

SUMMARY
Non cell-autonomous processes are thought to play critical roles in the cellular maintenance of the healthy and diseased brain but mechanistic details remain unclear. We report that the interruption of a non-cell autonomous mode of sonic hedgehog (Shh) signaling originating from dopaminergic neurons causes progressive, adult-onset degeneration of dopaminergic, cholinergic, and fast spiking GABAergic neurons of the mesostriatal circuit, imbalance of cholinergic and dopaminergic neurotransmission, and motor deficits reminiscent of Parkinson’s disease. Variable Shh signaling results in graded inhibition of muscarinic auto-receptor- and GDNF- expression in the striatum. Reciprocally, graded signals that emanate from striatal cholinergic neurons and engage the canonical GDNF receptor Ret inhibit Shh expression in dopaminergic neurons. Thus, we discovered a novel mechanism for neuronal subtype specific and reciprocal communication that is essential for neurochemical and structural homeostasis in the nigrostriatal circuit. These results provide integrative insights into non cell-autonomous processes likely at play in neurodegenerative conditions such as Parkinson’s disease.
INTRODUCTION
Neuronal computations in the basal ganglia rely on correlated changes in the activity of mecenphalic dopamine (DA) neurons and striatal acetylcholine (ACh) - and fast-spiking (FS) GABAergic neurons, which result from reciprocal pre- and postsynaptic interactions (Threlfell et al., 2010). The concerted activity of DA neurons and ACh and FS interneurons gate glutamatergic input from the cerebral cortex and thalamus onto medium spiny projection neurons (MSN) allowing the translation of thought into action (Figure 1) (Bolam et al., 2006). These neuronal subtypes form cartridges of a repetitive mesostriatal circuit in which each of the numerous MSN contributes to only few units, but each of the much fewer DA, ACh and FS neuron participates in several 100 units (Bolam et al., 2006). The phylogenetic conservation of circuit architecture (Reiner, 2010) suggests that the relative proportions of the constituent neurons of the mesostriatal circuit are important for proper circuit function. This view is supported by the pathophysiological finding that chorea, parkinsonism, and tics are associated with a loss of specific mesostriatal constituent neuronal subtypes such as MSN, DA and FS neurons, respectively (DeLong and Wichmann, 2009).
Figure 1. Mesostriatal Circuitry.
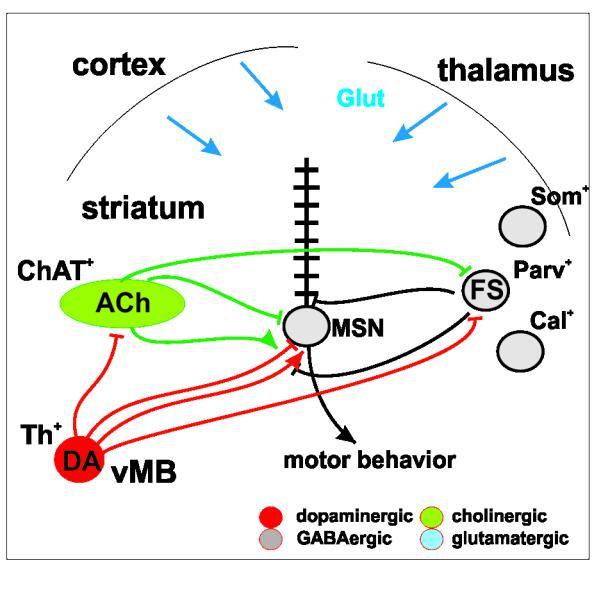
Striatal interneurons and MSNs receive dopaminergic projections. ACh and FS interneurons project to striatal neuronal subtypes and dopaminergic projections. (Glut: glutamine; GABA: γ-aminobutyric acid; ACh: acetylcholine; DA: dopamine; ChAT: Choline-Acetyl Transferase; Th: Tyrosine Hydroxylase; FS: “fast spiking”; Parv: Parvalbumin; Som: Somatostatin; Cal: Calretinin; vMB: ventral midbrain; MSN: medium spiny projection neurons.)
The mechanisms maintaining cellular and neurochemical homeostasis in the mature mesostriatal system in the healthy brain are not fully elucidated, but signaling by neurotrophic factors has emerged as a likely process. For example, the glial cell line-derived neurotophic factor (GDNF) protects catecholaminergic neurons from toxic insults, induces fiber outgrowth and is required for catecholaminergic neuron survival in the adult brain (Lin et al., 1993; Pascual et al., 2008). GDNF signaling can also act as a neuromodulator of dopaminergic signaling through the regulation of the quantal size of DA release (Pothos et al., 1998; Wang et al., 2001)). Despite the implication of GDNF in DA neuron maintenance and function, which motivated several clinical trials of GDNF-based therapies in Parkinson’s disease (PD) (Rangasamy et al., 2010), the regulation of GDNF expression in the healthy adult brain remains ill-defined.
Circumstantial evidence also implicates the secreted glycoprotein Sonic Hedghog (Shh), in the maintenance of the mesostriatal circuit since exogenously supplied Shh to the adult striatum increases the resilience of mesencephalic DA neurons to MPTP and 6-OHDA, neurotoxins used to model PD related DA neuron loss in animals (Dass et al., 2005; Tsuboi and Shults, 2002). In these paradigms, the neuroprotective effect of Shh on mesencephalic DA neurons is comparable to that observed with striatal delivery of GDNF (Dass et al., 2002). In vitro, Shh synergizes with neural growth factor (NGF) in providing trophic support to basal forebrain-derived, post-natal ACh neurons (Reilly et al., 2002). Despite these observations, it is not clear whether there is a functionally relevant source of Shh that could act in the mature mesostriatal system and if so, which cell types would communicate by Shh signaling.
Here, we present evidence for reciprocal, trophic factor signaling between mesencephalic DA- and striatal ACh- and FS- neurons. We show that DA neurons utilize Shh to signal to ACh and FS interneurons in the striatum where it regulates extracellular ACh tone, expression of GDNF, and maintenance of these neurons. Conversely, Shh expression by DA neurons is repressed by signals which originate from ACh neurons and engage the canonical GDNF receptor Ret on DA neurons. The conditional ablation of Shh from DA neurons results in a progressive model of PD with face, construct and predictive validity. Thus, our results shed light into new aspects of the chemical neuroanatomy of the basal ganglia which may have far-reaching implications for the understanding of the physiopathology and the treatment of movement disorders such as PD.
RESULTS
Shh expression by DA neurons is necessary for the long-term maintenance of mesencephalic DA neurons
To examine whether Shh-mediated signaling occurs among neurons of the mesostriatal circuit, we first visualized expression of Shh in the adult brain using mice heterozygous for a conditional, gene expression tracer allele of Shh (Shh-nLZC/+; Figure S1A). We observed Shh expression by all tyrosine hydroxylase-positive (Th+) neurons in the substantia nigra pars compacta (SNpc; Figures 2A, B), the ventral tegmental area (VTA, Figure 2A), and the retro rubral field (RRF, data not shown) along the entire rostro-caudal axis of these nuclei at 3 months of age (100 ± 0%, 683 cells, n=2). We did not observe Shh expression by Th+ neurons of the diencephalon or olfactory bulb or by cells in the striatum (data not shown).
Figure 2. Ablation of Shh from DA neurons results in non-cell autonomous DA neuron degeneration.

(A) X-Gal labeled coronal section of the vMB of a Shh-nLZC/+ mouse.
(B) 100 +/− 0 % of Th+ dopaminergic neurons in the mesencephalon express Shh at p90 (for all quantitations herein, data are represented as mean +/− s.e.m.; 683 cells, 2 mice).
(C) Th staining of VTA and SNpc is inconspicuous at one month but diminished at 12 months of age in Shh-nLZC/C/Dat-Cre mice.
(D) Progressive reduction in the numbers of Th+ neurons in the SNpc in Shh-nLZC/C/Dat-Cre mice. (*p < 0.05, unpaired t-test; n=7-8/genotype/age, 12 sections per subject with a 5 section interval).
