

Abstract
The affinity and efficacy at four subtypes (A1, A2A, A2B and A3) of human adenosine receptors (ARs) of a wide range of 2-substituted adenosine derivatives were evaluated using radioligand binding assays and a cyclic AMP functional assay in intact CHO cells stably expressing these receptors. Similar to previous studies of the N6-position, several 2-substituents were found to be critical structural determinants for the A3AR activation. The following adenosine 2-ethers were moderately potent partial agonists (Ki, nM): benzyl (117), 3-chlorobenzyl (72), 2-(3-chlorophenyl)ethyl (41), and 2-(2-naphthyl)ethyl (130). The following adenosine 2-ethers were A3AR antagonists: 2,2-diphenylethyl, 2-(2-norbornan)ethyl, R- and S-2-phenylbutyl, and 2-(2-chlorophenyl)ethyl. 2-(S-2-Phenylbutyloxy)a-denosine as an A3AR antagonist right-shifted the concentration–response curve for the inhibition by NECA of cyclic AMP accumulation with a KB value of 212 nM, which is similar to its binding affinity (Ki = 175 nM). These 2-substituted adenosine derivatives were generally less potent at the A1AR in comparison to the A3AR, but fully efficacious, with binding Ki values over 100 nM. The 2-phenylethyl moiety resulted in higher A3AR affinity (Ki in nM) when linked to the 2-position of adenosine through an ether group (54), than when linked through an amine (310) or thioether (1960). 2-[2-(l-Naphthyl)ethyloxy]adenosine (Ki = 3.8 nM) was found to be the most potent and selective (>50-fold) A2A agonist in this series. Mixed A2A/A3AR agonists have been identified. Interestingly, although most of these compounds were extremely weak at the A2BAR, 2-[2-(2-naphthyl)ethyloxy]adenosine (EC50 = 1.4 µM) and 2-[2-(2-thienyl)-ethyloxy]adenosine (EC50 = 1.8 (M) were found to be relatively potent A2B agonists, although less potent than NECA (EC50 = 140 nM).
Keywords: Adenosine receptors, Purines, Nucleosides, GPCR, Efficacy, Structure–activity relationships
1. Introduction
Extracellular adenosine acts as a local modulator at four subtypes of receptors (A1, A2A, A2B, and A3), which are involved in numerous physiological and pathophysiological processes [1]. For example, adenosine attenuates the effects of ischemia in the heart and brain. Acting through the A2A adenosine receptor (AR), it suppresses prolonged inflammation [2] and causes vasodilation and inhibits platelet aggregation, thus increasing the amount of oxygen available to an organ under stress. Adenosine agonists selective for the A3AR are of interest as cerebroprotective [3], cardioprotective [4, 5], and anticancer [6] agents.
Recently, we have characterized structure–efficacy relationships for adenosine derivatives as agonists at the A3AR. The intrinsic efficacy of adenosine derivatives in activation of the A3AR is more variable than at other subtypes [7–10]. Specific groups placed at the N6-position and on the ribose moiety have reduced or completely abolished the ability to activate this receptor, while maintaining high binding affinity. Thus, it has been possible to design nucleoside-based antagonists [11], which in many cases are selective for both the human and rat A3ARs. Such A3AR antagonists include adenosine derivatives bearing: steric constraints of the ribose moiety or its 5′-amide modification [11], N6-groups such as 2,2-diphenylethyl and cyclopropyl [12], and the combination of substituted N6-benzyl groups and various small substituents at the adenine 2-position (such as chloro, cyano, and methoxycarbonyl) [11, 13]. A radically altered analogue in which the properly functionalized adenine moiety was shifted from the 1′-position to the 4′-carbon proved to be an A3AR selective antagonist [9].
The aim of the present study is to expand knowledge of the structure activity relationships at the A3AR and at other subtypes, both in relation to binding affinity and intrinsic efficacy, of adenosine derivatives modified in the 2-position. In this manner, it will be possible to design new, selective A3AR agonists, partial agonists, and antagonists based on nucleoside structures. Derivatives of adenosine modified at the 2-position of the adenine ring have been studied in both binding and/or functional assays at the four AR subtypes. For this purpose, we have expressed the human ARs stably in Chinese hamster ovary (CHO) cells [14]. Most of these analogues are 2-ether substituted adenosine derivatives, which have been previously evaluated at the rat A1 and A2AARs but not at the four human subtypes in a systematic manner [15–20]. From previous studies [7, 13, 21] and in greatly expanded form in the present study, it is clear that the intrinsic efficacy of adenosine derivatives at the A3AR is dependent on structural changes at both the N6-position and the 2-position. The intrinsic efficacy at the A2AAR tended to be insensitive to the same structural changes. Additionally, here we have identified several substituents at the 2-position that contribute significantly to the A2BAR activity.
2. Materials and methods
2.1. Materials
[125I]N6-(4-Amino-3-iodobenzyl)adenosine-5′-N6-methyluronamide ([125I]I-AB-MECA; 2000 Ci/mmol), [3H]R-PIA (R-N6-[phenylisopropyl]adenosine, 34 Ci/mmol), [3H]CG-S21680 (2-[p-(2-carboxyethyl)phenylethylamino]-5′-N6-ethylcarboxamido-adenosine, 47 Ci/mmol) and [3H]cyclic AMP (40 Ci/mmol) were from Amersham Pharmacia Biotech (Buckinghamshire, UK). NECA, CGS21680, CPA, and R-PIA were purchased from Sigma-RBI (St. Louis, MO). Most of the 2-substituted adenosine derivatives examined were the kind gift of Dr. Ray A. Olsson (University of South Florida) and Dr. JohnW.Daly (NIDDK). Compound 24 was prepared as described [37]. Other chemicals were from standard commercial sources and of analytical grade.
2.2. Chemical synthesis
General synthetic procedure for substitution at the 2-position of adenosine (for compounds 21 and 44).
