

SUMMARY
Protease inhibitor discovery has focused almost exclusively on compounds that bind to the active site. Inhibitors targeting protease exosites, regions outside of the active site that influence catalysis, offer potential advantages of increased specificity but are difficult to systematically discover. Here we describe an assay suitable for detecting exosite-targeting inhibitors of the metalloproteinase anthrax lethal factor (LF) based on cleavage of a full length mitogen-activated protein kinase kinase (MKK) substrate. We used this assay to screen a small molecule library, and then subjected hits to a secondary screen to exclude compounds that efficiently blocked cleavage of a peptide substrate. We identified a compound that preferentially inhibited cleavage of MKKs compared with peptide substrates and could suppress LF-induced macrophage cytolysis. This approach should be generally applicable to the discovery of exosite-targeting inhibitors of many additional proteases.
INTRODUCTION
Anthrax is caused by infection with the encapsulated spore-forming gram-positive bacterium Bacillus anthracis (Spencer, 2003). While the most common form of the disease is a curable localized cutaneous infection, inhalation of anthrax spores gives rise to a rapidly progressing, highly fatal systemic disease. Fatality associated with inhalational anthrax has been attributed to two plasmid-encoded secreted protein toxins, edema toxin (EdTx) and lethal toxin (LeTx), which kill experimental animals upon injection (Moayeri and Leppla, 2009). Anthrax toxin inhibitors have been proposed as drugs that could be used in combination with conventional antibiotics, which alone have a poor success rate (~30%) against inhalational anthrax (Burnett et al., 2005; Holty et al., 2006).
LeTx has drawn particular attention due to its key role in promoting anthrax virulence. LeTx consists of two proteins: a pore-forming subunit, protective antigen (PA), which delivers an enzymatic subunit, lethal factor (LF), into the cytosol of host cells (Turk, 2007). LF is a zinc-dependent metalloproteinase that specifically cleaves and inactivates host mitogen activated kinase kinases (MKKs) at sites near their N-termini. LF cleavage disrupts interactions between MKKs and their MAPK substrates, thereby terminating MAPK signaling essential for proper cell function and survival (Duesbery et al., 1998; Vitale et al., 2000; Vitale et al., 1998). LeTx functionally impairs cells of the immune and vascular systems, allowing spread of the disease and directly causing pathology (Baldari et al., 2006; Moayeri and Leppla, 2009).
Many approaches taken to neutralize LeTx in vivo have involved blocking its uptake into cells, for example by inhibiting the LF-PA interaction or passive immunization with antibodies to PA, and these approaches have proven effective in animal models of toxemia and infection (Forino et al., 2005; Maynard et al., 2002; Mourez et al., 2001; Pini et al., 2006; Rai et al., 2006; Wild et al., 2003). However, because of prior success in therapeutic targeting of proteases, much effort has been directed to the identification of LF inhibitors (Turk, 2008). Numerous compounds that inhibit LF and block its biological activity have been discovered through modification of known metalloproteinase inhibitor scaffolds, fragment-based drug discovery, computational docking, and HTS using peptide substrates (Forino et al., 2005; Fridman et al., 2005; Lee et al., 2004; Min et al., 2004; Panchal et al., 2004; Schepetkin et al., 2006; Shoop et al., 2005; Tonello et al., 2002; Turk et al., 2004). These approaches, while successful, are strongly biased towards the identification of compounds targeting the LF active site. Such active site-directed inhibitors have clear drawbacks, such as a tendency to cross-react with related proteases. Such potential off target effects are particularly worrisome in the case of anthrax, since host matrix metalloproteinases can mediate defense against bacterial infections (Li et al., 2004; Renckens et al., 2006; Wilson et al., 1999).
An alternative strategy to develop protease inhibitors is to target exosites, generally defined as regions outside of the active site that are important for catalysis (Bock et al., 2007; Overall, 2002). A major function of exosites is to bind substrates at sites separate from the cleavage site, thus promoting proteolysis by increasing affinity for the substrate. Because LF is thought to harbor an exosite required for efficient proteolysis of MKKs (Chopra et al., 2003), we hypothesized that compounds targeting this exosite would provide a means to develop specific inhibitors of LF that could neutralize its biological activity. Previous approaches to protease exosite targeting have included screening phage display libraries for high affinity peptides, raising monoclonal antibodies against known exosite regions, screening small molecule libraries using model substrates that require exosite interactions, and serendipitous discovery of exosite-blocking compounds (Bjorklund et al., 2004; Dennis et al., 2000; Hardy et al., 2004; Lauer-Fields et al., 2009; Mikkelsen et al., 2008; Silhar et al., 2010). Here we describe a high-throughput screen to identify LF inhibitors using a full length MKK, rather than a short peptide, as a substrate. In addition to classical active site-binding compounds, such a screen is capable of identifying molecules that target the exosite. By this approach we have identified a small molecule that preferentially inhibits cleavage MKK protein substrates over short peptide substrates and blocks LF cytotoxicity. These results validate our approach to the discovery of exosite-targeting protease inhibitors.
