | gefitinib |
| 34 | 56 | F | NSC | Del19 | 0 | 0 | 1 | 0 | − | Y | NA | gefitinib |
| 35 | 54 | F | NSC | Del19 | 0 | 0 | 0 | 0 | + | N | NA | gefitinib |
Table 3.
Summary of resistance biomarker scoring in the paired specimens analyzed in Table 2.
| Marker | # positive |
% of evaluable |
ConcurrentT790M | Concurrent MET |
|---|---|---|---|---|
| AXL | (7/35) | 20 | 2 | 0 |
| GAS6 | (7/28) | 25 | 1 | 0 |
| T790M | (8/28) | 29 | - | 3 |
| MET | (6/31) | 19 | 3 | - |
| Vimentin | (2/10) | 20 | 0 | 0 |
Figure 5. AXL upregulation occurs in human EGFR-mutant NSCLCs with EGFR TKI acquired resistance.
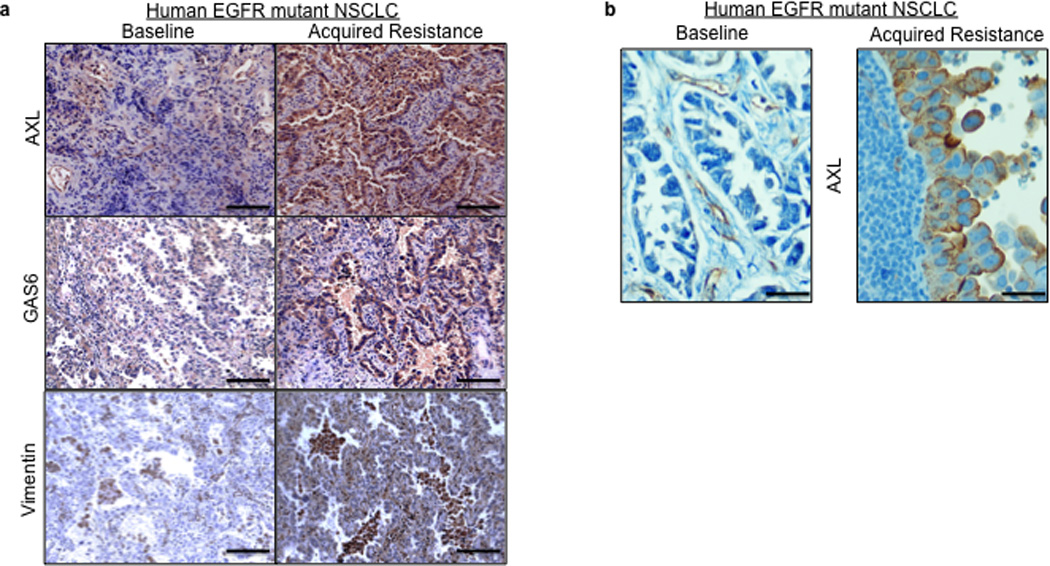
a–b, Representative expression of the indicated proteins by IHC in (a) case 7 and (b) case 9 shown in Table 2. IHC staining for AXL and GAS6 was scored as shown in Supplemental Figure 14. Vimentin IHC staining was scored using an established, clinically validated protocol26and EGFR T790M and MET amplification were assessed by sequencing and FISH, respectively, using established assays. Scale bars indicate in (a) 100 µM and (b) 10 µM.
Our analysis of the clinical specimens indicates that some EGFR mutant NSCLCs with EGFR TKI acquired resistance can harbor multiple mechanisms of resistance. Consistent with prior studies,13 we detected the EGFR T790M mutation in 50% of cases that also harbored MET amplification (Tables 2–3). Furthermore, we found co-occurrence of EGFR T790M and increased AXL and GAS6 in a minority of cases (Tables 2–3). In contrast, we did not detect MET amplification in any resistance specimen that had increased AXL or GAS6 expression (Tables 2–3). Coupled with the preclinical data, our findings suggest that activation of AXL and MET may be mutually exclusive mechanisms of acquired EGFR TKI resistance in NSCLCs.