(E) Progressive reduction in the numbers of Th+ neurons in the VTA in Shh-nLZC/C/Dat-Cre mice. (*p < 0.05, unpaired t-test (n=5/genotype/age, 12 sections per subject with a 5 section interval).
(F) Striatal Th+ nerve fiber density in Shh-nLZC/C/Dat-Cre mice (*p < 0.05 unpaired t-test, n = 10-11/group).
(G) Alterations in gene expression in the vMB of 4 weeks and 12 months old Shh-nLZC/C/Dat-Cre mice. (For all quantitative gene expression analysis herein data is expressed as relative fold change of experimental over control conditions with down-regulation shown as red bars and up-regulation shown as green bars;* p < 0.01, ** p < 0.001 two tailed t-test, n=5/genotype).
(H) Th staining of VTA and SNpc is inconspicuous at 18 months of age in SmoC/C/Dat-Cre mice.
(I) Comparative quantification of DA and non-DA neurons in the SNpc and VTA of 12 and 18 months old Shh-nLZC/C/Dat-Cre and SmoC/C/Dat-Cre mice resp. (*p < 0.05 unpaired t-test; n = 6-7/group).
To determine whether Shh signaling within the mesostriatal circuit is of physiological relevance, we selectively ablated Shh expression from DA neurons mediated by Cre activity expressed from the DA transporter locus (Dat-Cre; all mouse strains used in this study are referenced in extended experimental procedures). Shh-nLZC/C/Dat-Cre mutant animals were born alive and mobile with expected Mendelian frequency and no overt structural or motor signs at the end of postnatal development compared to Shh-nLZC/+/Dat-Cre control littermates (Figures S1 and S2, Table ST 1; Supplemental results A, B; for all comparative analyses herein littermates double heterozygous for Shh-nLZC/+and Dat-Cre served as controls).
Unbiased sterological cell counting of Th+ and Th− neurons in the SNpc and VTA revealed an adult-onset, progressive degeneration of DA neurons that plateaus at ~40% in Shh-nLZC/C/Dat-Cre mice compared to controls at 8 months of age (Figure 2C-F, I). No change in the number of glutamate dehydrogenase (GAD67) positive cells was observed in the SN pars reticulata of Shh-nLZC/C/Dat-Cre mice at 16 months of age (Figure S3A). Striatal Th+-fiber density was normal at 1 month of age, increased at 8 months and decreased at 12 months of age in Shh-nLZC/C/Dat-Cre mice compared to controls (Figure 2F). Gene expression analysis of DA markers in the ventral midbrain (vMB) revealed a down-regulation of Th, Dat, and DA receptor-2 (DaR2) at 5 weeks of age, which then returned to normal levels by 12 months in Shh-nLZC/C/Dat-Cre compared to controls (Figure 2G; all genes probed herein are listed in supplemental information Table ST2). The expression of the vesicular monoamine transporter-2 (vMat2) appeared normal at 5 weeks, but was diminished at 12 months. The activator of endoplasmic reticulum stress, Xbp1 and of the anti-oxidant enzyme, glutathione-peroxidase-1 (Gpx1) were up-regulated in Shh-nLZC/C/Dat-Cre animals at 5 weeks but not at 12 months of age (Figure 2G) indicative of the activation of physiological cell stress responses in the vMB in the absence of Shh expression in young adult mutant mice. In further support of a protracted dopaminergic cell syndrome in which neuronal degeneration is only the final step, we found progressive alterations in somato-dendritic and striatal DA content and deficits in amphetamine elicited DA release in Shh-nLZC/C/Dat-Cre mice (supplemental results C and Figures S3B – D).
Is the observed dopaminergic phenotype a cell autonomous effect of the interruption of Shh signaling? Shh can bind to several co-receptors which in turn facilitate the relief of repression of the serpentine transmembrane protein Smo by Ptc1 or Ptc2 (Izzi et al., 2011))To distinguish autocrine from paracrine Shh signaling, we analyzed the expression of the Shh co-receptors Ptc1 and Ptc2 and the phenotype of animals with a tissue restricted ablation of Smo from DA neurons, which we produced using the same Dat-Cre allele with which we also achieved the tissue specific ablation of Shh. We did not find evidence for the expression of Ptc1 in DA neurons utilizing a gene expression tracer mouse line (Ptc1-nLZ) or Ptc2 by in situ hybridization, consistent with public gene expression data information (Gensat, http://www.gensat.org.; data not shown). SmoC/C/Dat-Cre mutant animals were born alive and mobile with expected Mendelian frequency and no overt structural or motor signs through adulthood compared to SmoC/+/Dat-Cre control littermates (data not shown). Unbiased sterological cell counting of Th+ and Th− neurons in the SNpc and VTA of 18 months old SmoC/C/Dat-Cre mutants and SmoC/+/Dat-Cre littermate controls did not reveal DA neuron loss in the SNpc or VTA (Figure 2H, I). Collectively, the analyses of the vMB demonstrates a progressive structural and functional deterioration of the mesencephalic DA system in the absence of DA neuron produced Shh, whose cardinal cellular and functional aspects results from a non-cell autonomous mode of Shh signaling.
DA neuron expressed Shh acts as a DA neuron protectant
Dat-Cre mediated ablation of the conditional Shh allele is ~80 % complete in the SNpc at 2 months of age creating an experimentally induced heterogeneity among DA neurons (Figure S2A). We therefore explored whether the progressive nature of the phenotype might be caused by slowly continuous Cre-mediated deletion of Shh alleles over time in adult Shh-nLZC/C/Dat-Cre mutant animals. In contrast to this prediction, however, we observed an increase in the relative numbers of Shh expressing DA neurons among all DA neurons of the SNpc from ~20% at 2 months of age to ~37% at 12 months of age in Shh-nLZC/C/Dat-Cre mutants (Figure 3A, B). We also observed that the soma size of Shh expressing DA neurons was larger than of DA neurons that lost Shh expression in Shh-nLZC/C/Dat-Cre mice at 12 months of age (Figure 3: C). These observations demonstrate that: (1) accumulation of Shh null alleles by continuous Cre activity is insignificant after 8 weeks of age, and (2) Shh expressing DA neurons have a selective survival advantage over DA neurons in which Cre mediated Shh ablation occurred. Thus, these results provide genetic evidence that Shh signaling originating specifically from DA neurons confers a competitive survival advantage among mesencephalic DA neurons in vivo.
Figure 3. Shh expression confers a survival advantage to dopaminergic neurons.

(A) Shh expressing DA neurons in Shh-nLZC/C; Dat-Cre mice at 12 months of age.
(B) Incidence of Shh expression in the SNpc in Shh-nLZC/C; Dat-Cre mice at 1 and 12 months of age (*p < 0.05, unpaired t-test, n=12/group, 100 cells/subject).
(C) Somatic area of Th+, Shh− - and Th+, Shh+ cells in the SNpc of Shh-nLZC/C; Dat-Cre mice (*p < 0.05, single comparison by Mann-Whitney U test; box-whisker plots for median ± 95% CI bar, 25-75 percentiles box, n=4, 20 cells/subject).
Dopamine substitution and cholinergic antagonist treatment normalize progressive gait and locomotion disturbances in Shh-nLZC/C/Dat-Cre mice
Next, we assessed the functional significance of the progressive structural and neurochemical alterations of the mesencephalic DA system in Shh-nLZC/C/Dat-Cre mice by longitudinal analyses of elicited and spontaneous motor performance.