RESULTS
A screen for small molecules that inhibit LF cleavage of a full length protein substrate
We developed an LF protease assay suitable for HTS that takes advantage of the N-terminal location of its cleavage sites within MKKs. Proteolysis of an MKK conjugated to a fluorophore at its N-terminus liberates a low molecular weight fluorescent peptide (Figure 1). The labeled fragment can be distinguished from full length MKK by a change in fluorescence polarization (FP). In the absence of an inhibitor, LF cleavage produces a low FP signal, while a hit compound would prevent cleavage and thus provide a high FP signal.
Figure 1. Identification of compounds that inhibit LF cleavage of full a length MKK using a FP based screen.

(A) Assay scheme. In the absence of inhibitors, MKK6 is cleaved, releasing a small fluorescent fragment having decreased FP. In the presence of an inhibitor, higher quantities of full length MKK6 provide an increased FP signal.
(B) Scheme for purification and N-terminal TGase-mediated labeling of MKK6.
(C) SDS-PAGE analysis of LF-cleaved N-terminally labeled MKK6. Fluorescein fluorescence (top panel) and total protein (bottom panel) is shown for 2 µM labeled MKK6 treated with the indicated concentrations of LF for 30 min at RT.
(D) Z’ values for each plate from the primary screen of 2835 molecules.
(E) Hit identification. Thirteen compounds showed greater than 30% inhibition relative to control reactions run in the absence of added compound.
See also Figure S1.
We used MKK6 as a substrate, since we could produce large quantities of the full length protein, sufficient for HTS, in bacteria. As previously demonstrated for MKK1 (Chopra et al., 2003), we found that efficient proteolysis of MKK6 by LF required its intact kinase domain, indicating that cleavage is exosite-dependent (Supplemental Figure S1). We engineered an efficient acceptor site for the enzyme transglutaminase (TGase) that had previously been identified by phage display (Sugimura et al., 2006) into the N-terminus of MKK6 (Figure 1B). This enabled TGase-catalyzed conjugation of fluorescein cadaverine close to the protein N-terminus. Labeling occurred to high stoichiometry (78 ± 8% among four separate labeling reactions) and was specific to the intended site, as LF cleavage decreases the abundance of fluorescent MKK6 and produces a non-fluorescent cleavage product (Figure 1C).
Optimization of LF (20 nM) and MKK6 (125 nM) concentrations in a 384-well plate assay format provided a Z’ value of 0.61 suitable for HTS (Zhang et al., 1999). We confirmed that the known peptide hydroxamate inhibitor GM6001 (Turk et al., 2004) scored as a hit in this assay. We then used the assay to screen a collection of 2,835 small molecules and natural product extracts. Among LF inhibitors identified from this screen was epiafzelechin, a compound structurally similar to green tea flavonoids previously described as LF inhibitors (Dell'Aica et al., 2004). We identified 13 primary hits overall at a threshold of 30% inhibition (Figure 3B). Hit compounds were subjected to a secondary screen using a short fluorogenic peptide substrate to exclude active site-targeting inhibitors. Lastly, hits that showed dose-dependent inhibition in the primary assay were confirmed using an SDS-PAGE mobility shift assay for MKK6 cleavage to rule out compound interference. By these criteria, stictic acid (StA, compound 1 in Table 1), a depsidone natural product from lichens (Lawrey, 1986), was the only hit found to specifically inhibit MKK6 cleavage.
Figure 3. Depsidones neutralize LeTx activity on cultured cells.

(A) RAW264.7 cells were pretreated with either DMSO carrier or 400 µM of the indicated compound and then incubated with 0.03 µg/ml LF and 0.5 µg/ml PA. After 1h, cell lysates were extracted and analyzed by immunoblotting to detect MKK3 cleavage. Vinculin levels were detected by immunoblotting the same samples as a loading control.
(B) RAW264.7 cells were pretreated with either DMSO carrier or 400 µM of the indicated compound followed by various concentrations of LF in the presence of 0.5 µg/ml PA. After 4h, cell viability was determined by MTT assay.
Table 1.
Lichen compounds and their potency for LF inhibition.
| Compound | Name | Structure | IC50 (µM), MKK6 cleavage |
|---|---|---|---|
| 1 | Stictic acid (StA) | 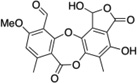 |
24 ± 3 |
| 2 | Salazinic acid | 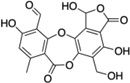 |
42 ± 5 |
| 3 | Parellic acid | 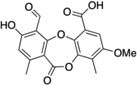 |
125 ± 20 |
| 4 | Gangaleoidin | 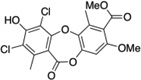 |
>250 |
| 5 | Physodic acid | 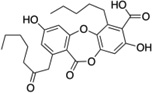 |
>250 |
| 6 | Lecanoric acid | 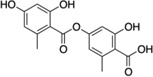 |
No inhibition |
| 7 | Atranorin | 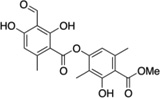 |
No inhibition |
| 8 | Reduced stictic acid |  |
>250 |
| 9 | Reduced parellic acid | 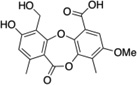 |
>250 |
Characterization of depsidone inhibitors of LF
The StA structure comprises substituted benzaldehyde and benzofuranone groups flanking a central 7-membered lactone ether ring. To identify specific features required for LF inhibition, an initial series of six commercially available StA analogs (Table 1) were obtained and evaluated for their ability to block MKK6 cleavage in the SDS-PAGE mobility shift assay (Figure 2A). While two of the compounds, salazinic acid (2) and parellic acid (3), were active as inhibitors, the remaining analogs had no observable activity. Inhibition by 3 indicates that the furanone ring is dispensable for inhibition. However, lack of inhibition by 6 and 7 suggests that the central 7-membered lactone ring is essential. We also noted that compounds 4 and 5, which lack the free aldehyde group, failed to inhibit LF. To formally evaluate the importance of the aldehyde, we synthesized and tested reduced forms of 1 and 3 (compounds 8 and 9, respectively). For both compounds, reduction of the aldehyde to an alcohol diminished but did not completely eliminate LF inhibition, suggesting that the aldehyde is important but not absolutely essential for activity. We observed no detectable inhibition of MKK6 cleavage by o-anisaldehyde, a simple benzaldehyde, at concentrations up to 2 mM (not shown), indicating that LF inhibition by depsidones is not due to non-specific reactivity of the aldehyde group.