Discussion
We have identified activation of the AXL kinase as a novel mechanism of acquired EGFR TKI resistance in EGFR-mutant NSCLCs through an integrated analysis of human EGFR-mutant NSCLC tumor models and one of the largest clinical cohorts of paired EGFR-mutant NSCLC specimens from EGFR TKI treated patients reported to date. Our analysis of the clinical specimens shows that AXL upregulation is the second most common mechanism of EGFR TKI acquired resistance (after EGFR T790M) in EGFR-mutant NSCLCs that has been validated using primary human data. The frequency of EGFR T790M in the EGFR TKI resistant samples in this cohort is lower than previously reported.9,12,13,14 Given that the sequencing assay we used to detect EGFR T790M is standard and validated, this likely represents either distinct biology of EGFR TKI resistance in the setting of AXL or GAS6 upregulation or that there may be greater variability in the frequency of EGFR T790M in EGFR TKI resistant lung cancers than previously described. Importantly, we found upregulation of AXL in patients that developed resistance to both EGFR TKIs (erlotinib and gefitinib) that are clinically approved for use in NSCLC patients worldwide. Our findings are thus relevant to the vast majority of EGFR-mutant NSCLC patients treated with an EGFR TKI.
The data suggest that activation of AXL can occur through its overexpression as well as through upregulation of its ligand GAS6 in the setting of EGFR TKI resistance in EGFR-mutant NSCLCs. This observation is consistent with recent work showing that both upregulation of MET and its ligand HGF can promote EGFR TKI resistance in some EGFR-mutant NSCLCs.10 Our data showing that in some ER cell lines GAS6 is not required for erlotinib resistance are consistent with earlier work demonstrating that AXL overexpression can promote downstream signaling and induce transformation in the absence of GAS6 expression.24 Further work will be necessary to fully elucidate the mechanisms by which GAS6 might promote EGFR TKI resistance through activation of AXL and whether somatic alterations (amplifications, rearrangements, point mutations) in AXL or GAS6 occur in human EGFR-mutant NSCLCs.
We found that in some cases AXL upregulation occurred in the context of an apparent EMT and that an EMT-associated transcriptional program involving upregulation of vimentin may, in part, drive AXL overexpression in EGFR mutant lung cancer cells with acquired EGFR TKI resistance. The data are consistent with prior studies in which vimentin upregulation was associated with AXL overexpression in breast cancer cells.17 Although other mechanisms of AXL activation likely exist and will need to be investigated, our data are consistent with a model in which AXL may mediate acquired EGFR TKI resistance in the setting of an EMT in EGFR-mutant NSCLCs. AXL-overexpressing HCC827 ER cells exhibited increased migration and adhesion, properties associated with EMT and the METastatic behavior of tumor cells. These findings in EGFR-mutant NSCLC cells with erlotinib acquired resistance are consistent with prior work showing that overexpression of AXL is associated with increased METastasis and worse prognosis in several cancers.15 Yet our studies uncover a distinct and specific role for AXL upregulation in acquired resistance to EGFR TKI treatment in EGFR-mutant NSCLC patients. Our data therefore extend current knowledge of the role of AXL as a general prognostic biomarker in human cancer and demonstrate for the first time that AXL is a biomarker of acquired resistance to EGFR-targeted therapy in NSCLC patients.
Our observation that multiple mechanisms of resistance may contribute to EGFR TKI treatment resistance in EGFR mutant NSCLC patients is consistent with other recent studies.13,14 Our data identifying AXL as a mechanism of EGFR TKI resistance enhance our understanding of tumor heterogeneity and the molecular mechanisms governing the evolution of resistance to molecularly targeted therapy in cancer patients. Further work will be necessary to determine the degree to which AXL activation may cooperate with other genetic and genomic alterations to induce resistance to EGFR inhibitors and other molecularly targeted therapies in lung and other cancers.