Adenosine derivatives were synthesized using the general method of Ueeda et al. [15]. A solution of appropriate alcohol or thiol (0.35 mmol) in 5 ml of dry 1,2-dimethoxyethane was cooled to 0 °C in an ice bath. To this solution was added 2.5 M n-BuLi (0.13 ml, 0.32 mmol), the reaction mixture was stirred for 1 h at 0 °C. The protected nucleoside 2-chloro-2′,3′-O-(ethoxymethylidene) adenosine (25 mg, 0.07 mmol) was added in one portion. The reaction mixture was refluxed for 4 days, at which time HPLC showed that starting material had almost completely disappeared.
The solvent was removed in vacuo, and a solution of the residue in 10 ml water was extracted with ethyl acetate (4 × 20 ml). The combined extracts were dried over Na2SO4 and evaporated in vacuo. The residue was purified with preparative thin layer chromatography (PTLC, silica gel) with the mobile phase consisting of mixtures of methanol (3% for 21 or 5% for 44, by volume) and chloroform. Fractions containing products were concentrated and dissolved in methanol–water. Acetic acid or trifluoroacetic acid was added and the solution refluxed until HPLC showed that the nucleoside was completely deblocked. The solution was adjusted to pH 9 with NH3–EtOH and was refluxed for 30 min. The solvent was removed, and the residue was purified with PTLC, with the mobile phase consisting of mixtures of methanol (20% for 21 or 10% for 44, by volume) and chloroform, to afford the corresponding product (overall yield 25–34% in two steps). Proton NMR and mass spectra were consistent with the assigned structures.
2.3. Cell culture and membrane preparation
The CHO cells stably expressing recombinant ARs were cultured in DMEM and F12 (1:1) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 µg/ml streptomycin, 2 µmol/ml glutamine and 800 µg/ml geneticin. After harvest and homogenization, cells were centrifuged at 500 × g for 10 min, and the pellet was re-suspended in 50 mM Tris–HCl buffer (pH 7.4) containing 10 mM MgCl2, 1 mM EDTA. The suspension was homogenized with an electric homogenizer for 10 s, and was then re-centrifuged at 20,000 × g for 20 min at 4 °C. The resultant pellets were resuspended in buffer in the presence of 3 units/ml adenosine deaminase, and the suspension was stored at −80 °C until the binding experiments. The protein concentration was measured using the Bradford assay [22].
2.4. Binding assays
For A3AR binding assays [23], each tube contained 100 ml of membrane suspension, 50 µl of [125I]I-AB-MECA (final concentration 0.5 nM), and 50 µl of increasing concentrations of compounds in Tris–HCl buffer (50 mM, pH 7.4) containing 10 mM MgC2. Nonspecific binding was determined using 10 (M NECA. The mixtures were incubated at 25 °C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandell, Gaithersburg, MD). Filters were washed three times with ice-cold buffer. Radioactivity was determined in a Beckman 5500B γ-counter. The binding of [3H]R-PIA to A1 receptors and the binding of [3H]CGS21680 to A2A receptors were as previously described [8].
2.5. Cyclic AMP accumulation assay
Intracellular cyclic AMP levels were measured with a competitive protein binding method [24]. CHO cells expressing four subtypes of recombinant ARs were harvested by trypsinization. After resupension in medium, cells were planted in 24-well plates in 0.5 ml medium. After 24 h, the medium was removed and cells were washed three times with 0.5 ml DMEM, containing 50 mM HEPES, pH 7.4. Cells were then treated with agonists and/or test compounds in the presence of rolipram (10 (M) and adenosine deaminase (3 units/ml). In the case of A1 and A3ARs, after 45 min forskolin (10 (M) was added to the medium, and incubation was continued an additional 15 min. The reaction was terminated by removing the medium, and cells were lysed upon the addition of 200 (L of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20 °C. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 µL of the cell lysate, and 30 µL 0.1 M HCl or 50 µL of cyclic AMP solution (0–16 pmol/200 µL for standard curve). Bound radioactivity was separated by rapid filtration through Whatman GF/C filters and washed once with cold buffer. Bound radioactivity was measured by liquid scintillation spectrometry.
2.6. Statistical analysis
Binding and functional parameters were estimated using Prism software (GraphPAD). IC50 values obtained from competition curves were converted to Ki values using the Cheng–Prusoff equation [25]. For an antagonist, Schild analysis was carried out as reported [26]. Data were expressed as mean ± standard error.
3. Results
3.1. Nucleoside structures examined
The classes of compounds examined included 2-alkyloxy ethers 1–18, 2-alkylaryl derivatives 19–46 including mainly ethers, however, an amino derivative 22 and a thioether 23 were included for comparison, two 5′,8-cyclo analogues 47 and 48, and four standard reference AR agonists 49–52. Although 47 and 48 were not substituted at the 2-position, they were included in this study, since conformational constraint in the ribose region has previously formed the basis of AR selectivity [27], and in these analogues the 2-position remains available for further derivatization.
Adenosine derivatives synthesized for this study were prepared by nucleophilic substitution by the appropriate alkoxy ion of the 2-chloro substituent in a ribose-protected derivative of 2-chloroadenosine, using methods similar to those reported previously [16].
3.2. Assays of binding and activation of ARs
Binding at the A1, A2A, and A3ARs was carried out using standard agonist radioligands [8, 23] in membranes of transfected CHO cells [14]. The activation of A1 and A3ARs (Gi-coupled) by the 2-substituted adenosine derivatives (Table 1) was examined by measuring the inhibition of forskolin-stimulated cyclic AMP accumulation in intact CHO cells stably expressing these receptors. The activation of A2A and A2BARs (Gs-coupled) in stably transfected CHO cells was measured in the absence of forskolin. The efficacy of each of these adenosine derivatives was evaluated at a fixed concentration of 10 (M and expressed as a percentage of the effect of a reference (full) agonist. In some cases, full concentration–response curves were measured. For compounds that bound weakly at a given AR, no indication of intrinsic efficacy was readily obtainable, however for the many examples of high affinity binding the degree of activation at 10 (M served as an approximate measure of intrinsic efficacy.
Table 1.