Figure 2. LF inhibition by lichen compounds.

(A) Structure-activity profile of StA analogs. Compounds (250 µM) were mixed with LF (25 nM) and MKK6 (1 µM) for 30 min at room temperature prior to electrophoresis. (B) Inhibitor titration for the 3 active compounds, performed as in panel A with the indicated concentrations of compound.
(C) Quantified inhibition curves. The average ± standard deviation is shown for three separate experiments performed as shown in panel B.
(D) Depsidones are weak inhibitors of LF cleavage of a peptide substrate. Initial rates of peptide cleavage were measured by continuous fluorometric assay in the presence of compounds 1, 2, 3 (at 100 µM) or GM6001 (GM, 50 µM). The percentage activity relative to the vehicle control ± standard deviation is shown for data collected in triplicate.
(E) Depsidones inhibit LF cleavage of MKK6 competitively. LF was incubated with varying concentrations of MKK6 in the presence of the indicated concentrations of compound 2. The double reciprocal plot displaying the data has lines intersecting at the y-axis, consistent with competitive inhibition.
(F) Inhibition of MKK3 and MKK4 cleavage by depsidones. RAW264.7 cell lysate was incubated with compounds 1 – 3 (250 µM) followed by LF (0.03 µg/mL) for 30 min at RT. MKK3 and MKK4 cleavage was analyzed by immunoblotting.
See also Figure S2.
Of all compounds tested, StA was the most potent, inhibiting cleavage of MKK6 with an IC50 of 24 ± 3 µM, while 2 and 3 had decreased potency (Figure 2 and Table 1). While compound interference prevented us from performing a full dose response, each compound only weakly inhibited cleavage of a peptide substrate at the highest concentration tested (100 µM, Figure 2D). For comparison we used both the protein and peptide cleavage assays to evaluate the active site inhibitor GM6001. GM6001 inhibited LF cleavage of MKK6 with an IC50 value of 45 µM, similar potency to StA (Table 1 and Figure S2). However, unlike StA, GM6001 more potently inhibited peptide cleavage (IC50 = 9 µM). The weaker potency of the standard active site-binding inhibitor observed with the MKK substrate compared with the peptide substrate may be because accurate quantification in the SDS-PAGE assay required overdigestion of the substrate (non-initial velocity conditions). Nonetheless, the observation that depsidones blocked cleavage of an MKK substrate more potently than they do a peptide substrate underscores that they work by mechanism distinct from that of a conventional inhibitor. While accurate kinetic analysis of the mode of inhibition was hampered by the gel-based cleavage assay, we found that compound 2 reproducibly increased KM with no effect on Vmax (Figure 2E). These data suggest that depsidones act as competitive inhibitors of LF, consistent with disruption of the LF-MKK6 interaction through exosite binding.
Because we had found depsidones to be substrate-specific inhibitors of LF, we further assessed the ability of these compounds to block cleavage of MKKs other than MKK6. Compounds 1, 2 and 3 each inhibited cleavage of both MKK3 and MKK4 in lysates from RAW264.7 cells, used as a source of endogenous MKKs (Figure 2F). These experiments indicate that depsidones are general inhibitors of MKK protein cleavage by LF.
Depsidone inhibitors neutralize LeTx activity in cultured cells
To assess the capacity of the new class of LF inhibitors to neutralize the biological activity of LeTx, we examined the effect of depsidones on MKK cleavage and cell viability in LeTx-treated macrophage cells. RAW264.7 cells were pretreated with compounds 1, 2 or 3 prior to adding LeTx. Cells were harvested 1 hour after treatment, a time subsequent to LF uptake but prior to cell death. Immunoblot analysis (Figure 3A) showed that all three depsidones decreased MKK3 cleavage in intact cells. In separate experiments, we preincubated cells with compounds before adding varying amounts of LF in the presence of a fixed PA concentration. Four hours later, we assessed cell viability by MTT assay. In the absence of compounds, LF causes dose-dependent killing of these cells (Figure 3B). Each of the depsidones capable of inhibiting LF (1, 2 and 3) provided partial protection against LeTx-induced cell death, even at LF concentrations causing complete killing. By contrast, the inactive ring-opened analogs 6 and 7 failed to inhibit cell death. These experiments indicate that depsidone LF inhibitors display target-specific biological activity, and may therefore be candidates for further development into anthrax therapeutics.