Our data show that the kinase activity of AXL is required for erlotinib resistance in EGFR-mutant NSCLC tumor models. This observation provides strong rationale for the development and testing of AXL kinase inhibitors for clinical use in EGFR-mutant NSCLC patients to either prevent or overcome EGFR TKI acquired resistance. The preclinical data also suggest that activation of multiple pathways including MAPK, AKT and NF-kB may promote EGFR TKI resistance downstream of AXL upregulation. This is consistent with prior work showing that AXL can drive the growth of cancer cells through activation of each of these pathways.15,16,25 Further work will be necessary to determine which of these or other signaling pathways is most critical for AXL-driven EGFR TKI resistance in EGFR-mutant NSCLCs. Such studies may identify additional points for therapeutic intervention in the setting of EGFR TKI acquired resistance driven by AXL in EGFR-mutant NSCLC patients. Based on our data, we propose that inhibition of AXL signaling may enhance responses to EGFR TKI treatment in appropriately selected EGFR-mutant NSCLC patients.
Online Methods
Cell lines and reagents
The human lung cancer cell lines were purchased from the American Type Culture Collection except for H3255, PC-9 cells which were generously provided by Susumu Kobayashi (Beth Israel Deaconess Medical Center, Boston, MA). Cells were grown in RPMI 1640 supplemented with 10% FBS and 1× Antibiotic/Antimycotic (Invitrogen) and were in the logarithmic growth phase at the initiation of all experiments. Erlotinib, XL880 (GSK1363089), PHA665752 and MP-470 were purchased from Selleck Chemicals (California, USA). Drugs were dissolved in DMSO at 10 mM and stored at −20°C. The final DMSO concentration in all experiments was <0.1% in medium. All antibodies were purchased from Cell Signaling (Boston, MA) except the anti-AXL and anti-phospho-AXL and anti-GAS6 antibodies, which were purchased from R&D Systems (Minneapolis, MN).
Establishment of erlotinib-resistant subclones
Cells were exposed to increasing concentrations of erlotinib every 3 weeks from 1, 3, 5, 7, and so on until 15 µM over a 5 months period. Single-cell cloning was performed by the use of cloning cylinders and erlotinib-resistant subclones were successfully expanded with 10% fetal bovine serum culture medium containing 1 µM erlotinib. The genetic identity of each subclone with the parental cells was verified by STR (short tandem repeat) analysis conducted at Johns Hopkins University (Baltimore, MD, USA) or the Korean Cell Line Bank (http://cellbank.snu.ac.kr) according to established protocols.
Cell growth and viability assays
Cells were seeded at a density of 3000 cells/well in 96-well plates in RPMI 1640 containing 10% FBS overnight, then treated with respective agents for an additional 3 days. Viable cell numbers were determined using CellTiterGLO or MTS assay kit, as indicated in the Figure legends, according to the manufacturer's protocols (Promega, Madison, WI, USA). Each assay consisted of 3 replicate wells and was repeated at least twice. Data were expressed as the percentage survival of control calculated from the absorbance, corrected for background.
Western blot analysis
Cells were serum starved overnight and whole cell lysates were prepared using 10% TCA lysis or RIPA buffer and clarified by centrifugation. Proteins were separated by 10% SDS-PAGE gel and transferred onto PVDF membranes (Invitrogen) for Western blot analysis. After primary antibody incubation overnight, washing and incubation with secondary antibodies, blots were developed with a chemiluminescence system (Pierce).
Gene expression profiling
Gene expression microarray profiling was performed in triplicate following RNA isolation from cells or xenograft tumors using the Qiagen RNeasy kit according to the manufacturer’s instructions using either AffyMETrix U1333 2.0 plus arrays (ER1, ER2) and analyzed as described previously27,25 or Illumina Human HT12- v3 arrays (ER3) and analyzed by Illumina BeadStudio Gene Expression Module v3.2. Bioinformatics analysis was performed and genes were filtered to include genes with differential expression based on setting a threshold of 3 fold change and a false discovery rate (FDR) threshold at 0.1 for the comparison of gene expression in the ER cell lines to the parental (vehicle treated) cells.