Binding affinities of adenosine derivatives at human A1, A2A, and A3ARs expressed in CHO cells (expressed as Ki value or percent displacement at 10 (M) and maximal agonist effects at 10 (M (% of full agonist) at the ARs
| Compound | 2-Substitution |
Ki at A1AR (nMa) (% activation) |
Ki at A2AAR (nMa) (% activation) |
% activation at A2BARa (and % inhibition), unless noted |
Ki at A3AR (nMa) (% activation) |
|---|---|---|---|---|---|
| 2-Alkoxy analogues | |||||
| 1 | CH3O | 155 ± 32 (119) | 970 ± 310 (78.6) | −2.7 ± 5.3 (−0.2) | 156 ± 37 (75.2 ± 5.1) |
| 2 | CH3CH2O | 2640 ± 540 (81.3) | 360 ± 139 (92.4) | −0.8 ± 1.9 (−6.7) | 568 ± 205 (99.1 ± 4.2) |
| 3 | (CH3)2CHO | 16% (15.9) | 927 ± 204 (87.5) | 0.4 ± 2.9 (−3.2) | 457 ± 154 (101 ± 5) |
| 4 | (CH3)2CHCH2O | 4410 ± 1150 (44.6) | 222 ± 89 (93.2) | −1.2 ± 8.8 (−3.4) | 84.2 ± 8.4 (108 ± 10) |
| 5 | (CH3CH2)2CHCH2O | 42% (1.0) | 45.8 ± 22.1 (90.0) | −4.4 ± 4.2 (2.0) | 336 ± 116 (4.9 ± 5.2) |
| 6 | Cyclohexyl-CH2O | 3350 ± 390 (27.5) | 342 ± 22 (92.8) | −0.9 ± 2.3 (−4.7) | 143 ± 32 (27.1 ± 4.9) |
| 7 | (CH3)2CH(CH2)2O | 3560 ± 1120 (29) | 37.7 ± 4.9 (92.4) | −0.6 ± 1.8 (−8.4) | 81.1 ± 9.0 (96.3 ± 3.0) |
| 8 | Cyclohexyl-(CH2)2O | 36% (27.8) | 579 ± 250 (102) | −4.6 ± 8.8 (0.4) | 578 ± 182 (51.6 ± 3.4) |
| 9 | 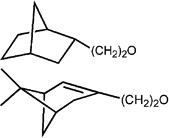 |
3590 ± 670 (0) | 137 ± 31 (98.6) | −0.1 ± 2.6 (2.8) | 149 ± 45 (−3.5 ± 9.0) |
| 10 | 3350 ± 720 (25.9) | 21.2 ± 12.7 (97.7) | −0.5 ± 4.4 (2.8) | 341 ± 132 (−0.2 ± 5.9) | |
| 11 | (CH3)2CH(CH2)3O | 3700 ± 790 (18.3) | 77.8 ± 20.9 (96.4) | −1.7 ± 2.8 (0.0) | 105 ± 31 (49.6 ± 8.1) |
| 12 | Cyclohexyl-(CH2)3O | 1730 ± 330 (33.5) | 92.0 ± 52.5 (105) | −2.5 ± 4.6 (2.2) | 83.3 ± 8.4 (21.2 ± 11.8) |
| 13 | Cyclohexyl-(CH2)4O | 883 ± 99 (109) | 291 ± 73 (98.5) | −0.27 ± 2.8 (−0.5) | 105 ± 13 (12.7 ± 1.5) |
| 14 | CH3(CH2)4O | 2430 ± 620 (48.7) | 6.9 ± 1.3 (94.1) | −2.2 ± 2.6 (9.9) | 222 ± 68 (92.8 ± 13.4) |
| 15 | CH3C≡C–(CH2)2O | 583 ± 78 (49.8) | 63.2 ± 50.3 (95.9) | 1.4 ± 3.7 (−7.9) | 90.2 ± 36.2 (73.4 ± 4.4) |
| 16 | CH3(CH2)5O | 1830 ± 340 (11.8) | 156 ± 89 (100) | 12.6 ± 0.5 (−6.5) | 124 ± 28 (43.7 ± 6.3) |
| 17 | (CH3)2C=CH(CH2)2CH(R-CH3)(CH2)2O | 34% (23.6) | 382 ± 100 (91.3) | 0.3 ± 3.4 (−5.0) | 431 ± 130 (−1.5 ± 1.5) |
| 18 | (CH3)2C=CH(CH2)2CH(S-CH3)(CH2)2O | 4550 ± 950 (21.3) | 78.3 ± 10.3 (90.2) | −0.4 ± 1.7 (−1.7) | 74.4 ± 22.5 (31.5 ± 6.4) |
| 2-Aryl- and arylalkyloxo (or amino) | |||||
| 19 | Phenyl-O | 5140 ± 1110 (57.6) | 44 ± 12 (100) | 0.7 ± 1.8 (−0.9) | 364 ± 96 (32.2 ± 3.5) |
| 20 | Benzyl-O | 642 ± 79 (11.5) | 585 ± 155 (85.1) | −3.3 ± 0.6 (9.5) | 117 ± 8 (16.9 ± 3.9) |
| 21 | 3-Chlorobenzyl-O | 27.4 ± 3.9 (46) | 228 ± 66 | 7.4 ± 2.1 | 71.6 ± 24.6 (16.2 ± 3.8) |
| 22 | Phenyl-(CH2)2O | 221 ± 57 (112) | 9.3 ± 2.9 (99.6) | 3490 ± 1490c (0.0) | 54.2 ± 14.3 (70.7 ± 2.7) |
| 23 | Phenyl-(CH2)2NH | 530 ± 88 (70.5) | 62.0 ± 17.6 (105) | −1.7 ± 3.1 (−6.9) | 310 ± 163 (72.0 ± 3.2) |
| 24 | Phenyl-(CH2)2S | 3700 ± 770 | 590 ± 260 | ND | 1960 ± 310 |
| 25 | 2-Methylphenyl-(CH2)2O | 396 ± 83 (74.6) | 17.4 ± 7.4 (110) | −1.0 ± 2.7 (3.8) | 214 ± 47 (7.3 ± 6.0) |
| 26 | 3-Methylphenyl-(CH2)2O | 295 ± 8 (106) | 41.6 ± 22.0 (98.4) | 15.3 ± 2.5 (0.6) | 242 ± 55 (70.9 ± 3.9) |
| 27 | 4-Methylphenyl-(CH2)2O | 1250 ± 250 (61.8) | 118 ± 95 (98.3) | 12.1 ± 1.9 (−12.2) | 470 ± 81 (95.5 ± 15.1) |