DISCUSSION
Exosites were first identified in proteases in the coagulation cascade as binding sites for substrates and allosteric modulators (Bock et al., 2007). To date derivatives of the leech anticoagulant protein hirudin, which binds both the exosite and the active site in a bivalent manner, remain the only clinically approved exosite-targeting protease inhibitors (Di Nisio et al., 2005). Interest in targeting exosites for other proteases has arisen from the failure to develop highly selective inhibitors for important therapeutic targets (Drag and Salvesen, 2010; Kornacker et al., 2005; Matheson and Goa, 2000). While the identification of conventional active site inhibitors can rely on derivatization of scaffolds that target a given class of proteases (i.e. peptide hydroxamates for metalloproteinases), discovery of exosite targeting inhibitors has not been straightforward. Standard HTS using peptide substrates can identify allosteric inhibitors that bind to exosites, and several classes of polyphenolic compounds have been identified as non-competitive LF inhibitors (Min et al., 2004; Numa et al., 2005). However, allosteric inhibitors discovered in this manner must by definition influence the enzyme active site. By contrast, identification of inhibitors that bind substrate-recruiting exosites cannot be identified using short peptide substrates. Such inhibitors work by blocking a protein-protein interaction and may be regarded as difficult to discover by HTS, even with the appropriate assay. However, substrate recruitment sites in protein modifying enzymes tend to be grooves or pockets that bind to short peptide structures, interactions more amenable to disruption with small molecules than those involving large interaction surfaces (Arkin and Whitty, 2009). Indeed, the recent serendipitous discovery of small molecules that disrupt exosite interactions with the botulinum neurotoxin metalloproteinase supports the feasibility of therapeutic exosite targeting (Silhar et al., 2010).
The identification of such exosite targeting protease inhibitors however requires novel approaches distinct from conventional HTS. Exosite-binding peptides for proteases have been selected from phage display libraries (Dennis et al., 2000; Dennis et al., 2001; Kornacker et al., 2005) and from known exosite-binding regions of substrates (Lauer-Fields et al., 2008). In addition, exosite-blocking monoclonal antibodies have been produced for several proteases (Atwal et al., 2011; Mikkelsen et al., 2008). However, peptide and antibody-based therapeutics have undesirable pharmacology, and particularly for an intracellular target, small molecule inhibitors are advantageous. A fragment-based screening method using mass spectrometry called “tethering” has been used to identify allosteric small molecule inhibitors of caspases that bind through novel sites, though the approach requires the presence of reactive amino acids at the exosite (Hardy et al., 2004). We have taken an alternative approach using HTS with an exosite-dependent substrate. Fields and co-workers identified a substrate-selective matrix metalloproteinase-13 inhibitor by HTS using model triple helical peptide substrates whose cleavage is exosite-dependent (Lauer-Fields et al., 2009). However, to our knowledge our study is the first report of a high throughput screen using a full length protease substrate. While the FP assay used requires that cleavage occur close to one terminus of the substrate, FRET or LRET-based assays without such restrictions could also be generated by dual labeling of the substrate (Yang and Yang, 2009).
The sole MKK-specific LF inhibitor identified in our primary screen was the depsidone StA. Depsidones and related compounds are lichen secondary metabolites that are thought to provide protection against UV light, pollutants, oxidative stress and harmful microbes (Molnar and Farkas, 2010). Lichen metabolites have attracted some attention as potential therapeutics. For example depsidones were discovered through HTS as moderate HIV integrase inhibitors and have also been found to induce apoptosis of a prostate cancer cell line (Neamati et al., 1997; Russo et al., 2006). Lichen compound activity in these assays follows a distinct structure-activity relationship in comparison to LF inhibition, suggesting that it should be possible to identify depsidone analogs with specificity for LF.
In addition to offering a novel scaffold for LF inhibition, depsidones may also find use as structural probes to characterize the LF exosite. Despite its importance for MKK cleavage, specific residues comprising the LF exosite remain to be discovered. The depsidone binding site may overlap with the LF exosite, or alternatively may allosterically influence exosite interactions. Crystallography of LF in complex with active site inhibitors uncovered critical details of cleavage site recognition (Turk et al., 2004). Similarly we expect that structural characterization of an LF-depsidone complex will provide insight into the LF exosite. The importance of the aldehyde group for depsidone activity suggests that it may form a covalent complex with LF. Identification of the residue modified by the compounds could thereby provide a means to map the depsidone binding site.
SIGNIFICANCE
Proteolytic enzymes have emerged as important drug targets. While conserved features within protease active sites have been exploited in the development of mechanism-based inhibitors for all protease classes, specific targeting of individual proteases remains a formidable challenge. Here we have described a systematic approach to target exosites, which are generally unique to individual proteases, as a means to identify specific inhibitors. Using this approach we have identified substrate specific inhibitors of anthrax lethal factor, a known virulence factor and potential target for anthrax therapeutics. These molecules provide an important research tool, both in the manipulation of LF activity in experimental systems and as structural probes for better understanding exosite interactions. Our approach should be generally useful for discovering exosite probes for other proteases as well.
