

Abstract
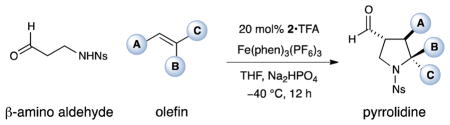
A new method to rapidly generate pyrrolidines via a SOMO-activated enantioselective (3 + 2) coupling of aldehydes and conjugated olefins has been accomplished. A radical-polar crossover mechanism is proposed wherein olefin addition to a transient enamine radical cation and oxidation of the resulting radical furnishes a cationic intermediate which is vulnerable to nucleophilic addition of a tethered amine group. A range of olefins, including styrenes and dienes, are shown to provide stereochemically complex pyrrolidine products with high chemical efficiency and enantiocontrol.
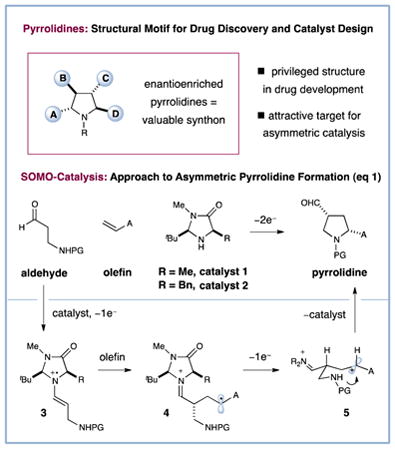
The pyrrolidine ring system is central to a broad range of bioactive natural products,1 is increasingly present in pharmaceutical agents,2 and recently has become ubiquitous in catalysis, finding use as organocatalysts as well as ligands for a wide range of metal-mediated enantioselective protocols.3 Not surprisingly, this high value heterocyclic framework has become an attractive target for new reaction invention,4 with (3 + 2) cycloadditions5 and the aza-Cope Mannich rearrangement6 providing elegant solutions to access stereochemically complex variants. Recently, we sought to develop a new catalytic approach to pyrrolidines via the modular combination of β-amino aldehydes and simple olefins using an enantioselective SOMO-activation/ cycloaddition mechanism. Herein we describe the successful execution of these ideals and demonstrate a simple yet powerful pyrrolidine forming reaction that we hope will be of significant utility to practitioners of medicinal agent synthesis and asymmetric catalysis.
 |
Design Plan
Within the last few years, our laboratory has introduced a new mode of activation termed SOMO-catalysis that has enabled the direct enantioselective allylic alkylation,7 enolation,8 vinylation,9 nitro-alkylation,10 and carbo-oxidation11 of aldehydes, as well as a new (4 + 2) cycloaddition to generate complex cyclohexyl rings.12 Recently, we questioned if this radical-polar crossover mechanism might be employed to design a (3 + 2) cycloaddition to enantioselectively build complex pyrrolidine rings.13,14 As outlined in equation 1, we hypothesized that exposure of β-amino aldehydes to SOMO-activation using imidazolidinone catalyst 1 and an oxidant would transiently form the radical cation 3, which should rapidly engage an olefinic substrate in an enantioselective coupling to deliver the alkyl radical 4. Oxidative radical-polar crossover would then furnish a carbocation that should trigger a stereoselective nitrogen addition–ring closure to deliver a complex pyrrolidine motif. In accord with previous SOMO-activation studies,7–12 we presumed that high levels of enantiocontrol would be achieved on the basis of 3π-electron geometry control in catalyst-substrate adduct 3 and selective benzyl group shielding of the radical cation Si-face on the same system. As an important design criteria, we recognized that this new pyrrolidine forming reaction should be possible using readily accessible starting materials, such as simple olefins and β-amino aldehydes.