| 28 | 2-Methyloxyphenyl-(CH2)2O | 490 ± 114 (111) | 274 ± 142 (94.2) | 7.4 ± 1.3 (0.6) | 940 ± 354 (3.6 ± 6.7) |
| 29 | 3-Methyloxyphenyl-(CH2)2O | 246 ± 34 (114) | 32.1 ± 1.6 (103) | 0.4 ± 4.1 (0.0) | 231 ± 53 (85.1 ± 5.8) |
| 30 | 4-Methyloxyphenyl-(CH2)2O | 288 ± 22 (109) | 64.3 ± 7.8 (97.6) | 11.3 ± 1.8 (−4.4) | 105 ± 20 (91.1 ± 9.2) |
| 31 | 3,4-Dimethyloxyphenyl-(CH2)2O | 469 ± 118 (101) | 30.3 ± 2.8 (99.8) | −1.4 ± 2.1 (6.2) | 863 ± 313 (66.5 ± 3.7) |
| 32 | 2-Fluorophenyl-(CH2)2O | 331 ± 22 (18.9) | 58.1 ± 24.9 (99.1) | 16.6 ± 2.5 (0.3) | 77.8 ± 13.5 (44.5 ± 5.1) |
| 33 | 4-Fluorophenyl-(CH2)2O | 467 ± 100 (115) | 56.8 ± 16.3 (97.9) | 17.3 ± 3.1 (−7.2) | 112 ± 16 (73.4 ± 5.2) |
| 34 | 2-Chlorophenyl-(CH2)2O | 366 ± 33 (31.8) | 17.9 ± 6.1 (94.9) | 1.0 ± 3.4 (6.9) | 144 ± 22 (1.4 ± 2.7) |
| 35 | 3-Chlorophenyl-(CH2)2O | 372 ± 116 (85.3) | 11.5 ± 5.3 (96.7) | 28.0 ± 4.9 (2.9) | 41.0 ± 7.8 (31.0 ± 7.0) |
| 36 | 4-Chlorophenyl-(CH2)2O | 331 ± 51 (113) | 58.5 ± 8.0 (97.4) | 17.5 ± 2.8 (5.7) | 116 ± 23 (69.0 ± 6.3) |
| 37 | R-2-Phenylbutyl-O | 28% (24.5) | 503 ± 98 (97.7) | −0.2 ± 6.0 (4.6) | 201 ± 61 (−1.6 ± 3.8) |
| 38 | S-2-Phenylbutyl-O | 4780 ± 990 (16.0) | 26.9 ± 6.9 (96.0) | 0.4 ± 5.6 (−5.5) | 175 ± 31 (−3.9 ± 3.9) |
| 39 | 2-(1-Naphthyl)ethyl-O | 220 ± 18 (101) | 3.8 ± 1.4 (102) | −2.1 ± 5.1 (−3.7) | 205 ± 19 (12.8 ± 5.9) |
| 40 | 2-(2-Naphthyl)ethyl-O | 141 ± 51 (102) | 16.1 ± 7.0 (105) | 1440 ± 70c | 130 ± 8 (45.1 ± 8.5) |
| 41 | 2,2-Diphenylethyl-O | 39% (10.4) | 310 ± 119 (97.5) | −1.8 ± 5.6 (0.8) | 53.6 ± 10.4 (−0.0 ± 0.6) |
| 42 | 2-(2-Thienyl)ethyl-O | 174 ± 20 (112) | 10.9 ± 4.8 (105) | 1780 ± 260c | 93.3 ± 16.8 (79.7 ± 4.5) |
| 43 | 2-(3-Thienyl)ethyl-O | 280 ± 72 (117) | 13.3 ± 4.1 (106) | 8.9 ± 5.5 (−2.0) | 101 ± 34 (61.6 ± 15.1) |
| 44 | trans-2-Phenylcyclopropyl-O | 367 ± 27 (19.1) | 2050 ± 700 | 1.2 ± 2.3 | 292 ± 46 (0.4 ± 1.6) |
| 45 | 4-Phenylbutyl-O | 1100 ± 230 (24.6) | 243 ± 166 (103) | −0.4 ± 2.3 (−7.8) | 251 ± 80 (19.3 ± 6.2) |
| 46 | 5-Phenylpentyl-O | 700 ± 126 (104) | 249 ± 54 (101) | 6.2 ± 3.6 (5.4) | 429 ± 159 (9.8 ± 8.8) |
| 5′,8-Cyclo analogues | Name | ||||
| 47 | Cyclo-CPAb | 418 ± 62 (117) | 12% (11.7) | −0.6 (−2.9) | 25% (11.7 ± 0.3) |
| 48 | Cyclo-R-PIAb | 49% (26.3) | 18% (27.6) | 4.5 (−9.6) | 27% (1.7 ± 1.7) |
| Reference compounds | Name | ||||
| 49 | CPA | 1.8 ± 0.5 (112) | 820 ± 216 (98) | 8.6 ± 2.9 (0.0) | 72 ± 12 (99 ± 6) |
| 50 | R-PIA | 2.0 ± 0.3 (101) | 884 ± 188 (101) | 1680 ± 498c | 8.7 ± 0.9 (102 ± 6) |
| 51 | CGS21680 | 1570 ± 460 (99) | 8.8 ± 1.6 (100) | 4.7 ± 1.8 (0.0) | 114 ± 16 (98 ± 5) |
| 52 | NECA | 6.8 ± 2.4 (100) | 2.2 ± 0.6 (100) | 140 ± 19c (0.0) | 16.0 ± 5.4 (100) |
At the A2BAR, EC50 values or the percent stimulation at 1 (M (and in parentheses percent inhibition at 1 (M of the effects of 100 nM NECA) are shown.
All experiments were performed using adherent CHO cells stably transfected with cDNA encoding a human adenosine receptor. Percent activation of the human A1 A2A and A3AR was determined at 10 µM. Binding at A1, A2A and A3ARs was carried out as described in Section 2. The A1 and A2AAR activation results were expressed as the mean values from two separate experiments, while the A3AR activation results were from three separate experiments. The Ki and EC50 values from the present study are means ± S.E.M., N = 3–5.