Analysis of horizontal activity profiles of freely locomoting mice in an open field let us define successive phases of progressive changes in locomotion of Shh-nLZC/C/Dat-Cre mice compared to control litter mates: Locomotion activity was normal up to 6 weeks (phase I), reduced by ~35% at 2-5 months (phase II), increased by ~60% at 7-12 months (phase III), inconspicuous at 16 months (phase IV), but then rapidly deteriorating leading first to pelvic dragging, followed by partial hind limb paralysis and premature death at about 18 months (phase V; Figure 4A). The switch from relative hypo-activity to relative hyper-activity of Shh-nLZC/C/Dat-Cre mice compared to control littermates manifested with high temporal specificity around 6 months of age (Figure 4B). The inversion in relative locomotion activity coincided with a switch from a 3-fold reduction to a 30 % increase in striatal DA content (Figure S3C, supplemental results C) and a relative increase in the frequency of bouts of locomotion from phase II to phase III in Shh-nLZC/C/Dat-Cre mice compared to controls (data not shown).
Figure 4. Abnormalities in locomotion and gait dynamics in the absence of Shh expression by DA neurons.

(A) Horizontal activity in an “Open Field” as a function of age (*p < 0.05, repeated measures ANOVA follow by Tukey’s post hoc test; n = 10/group).
(B) High temporal predictability of transition from phase II to phase III (n=83-100; 23 litters; p<0.000001, phase x genotype ANOVA)
(C) Increase in stride length variability (CV), reduction in brake/stride ratio, and increase in absolute paw angle during phase III in Shh-nLZC/C/Dat-Cre (n=5; p<0.01; ANOVA followed by Tukey’s test).
(D) Normalization of stride length CV, braking time and paw angle by L-DOPA (sc., 20 mg/kg) and/or THP (sc., 3 mg/kg; n=8/genotype; *p < 0.05, genotype x time ANOVA for repeated measures followed by Tukey’s post-hoc test.
We next investigated gait dynamics by ventral plane videography of Shh-nLZC/C/Dat-Cre mice and controls of 3 to 16 months of age walking on a translucent treadmill. During phase III, the gait length coefficient of variability was increased ~30% at 10 months of age, and the time allotted for braking in each stride was shortened by 40% and the paw angle was increased by 50% at 11 months of age of Shh-nLZC/C/Dat-Cre mice relative to controls (Figure 4C; and supplemental results D; see supplemental Table ST1 for all indices of gait analyzed). We then tested whether drugs efficacious in the management of PD, would modify the locomotion abnormalities of Shh-nLZC/C/Dat-Cre mice. We systemically injected L-DOPA (dopamine precursor, Trihexylphenidate (THP; muscarinic antagonist), or vehicle 30 min prior to the analysis of locomotion in 12 month old Shh-nLZC/C/Dat-Cre and control mice. The increased variability in stride length was normalized by L-DOPA and THP, brake stride ratios were normalized by THP but not L-DOPA, and alterations in paw angles were normalized by L-DOPA, but not THP, (Figure 4D). Taken together, our behavioral studies revealed a dynamic and progressive locomotion phenotype whose pharmacological responsiveness suggests underlying alterations in the functional balance of dopaminergic and cholinergic neurotransmission.
Ablation of Shh from DA neurons results in progressive cellular and functional corruption of the adult striatum
Similarily to BDNF, which supports survival of cortical-striatal neurons (Baquet et al., 2004), Shh can also be transported anteriorgradely through axons (Therond, 2012) . Because of the lack of evidence for an autocrine mechanism for Shh dependent support of DA neurons we therefore hypothesized that Shh signaling from dopaminergic projections to striatal targets might be of relevance to the maintenance of DA-neurons.
We found that ~25 % of all Ptc1+ cells in the striatum were neurons (Figures 5A-C) and that 6% of all striatal neurons co-express Ptc1 (Figure 5F). Conversely, 100% of all ACh neurons and 98% of all FS interneurons express Ptc1 (Figures 5D-F), consistent with the relative prevalence of ACh and FS neurons among all striatal neuronal subtypes (Bolam et al., 2006). Hence, our expression data suggested that mesencephalic DA neurons could communicate by Shh signaling selectively with all ACh and FS neurons, and non-neuronal cells among their projection targets in the adult striatum.
Figure 5. Neuronal subtype specific and progressive degeneration in the striatum in absence of Shh signaling from mesencephalic DA neurons.

(A) X-Gal labeled coronal section of striatum of a Ptc1-nLZ mouse.
(B) Ptc1 expression in neuronal and non-neuronal cells of the striatum.
(C) 25 +/− 5.2 % of Ptc1+ striatal cells are neurons (n=612 cells, 3 mice).
(D) Ptc1 expression in striatal ACh neurons.
(E) Ptc1 expression in striatal FS- neurons.
(F) 6 +/− 2.2 % of striatal neurons express Ptc1 (n=420 cells, 3 mice); 100 +/− 0 % of ACh-neuron express Ptc1 (n=140 cells, 3 mice); 98 +/− 2.0 % of FS-neurons express Ptc1 (n=150 cells, 3 mice).
(G) Loss of Ach-neurons is most pronounced in lateral areas of the dorsal striatum (circled in red) in Shh-nLZC/C/Dat-Cre mice.
(H) Quantification of striatal cell types by heterochromatin pattern (Figure S6A, B) and nuclear size (Figure S6C) in Shh-nLZC/C/Dat-Cre. (*p < 0.05, **p < 0.001, t-test (10 striatal slices/subject, n=7 mice per genotype).
(I) Progressive reduction in the numbers of ChAT+ neurons in the striatum in Shh-nLZC/C/Dat-Cre mice (p < 0.05, unpaired t-test (n=6-7/group, 12 sections per subject with a 5 section interval).
(K) Progressive reduction in the numbers of Parv+ FS neurons in the striatum in Shh-nLZC/C/Dat-Cre mice. (*p < 0.05, unpaired t-test (n=4/group, 12 sections per subject with a 5 section interval).
(L) Reduced extracellular ACh concentration in the striatum in 8 month old Shh-nLZC/C/Dat-Cre mice (** p < 0.001, unpaired t-test (n = 8/genotype, 4 samples/subject).
(M) Quantification of changes in gene expression in the striatum of 4 weeks and 12 months old Shh-nLZC/C/Dat-Cre mice (1 & 2) and after unilateral injection of increasing concentrations of SAG or cyclopamine (* p < 0.01, ** p < 0.001, two tailed t-test; n=5/genotype or treatment).
In Shh-nLZC/C/Dat-Cre mice compared to controls at 6 months of age we observed a reduction in the number of ChAT+ neurons in the striatum which was most pronounced in lateral/anterior aspects of the dorsal striatum (Figures 5G; SA-C). ACh and FS interneurons make up together only ~6% of total striatal neurons (Figure 5F) and are locally projecting. These attributes make it impossible to distinguish neuronal loss from down-regulation of phenotypic marker expression by the quantitation of the total number of neurons or the exploitation of specific projection patterns. However, the main striatal cell populations can be identified based on cell type specific perinuclear staining patterns which can be visualized by the DNA intercalating dye ToPro3 (Figures S5A-C). Based on this technique, we found a ~40% reduction in the numbers of ACh and FS interneurons but no change in the numbers of calretinin and somatostatin interneurons, MSN, and non-neuronal cells in 4-month-old Shh-nLZC/C/Dat-Cre mice compared to controls (Figures 5H). ACh and FS neurons as a group exhibit the largest nuclei and can therefore be distinguished from all other striatal cells by a nuclear circumference larger than 28 μm (Figure S5C). This second approach, revealed a ~60% reduction of the numbers of striatal cells with nuclear circumference larger than 28 μm in 12-month-old Shh-nLZC/C/Dat-Cre mice compared to controls (Figure 5H). Stereological quantitation of ACh and FS neurons using ChAT and parvalbumin immunohistochemistry revealed an adult-onset, progressive reduction in the numbers of ChAT+ and parvalbumin+ cells, which plateaus at 8 months of age at ~50% and ~40%, respectively (Figures 5I, K). The kinetics of striatal ACh and mesencephalic DA neuron degeneration was correlated (R2=0.98; p<0.006; Figures 5I; 2D). Consistent with the activation of physiological cell stress response pathways in ACh and FS neurons prior to neurodegeneration, we found increased expression of the luminal endoplasmic reticulum (ER) protein BiP/Grp78 by large bodied cells in the striatum by mRNA in situ hybridization at 5 weeks of age which then becomes more pronounced at 12 months of age (Figure S6A-D). Thus, in aggregate, the analysis of perinuclear staining pattern, nuclear size, and cell type specific marker gene expression, demonstrate a cell type selective, adult-onset, progressive degeneration of ACh and FS neurons in the striatum in the absence of Shh expression by mesencephalic DA neurons that is correlated with the degeneration of DA neurons themselves.