Results
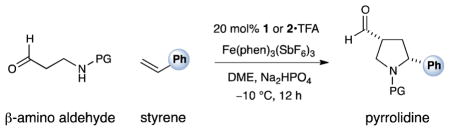
As revealed in Table 1, the proposed design plan was rendered successful via the exposure of a variety of N-protected β-amino aldehydes to styrene and two equivalents of Fe(III)trisphenanthroline in the presence of amine catalysts 1 and 2. Notably, high levels of enantiocontrol could be achieved using various protecting groups on the aldehydic nitrogen; however, the degree of diastereocontrol ranged from poor with Cbz to synthetically useful with nosyl (Table 1, entries 1–5, 62–86% yield, 1.2–6:1 dr, 82–90% ee). Interestingly, the more electronwithdrawing amine substituents (Cbz < Ts < Ns) clearly provide increased diastereocontrol, suggesting a more ordered (later) transition state with less nucleophilic tethered amines. Finally, changing the methyl group of the catalyst 1 to the more traditional benzyl-substituted imidazolidinone 2 further increased the relative stereoselectivities (Table 1, entry 6, 92% ee, 9:1 dr), a significant outcome given the propensity of five-membered ring cyclizations to be less diastereoselective than their six- and seven-membered counterparts.15
Table 1.
Effect of Amine Protecting Group and Catalyst Structure
| ||||||
|---|---|---|---|---|---|---|
| entry | PG | equiv. ald. | cat. (R) | % yielda | drb | %eec |
| 1 | Cbz | 3.0 | 1 (Me) | 66 | 1.2:1 | 82 |
| 2 | Boc | 3.0 | 1 (Me) | 62 | 1.4:1 | 86 |
| 3 | Ts | 3.0 | 1 (Me) | 86 | 4:1 | 90 |
| 4 | Ns | 3.0 | 1 (Me) | 79 | 6:1 | 90 |
| 5 | Ns | 1.5 | 1 (Me) | 75 | 6:1 | 90 |
| 6 | Ns | 1.5 | 2 (Bn) | 77 | 9:1 | 92 |
Isolated yield.
Determined by 1H NMR analysis of crude reaction or isolated mixture.
Determined by chiral HPLC analysis of corresponding alcohol; absolute stereochemistry assigned by X-ray crystal structure or by analogy.
Boc = t-butoxycarbonyl, Cbz = carboxybenzyl, Ts = 4- toluenesulfonyl, Ns = 4-nitrophenylsulfonyl.
With the optimized conditions in hand, we next examined the scope of the olefinic component. As shown in Table 2, a range of electron rich (entries 2, 4 and 6, 71–75% yield, 7–10:1 dr, 89– 92% ee) and electron poor (entries 7 and 8, 61–81% yield, 5–8:1 dr, 89–91% ee) styrenes are viable in this new cycloaddition reaction. Moreover, the enantioselectivity and efficiency are maintained for the o-, m-, and p-bromostyrenes (entries 1, 3 and 5, 72–76% yield, 93–94% ee), all useful substrates for further diversification in metal-catalyzed cross coupling protocols. To our delight, varying the olefin substitution pattern is readily achieved. For example, the use of α-methyl styrene provides tertiary amino substituted pyrrolidines with excellent enantiocontrol (entry 9, 92% ee) and in 3:1 diastereocontrol (presumably due to the decreased preference for a pseudo-equatorial phenyl group in the cyclization transition state). The diastereoselectivity improves significantly, to >20:1, when using trans-β-methyl styrene to stereoselectively build the pyrrolidine framework with three contiguous stereocenters (entry 10, 50% yield, 96% ee).
Table 2.
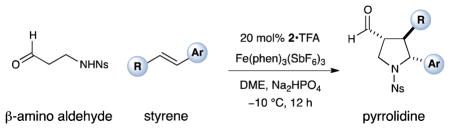
| |||
|---|---|---|---|
| 1 |
 72% yield, 9:1 dr, 93% ee |
2 |
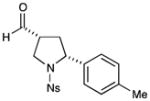 71% yield, 7:1 dr, 91% ee |
| 3 |
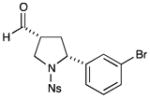 76% yield, 8:1 dr, 93% ee |
4 |
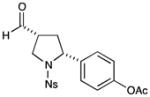 73% yield, 8:1 dr, 92% ee |
| 5 |
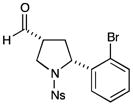 76% yield, 3:1 dr, 94% ee |
6 |
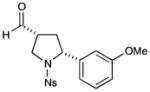 75% yield, 10:1 dr, 89% ee |
| 7 |
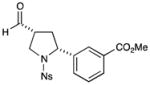 61% yield, 5:1 dr, 89% ee |
8 |
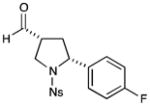 81% yield, 8:1 dr, 91% ee |
| 9c |
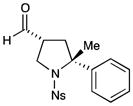 67% yield, 3:1 dr, 92% ee |
10c |
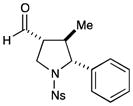 50% yield, >20:1 dr, 96% ee |
Results listed as product, yield, diastereomeric ratio (dr), enantiomeric excess (% ee).