Structures shown in Fig. 4; ND: not determined.
EC50 (nM) for activation of the A2BAR.
3.3. Affinity and potency at the A3AR
Various 2-substituents were found to be critical structural determinants for activation of the A3AR. Less than maximal efficacy or lack of efficacy was frequently observed for the 2-modified adenosine derivatives at the A3AR. The following adenosine 2-ethers were moderately potent partial agonists (Ki): benzyl 20 (117 nM), 2-(3-chlorophenyl)ethyl 35 (41 nM), and 2-(2-naphthyl)ethyl 40 (130 nM). Fig. 1 shows the concentration–response curves for inhibition of cyclic AMP accumulation by a variety of 2-position ethers, indicating the variation in efficacy.
Fig. 1.

Inhibition of forskolin-stimulated cyclic AMP production in CHO cells stably transfected with the human A3AR, induced by various agonists. All experiments were performed in the presence of 10 µM rolipram and 3 units/ml adenosine deaminase. Forskolin (10 (M) was used to stimulate cyclic AMP levels. The data shown were from one experiment performed in duplicate and are typical of three independent experiments giving similar results. EC50 values were (nM): NECA, 20.0 ± 4.5; 2-isobutyloxy derivative 4, 150 ± 32; 2-S-(3,7-dimethyl)oct-6-enyloxy derivative 18, 187 ± 66; 2-benzyloxy derivative 20, 160 ± 40; 2-S-(2-phenyl)butyloxy derivative 38, not applicable.
The following adenosine 2-ethers were A3AR antagonists: 2,2-diphenylethyl 41, R-2-phenylbutyl 37, S-2-phenylbutyl 38, 2-(2-chlorophenyl)ethyl 34 and 2-nor-zbornylethyl 9. 2-(S-2-Phenylbutyloxy)adenosine 38 right-shifted the concentration–response curve for the inhibition by NECA of cyclic AMP accumulation with a KB value of 212 nM (Fig. 2) calculated by Schild analysis [26], which is similar to its A3AR affinity (Ki = 175 nM) determined in the binding assay.
Fig. 2.

Antagonism by compound 38 of the inhibition of cyclic AMP production elicited by NEC in CHO cells stably transfected with the human A3AR (A) and Schild analysis of the data (B). The experiment was performed in the presence of 10 µM rolipram and 3 units/ml adenosine deaminase. Forskolin (10 (M) was used to stimulate cyclic AMP levels. The level of cAMP corresponding to 100% was 220 ± 30 pmol ml−1. The KB value for compound 38 was calculated to be 212 nM.
Among small 2-alkyloxy groups, efficacy at the A3AR remained nearly full with increasing size until the branched hexyl derivative 5. Branching of the O-hexyl group more distally in 11 did not alter affinity, but increased efficacy. The straight-chain pentyl 14 and hexyl 16 ether derivatives were full and partial agonists, respectively. In the series of O-alkylcyclohexyl derivatives of varying chain length 6, 8, 12, and 13, all were partial agonists at the A3AR with similar affinity (100–600 nM), but with efficacy diminishing as chain length increased beyond two methylenes. A diastereomeric pair of branched decenyl ethers 17 and 18 differed greatly in both affinity (see below) and efficacy at the A3AR.
The series of phenyl 19 and phenylalkyl (20, 22, 45, and 46) ethers showed high A3AR efficacy only at intermediate length, i.e. the 2-phenylethyl ether 22, which also demonstrated the highest A3AR affinity among the five analogues with a Ki value of 54 nM. Nevertheless, 22 was not selective for the A3AR. The benzyl ether 20 was modestly potent as a partial agonist at the A3AR and slightly selective (5–6-fold) in comparison to A1 and A2AARs. The 3-chlorobenzyl ether 21 was approximately equipotent to 20 at the A3AR with a Ki value of 71.6 nM, but 23-fold more potent in A1AR binding. The 2-phenylethyl amino derivative 23 was less potent than corresponding O-ether 22 at all AR subtypes, with Ki values at the A3AR of 310 and 54 nM, respectively. The corresponding 2-phenylethylthio derivative 24 was less potent at three AR subtypes. Thus, the 2-O-ether linkage was selected for further modification.
Numerous modifications were included on the 2-phenylethyl moiety of ether 22, including phenyl substitution with methyl 25–27, methyloxy 28–31, and halo 32–36. In a number of these cases (25, 28, and 34), the 2-substitution of the phenyl ring resulted in the greatest reduction of A3AR efficacy. Methoxy substitution at both 3- and 4-positions of the phenyl ring in 31 reduced A3AR affinity in comparison of substitution of either position alone (i.e. 29, 30). Substitution of the phenylethyl ring with groups having electron withdrawing character (i.e. F) versus donating (i.e. methoxy or methyl) had little effect on A3AR affinity, however the efficacy of 3- and 4-substituted halo derivatives tended to be less than the corresponding methoxy or methyl derivatives. Also, the phenyl ring was substituted with other ring systems, as in 39, 40, and 42–44, all of which had reduced efficacy at the A3AR. Substitution at the 2-position of the alkyl (ethyl) group of 22 with ethyl (the diastereomers 38 and 38) or with phenyl 41 completely abolished efficacy at the A3AR.
3.4. Affinity and potency at the A1AR
These 2-substituted adenosine derivatives were generally less potent at the A1AR than at the A3AR with binding Ki values over 100 nM. Among the most potent derivatives at the (Ki < 200 nM) were 1, 40, 42 and 44. Among the two 5′,8-cyclo derivatives 47 and 48, only moderate affinity was observed for the N6-cyclopentyl analogue 47 at the A1AR. Compound 47 was a somewhat selective A1AR agonist.
Those 2-modified compounds with high to moderate affinity at the A1AR tended to be fully efficacious at that subtype. However, the alkynyl ether 15, 2-phenylethyl amine 23, 2-methoxyphenylethyl ether 25, 2-fluorophenylethyl ether 32, and 2-chlorophenylethyl ether 34 were not fully efficacious at the A1AR.