EXPERIMENTAL PROCEDURES
Materials
Recombinant LF and PA were from List Biological Laboratories. StA was obtained from Sigma-Aldrich. Compounds 4, 6 and 7 were obtained from Chromadex. Compounds 2 (NSC 87509), 3 (NSC 92186) and 5 (NSC 5916) were obtained from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov). Compound 3 for chemical modification was obtained from MP Biomedical.
FP assay and inhibitor screening
Production of N-terminally labeled MKK6 for HTS is described in Supplemental Information. Screens were conducted at the Yale Small Molecule Discovery Center using a Tecan Aquarius robot in combination with a Freedom EVO Workstation. A V&P Scientific 384-pin tool transferred small molecules (10 mM stock) from library plates into black 384-well assay plates containing assay buffer (20 mM HEPES pH 7.4, 0.1 mg/ml BSA, 10 mM CaCl2, 0.01% Brij35). Assays were performed in a total volume of 20 µL with final concentrations of 10 µM compound, 125 nM MKK6, and 20 nM LF. FP was read after 60 min incubation at RT on a Wallac Envision plate reader equipped with fluorescein filters. Each assay plate included reactions without added compounds and reactions without lethal factor to set high and low boundaries for FP signals, respectively. Z′ analysis (Zhang et al., 1999) was done from 32 maximum control values (c+) and 32 minimum controls (c−) on each plate. Assays were consistently done with Z′ factors of 0.6, higher than the minimum 0.5 considered robust. Screening was performed against 5 libraries: NIH Clinical Collection 1 (450 compounds), the Yale Compound Repository (a collection of 250 compounds synthesized by Yale chemists), the Gen-Plus (960 compounds) and Pure Natural Products (800 compounds) collections from MicroSource Discovery Systems, and the Yale Rainforest Collection (393 bacterial or fungal extracts).
SDS-PAGE-based cleavage assay
LF (25 nM in assay buffer) was mixed with compound at the indicated concentration, and the reaction was initiated by adding unlabeled MKK6 to 1 µM and stopped after 30 min at RT by adding 4X SDS-PAGE loading buffer. For determination of IC50 values, reaction was performed with compound concentrations ranging from to 7.8 µM to 250 µM in two-fold increments. Samples were subjected to SDS-PAGE (10% acrylamide), stained with Coomassie, scanned with a flatbed scanner, and analyzed using Quantity One (BioRad) or ImageJ (National Institutes of Health) software to quantify band intensities. The extent of inhibition was determined by measuring the intensity of the band corresponding to the cleavage product in the absence or presence of compound. IC50 values were determined by fitting to the following equation: % inhibition = 100*[I]/(IC50 + [I]). Reported values are the average ± standard deviation of three separate measurements. To determine the mode of inhibition, LF (10 nM) was incubated with a range of MKK6 concentrations flanking the KM value (two-fold increments from 0.5 µM to 16 µM) in the presence at varying inhibitor concentration. Samples were diluted so that an equal amount of MKK6 was run in each lane of an SDS-polyacrylamide gel, and the extent of cleavage was quantified as described above.
Peptide cleavage assay
Cleavage was assayed using the LF optimized fluorescent substrate Mca-AKVYPYPME-Dnp (Turk et al., 2004), where Mca is the fluorophore 7-methoxycoumarin-4-acetic acid and Dnp is the quencher 2,4-dinitrophenyldiaminopropionic acid. LF (100 nM) in 20 mM HEPES, pH 7.4, 0.1 mg/ml BSA and 0.1% Brij35 was incubated with 10 µM peptide in the presence of compound or vehicle (1% DMSO), and fluorescence was monitored at excitation and emission wavelengths of 325 nm and 393 nm, respectively. Cleavage rates were calculated from kinetic measurements taken once per min during the initial rate period. Assays were performed in triplicate.
Assay of MKK cleavage in cell lysates
RAW264.7 cells were cultured in DMEM containing 10% fetal bovine serum. Cells were grown to confluence in 10 cm plates, washed twice with ice-cold phosphate-buffered saline and extracted into RAW cell lysis buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 0.5% NP40, 10 µg/ml leupeptin, 2 µg/ml pepstatin A, 10 µg/ml aprotinin, 1 mM DTT and 1 mM PMSF) and cleared by centrifugation (10 min at 16,000 × g). Clarified lysates were mixed with DMSO or compound to 250 µM, and LF was immediately added to 50 nM prior to incubation for 30 min at RT. Reactions were subjected to SDS-PAGE followed by immunoblotting with anti-MKK3 and anti-MKK4 antibodies (Cell Signaling Technology, dilution 1:2000).
Inhibition of MKK cleavage in intact cells
RAW264.7 cells were plated in 6-well dishes at 1.2 × 107 cells per well and allowed to recover overnight. After exchanging to fresh media, cells were preincubated for 30 min with 400 µM compound or 4% DMSO control. PA (0.5 µg/mL) and LF (0.01 µg/mL) were then added, and plates were incubated at 37 °C for 1 h. Cells were then washed once with ice-cold phosphate-buffered saline and then extracted into RAW cell lysis buffer. Lysates were cleared by centrifugation and subjected to SDS-PAGE followed by immunoblotting with anti-MKK3 or anti-vinculin antibody as a loading control.