Diastereomeric ratio, % ee determined as in Table 1.
Reaction conducted at −20 °C using THF and Fe(phen)3(PF6)3.
Given the importance of heteroaryl-substituted pyrrolidines in medicinal chemistry, we recognized an attractive prospect might be the replacement of styrene with vinyl heteroaromatics in this protocol (Table 3). As representative examples, both benzyl pyrazole (entry 1, 75% yield, 3:1 dr, 91% ee) and isoxazole (entry 2, 55% yield, 6:1 dr, 95% ee) were successfully incorporated in this manner. Importantly, this process is not limited to vinyl arenes. A full range of electron neutral dienes was also competent in this transformation (entries 3–6, 62–85% yield, 1.5–4:1 dr, 87– 92% ee), forming both bicyclic structures and fully substituted carbon stereocenters. Enantioselectivities and yields remained high in all cases; however, diasterocontrol was observed to be moderate relative to other olefinic substrates.
Table 3.
| |||
|---|---|---|---|
| 1c |
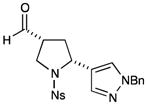 75% yield, 3:1 dr, 91% ee |
2c,d |
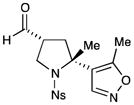 55%yield,6:ldr, 95% ee |
| 3 |
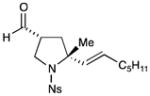 85% yield, 2:1 dr, 92% ee |
4e |
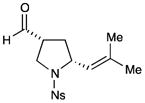 77% yield, 4:1 dr, 89% ee |
| 5d |
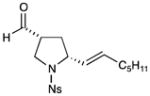 62% yield, 3:1 dr, 87% ee |
6 |
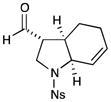 74% yield, 3:2 dr, 92% ee |
Results listed as product, yield, diastereomeric ratio (dr), enantiomeric excess (% ee).
Diastereomeric ratio, % ee determined as in Table 1.
Reaction conducted with DME as solvent.
Reaction conducted at −20 °C.
Reaction conducted at −30 °C.
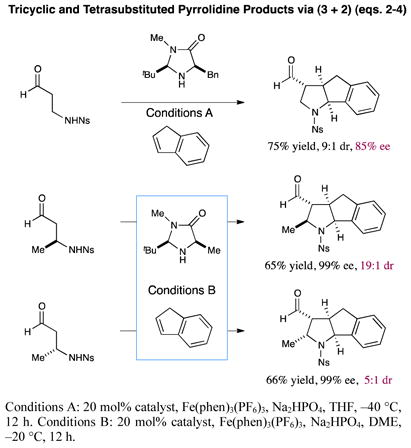 |
Finally, the use of indene as the π-coupling partner (eq. 2) allows the production of unique tricyclic pyrrolidines in high yield and diastereoselectivity (75% yield, 9:1 dr, 85% ee). Perhaps even more notable, the use of optically active β-methyl β-amino aldehydes has enabled the construction of all four carbon stereocenters on the pyrrolidine ring while achieving excellent levels of disatereocontrol (eqs. 3 and 4, 65–66% yield, ≥99% ee, 5–19:1 dr). Importantly, in these cases catalyst control appears to dominate for the formation of the formyl-bearing stereocenter, as well as the amine-carbocation cyclization event. As a result, various pyrrolidine isomers can be formed enantioselectively via the judicious choice of substrate and catalyst antipodes.