Quantitative real time RT–PCR
For the validation of genes identified by gene expression profiling, quantitative real-time RT–PCR was performed on RNA isolated from NSCLC cells. Total RNA was collected from cultured cells using PureLink Micro-to-Midi Total RNA Purification kit (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized with SuperScript III reverse transcriptase with the use of oligo(dT) primers (Invitrogen) and RT-PCR was performed by using LightCycler with Syber green probes (Roche) using the following variables: denaturation at 95°C for 10 min, followed by 45 cycles of amplification(95°C 10 s, 60°C 10 s, and 72°C 15 s), and cooling to 40°C at a transition rate of 20°C/s. Levels of glyceraldehyde-3-phosphate dehydrogenase or actin expression were used as internal reference to normalize input cDNA. Ratios of level of each gene to as compared to reference standard were then calculated. Sequences for the primers used for AXL and GAS6 Q RT-PCR are shown in Supplementary Table 4. Primers and probes used for EGFR and MET sequencing and FISH, respectively, were as previously published.13,28
siRNA knockdown
Knockdown of AXL, MET and vimentin was performed using specific single or pooled siRNAs, as indicated, targeting the indicated genes purchased from Dharmacon RNAi Technologies (Thermo Scientific, Rockford, IL). SiGENOME non-targeting siRNAs served as negative controls. Introduction of siRNA was performed with DharmaFect1 reagent according to the manufacturer’s instructions. Efficiency of knockdown at different times or dose points was assessed by Q RT PCR or western blotting on cell lysates.
AXL cDNA and overexpression studies
The AXL gene with an HA tag at the C-terminus was amplified by overlapping PCR using cDNA generated from ER3 cells and cloned into pcDNA3.1 (+) vector. Amino acid changes (K567R or L620M) were introduced using the Strategene QuickChange Mutagenesis Kit, according to the manufacturer’s protocol. The accuracy of the constructs was confirmed by DNA sequencing. Cells were transfected with the AXL-expressing vectors or empty vector as a control by using FuGENE HD reagent according to the manufacturer's protocol (Roche Applied Sciences, Indianapolis, IN, USA). Stable HCC827 subclones resistant to the selection antibiotic, 500µg/ml G418 were generated 24 hours post-transfection. Empty-vector expressing HCC827 cells were generated as a control. AXL protein expression was detected by using anti-HA probe (Santa Cruz Biotechnology).
Migration and adhesion studies
180,000 cells were plated onto 3.5 cm cell culture dishes. At 30 minutes, the dishes were washed and cells were fixed and stained for counting. Average cell numbers of 5 random 100× fields under light microscope are presented with ± SEM. Migration: 10,000 cells in 200 µl medium without FBS were placed in the upper chamber of transwells(6.5 mm diameter, 8 µm pore size polycarbonate membrane, Corning), and the lower chamber filled with 1 ml of medium with 10% FBS supplemented with or without the indicated drug treatments. After incubation for 16 hours, non-migrating cells were removed with cotton swabs, and the cells that migrated into the lower surface of the filters were stained with crystal violet. Cells were counted under a microscope, and triplicate results are expressed as means ± SEM.
shRNA knockdown
For the shRNA experiments each retroviral and lentiviral shRNA was packaged in 293FT cells according to the manufacturer’s instructions. Indicated cell lines were spin infected with 1 ug/ml polybrene and 48h after infection were selected with 2 µg/ml of puromycin for 48h. Target gene expression was measured by either Q RT PCR or western blot 48–72h later.
Tumorigenesis assays
Human lung cancer cells (as indicated) were injected subcutaneously into the flanks of CB17 SCID mice (Taconic). Tumor-bearing mice (tumor size 200–500 mm3) were randomized to treatment with vehicle (DMSO/CMC) or erlotinib HCL 12.5 mg/kg/day intraperitoneally. Tumor measurements were made on days indicated and expressed as described in the Figure legends. All animal experiments were performed in compliance with the guidelines of the Research Animal Resource Center of the Memorial Sloan-Kettering Cancer Center.