Cytotoxicity assay
RAW264.7 cells were plated in 96-well dishes at 4 × 105 cells per well and allowed to recover for 16 h, after which the medium was removed and replaced with fresh complete medium (100 µl per well) containing 400 µM compound or 4% DMSO vehicle control. After 30 min, PA (0.5 µg/mL) and/or LF (0.001–0.3 µg/mL) were added and incubation continued for an additional 4 h. To assay viability, 10 µl of 5 mg/ml MTT in PBS was added to each well, and incubation was continued for 2 h before aspirating the supernatant and extracting with 0.1 M HCl in isopropanol. Absorbance at 570 nm with a background correction at 690 nm was determined in an absorbance plate reader. Experiments were done in triplicate and repeated three independent times for each of the inhibitors tested. The results are presented as the average ± standard deviation.
Supplementary Material
HIGHLIGHTS.
Development of a novel fluorescence polarization assay to monitor cleavage of a full length protein substrate of the anthrax lethal factor metalloproteinase
Identification of a family of lichen-derived natural products as exosite-targeting inhibitors of LF through high throughput screening
Exosite-dependent inhibitors of LF protect macrophages from lethal factor cytotoxicity
ACKNOWLEDGMENTS
We thank the following staff at the Yale Small Molecule Discovery Center: Janie Merkel for helpful discussions and advice, Michael Salcius and Laura Abriola for small molecule screening, and Jay Schneekloth for synthesis of modified depsidones. We are grateful to Sangwon Lee and Ya Ha for advice and technical assistance with this project. A.B.G. and C.C. were supported by training grants from the National Institutes of Health (T32 GM007324 and T32 CA009085, respectively). This work was supported by National Institutes of Health Grant R21 NS071532 to B.E.T.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Arkin MR, Whitty A. The road less traveled: modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr. Opin. Chem. Biol. 2009;13:284–290. doi: 10.1016/j.cbpa.2009.05.125. [DOI] [PubMed] [Google Scholar]
- Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y, Heise CE, Hoyte K, Luk W, Lu Y, et al. A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci. Transl. Med. 2011;3:84ra43. doi: 10.1126/scitranslmed.3002254. [DOI] [PubMed] [Google Scholar]
- Baldari CT, Tonello F, Paccani SR, Montecucco C. Anthrax toxins: a paradigm of bacterial immune suppression. Trends Immunol. 2006;27:434–440. doi: 10.1016/j.it.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Bjorklund M, Heikkila P, Koivunen E. Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J. Biol. Chem. 2004;279:29589–29597. doi: 10.1074/jbc.M401601200. [DOI] [PubMed] [Google Scholar]
- Bock PE, Panizzi P, Verhamme IM. Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost. 2007;5(Suppl. 1):81–94. doi: 10.1111/j.1538-7836.2007.02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett JC, Henchal EA, Schmaljohn AL, Bavari S. The evolving field of biodefence: therapeutic developments and diagnostics. Nat. Rev. Drug Discov. 2005;4:281–297. doi: 10.1038/nrd1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra AP, Boone SA, Liang X, Duesbery NS. Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J. Biol. Chem. 2003;278:9402–9406. doi: 10.1074/jbc.M211262200. [DOI] [PubMed] [Google Scholar]
- Dell'Aica I, Dona M, Tonello F, Piris A, Mock M, Montecucco C, Garbisa S. Potent inhibitors of anthrax lethal factor from green tea. EMBO Rep. 2004;5:418–422. doi: 10.1038/sj.embor.7400118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MS, Eigenbrot C, Skelton NJ, Ultsch MH, Santell L, Dwyer MA, O'Connell MP, Lazarus RA. Peptide exosite inhibitors of factor VIIa as anticoagulants. Nature. 2000;404:465–470. doi: 10.1038/35006574. [DOI] [PubMed] [Google Scholar]
- Dennis MS, Roberge M, Quan C, Lazarus RA. Selection and characterization of a new class of peptide exosite inhibitors of coagulation factor VIIa. Biochemistry. 2001;40:9513–9521. doi: 10.1021/bi010591l. [DOI] [PubMed] [Google Scholar]
- Di Nisio M, Middeldorp S, Buller HR. Direct thrombin inhibitors. N. Engl. J. Med. 2005;353:1028–1040. doi: 10.1056/NEJMra044440. [DOI] [PubMed] [Google Scholar]
- Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010;9:690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- Forino M, Johnson S, Wong TY, Rozanov DV, Savinov AY, Li W, Fattorusso R, Becattini B, Orry AJ, Jung D, et al. Efficient synthetic inhibitors of anthrax lethal factor. Proc. Natl. Acad. Sci. USA. 2005;102:9499–9504. doi: 10.1073/pnas.0502733102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman M, Belakhov V, Lee LV, Liang FS, Wong CH, Baasov T. Dual effect of synthetic aminoglycosides: antibacterial activity against Bacillus anthracis and inhibition of anthrax lethal factor. Angew. Chem. Int. Ed. Engl. 2005;44:447–452. doi: 10.1002/anie.200462003. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Lam J, Nguyen JT, O'Brien T, Wells JA. Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. USA. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holty JE, Bravata DM, Liu H, Olshen RA, McDonald KM, Owens DK. Systematic review: a century of inhalational anthrax cases from 1900 to 2005. Ann. Intern. Med. 2006;144:270–280. doi: 10.7326/0003-4819-144-4-200602210-00009. [DOI] [PubMed] [Google Scholar]
- Kornacker MG, Lai Z, Witmer M, Ma J, Hendrick J, Lee VG, Riexinger DJ, Mapelli C, Metzler W, Copeland RA. An inhibitor binding pocket distinct from the catalytic active site on human beta-APP cleaving enzyme. Biochemistry. 2005;44:11567–11573. doi: 10.1021/bi050932l. [DOI] [PubMed] [Google Scholar]
- Lauer-Fields JL, Minond D, Chase PS, Baillargeon PE, Saldanha SA, Stawikowska R, Hodder P, Fields GB. High throughput screening of potentially selective MMP-13 exosite inhibitors utilizing a triple-helical FRET substrate. Bioorg. Med. Chem. 2009;17:990–1005. doi: 10.1016/j.bmc.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer-Fields JL, Whitehead JK, Li S, Hammer RP, Brew K, Fields GB. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J. Biol. Chem. 2008;283:20087–20095. doi: 10.1074/jbc.M801438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrey JD. Biological role of lichen substances. Bryologist. 1986;89:111–122. [Google Scholar]
- Lee LV, Bower KE, Liang FS, Shi J, Wu D, Sucheck SJ, Vogt PK, Wong CH. Inhibition of the proteolytic activity of anthrax lethal factor by aminoglycosides. J. Am. Chem. Soc. 2004;126:4774–4775. doi: 10.1021/ja0495359. [DOI] [PubMed] [Google Scholar]
- Li CK, Pender SL, Pickard KM, Chance V, Holloway JA, Huett A, Goncalves NS, Mudgett JS, Dougan G, Frankel G, et al. Impaired immunity to intestinal bacterial infection in stromelysin-1 (matrix metalloproteinase-3)-deficient mice. J. Immunol. 2004;173:5171–5179. doi: 10.4049/jimmunol.173.8.5171. [DOI] [PubMed] [Google Scholar]
- Matheson AJ, Goa KL. Desirudin: a review of its use in the management of thrombotic disorders. Drugs. 2000;60:679–700. doi: 10.2165/00003495-200060030-00012. [DOI] [PubMed] [Google Scholar]
- Maynard JA, Maassen CB, Leppla SH, Brasky K, Patterson JL, Iverson BL, Georgiou G. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat. Biotechnol. 2002;20:597–601. doi: 10.1038/nbt0602-597. [DOI] [PubMed] [Google Scholar]
- Mikkelsen JH, Gyrup C, Kristensen P, Overgaard MT, Poulsen CB, Laursen LS, Oxvig C. Inhibition of the proteolytic activity of pregnancy-associated plasma protein-A by targeting substrate exosite binding. J. Biol. Chem. 2008;283:16772–16780. doi: 10.1074/jbc.M802429200. [DOI] [PubMed] [Google Scholar]
- Min DH, Tang WJ, Mrksich M. Chemical screening by mass spectrometry to identify inhibitors of anthrax lethal factor. Nat. Biotechnol. 2004;22:717–723. doi: 10.1038/nbt973. [DOI] [PubMed] [Google Scholar]
- Moayeri M, Leppla SH. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 2009;30:439–455. doi: 10.1016/j.mam.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar K, Farkas E. Current results on biological activities of lichen secondary metabolites: a review. Z. Naturforsch. C. 2010;65:157–173. doi: 10.1515/znc-2010-3-401. [DOI] [PubMed] [Google Scholar]
- Mourez M, Kane RS, Mogridge J, Metallo S, Deschatelets P, Sellman BR, Whitesides GM, Collier RJ. Designing a polyvalent inhibitor of anthrax toxin. Nat. Biotechnol. 2001;19:958–961. doi: 10.1038/nbt1001-958. [DOI] [PubMed] [Google Scholar]
- Neamati N, Hong H, Mazumder A, Wang S, Sunder S, Nicklaus MC, Milne GW, Proksa B, Pommier Y. Depsides and depsidones as inhibitors of HIV-1 integrase: discovery of novel inhibitors through 3D database searching. J. Med. Chem. 1997;40:942–951. doi: 10.1021/jm960759e. [DOI] [PubMed] [Google Scholar]
- Numa MM, Lee LV, Hsu CC, Bower KE, Wong CH. Identification of novel anthrax lethal factor inhibitors generated by combinatorial Pictet-Spengler reaction followed by screening in situ. Chembiochem. 2005;6:1002–1006. doi: 10.1002/cbic.200500009. [DOI] [PubMed] [Google Scholar]