In summary, a new protocol to rapidly construct enantioenriched pyrrolidines using β-amino aldehydes and π-nucleophilic olefins has been achieved using organo-SOMO catalysis in a formal cycloaddition cascade. As a critical design feature, the starting materials and catalysts are commercial materials or easily prepared, allowing rapid and modular production of a variety of pyrrolidine cores. We anticipate that this transformation will be valuable for catalyst development, natural product synthesis, and in the identification of new medicinal agents. Given this conceptual advance, future work will focus on the development of alternative oxidants of lower molecular weight for this protocol.
Supplementary Material
Acknowledgments
FGF thanks CNPq-Brazil for a postdoctoral fellowship. Financial support was provided by NIHGMS (R01 01 GM078201-06) and kind gifts from Merck, Amgen, and Abbott.
Footnotes
Supporting Information Available. Experimental procedures, structural proofs, and spectral data for all new compounds are provided (PDF).
References
- 1.For reviews containing recent syntheses of pyrrolidine-containing alkaloids see: Donohoe TJ, Bataille CJR, Churchill GW. Annu Rep Prog Chem, Sect B. 2006;102:98.Felpin FX, Lebreton J. Eur J Org Chem. 2003;19:3693.For a review with examples of biologically active pyrrolidines inspired by natural products, see: Galliford CV, Scheidt KA. Angew Chem Int Ed. 2007;46:8748. doi: 10.1002/anie.200701342.
- 2.For some recent examples of biologically active pyrrolidines, see: Hensler ME, Bernstein G, Nizet V, Nefzi A. Bioorg Med Chem Lett. 2006;16:5073. doi: 10.1016/j.bmcl.2006.07.037.Li X, Li J. Mini-Rev Med Chem. 2010;10:794. doi: 10.2174/138955710791608334.Lexa KW. Proteins. 2011;79:2282. doi: 10.1002/prot.23054.For recent studies of pyrrolidines as peptidomimetics, see: Whitby LR, Ando Y, Setola V, Vogt PK, Roth BL, Boger DL. J Am Chem Soc. 2011;133:10184. doi: 10.1021/ja201878v.Raghuraman A, Ko E, Perez LM, Ioerger TR, Burgess K. J Am Chem Soc. 2011;133:12350. doi: 10.1021/ja2033734.
- 3.For a recent survey of chiral amine organocatalysis, see: Zhang S, Wang W. In: Privileged Chiral Ligands and Catalysts. Zhou Q-L, editor. Wiley-VCH; Weinheim, Germany: 2011. pp. 409–439.Watson AJB, MacMillan DWC. Enantioselective Organocatalysis Involving Iminium, enamine SOMO and Photoredox Activation. In: Ojima I, editor. Catalytic Asymmetric Synthesis. 3. Wiley & Sons; Hoboken, NJ: 2010. pp. 39–57.For a review with examples of chiral amine ligands, see: Caputo CA, Jones ND. Dalton Trans. 2007:4627. doi: 10.1039/b709283k.
- 4.For a review of recent enantioselective approaches, see: Companyo X, Alba A-N, Rios R. In: Targets in Heterocyclic Systems. Attanasi OA, editor. Vol. 13. Royal Society of Chemistry; London: 2009. pp. 147–174.For a review of recent organocatalytic approaches to cyclic products including pyrrolidines, see: Myano A, Rios R. Chem Rev. 2011;111:4703. doi: 10.1021/cr100348t.