Combination index (CI) analysis
The combination effects were evaluated with the MTT assay at a 1:1 ratio (erlotinib (µM):XL880 (µM) and erlotinib (µM):PHA665752 (µM)) in HCC827ER cells. The fraction affected (Fa) (i.e., Fa of 0.25 is equivalent to 75% viable cells) and CI values processed using CalcuSyn software (Biosoft, Cambridge, UK).CI values of less than 1, 1, and greater than 1 were taken as synergism, additive effect, and antagonism, respectively.
Immunoprecipitation
For immunoprecipitation, 1 mg protein from total lysates was mixed with 1 µg of anti-AXL (Santa Cruz, CA), and incubated on ice for 2 h. Protein G Sepharose Fast Flow (GE Healthcare, Little Chalfont, Buckinghamshire, UK) was added to the mixture and incubated overnight at 4°. Immune complexes were pelleted by centrifugation at 3000 rpm for 5 min, washed twice with lysis buffer, and resuspended in 70 µl of SDS-PAGE sample buffer. Subsequently, immune complexes were probed with anti-AXL and phosphotyrosine antibodies(Santa Cruz, CA).
Sequencing of AXL, EGFR, K-Ras and B-Raf
Sequencing of full length of AXL and EGFR was performed by standard Methods using the primer sequences listed Supplementary Table 5. Sequencing of exon 2 of K-Ras and exons 11 and 15 of BRAF was performed on genomic DNA using validated primers and protocols.13
IHC on NSCLC clinical specimens
All specimens were acquired from patients under the auspices of IRB-approved clinical protocols at each hospital in which informed consent was obtained and were formalin-fixed, paraffin-embedded tumour tissues (FFPE), that were examined to ensure >75% tumor infiltration, and stained with haematoxilin/eosin and assessed by a thoracic pathologist at MSKCC (New York, NY, USA), As an Medical Center (Seoul, South Korea), USP Dexeus University Institute (Barcelona, Spain). IHC was performed in the Core Research IHC Laboratory at each institution on FFPE sections, as previously described29, using the indicated antibodies: Human AXL antigen - affinity-purified polyclonal antibody, R&D systems, catalog #AF154AXL ; Human GAS6 - affinity-purified polyclonal antibody, R&D systems, catalog #AB885, Human vimentin - affinity-purified polyclonal antibody, Cell Signaling Technologies, catalog #3932. Expression was examined by standard immunohistochemistry using 4um thick sections of matched, paired specimens. Deparaffinized tissue sections were stained following the manufacturer’s protocol at a dilution of 1:500 on a Ventana Dixcovery XT automated stainer. EGFR T790M mutation and MET amplification were analyzed using previously described sequencing and FISH protocols,13 respectively, and vimentin expression by IHC using established MET hods and clinical scoring criteria.26
Supplementary Material
Acknowledgments
Acknowledgements/Funding. The authors thank Dr. KevanShokat (UCSF) for assistance with structural modeling of the AXL gatekeeper mutation and Dr. William Polkinghorn, John Wongvipat and Elton Chan for advice and technical assistance. This work was supported by Korean Health Technology R&D Project Grant A102059 (to J.C.L), NIH K08 1K08CA154787, a Uniting Against Lung Cancer Research Award, and a National Lung Cancer Partnership Young Investigator Award (to T.G.B.), a grant from the La Caixa Foundation(to R.R.), an American Cancer Society Research Scholar Grant RSG-08-303-01 (to B.H.), and by NIH P01 CA129243 (to M.L.).
Footnotes
Author Contributions. ZZ (performed in vitro experiments), JCL(performed in vitro experiments and analysis of clinical specimens), LL (performed in vitro experiments), VO (analysis of clinical specimens), VA (performed in vitro experiments), TL (performed in vitro experiments), MAR (performed in vitro experiments), XW (performed in vitro experiments), ADL (performed in vitro experiments), JKR (performed in vitro experiments), YJC (performed in vitro experiments), CMC (performed in vitro experiments), YSL (performed in vitro experiments), SWK (performed in vitro experiments), DHL (performed in vitro experiments), JSL (performed in vitro experiments), SJJ (analyzed clinical specimens), YSP (analyzed clinical specimens), MA (analyzed clinical data), CC (analyzed clinical specimens), PM (performed in vitro and in vivo experiments), TGB (performed in vitro and in vivo experiments and analyzed clinical data). ML, TB, VM, CS, PM, MT, RR, BH, TGB each analyzed in vitro and in vivo data. BH, TGB wrote the paper.