We next examined steady state, extracellular ACh levels in the striatum by in vivo dialysis. Despite of a mere loss of ~50% ACh neurons, we found a ~6-fold, reduction in basal levels of extracellular ACh in 8 month old Shh-nLZC/C/Dat-Cre mice compared to controls (Figure 5L). This observation suggests that surviving ACh neurons cannot functionally compensate for the reduction in their numbers in absence of Shh signaling from DA neurons.
To explore the molecular underpinnings of the inability of surviving ACh neurons to increase ACh production, we investigated the expression levels of candidate genes, which could inform about the neurophysiological status of the striatum before (at 5 weeks of age) and after (at 12 months of age) the onset of neurodegeneration. We found that the expression of striatal ChAT, vesicular acetylcholine transporter (vAChT) and GTPase regulator RGS4 were down-regulated while the expression of striatal muscarinic autoreceptor M2 was up-regulated in 5-week-old Shh-nLZC/C/Dat-Cre mice compared to controls (Figure 5M(1)). The expression of ChAT, M2, and RGS4 was further distorted at 12 months of age while the expression of vAChT returned to normal levels (Figure 5M(1)). Acetylcholine esterase (AChE) expression was unaltered at 5 weeks but reduced at 12 months of age consistent with the observed reduction of striatal ACh levels in older animals (Figure 5M(1)). ACh tone in the striatum is in part regulated by muscarinic autoreceptors M2 and M4 whose functions in turn are negatively modulated by the GTPase accelerator RGS4 (Ding et al., 2006). Thus, the observed up-regulation of M2 and down-regulation of RGS4 gene expression indicates an enhancement of cholinergic auto-receptor function in surviving ACh neurons in the striatum of Shh-nLZC/C/Dat-Cre mice consistent with the observed reduction in striatal cholinergic tone.
In contrast to the situation in ACh neurons, parvalbumin gene expression was strongly reduced at 5 weeks of age, but reached normal levels at 12 months suggesting a compensatory up-regulation by surviving FS neurons (Figure 5M(2)). General GABAergic marker and DA receptor gene expression were not affected at 5 weeks but DA receptors D1-D4, DARP32, and Gad1 were down-regulated, while DA receptor interacting protein (D-IP) was up-regulated at 12 months of age (Figure 5M(2)) suggesting that GABAergic neuronal subtypes in addition to FS neurons in the striatum become phenotypically involved subsequently to ACh and FS neurons.
In support of a direct control of gene expression by Shh signaling in the striatum, we found that the transcription factor Gli3, whose expression is inhibited by Shh signaling (Ulloa and Briscoe, 2007 ), is up-regulated by the Smo - antagonist cyclopamine (Chen et al., 2002) and down-regulated by the Smo - agonist “SAG” (Frank-Kamenetsky et al., 2002) when injected into the adult striatum of C57Bl/6 wt mice (Figure 5M(3)). M2 expression was also acutely and dose-dependently increased by cyclopamine and decreased by SAG injection into the striatum, indicating that Shh signaling impinges directly on the regulation of cholinergic tone in the healthy striatum (Figure 5M(3)).
Thus, the absence of Shh signaling originating from DA-neurons elicits a sequential structural and functional corruption of the striatum which begins with cell physiological alterations in ACh and FS neurons and culminates in a progressive, adult-onset degeneration of ACh and FS neurons without compensatory adaptations of surviving ACh- neurons.
DA neuron-derived Shh maintains the cellular source of GDNF but represses its transcription in the striatum
GDNF is expressed by ACh and FS interneurons (Hidalgo-Figueroa et al., 2012). Consistent with the Shh-dependent maintenance of ACh and FS neurons, we found a progressive reduction in GDNF mRNA and protein expression, and an up-regulation of the canonical receptor Ret and its co-receptor Gfrα1, which bind all members of the GDNF family of ligands, in the striatum of Shh-nLZC/C/Dat-Cre mice compared to controls (Figure 6A, B). The progressive reduction in striatal GDNF tissue content correlated with the progressive degeneration of ACh neurons in Shh-nLZC/C/Dat-Cre mice (Figures 6B and 2D; R2=0.95, p<0.02 for ACh neurons). We then utilized the cholinotoxin AF64a, which causes reversible inhibition of ACh neurons at moderate concentrations and ACh neuron degeneration at higher concentration (Sandberg et al., 1984) to assess the relative contribution of ACh neurons over other sources to GDNF production in the striatum. We found that unilateral striatal injections of moderate concentrations of AF64a led to a ~30% reduction in striatal GDNF protein content over vehicle injected controls 36 h after toxin application in 4-month-old C57Bl/6 wt animals (Figure 6C). Together, these experiments demonstrate that Shh signaling originating from mesencephalic DA neurons contribute to the long-term maintenance of striatal GDNF production through trophic support of striatal ACh and FS neurons.
Figure 6. Shh signaling inhibits GDNF expression in the striatum.

(A) Changes in GDNF, Ret and Gfra1 gene expression in the striatum of 4 weeks and 12 months old Shh-nLZC/C/Dat-Cre mice (* p < 0.01, ** p < 0.001, two tailed t-test; n=5/genotype).
(B) Quantification of striatal GDNF tissue content as a function of age in Shh-nLZC/C/Dat-Cre mice (* p<0.01, unpaired t-test; n=10-11/group).
(C) Injection of AF64a reduces GDNF tissue content. (37 +/− 3 %; * p<0.05, unpaired t-test, n=12-14/group).
(D) Experimental design: Comparative analysis of GDNF expression upon unilateral injection of SAG or cyclopamine into the striatum of wt mice, or 6-OHDA into the mFB of GDNF-LZ+/− mice.
(E) SAG decreases GDNF mRNA expression (* p < 0.05, ANOVA followed by Tukey’s post hoc Test; n=5/group/drug concentration).
(F) Injection of cyclopamine into the striatum or 6-OHDA into the mFB increases GDNF mRNA expression (* p < 0.05, ANOVA followed by Tukey’s post hoc Test; n=5/group/drug concentration; the up-regulation caused by cyclopamine was dose dependent).
(G) Experimental design: Comparative analysis of gene expression in the vMB and striatum upon unilateral injection of AF64a into the PPTg.
(H) Quantification of turning bias 36 h post unilateral injection of AF64a into the PPTg in Shh-nLZC/C/Dat-Cre mice (* p < 0.05, post hoc test after ANOVA, n=5/genotype).
(I, K) Quantification of changes in gene expression in the vMB (I) or striatum (K) upon AF64a injection into the PPTg of Shh-nLZC/C/Dat-Cre and control mice (** p < 0.001, AF64a x genotype ANOVA followed by Tukey’s post hoc Test, (n=5/group/genotype).
The analysis of the long-term effects of the chronic absence of Shh signaling from DA neurons does not inform about whether Shh signaling plays a role in the transcriptional regulation of striatal GDNF expression in the absence of physiological cell stress and/or neurodegeneration. We therefore examined whether Shh signaling regulates striatal GDNF gene expression acutely by unilaterally injecting SAG or cyclopamine in 8-week-old C57/Bl6 male mice (Figure 6D). Comparative qRT-PCR analysis revealed a SAG specific reduction in GDNF mRNA and a dose-dependent, cyclopamine specific increase in GDNF mRNA 30 h after injection (Figure 6E, F), demonstrating that GDNF expression in the adult striatum is dynamically regulated by Shh signaling . Consistent with the inhibition of GDNF expression by Shh signaling originating from DA neurons we observed an up-regulation of GDNF in the striatum upon the interruption of the mesostriatal pathway by the unilateral injection of 6-OHDA into the medial forebrain bundle (mFB) of GDNF-LZ mice (Figure 6F). Together with the finding that systemic injections of the dopaminergic toxin MPTP results in the transient up-regulation of striatal GDNF expression (Hidalgo-Figueroa et al., 2012), our results suggested that the relevant Shh signal for the regulation of GDNF expression in vivo could come from the vMB.