3.5. Affinity and potency at the A2AAR
Consistent with previous studies [15, 16], the 2-substituted ether derivatives of adenosine were generally fully efficacious at the A2AAR. A number of substitutions at the 2-position, which were previously found to contribute to the affinity for the rat A2AAR [20, 28, 29], were also demonstrated to be important for the affinity and selectivity at the human A2AAR homologue. A single substitution at the 2-position might contribute significantly to both A2AAR affinity and selectivity. For example, 2-[2-(l-naphthyl) ethyloxy]adenosine 39 was found to be the most potent (Ki = 3.8 nM) and selective (>50-fold) A2A agonist in this series. Thus, 39 was roughly as potent as an A2AAR agonist as the nonselective agonist NECA 52. The 2-naphthyl isomer 40 was also potent at the A2AAR.
3.6. Potency at the A2BAR
All of nucleosides were tested at a fixed concentration for the ability to stimulate or inhibit adenylate cyclase mediated by the A2BAR, and activation curves were determined for only a few compounds (Fig. 3). Since an agonist radioligand for the A2BAR is not yet available, the binding affinities of these nucleosides at this subtype have not been determined. Interestingly, although most of these compounds were extremely weak at the A2BAR, 2-[2-(2-naphthyl)ethyloxy]adenosine 40 (EC50 = 1.44 (M) and 2-[2-(2-thienyl)ethyloxy]adenosine 42 (EC50 = 1.78 (M) were found to be moderately potent A2BAR agonists, although less potent than NECA (EC50 = 140 nM). Another 2-ether having substantial potency at the A2BAR was the 2-phenylethyl ether 22, which displayed an EC50 value of 3.49 (M. At the A2AAR 39 was a more potent agonist than 40, while the opposite order was seen at the A2BAR.
Fig. 3.

Stimulation of cyclic AMP production elicited by NECA and compounds 39 and 40 in CHO cells stably transfected with the human A2BAR. The experiment was performed in the presence of 10 (M rolipram and 3 units/ml adenosine deaminase. The data shown were from one experiment performed in duplicate and are typical of three independent experiments giving similar results. The EC50 values were listed in Table 1.
4. Discussion
Although in the present study we have not identified highly selective ligands for a given AR, we have found that substitution at the 2-position greatly modulates the pharmacological characteristics at the A3AR. Thus, the aim of the study has been partially achieved, and in future studies, specific 2-ether groups identified here may be studied in combination with other modifications of adenosine known to provide subtype selectivity. Until present, the modifications of the 2-position to be included in A3AR selective agonists have been somewhat limited (e.g. chloro, methylthio, and alkynyl groups). The SAR of alkynyl groups at the 2-position based on affinity at several subtypes of ARs has been explored [21, 29].
Several of the nucleosides studied here were found to have Ki values at the A3AR of approximately 50 nM and compared favorably in selectivity to other analogues of adenosine in which only single sites have been modified [11–13, 21]. For example, compound 41 was >100-fold selective for the A3AR in comparison to both the A1 and A2BARs. Alkyl ethers 4 and 7 were moderately selective full agonists of the A3AR in comparison to the A1AR. Compounds 13 and 20 were slightly selective for the A3AR in comparison to the A1AR and other subtypes.
Similar to previous studies of the N6-position [8] and the ribose moiety [9, 30, 31], structural determinants at the 2-position of adenosine have been found to be critical for A3AR recognition and activation. Fig. 4 shows the structures of selected analogues having A3AR selectivity in comparison to at least two other AR subtypes and/or reduced efficacy at the A3AR and the 5′,8-cyclo analogues (47 and 48). A striking dependence of the A3AR efficacy on relatively minor structural changes of the 2-ether group, for example, substitution of 2-O-phenylalkyl ethers, is illustrated. The benzyl ether 20 is a low efficacy partial agonist. Homologation to give 22 restored most of the ability to activate the A3AR, however simple substitution of this ring in 34 and 35 or additional aryl substitution in 37, 39, and 41 greatly reduced the intrinsic efficacy. The alkyl ether 7 is structurally related to 18, however, the efficacy at the A3AR was greatly reduced upon chain elaboration.
Fig. 4.

Representative adenosine derivatives examined in this study. Compounds having A3AR selectivity in comparison to at least two other AR subtypes (4, 7, 35, 37, and 41) and/or reduced efficacy (2-O-benzyl ether 20 and others) at the A3AR are included. A striking dependence of the efficacy on the position of substitution of 2-O-phenylethyl ethers (22 in comparison to 34, 35, 37, 37, and 41) is illustrated. The full agonism of alkyl ethers (4 and 7) is in contrast to the branched decenyl ether 18, which is a partial agonist. 5′,8-Cyclo analogues (47 and 48) of known adenosine agonists CPA and R-PIA are also shown. The percent of maximal activation of the human A3AR at 10 (M is indicated, where applicable.
In binding experiments, typically these 2-substituted nucleosides displaced AR radioligand binding with the following order of potency: A2A > A3 > A1. The benzyl ether 20 displayed selectivity (5–6-fold) for the A3AR. Mixed A2A/A3AR agonists have been identified. For example, 7 was a mixed agonist with full A3AR intrinsic efficacy. Compounds 11, 12, and 18 were mixed agonists, but with partial intrinsic efficacy at the human A3AR. Finally, the 2-norbornanethyl ether 9, the branched decenyl ether 17, and the 2-phenylbutyl ethers 37 and 38 were full agonists at the A2AAR and antagonists at the human A3AR, with affinities roughly matched at the two subtypes and having selectivity over the A1 and A2B ARs. Nucleosides with combined action in activating the A2AAR and antagonizing the A3AR may be of use in treating inflammation. Several arylethyloxy substituents at the 2-position were found to contribute significantly to the A2BAR activity, for which there are not yet any selective agonists although the SAR has been explored [32, 33].
In the absence of a physically-determined structure for the A3AR, the use of molecular modeling [12, 13, 34] will be necessary to understand the structural basis for the loss of efficacy associated with certain 2-ether groups, such as benzyl, 2,2-diphenylethyl, and S-2-phenylbutyl. There were parallels between the effects on A3AR binding and activation of the same substitution at either the 2-position (in the form of an ether) or at the N6-position (in the form of a secondary arylamine). For example, 2-(2,2-diphenylethyloxy)-adenosine and N6-(2,2-diphenylethyl)adenosine were both antagonists of the A3AR [12].