Competing Financial Interests. The authors declare no competing financial interests.
References
- 1.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 2.Pao W, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 4.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. d. [DOI] [PubMed] [Google Scholar]
- 5.Janne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discov. 2009;8:709–723. doi: 10.1038/nrd2871. [DOI] [PubMed] [Google Scholar]
- 6.Gazdar AF. Personalized medicine and inhibition of EGFR signaling in lung cancer. The New England journal of medicine. 2009;361:1018–1020. doi: 10.1056/NEJMe0905763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 8.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelman JA, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 10.Turke AB, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bivona TG, et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–526. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arcila ME, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:1169–1180. doi: 10.1158/1078-0432.CCR-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bean J, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sequist LV, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3002003. 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linger RM, Keating AK, Earp HS, Graham DK. Taking aim at Mer and AXL receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin Ther Targets. 2010;14:1073–1090. doi: 10.1517/14728222.2010.515980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keating AK, et al. Inhibition of Mer and AXL receptor tyrosine kinases in astrocytoma cells leads to increased apoptosis and improved chemosensitivity. Mol Cancer Ther. 2010;9:1298–1307. doi: 10.1158/1535-7163.MCT-09-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vuoriluoto K, et al. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing AXL expression in breast cancer. Oncogene. 2011;30:1436–1448. doi: 10.1038/onc.2010.509. [DOI] [PubMed] [Google Scholar]
- 18.Suda K, et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6:1152–1161. doi: 10.1097/JTO.0b013e318216ee52. [DOI] [PubMed] [Google Scholar]
- 19.Ye X, et al. An anti-AXL monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene. 2010;29:5254–5264. doi: 10.1038/onc.2010.268. [DOI] [PubMed] [Google Scholar]
- 20.Qian F, et al. Inhibition of tumor cell growth, invasion, and MET astasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer research. 2009;69:8009–8016. doi: 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- 21.Yun CH, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature reviews. Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 23.Jackman D, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:357–360. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burchert A, Attar EC, McCloskey P, Fridell YW, Liu ET. Determinants for transformation induced by the AXL receptor tyrosine kinase. Oncogene. 1998;16:3177–3187. doi: 10.1038/sj.onc.1201865. [DOI] [PubMed] [Google Scholar]
- 25.Tai KY, Shieh YS, Lee CS, Shiah SG, Wu CW. AXL promotes cell invasion by inducing MMP-9 activity through activation of NF-kappaB and Brg-1. Oncogene. 2008;27:4044–4055. doi: 10.1038/onc.2008.57. [DOI] [PubMed] [Google Scholar]
- 26.Azumi N, Battifora H. The distribution of vimentin and keratin in epithelial and nonepithelial neoplasms. A comprehensive immunohistochemical study on formalin- and alcohol-fixed tumors. American journal of clinical pathology. 1987;88:286–296. doi: 10.1093/ajcp/88.3.286. [DOI] [PubMed] [Google Scholar]
- 27.Chitale D, et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;28:2773–2783. doi: 10.1038/onc.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosell R, Wei J, Taron M. Circulating MicroRNA Signatures of Tumor-Derived Exosomes for Early Diagnosis of Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2009;10:8–9. doi: 10.3816/CLC.2009.n.001. [DOI] [PubMed] [Google Scholar]
- 29.Brevet M, Arcila M, Ladanyi M. Assessment of EGFR mutation status in lung adenocarcinoma by immunohistochemistry using antibodies specific to the two major forms of mutant EGFR. The Journal of molecular diagnostics : JMD. 2010;12:169–176. doi: 10.2353/jmoldx.2010.090140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.