Guided by these results, we tested whether Shh produced specifically by DA neurons acutely regulates the expression of GDNF in the mesostriatal system in vivo. The penduncolo pontine tegmental nucleus (PPTg) provides excitatory, nicotinic receptor mediated cholinergic input to mesencephalic DA neurons (Futami et al., 1995) (Figure 6G). Similar to previous observations upon the excitotoxic ablation of PPTg neurons (Dunbar et al., 1992), we found that unilateral injection of the cholinotoxin AF64a into the PPTg of 2-month-old Shh-nLZC/C/Dat-Cre- or control mice elicited a controlateral turning bias consistent with reduced cholinergic stimulation of ipsilateral DA neurons (Figures 6H) (Lester et al., 2010). Comparative qRT-PCR analysis of dopaminergic markers between the ipsilateral and controlateral vMB revealed a transcriptional up-regulation of Th and a down-regulation of the DA autoreceptor DaR2 ipsilateral in both genotypes, suggesting an adaptive up-regulation of mesencephalic DA signaling upon AF64a injection into the PPTg (Figure 6I). AF64a injection into the PPTg also resulted in a ~10-fold up-regulation of Shh transcription in the ipsilateral vMB compared to the controlateral control (Figure 6I). Thus, the comparative analysis of Shh-nLZC/C/Dat-Cre mice, which are unable to express functional Shh, and of Shh-nLZC/+/Dat-Cre control animals allows the distinction of Shh-dependent and independent regulation of gene expression in the experimentally undisturbed striatum that occurs in response to AF64a injection into the PPTg.
Utilizing this experimental paradigm, we found that the expression of ChAT and vAChT in the ipsilateral striatum were down-regulated to similar extent, regardless of Shh expression by DA neurons compared to the contralateral striatum (Figure 6K). In contrast, we observed a ~4-fold down-regulation of GDNF expression upon AF64a injection into PPTg of control mice, i.e. mice that produce Shh in DA neurons, but not of Shh-nLZC/C/Dat-Cre mutant animals (Figure 6K). These results provide genetic evidence that increased Shh signaling specifically originating from mesencephalic DA neurons results in the repression of GDNF transcription in the striatum.
Cholinergic neurons of the striatum regulate Shh expression in mesencephalic DA neurons
The observed up-regulation of Shh expression by DA neurons upon neurotoxic insult to the PPTg suggested that expression of Shh by DA neurons is not static but could be regulated by cell extrinsic signals. We therefore explored the dynamic range of Shh expression and whether the striatum in addition to the PPTg might be a source of signals that could contribute to the regulation of Shh expression by DA neurons.
We first investigated whether the acute interruption of mesostriatal communication by unilateral injection of 6-OHDA into the mFB of C57BL/6 wt- and GDNF-LZ-mice alters Shh expression by DA neurons. 30 h after toxin injection, we observed contralateral turning biases in wt and GDNF-LZ animals consistent with reduced DA signaling to the striatum (Figure 7A, B). In these animals, we found an up-regulation of Shh expression in the vMB ipsilateral to the toxin injection. These results suggested that the striatum could be a source of signals that inhibit Shh expression in the vMB (Figure 7C).
Figure 7. Shh expression by mesencephalic DA neurons is repressed by signals emanating from ACh-neurons.

(A) Experimental design: Unilateral injection of AF64a into the striatum of 6-OHDA in the mFB results in ipsi-lateral or contro-lateral, resp., turning.
(B) Quantification of turning bias upon injection of 6-OHDA into the mFB of wt- or GDNF-LZ+/− mice (*p <0.01, AF64a dose vs. vehicle, ANOVA followed by Tuckey’s post hoc test (n=5/dose).
(C) Up-regulation of Shh gene expression in the vMB bias upon injection of 6-OHDA into the mFB of wt- or GDNF-LZ+/− mice, or AF64a into wt- mice (*p< 0.05, ANOVA followed by Tuckey’s post hoc test; n=5/condition).
(D) Quantification of turning bias upon injection of AF64a into wt- mice (*p <0.01, AF64a dose vs. vehicle, ANOVA followed by Tuckey’s post hoc test (n=5/dose).
(E) Dose dependent up regulation of Shh gene expression in the vMB bias upon injection AF64a into wt- mice (*p< 0.05, ANOVA followed by Tuckey’s post hoc test; n=5/condition).
(F) Shh expression in the vMB is activated in Shh-nLZC/C/Dat-Cre and RetC/C/Dat-Cre mice (5′: Shh exon 1 amplimer, 3′: Shh exon 2,3 amplimer, n=4-6/ genotype and age, *p <0.01).
Use of the cholinotoxin AF64a afforded us to test next whether signals emanating from striatal ACh neurons could contribute to the repression of Shh transcription in DA neurons. 30 h post unilateral striatal injection of AF64a into wt C57B/6 mice, we observed a dose-dependent ipsilateral turning bias consistent with a graded increase in striatal motor output due to a progressive, AF64a induced, inhibition of cholinergic activity (Figure 7A, D) (Lester et al., 2010). In the ipsilateral vMB of these animals, we found an AF64a dose-dependent, step-wise, up-regulation of Shh transcription correlated with the graded turning bias (R2= 0.79, p < 0.001; Figure 7E). The up-regulation of Shh expression was transient and returned to control levels by 6 days post toxin injection (Figure 7E). In addition to Shh, we also found AF64a dose-dependent alterations in the expression of dopaminergic markers that resembled the distortions seen in Shh-nLZC/C/Dat-Cre mice (Supplemental Results D, Figure S3E) indicating that altered signaling by cholinergic neurons contributes to the dopaminergic cell syndrome that we observe in the absence of Shh signaling from DA neurons (Figure 2).
These findings suggested that the transcription of Shh in DA neurons is activated in Shh-nLZC/C/Dat-Cre mice. Since the design of the Shh-nLZC/C allele leaves intact the promoter and most transcriptional enhancer regions of the native Shh locus after Cre mediated recombination we devised a qPCR based assay to test whether Shh expression was activated in DA neurons before and after ACh neuron degeneration in Shh-nLZC/C/Dat-Cre mice (for technical details see Supplemental information A and Figure S1). We found a ~5-fold and ~4-fold increase in the transcription of the 5′-end of the Shh mRNA which can be expressed from the truncated Shh locus in Shh-nLZC/C/Dat-Cre at 4 weeks and 12 months of age, respectively. Thus, Shh-nLZC/C/Dat-Cre animals cease to produce signals that otherwise inhibit Shh expression by mesencephalic DA neurons in the undisturbed brain (Figure 7F).
The identification of the receptor(s) on DA neurons that transmit the signals that impinge on the regulation of Shh expression can inform on the nature of the signals. We noted that the tissue specific ablation of the canonical receptor Ret, which can bind all members of the GDNF family of ligands, from DA neurons utilizing the same Dat-Cre allele also employed in the present study, resulted in alterations in dopaminergic marker gene expression, deficits in elicited DA release and late-onset, progressive DA neuron degeneration, (RetC/C/Dat-Cre mice) (Kramer et al., 2007). We tested whether also the expression of of Shh is alteredin the vMB of RetC/C/Dat-Cre mice. We found a ~6-fold up-regulation of Shh expression in RetC/C/Dat-Cre mice compared to litter controls at 3 months of age prior to observable neurodegeneration (Figure 7F). Taken together, our studies provide pharmacological and genetic evidence that ACh neurons of the striatum produce signals, which engage the canonical GDNF receptor Ret on DA neurons and repress the expression of Shh, and regulate the expression of multiple other genes in DA neurons.