Also, in the series of 2-phenylalkylethers, the benzyl ether had more favorable affinity particularly at the A3AR in comparison to other subtypes and was a partial agonist. The same was observed for N6-benzyladenosine derivatives [11]. This raises the possible explanation, already invoked by Olsson and coworkers [16] prior to any knowledge of the receptor binding site, that these two substitutions might be overlayed in their receptor-bound positions [13, 34]. However, the trans-2-phenylcyclopropyl group at the N6-position resulted in high A3AR affinity as an agonist, while at the 2-ether position (44), only moderate affinity was observed with no activation of the A3AR.
Thus, we have demonstrated that affinity at the A3AR may be enhanced by modifying the nucleoside in a systematic fashion at a position that was not previously explored for ether groups. The 2-ether modification of adenosine, previously known as a means of enhancing potency at the A2AAR, has now been shown to variably enhance potency at the A3AR. Typically, adenosine 2-benzyl and 2-phenylethyl ethers showed favorable binding affinity at the A3AR and, depending on substitution, ranged from agonists to partial agonists to antagonists at this receptor. The 2-substituted adenosine derivatives examined in this study were generally less potent at the A1AR in comparison to the A3AR, but fully efficacious. However, a single substitution at the 2-position could also lead to a potent and selective A1AR agonist. For example, compound 21 is somewhat selective for the A1AR. Mixed A2A/A3AR agonists have been identified. Additionally, we have identified several substituents at the 2-position that contribute significantly to the A2BAR activity.
In conclusion, a number of novel structural determinants for the A3AR activation have been identified. Given the interest in A3AR agonists as antiischemic agents [35] and antagonists as antiglaucoma agents [36] and for other therapeutic applications, there is need for additional selective ligands to interact with the receptor. Presently, only one A3AR agonist (IB-MECA) is in clinical trials [6], and the drug-like properties of most of the analogues in this study are unexplored. Selective agonists and antagonists, especially those whose selectivity extend across species, are needed both as receptor probes for research and as clinical candidates. In some cases, nucleoside AR ligands of mixed selectivity (e.g. mixed A1/A3AR agonists for cardioprotection) would be desirable for a particular clinical application. Structural insights gained in the present study may now be extended to multiple substitutions of adenosine.
Acknowledgements
We are grateful to Prof. Ray A. Olsson and Dr. John Daly for the gift of adenosine agonists and for helpful discussions. We thank Dr. Gary Stiles (Duke University) for the gifts of CHO cells expressing the human and rat A3 receptors. L.M. thanks the Gilead Sciences (Foster City, CA) for financial support. We acknowledge the Guthrie cDNA Resource Center (www.cdna.org) as the source of the cDNA for the A2BAR.
Abbreviations
- AR
adenosine receptor
- CHO
Chinese hamster ovary
- CPA
N6-cyclopentyladenosine
- DMEM
Dulbeccos modified Eagles medium
- I-AB-MECA
N6-(4-amino-3-iodobenzyl)-5′-N-methylcarboxa-midoadenosine
- NECA
5′-N-ethylcarboxamidoadenosine
- PIA
N6-phenylisopropyladenosine
- PTLC
preparative thin layer chromatography
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 2.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 3.von Lubitz DKJE, Lin RC-S, Popik P, Carter MF, Jacobson KA. Adenosine A3receptor stimulation and cerebral ischemia. Eur J Pharmacol. 1994;263:59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu GS, Richards SC, Olsson RA, Mullane K, Walsh RS, Downey JM. Evidence that the adenosine A3receptor may mediate the protection afforded by preconditioning in the isolated rabbit heart. Cardiovasc Res. 1994;28:1057–1061. doi: 10.1093/cvr/28.7.1057. [DOI] [PubMed] [Google Scholar]
- 5.Strickler J, Jacobson KA, Liang BT. Direct preconditioning of cultured chick ventricular myocytes: novel functions of cardiac adenosine A2Aand A3receptors. J Clin Invest. 1996;98:1773–1779. doi: 10.1172/JCI118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fishman P, Bar-Yehuda S, Ohana G, Barer F, Ochaion A, Erlanger A, et al. An agonist to the A3adenosine receptor inhibits colon carcinoma growth in mice via modulation of GSK-3 beta and NF-kappa B. Oncogene. 2004;23:2465–2471. doi: 10.1038/sj.onc.1207355. [DOI] [PubMed] [Google Scholar]
- 7.van Tilburg EW, van der Klein PA, von Frijtag Drabbe Kiinzel J, de Groote M, Stannek C, Lorenzen A, et al. 5′-O-Alkyl ethers of N,2-substituted adenosine derivatives: partial agonists for the adenosine A1and A3receptors. J Med Chem. 2001;44:2966–2975. doi: 10.1021/jm001114o. [DOI] [PubMed] [Google Scholar]
- 8.Gao ZG, Blaustein JB, Gross AS, Melman N, Jacobson KA. N6-Substituted adenosine derivatives: selectivity, efficacy, and species differences at A3adenosine receptors. Biochem Pharmacol. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao ZG, Jeong LS, Moon HR, Kim HO, Choi WJ, Shin DH, et al. Structural determinants of efficacy at A3adenosine receptors: modification of the ribose moiety. Biochem Pharmacol. 2004;67:893–901. doi: 10.1016/j.bcp.2003.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao ZG, Jacobson KA. Partial agonists for A3adenosine receptors. Curr Top Med Chem. 2004;4:855–862. doi: 10.2174/1568026043450989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, et al. Structural determinants of A3adenosine receptor activation: nucleoside ligands at the agonist/antagonist boundary. J Med Chem. 2002;45:4471–4484. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tchilibon S, Kim SK, Gao ZG, Harris BA, Blaustein JB, Gross AS, et al. Exploring distal regions of the A3adenosine receptor binding site: Sterically-constrained N6-(2-phenylethyl)adenosine derivatives as potent ligands. Bioorg Med Chem. 2004;12:2021–2034. doi: 10.1016/j.bmc.2004.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohno M, Gao ZG, van Rompaey P, Tchilibon S, Kim SK, Harris BA, et al. Modulation of adenosine receptor affinity and intrinsic efficacy in adenine nucleosides substituted at the 2-position. Bioorg Med Chem. 2004;12:2995–3007. doi: 10.1016/j.bmc.2004.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, et al. Comparative pharmacology of human adenosine receptor subtypes— characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 15.Ueeda M, Thompson RD, Arroyo LH, Olsson RA. 2-Aralkoxyadenosines: potent and selective agonists at the coronary artery A2 adenosine receptor. J Med Chem. 1991;34:1340–1344. doi: 10.1021/jm00108a015. [DOI] [PubMed] [Google Scholar]