- Overall CM. Molecular determinants of metalloproteinase substrate specificity: matrix metalloproteinase substrate binding domains, modules, and exosites. Mol. Biotechnol. 2002;22:51–86. doi: 10.1385/MB:22:1:051. [DOI] [PubMed] [Google Scholar]
- Panchal RG, Hermone AR, Nguyen TL, Wong TY, Schwarzenbacher R, Schmidt J, Lane D, McGrath C, Turk BE, Burnett J, et al. Identification of small molecule inhibitors of anthrax lethal factor. Nat. Struct. Mol. Biol. 2004;11:67–72. doi: 10.1038/nsmb711. [DOI] [PubMed] [Google Scholar]
- Pini A, Runci Y, Falciani C, Lelli B, Brunetti J, Pileri S, Fabbrini M, Lozzi L, Ricci C, Bernini A, et al. Stable peptide inhibitors prevent binding of lethal and oedema factors to protective antigen and neutralize anthrax toxin in vivo. Biochem. J. 2006;395:157–163. doi: 10.1042/BJ20051747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai P, Padala C, Poon V, Saraph A, Basha S, Kate S, Tao K, Mogridge J, Kane RS. Statistical pattern matching facilitates the design of polyvalent inhibitors of anthrax and cholera toxins. Nat. Biotechnol. 2006;24:582–586. doi: 10.1038/nbt1204. [DOI] [PubMed] [Google Scholar]
- Renckens R, Roelofs JJ, Florquin S, de Vos AF, Lijnen HR, van't Veer C, van der Poll T. Matrix metalloproteinase-9 deficiency impairs host defense against abdominal sepsis. J. Immunol. 2006;176:3735–3741. doi: 10.4049/jimmunol.176.6.3735. [DOI] [PubMed] [Google Scholar]
- Russo A, Piovano M, Lombardo L, Vanella L, Cardile V, Garbarino J. Pannarin inhibits cell growth and induces cell death in human prostate carcinoma DU-145 cells. Anticancer Drugs. 2006;17:1163–1169. doi: 10.1097/01.cad.0000236310.66080.ed. [DOI] [PubMed] [Google Scholar]
- Schepetkin IA, Khlebnikov AI, Kirpotina LN, Quinn MT. Novel small-molecule inhibitors of anthrax lethal factor identified by high-throughput screening. J. Med. Chem. 2006;49:5232–5244. doi: 10.1021/jm0605132. [DOI] [PubMed] [Google Scholar]
- Shoop WL, Xiong Y, Wiltsie J, Woods A, Guo J, Pivnichny JV, Felcetto T, Michael BF, Bansal A, Cummings RT, et al. Anthrax lethal factor inhibition. Proc. Natl. Acad. Sci. USA. 2005;102:7958–7963. doi: 10.1073/pnas.0502159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhar P, Capkova K, Salzameda NT, Barbieri JT, Hixon MS, Janda KD. Botulinum neurotoxin A protease: discovery of natural product exosite inhibitors. J. Am. Chem. Soc. 2010;132:2868–2869. doi: 10.1021/ja910761y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer RC. Bacillus anthracis. J. Clin. Pathol. 2003;56:182–187. doi: 10.1136/jcp.56.3.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura Y, Hosono M, Wada F, Yoshimura T, Maki M, Hitomi K. Screening for the preferred substrate sequence of transglutaminase using a phage-displayed peptide library: identification of peptide substrates for TGase 2 and Factor XIIIa. J. Biol. Chem. 2006;281:17699–17706. doi: 10.1074/jbc.M513538200. [DOI] [PubMed] [Google Scholar]
- Tonello F, Seveso M, Marin O, Mock M, Montecucco C. Screening inhibitors of anthrax lethal factor. Nature. 2002;418:386. doi: 10.1038/418386a. [DOI] [PubMed] [Google Scholar]
- Turk BE. Manipulation of host signalling pathways by anthrax toxins. Biochem. J. 2007;402:405–417. doi: 10.1042/BJ20061891. [DOI] [PubMed] [Google Scholar]
- Turk BE. Discovery and development of anthrax lethal factor metalloproteinase inhibitors. Curr. Pharm. Biotechnol. 2008;9:24–33. doi: 10.2174/138920108783497604. [DOI] [PubMed] [Google Scholar]
- Turk BE, Wong TY, Schwarzenbacher R, Jarrell ET, Leppla SH, Collier RJ, Liddington RC, Cantley LC. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat. Struct. Mol. Biol. 2004;11:60–66. doi: 10.1038/nsmb708. [DOI] [PubMed] [Google Scholar]
- Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000;352:739–745. [PMC free article] [PubMed] [Google Scholar]
- Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 1998;248:706–711. doi: 10.1006/bbrc.1998.9040. [DOI] [PubMed] [Google Scholar]
- Wild MA, Xin H, Maruyama T, Nolan MJ, Calveley PM, Malone JD, Wallace MR, Bowdish KS. Human antibodies from immunized donors are protective against anthrax toxin in vivo. Nat. Biotechnol. 2003;21:1305–1306. doi: 10.1038/nbt891. [DOI] [PubMed] [Google Scholar]
- Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- Yang JY, Yang WY. Site-specific two-color protein labeling for FRET studies using split inteins. J. Am. Chem. Soc. 2009;131:11644–11645. doi: 10.1021/ja9030215. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.