DISCUSSION
Our results reveal that mesencephalic DA neurons express Shh throughout life and demonstrate that DA neuron-produced Shh is necessary for the long-term structural and functional maintenance of mesencephalic DA neurons. Our studies, however, did not uncover any evidence for an autocrine mode of Shh signaling. Instead, we demonstrate that DA neuron-produced Shh regulates the availability of the dopaminotrophic factor GDNF and cholinergic physiology within the striatum in two distinct ways: First, DA neuron-produced Shh is required for the structural and functional maintenance of FS and ACh interneurons of the striatum, which are prominent sources of GDNF in the adult basal ganglia. Second, signaling by DA neuron-produced Shh results in the transcriptional repression of GDNF and the regulation of expression of muscarinic autoreceptor signaling components. Taken together, our results reveal an entirely novel means by which mesencephalic DA neurons communicate to a subset of their striatal neuronal targets and regulate the cellular and neurochemical homeostasis in the mesostriatal circuit in the adult brain. We further provide in vivo evidence that signals engaging the canonical GDNF receptor Ret expressed specifically on DA neurons and originating from the striatum inhibit the transcription of Shh in DA neurons. Our findings are consistent with the existence of a reciprocal trophic factor signaling loop between DA neurons on one side and ACh and FS neurons on the other side and reveal that the regulation of expression of these factors has rheostat properties.
Reciprocal trophic factor signaling between mesencephalic DA- and striatal ACh- and FS-neurons neurons
To resolve the mechanism of action of Shh signaling in the mesostriatal circuit required us to reconcile two sets of seemingly contradictory observations: a) the apparent cell autonomous activity of Shh on DA neurons in the absence of evidence for autocrine signaling, and b) the reciprocal inhibition of expression of Shh and GDNF in the mesostriatal circuit while these factors are necessary for the trophic support of DA, and ACh and FS neurons.
The inefficiency of Cre-mediated recombination of the Shh allele created heterogeneity among DA neurons in regard of Shh expression, which allowed us to investigate whether Shh expression by DA neurons confers a cell survival advantage. Our results reveal a ~2-fold enrichment of Shh-expressing DA neurons during phenotype progression in Shh-nLZC/C/Dat-Cre mice, demonstrating that mostly Shh−/− DA neurons degenerate. Thus, our studies provide evidence for a neuroprotective function of DA neuron-expressed Shh on DA neurons in the adult mesencephalon and are consistent with findings that exogenously supplied Shh to the basal ganglia increases the resilience of mesencephalic DA neurons to neurotoxic insults (Dass et al., 2005; Tsuboi and Shults, 2002). Yet, DA neuron degeneration in the absence of Shh expression by DA neurons is unlikely a result of the interruption of a cell autonomous effect of Shh since: (1) we cannot find evidence for the expression of the Shh co-receptors Ptc1 and Ptc2 on mesencephalic DA neurons, and (2) the Dat-Cre mediated tissue restricted ablation of the obligate necessary Shh signaling component Smo from DA neurons does not phenocopy the Dat-Cre mediated tissue restricted ablation of Shh from DA neurons. Instead, we find that DA neuron-produced Shh modulates gene expression, physiology, and survival of striatal ACh and FS neurons, which, in turn, express the dopaminotrophic factor GDNF (Figure 8A) (Hidalgo-Figueroa et al., 2012). Thus, the most parsimonious explanation for the apparent cell autonomous protection of DA neurons by Shh expression is the possibility that individual cartridges of mesostriatal circuits act as autonomic units. In this scenario, neuronal circuits in which DA neurons have escaped Cre-mediated recombination of the Shh alleles will continue to supply Shh to support ACh and FS neurons and ACh and FS neurons will continue to supply GDNF to support DA neuron survival. This model is supported by the quantification of synaptic connectivity in the striatal microcircuit: while ACh, FS and DA neurons elaborate widespread arborisations, each neuron only contributes to a few hundred of the estimated two million mesostriatal circuits in the striatum (Bolam et al., 2006). Further support of a confinement of Shh action to the vicinity of Shh release sites comes from Loulier et al. (2005) who found strong expression in the adult striatum of the Hedgehog-interacting protein (Hhip), which inhibits Shh signaling by complexing to secreted Shh, likely further limiting the poor diffusion of Shh once secreted (Ulloa and Briscoe, 2007 ). Thus, a given DA neuron might be able to signal via Shh to only a few ACh and FS neurons and receive trophic support from the same neurons resulting in the appearance of cell autonomy.
Figure 8. Shh signaling from dopaminergic neurons controls structural and neurochemical homeostasis in the mesostriatal circuit.

(A) Shh and GDNF provide additional means of communication between DA, and ACh and FS neurons of the mesostriatal circuit.
(B) Rheostat properties of trophic factor signaling.
(C) Potential signaling pathways by which Shh originating from mesencephalic DA neurons determines the set-point of extracellular ACh tone in the striatum. (red arrows indicate direction of change observed in Shh-nLZC/C/Dat-Cre mice.)
(D) Absence of Shh signaling from DA neurons causes progressive structural and functional decay of the mesostriatal circuit.
Trophic support of ACh and FS neurons by DA neuron-produced Shh on one side and of DA neurons by ACh and FS neuron produced GDNF on the other side could be provided in a static manner or be induced in response to physiological needs. We observe transcriptional activation of Shh loci in the vMB upon (1) injection of the dopaminergic neurotoxin 6-OHDA into the mFB, (2) induction of cholinergic dysfunction by injection of the cholinotoxin AF64a into the striatum, (3) genetic ablation of the canonical GDNF receptor Ret from DA neurons, and (4) genetic reduction of Shh signaling from DA neurons to the striatum. Conversely, we find that the interruption of mesostriatal communication by the neurotoxin 6-OHDA or striatal injection of the Shh antagonist cyclopamine leads to an up-regulation of GDNF expression in the striatum, whereas striatal injection of the Shh agonist SAG or the pharmacological induced up-regulation of endogenous Shh signaling specifically from mesencephalic DA neurons results in the inhibition of GDNF expression in the striatum. Thus, ACh neurons, which are trophically dependent on Shh from DA neurons, are a source of graded inhibitory signals for the transcription of Shh by DA neurons. In a mirror arrangement, DA neurons that are supported by GDNF modulate the expression of GDNF in the striatum by graded Shh expression (Figure 8B).
While our data strongly implicates striatal GDNF in the regulation of Shh in DA neurons, our results do not exclude the possibility that other members of the GDNF family of ligands, all of which can signal via Ret and Gfra1, could contribute to the modulation of Shh expression in DA neurons.
The extent of relief of transcriptional inhibition of Shh expression by signals from ACh neurons is correlated with the degree of cholinergic dysfunction when averaged across DA neurons and has a dynamic range of 2 to 10 fold. This observation fits well with the established concentration-dependence and dynamic range of Shh signaling (Ulloa and Briscoe, 2007 ). Low levels of Shh signaling are necessary for tissue maintenance in the developing spinal cord. At higher concentrations Shh regulates in a concentration-dependent manner, gene expression mediated by either transcriptional repressor or activator forms of the Shh signaling components Gli-1, -2, and - 3. At these expression levels 1.8 fold alterations in the concentration of Shh results in distinct patterns of gene expression (Ulloa and Briscoe, 2007 ), suggesting that the dynamic range of 2 to 10 fold observed in our studies could result in several distinct physiological responses within ACh and FS neurons in response to Shh signaling.
Mutual trophic dependence combined with reciprocal inhibition of trophic factor expression must result in tight homeostatic control of Shh and GDNF expression and links the extent of Shh and GDNF signaling to the cell physiological status of DA, ACh and FS neurons. We thus propose that attenuation of gene expression in response to physiological stress in DA, or ACh and FS neurons will result in a corresponding reduction in the repression of either Shh or GDNF, respectively, in such a way that cells in need will receive increased trophic factor support. After regaining intra cellular homeostasis reactivated gene expression will reduce trophic factor production through normalization of trophic factor production in those cells that had suffered from a cell physiological insult and, in turn, will lead to a reduction in the expression of the corresponding trophic factor (Figure 8B).