- 16.Ueeda M, Thompson RD, Arroyo LH, Olsson RA. 2-Alkoxyadenosines: potent and selective agonists at the coronary artery A2 adenosine receptor. J Med Chem. 1991;34:1334–1339. doi: 10.1021/jm00108a014. [DOI] [PubMed] [Google Scholar]
- 17.Makujina SR, Olsson RA, Esinhart JD, Mustafa SJ. Structure-activity relationship of 2-(ar) alkoxyadenosines at the adenosine A2 receptor in coronary artery. Eur J Pharmacol. 1993;243:35–38. doi: 10.1016/0014-2999(93)90164-d. [DOI] [PubMed] [Google Scholar]
- 18.Cristalli G, Vittori S, Thompson RD, Padgett WL, Shi D, Daly JW, et al. Inhibition of platelet aggregation by adenosine receptor agonists. Naunyn Schmiedeberge Arch Pharmacol. 1994;349:644–650. doi: 10.1007/pl00004904. [DOI] [PubMed] [Google Scholar]
- 19.Daly JW, Padgett WL, Secunda SI, Thompson RD, Olsson RA. Structure-activity relationships for 2-substituted adenosines at A1and A2adenosine receptors. Pharmacology. 1993;46:91–100. doi: 10.1159/000139033. [DOI] [PubMed] [Google Scholar]
- 20.Kull B, Arslan G, Nilsson C, Owman C, Lorenzen A, Schwabe U, et al. Differences in the order of potency for agonists but not antagonists at human and rat adenosine A2Areceptors. Biochem Pharmacol. 1999;57:65–75. doi: 10.1016/s0006-2952(98)00298-6. [DOI] [PubMed] [Google Scholar]
- 21.Volpini R, Costanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, et al. N6-Alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3receptor and a starting point for searching A2Bligands. J Med Chem. 2002;45:3271–3279. doi: 10.1021/jm0109762. [DOI] [PubMed] [Google Scholar]
- 22.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 23.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. 125I-4-Aminobenzyl- 5′- N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3adenosine receptor. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 24.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 25.Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 26.Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobson KA, Ji d, Li AH, Melman N, Siddiqui MA, Shin KJ, et al. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J Med Chem. 2000;43:2196–2203. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutchison AJ, Williams M, de Jesus R, Yokoyama R, Oei HH, Ghai GR, et al. 2-(Arylalkylamino)adenosine-5′-uronamides: a new class of highly selective adenosine A2 receptor ligands. J Med Chem. 1990;33:1919–1924. doi: 10.1021/jm00169a015. [DOI] [PubMed] [Google Scholar]
- 29.Matsuda A, Shinozaki M, Yamiguchi T, Homma H, Nomoto R, Miyasaka T, et al. 2-Alkynyladenosines: A novel class of selective A2 receptor agonists with potential antihypertensive effects. J Med Chem. 1992;35:241–252. doi: 10.1021/jm00080a007. [DOI] [PubMed] [Google Scholar]
- 30.Jeong LS, Jin DZ, Kim HO, Shin DH, Moon HR, Gunaga P, et al. N6- Substituted D-4′-thioadenosine-5′-methyluronamides: potent and selective agonists at the human A3adenosine receptor. J Med Chem. 2003;46:3775–3777. doi: 10.1021/jm034098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim MH, Kim HO, Moon HR, Lee SJ, Chun MW, Gao ZG, et al. Design, synthesis and binding affinity of 3′-fluoro analogues of Cl-IBMECA as adenosine A3receptor ligands. Bioorg Med Chem Lett. 2003;13:817–820. doi: 10.1016/s0960-894x(03)00027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Zwart M, Link R, von Frijtag Drabbe Künzel JK, Cristalli G, Jacobson KA, Townsend-Nicholson A, et al. A screening of adenosine analogues on the human adenosine A2Breceptor as part of a search for potent and selective agonists. Nucleosides Nucleotides. 1998;17:969–986. doi: 10.1080/07328319808004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vittori S, Costanzi S, Lambertucci C, Portino FR, Taffi S, Volpini R, et al. A2Badenosine receptor agonists: synthesis and biological evaluation of 2-phenylhydroxypropynyl adenosine and NECA derivatives. Nucleosides Nucleotides Nucleic Acids. 2004;23:471–481. doi: 10.1081/ncn-120028340. [DOI] [PubMed] [Google Scholar]
- 34.Kim SK, Gao ZG, Van Rompaey P, Gross AS, Chen A, Van Calenbergh S, et al. Modeling the adenosine receptors: comparison of binding domains of A2Aagonist and antagonist. J Med Chem. 2003;46:4847–4859. doi: 10.1021/jm0300431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, et al. 3′-Aminoadenosine-5′-uronamides: discovery of the first highly selective agonist at the human adenosine A3receptor. J Med Chem. 2003;46:353–355. doi: 10.1021/jm0255724. [DOI] [PubMed] [Google Scholar]
- 36.Civan MM, Macknight AD. The ins and outs of aqueous humour secretion. Exp Eye Res. 2004;78:625–631. doi: 10.1016/j.exer.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 37.Fischer B, Boyer JL, Hoyle CHV, Ziganshin AU, Brizzolara AL, Knight GE, et al. Identification of potent, selective P2Y-purinoceptor agonists: structure activity relationships for 2-thioether derivatives of adenosine-5′-triphosphate. J Med Chem. 1993;36:3937–3946. doi: 10.1021/jm00076a023. [DOI] [PMC free article] [PubMed] [Google Scholar]