- 5.For reviews, see: Coldham I, Hufton R. Chem Rev. 2005;105:2765. doi: 10.1021/cr040004c.Husinec S, Savic V. Tetrahedron: Asymm. 2005;16:2047.Pandey G, Banerjee P, Gadre SR. Chem Rev. 2006;106:4484. doi: 10.1021/cr050011g.For a recent example and a comprehensive list of metal-catalyzed enantioselective methods to date, see: Yamashita Y, Imaizumi T, Kobayashi S. Angew Chem Int Ed. 2011;50:4893. doi: 10.1002/anie.201008272.and references therein. For methods using iminium catalysis and ylides, see: Vicario JL, Reboredo S, Badia, Carrillo L. Angew Chem Int Ed. 2007;46:5168. doi: 10.1002/anie.200700988.Ibrahem I, Rios R, Vesely J, Cordova A. Tetrahedron Lett. 2007;48:6252.Lin S, Deiana L, Zhao GL, Sun J, Cordova A. Angew Chem Int Ed. 2011;50:7624. doi: 10.1002/anie.201101966.For methods using thiourea catalysts and ylides, see: Enders D, Goddertz DP, Beceno C, Raabe G. Adv Synth Catal. 2010;352:2863.Liu YK, Liu H, Du W, Yue L, Chen YC. Chem Eur J. 2008;14:9873. doi: 10.1002/chem.200801410.Bai JF, Wang LL, Peng L, Guo YL, Ming JM, Wang FY, Xu XY, Wang LX. Eur J Org Chem. 2011:4472.Xie J, Yohsida K, Taksu K, Takemoto Y. Tetrahedron Lett. 2008;49:6910.For methods using Bronsted acid catalysis, see: Chen XH, Zhang WQ, Gong LZ. J Am Chem Soc. 2008;130:5652. doi: 10.1021/ja801034e.Liu WJ, Chen XH, Gong LZ. Org Lett. 2008;10:5357. doi: 10.1021/ol802177s.Chen X-H, Wei Q, Luo S-W, Xiao H, Gong L-Z. J Am Chem Soc. 2009;131:13819. doi: 10.1021/ja905302f.Cheng MN, Wang H, Gong LZ. Org Lett. 2011;13:2418. doi: 10.1021/ol200652j.We are aware of only one cascade formal cycloaddition that proceeds without the use of ylides: Li H, Zu L, Xie H, Wang J, Wang W. Chem Commun. 2008:5636. doi: 10.1039/b812464g.
- 6.For a recent review: Overman LE, Humphreys PG, Welmaker GS. Org React. 2011;75:747.For seminal publications in this area: Overman LE, Kakimoto M. J Am Chem Soc. 1979;101:1310.Overman LE, Kakimoto M, Okazaki M, Meier GP. J Am Chem Soc. 1983;105:6622.Overman LE, Okazaki M, Jacobsen EJ. J Org Chem. 1985;50:2403.
- 7.Beeson TD, Mastracchio A, Hong JB, Ashton K, MacMillan DWC. Science. 2007;316:582. [PubMed] [Google Scholar]
- 8.Jang H, Hong J, MacMillan DWC. J Am Chem Soc. 2007;129:7004. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]
- 9.Kim H, MacMillan DWC. J Am Chem Soc. 2008;130:398. doi: 10.1021/ja077212h. [DOI] [PubMed] [Google Scholar]
- 10.Wilson JE, Casarez AD, MacMillan DWC. J Am Chem Soc. 2009;131:11332. doi: 10.1021/ja904504j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham TH, Jones CM, Jui NT, MacMillan DWC. J Am Chem Soc. 2008;130:16494. doi: 10.1021/ja8075633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jui NT, Lee ECY, MacMillan DWC. J Am Chem Soc. 2010;132:10015. doi: 10.1021/ja104313x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cascade cycloaddition refers to a reaction that unites multiple reaction partners to form a cyclic product via a cascade mechanism, which is in accord with the IUPAC Gold Book definition of cycloaddition.
- 14.Murphy JA. The Radical-Polar Crossover Reaction. In: Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. Vol. 1. Wiley-VCH; Weinheim, Germany: 2001. p. 298. [Google Scholar]
- 15.For a review (a) and studies (b–d) of five-membered ring conformations, see: Legon AC. Chem Rev. 1980;80:231.Džakula Z, DeRider ML, Markley JL. J Am Chem Soc. 1996;118:12796.Mikhova BP, Ivanov PM, Spassov SL. J Mol Struct. 1992;265:225.Ohwada T, Okamoto I, Shudo K, Yamaguchi K. Tetrahedron Lett. 1998;39:7877.Concerning six-membered ring conformations in transition states, see: Zimmerman HE, Traxler MD. J Am Chem Soc. 1957;79:1920.Yang J. Six-Membered Transition States in Organic Synthesis. Wiley-Interscience; Hoboken, NJ: 2008. and references therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.