These results also imply that trophic factor expression cannot be maintained chronically at levels that are beneficial for the survival of DA, ACh and FS neurons once in distress. This reasoning points to multiple functions of GDNF and Shh signaling in the basal ganglia. Indeed GDNF signaling can regulate the quantal size of DA release of DA neurons (Pothos et al., 1998). Our studies reveal a corresponding function of Shh signaling on cholinergic neuro-transmission extending the symmetry of Shh and GDNF signaling from trophic interactions to neuromodulation within the nigro-striatal circuit.
Regulation of the balance of cholinergic and dopaminergic tone in the striatum by Shh signaling
Extracellular ACh tone in the striatum is variably regulated by DA neuron activity (Threlfell et al., 2010). However, dopaminergic activity does not exert its effects on ACh neurons exclusively through DA receptor signaling, but also through the regulation of the coupling of muscarinic autoreceptors to K+ and Ca2+ channels by altering the expression of “regulator of G-protein signaling” (RGS) (Ding et al., 2006). These findings raise the possibility that signaling molecules produced by DA neurons other than DA are involved.
Our experiments show that Shh signaling originating from DA neurons impinges onto the regulation of the set-point of ACh in the striatum through the transcriptional regulation of M2 and possibly by engaging a pathway that involves the Shh dependent inhibition of protein kinase A (PKA) (Ogden et al., 2008) which is an activator of RGS4 (Huang et al., 2007), whose inhibition increases the efficacy of muscarinic autoreceptor function (Ding et al., 2006) (Figure 8C).
Lowered cholinergic tone, a reduction of striatal GDNF levels due to progressive degeneration of ACh neurons, and a lack of functional adaptations by surviving ACh neurons should all influence physiology of surviving DA neurons in Shh-nLZC/C/Dat-Cre mice. Consistent with this expectation, we observe highly dynamic distortions in DA tissue content in both the vMB and the striatum. Hence, our results suggest that surviving DA neurons, but not surviving ACh neuron, are able to adapt their physiology dynamically in the face of progressive neurodegeneration and decreased ACh and GDNF/Ret signaling during early adulthood. However, by 10 months of age, we find the manifestation of discrete locomotion and gait disturbances, indicating that the progressive deterioration of the mesostriatal circuit surpasses the compensatory capacity of DA neurons in aged Shh-nLZC/C/Dat-Cre mice (Figure 8D).
Possible implications for understanding the etiology of diseases of the basal ganglia
Archetypes of basal ganglia models imply that an imbalance of cholinergic and dopaminergic signaling in the striatum is responsible for the hyper- and hypo-kinetic manifestations of progressive movement disorders such as PD (Obeso et al., 2010). Our work describes a novel mouse paradigm that recapitulates many of the key features of the progressive cellular, neurochemical, and functional pathologies observed in PD with apparent face-, construct- and predictive- validity since the functional phenotype that is associated with progressive neuronal loss can be ameliorated with DA supplementation or a muscarinic antagonist also used in the management of PD. Yet, the resemblance of the phenotype of Shh-nLZC/C/Dat-Cre mice with PD does not extend to the absolute direction of alterations in cholinergic tone. How can these findings be reconciled?
In PD, ACh tone is increased, while DA levels fall due to DA neuron degeneration (Wooten, 1990). Our experiments demonstrate that the loss of Shh signaling, which also must occur in PD due to DA neuron degeneration, decreases ACh secretion consistent with an observed increase in muscarinic autoreceptor expression. These divergent observations could be caused by an up-regulation of Shh production in still functioning DA neurons in PD. This notion is supported by several in vivo experiments described herein: we demonstrate that the transcription of Shh in DA neurons is strongly up-regulated upon (1) injection of 6-OHDA into the mFB, (2) induction of cholinergic dysfunction in the striatum, (3) induction of cholinergic dysfunction in the PPTg, and (4) the genetic ablation of part of the Shh locus which abrogates the production of functional Shh by DA neurons. Hence, our data are consistent with a scenario in which prior to reaching an advanced stage of PD, surviving DA neurons express elevated levels of Shh, which, in turn, cause an increase of ACh tone in the striatum mediated by a down-regulation of muscarinic autoreceptor efficacy.
Our data reinforce the rationale for supporting growth factor signaling as a disease modifying therapeutic strategy in basal ganglia diseases. However, the uncovered negative feedback regulation of endogenous growth factor expression within the mesostriatal circuit predicts that exogenously supplied trophic factors could inhibit endogenous expression of the same factors possibly curtailing the therapeutic benefit of this approach. Instead, our results point to the possibility that undercutting the negative feedback regulation of endogenous growth factor expression could result in therapeutically effective increases of trophic factor signaling within the basal ganglia.
EXPERIMENTAL PROCEDURES
Mouse strains
The Shh-nLZC allele was generated by homologous recombination in ES cells. Additional construction details, mouse strains and genotyping procedures are described in extended experimental procedures. All animal handling and procedures were approved by the Animal Care and Use Committee of Columbia University and performed in accordance with NIH guidelines.
Immunohistochemistry
Immunohistochemistry was performed on 16-100 μm cryostat-cut sections using primary and secondary antibodies listed in supplemental information. Images were acquired on a Zeiss LSM510 Meta confocal microscopes. Quantification of the size of populations of cells was estimated by the optical fractionator method described in supplemental experimental procedures.
Quantification of GDNF tissue content
Tissue levels of GDNF were measured by ELISA (GDNF Emax ImmunoAssay System; Promega, Madison, WI), according to the manufacturer’s protocol.
Quantification of gene expression
Total RNA from striatum and lateral vMB containing the entire SN and VTA was isolated (RNeasy Mini Kit; Qiagen) and reverse transcribed using oligo(dT) primers and the SuperScript First-Strand Synthesis System (Invitrogen), according to the manufacturers’ protocols. Relative changes in gene expression were quantified by rtPCR usin TaqMan gene expression assays (Applied Biosystems) with amplicons listed in supplemental Table 2 and calculated by the ΔΔCt method.
Neurochemical Analysis
Determination of the concentration of dopamine and acetylcholine and neurotoxicological challenges were performed as described in supplemental experimental procedures.
Locomotion analysis
Analysis of gait parametes by forced locomotion was performed by ventral plane videography (Digigait, Inc.) Spontaneous motor activity was measured in an open field arena using automatic tracking at 6 Hz by an EthoVision 3.1 system (Noldus Information Technology, Leesburg, VA). Derivation of acceleration and deceleration profiles and indices for turning bias and locomotion complexity is described in Extended Experimental Procedures.
Statistical analysis
The mean and SEM of values were calculated and the significance of all pair-wise comparisons was determined by two-tailed distribution homoscedastic Student’s t-test and by ANOVA, including a repeated measures factor when necessary. Follow up analysis between groups with multiple comparisons was by Tukey’s post-hoc test. Nonparametric data were analyzed by Mann-Whitney U test. For all box plots, the box includes data points between the 25th and 75th percentile of all values, with the line representing the median value. The lines and whiskers represent data between the 9th and 91st percentile and individual dots represent outlier points.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Elizabeth Gregorutti for technical assistance; B. Han, M Mendelsohn and J. Kirkland for help with mouse husbandry; S. Morton for antibody production and I Schieren for help with IT related issues. We thank Stephen Rayport, David Sulzer, Robert Burke, and Christopher Henderson for discussion and critical comments on the manuscript. SP was supported by the Parkinson’s Disease Foundation and the Thomas Hartman Foundation For Parkinson’s Research. AHK was supported NIH grant R21 NS056312-01A1, the American Parkinson’s Disease Association, The Michael J. Fox Foundation, The ALS Association and NYS Department of Health NYSTEM SDH CO24293.
Footnotes
SUPPLEMENTAL INFORMATION: Supplemental Data include supplemental results with 6 figures and one table, and extended experimental procedures with one table.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Bergman R, Graybiel A, Kimura M, Plenz D, Seung H, Surmeier D, Wickens J. Group Report: Microcircuits, Molecules, and Motivated Behavior - Microcircuits in the Striatum. In: Grillner S, Graybiel A, editors. Microcircuits. The MIT Press; Cambridge: 2006. pp. 165–189. [Google Scholar]
- Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dass B, Iravani MM, Huang C, Barsoum J, Engber TM, Galdes A, Jenner P. Sonic hedgehog delivered by an adeno-associated virus protects dopaminergic neurones against 6-OHDA toxicity in the rat. J Neural Transm. 2005;112:763–778. doi: 10.1007/s00702-004-0227-7. [DOI] [PubMed] [Google Scholar]
- Dass B, Iravani MM, Jackson MJ, Engber TM, Galdes A, Jenner P. Behavioural and immunohistochemical changes following supranigral administration of sonic hedgehog in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated common marmosets. Neuroscience. 2002;114:99–109. doi: 10.1016/s0306-4522(02)00214-2. [DOI] [PubMed] [Google Scholar]
- DeLong M, Wichmann T. Update on models of basal ganglia function and dysfunction. Parkinsonism Relat Disord. 2009;15(Suppl 3):S237–240. doi: 10.1016/S1353-8020(09)70822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Guzman JN, Tkatch T, Chen S, Goldberg JA, Ebert PJ, Levitt P, Wilson CJ, Hamm HE, Surmeier DJ. RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat Neurosci. 2006;9:832–842. doi: 10.1038/nn1700. [DOI] [PubMed] [Google Scholar]
- Dunbar JS, Hitchcock K, Latimer M, Rugg EL, Ward N, Winn P. Excitotoxic lesions of the pedunculopontine tegmental nucleus of the rat. II. Examination of eating and drinking, rotation, and reaching and grasping following unilateral ibotenate or quinolinate lesions. Brain Res. 1992;589:194–206. doi: 10.1016/0006-8993(92)91278-m. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetsky M, Zhang XM, Bottega S, Guicherit O, Wichterle H, Dudek H, Bumcrot D, Wang FY, Jones S, Shulok J, et al. Small-molecule modulators of Hedgehog signaling: identification and characterization of Smoothened agonists and antagonists. J Biol. 2002;1:10. doi: 10.1186/1475-4924-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futami T, Takakusaki K, Kitai ST. Glutamatergic and cholinergic inputs from the pedunculopontine tegmental nucleus to dopamine neurons in the substantia nigra pars compacta. Neurosci Res. 1995;21:331–342. doi: 10.1016/0168-0102(94)00869-h. [DOI] [PubMed] [Google Scholar]
- Hidalgo-Figueroa M, Bonilla S, Gutierrez F, Pascual A, Lopez-Barneo J. GDNF is predominantly expressed in the PV+ neostriatal interneuronal ensemble in normal mouse and after injury of the nigrostriatal pathway. J Neurosci. 2012;32:864–872. doi: 10.1523/JNEUROSCI.2693-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhou H, Mahavadi S, Sriwai W, Murthy KS. Inhibition of Galphaq-dependent PLC-beta1 activity by PKG and PKA is mediated by phosphorylation of RGS4 and GRK2. Am J Physiol Cell Physiol. 2007;292:C200–208. doi: 10.1152/ajpcell.00103.2006. [DOI] [PubMed] [Google Scholar]
- Izzi L, Levesque M, Morin S, Laniel D, Wilkes BC, Mille F, Krauss RS, McMahon AP, Allen BL, Charron F. Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev Cell. 2011;20:788–801. doi: 10.1016/j.devcel.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ER, Aron L, Ramakers GM, Seitz S, Zhuang X, Beyer K, Smidt MP, Klein R. Absence of Ret signaling in mice causes progressive and late degeneration of the nigrostriatal system. PLoS Biol. 2007;5:e39. doi: 10.1371/journal.pbio.0050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester DB, Miller AD, Blaha CD. Muscarinic receptor blockade in the ventral tegmental area attenuates cocaine enhancement of laterodorsal tegmentum stimulation-evoked accumbens dopamine efflux in the mouse. Synapse. 2010;64:216–223. doi: 10.1002/syn.20717. [DOI] [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Loulier K, Ruat M, Traiffort E. Analysis of hedgehog interacting protein in the brain and its expression in nitric oxide synthase-positive cells. Neuroreport. 2005;16:1959–1962. doi: 10.1097/01.wnr.0000187632.91375.81. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AH, Halliday G. Missing pieces in the Parkinson’s disease puzzle. Nat Med. 2010;16:653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- Ogden SK, Fei DL, Schilling NS, Ahmed YF, Hwa J, Robbins DJ. G protein Galphai functions immediately downstream of Smoothened in Hedgehog signalling. Nature. 2008;456:967–970. doi: 10.1038/nature07459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual A, Hidalgo-Figueroa M, Piruat JI, Pintado CO, Gomez-Diaz R, Lopez-Barneo J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat Neurosci. 2008;11:755–761. doi: 10.1038/nn.2136. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Davila V, Sulzer D. Presynaptic recording of quanta from midbrain dopamine neurons and modulation of the quantal size. J Neurosci. 1998;18:4106–4118. doi: 10.1523/JNEUROSCI.18-11-04106.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy SB, Soderstrom K, Bakay RA, Kordower JH. Neurotrophic factor therapy for Parkinson’s disease. Prog Brain Res. 2010;184:237–264. doi: 10.1016/S0079-6123(10)84013-0. [DOI] [PubMed] [Google Scholar]
- Reilly JO, Karavanova ID, Williams KP, Mahanthappa NK, Allendoerfer KL. Cooperative effects of Sonic Hedgehog and NGF on basal forebrain cholinergic neurons. Mol Cell Neurosci. 2002;19:88–96. doi: 10.1006/mcne.2001.1063. [DOI] [PubMed] [Google Scholar]
- Reiner A. The conservative evolution of the basal ganglia. In: Steiner H, Tseng K, editors. Handbook of Basal Ganglia Structure and Function. Academic Press & Elsevier, CA; New York: 2010. pp. 29–62. [Google Scholar]
- Sandberg K, Sanberg PR, Coyle JT. Effects of intrastriatal injections of the cholinergic neurotoxin AF64A on spontaneous nocturnal locomotor behavior in the rat. Brain Res. 1984;299:339–343. doi: 10.1016/0006-8993(84)90715-7. [DOI] [PubMed] [Google Scholar]
- Therond PP. Release and transportation of Hedgehog molecules. Curr Opin Cell Biol. 2012;24:173–180. doi: 10.1016/j.ceb.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Threlfell S, Clements MA, Khodai T, Pienaar IS, Exley R, Wess J, Cragg SJ. Striatal muscarinic receptors promote activity dependence of dopamine transmission via distinct receptor subtypes on cholinergic interneurons in ventral versus dorsal striatum. J Neurosci. 2010;30:3398–3408. doi: 10.1523/JNEUROSCI.5620-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K, Shults CW. Intrastriatal injection of sonic hedgehog reduces behavioral impairment in a rat model of Parkinson’s disease. Exp Neurol. 2002;173:95–104. doi: 10.1006/exnr.2001.7825. [DOI] [PubMed] [Google Scholar]
- Ulloa F, Briscoe J. Morphogens and the control of cell proliferation and patterning in the spinal cord. Cell Cycle. 2007;6:2640–2649. doi: 10.4161/cc.6.21.4822. [DOI] [PubMed] [Google Scholar]
- Wang CY, Yang F, He X, Chow A, Du J, Russell JT, Lu B. Ca(2+) binding protein frequenin mediates GDNF-induced potentiation of Ca(2+) channels and transmitter release. Neuron. 2001;32:99–112. doi: 10.1016/s0896-6273(01)00434-2. [DOI] [PubMed] [Google Scholar]
- Wooten G. Parkinsonism. In: Pearlman A, Collins R, editors. Neurobiology of Disease. Oxford University Press; New York: 1990. pp. 454–468. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.