

Abstract
Background
p38-alpha MAPK (p38) is a tumor suppressor often mutated in human cancers, but its specific role in colorectal cancer is not completely understood. Prior studies have found that p38 activity inhibits epithelial proliferation and promotes apoptosis in the intestine. Therefore, we sought to test the hypothesis that intestinal disruption of p38 would lead to increased tumorigenesis in the colon.
Methods
p38 was deleted in mice within the intestinal epithelium using a tamoxifen-inducible Cre system under control of the villin promoter [villin-Cre ERT2(+), MAPK14(f/f)]. An azoxymethane (AOM) and dextran sodium sulfate (DSS) protocol was used to drive intestinal tumor development. Tumor measurements were made using computer software from photographs of excised colon specimens.
Results
The number of mice that developed tumors was not statistically different when comparing wild type (WT) mice (7 of 14) to inducible, intestine epithelial-deleted p38 (ed-p38, 9 of 11) mice after AOM/DSS treatment (p=0.21). However, the ed-p38 mice developed significantly more tumors (3.7 vs 1.1, p=0.008) and nearly four-times the total tumor burden as WT mice (17.4 vs 4.8 mm2, p=0.03). WT and ed-p38 groups demonstrated a similar degree of colon inflammation.
Conclusions
Deletion of p38-alpha MAPK within the colonic mucosa leads to a hyperplastic state promoting greater tumor development. Since the severity of colitis was not augmented in mice with p38 deficiency, tumor development is likely mediated by impaired cell cycle regulation within the colonic epithelium. Manipulation of p38 activity may provide a novel treatment and/or prevention strategy in the management of colorectal cancer, particularly in the setting of inflammatory bowel disease.
Keywords: p38 MAPK, colorectal cancer, inflammatory bowel disease, colon tumor, inducible, enterocyte-specific knockout
Introduction
In the United States, colorectal cancer (CRC) is the second leading cause of cancer-related deaths1. In 2011 an estimated 141,210 people were diagnosed with CRC, while 49,380 people died from the disease2. This malignancy often results as a complication from inflammatory diseases of the colon, such as ulcerative colitis and Crohn’s disease3,4. As the extent and duration of each of these diseases increases, so too does the exposure of the colon to inflammation and the concomitant risk of CRC. Currently, there are four main modalities utilized to treat CRC including surgery, radiation, chemotherapy, and biological therapy. As our knowledge about the mechanisms of the progression of CRC grows, our armamentarium of chemo- and bio-therapeutic agents increases.
Recently, a new experimental model has been developed in rodents that mimics CRC progression in humans, especially in the setting of inflammatory bowel disease5. This tumorigenesis model uses a potent carcinogen and an agent that causes chronic inflammation of the colon to drive tumor development. The regimen uses a single intraperitoneal dose of the genotoxic carcinogen azoxymethane (AOM) followed by oral exposure to dextran sodium sulfate (DSS) in the animal’s drinking water to effectively and reliably induce colonic tumors6,7. This animal model represents a powerful tool for investigating the pathogenesis and chemoprevention of colitis-related colon cancer.
p38-alpha mitogen-activated protein kinase (p38 MAPK, p38) has many physiologic functions. p38 MAPKs are activated by environmental and genetic stresses and play a role in inflammation and tissue homeostasis by regulating cell proliferation, differentiation, and survival8–10. In addition, p38 MAPK is known to function as a tumor suppressor by negatively regulating cell cycle progression via several mechanisms, including down-regulating cyclins, up-regulating cyclin-dependent kinase inhibitors and modulating the tumor suppressor p5311,12. p38 also regulates induction of apoptosis after cellular stress8. Indeed, chemotherapeutic drugs have been found to stimulate apoptosis in a p38-dependent fashion13,14.
p38 MAPK has been shown to play an important role in tumorigenesis. In cell culture experiments, p38 deletion in fibroblasts resulted in augmented proliferative rates and increased formation of subcutaneous tumors15. Furthermore, several negative regulators of p38 MAPK signalling have been demonstrated to be overexpressed in human tumors and cancer cell lines16,17. Sequencing studies have also identified somatic mutations in the p38 MAPK pathway in several human tumors18. In addition, hepato-cellular carcinomas have been found to have lower p38 MAPK activity with an inverse correlation between tumor size and p38 activity19. Mouse models have shown p38 MAPK deficiency increases liver and lung tumor development20,21.
Our lab has recently developed an inducible, enterocyte-specific p38-alpha-deleted mouse model22. We have found that p38 MAPK is efficiently deleted in all enterocytes including the colonic epithelium. Before the development of this temporal-spatial knockout model, it was difficult to study the effects of p38 deletion on the gut as global p38 MAPK deletion is embryonic lethal20,23. Moreover, animals with p38 knocked-out constitutively from the intestinal mucosa do not breed well and have small litters24. Our findings in mice with enterocyte-specific p38 deletion are consistent with its role as a tumor suppressor. Mice that lack p38 display increased proliferation and decreased apoptosis in the crypt cells of the small intestine22. Our results mirror work recently published by Otsuka et al who found that a constitutive, intestine-specific deletion of p38 led to a hyperproliferative state in the crypt compartment24.
The purpose of the present experiments was to determine the effect of p38 deletion on tumorigenesis. Specifically, we hypothesized that inactivating p38 in enterocytes would increase the development of colon tumors in the setting of chronic inflammation. We found that p38 MAPK deletion in the colonic epithelium results in an increased propensity for tumor formation. These findings may offer important translational implications for the diagnosis and treatment of colorectal cancers in humans, particularly in the setting of inflammatory bowel disease.
Methods
Animals
The protocol for this study was approved by the Washington University Institutional Animal Care and Use Committee (Protocols #20070145 and #20100103, Washington University School of Medicine, St Louis, MO). All mice were bred and housed in a standard vivarium with a 12-hour day-night cycle. Inducible, intestine epithelial-deleted p38-alpha mice (ed-p38 mice) were generated by crossing mice with a tamoxifen-inducible Cre-fusion protein under control of the villin promoter (obtained via generous donation from Sylvie Robine, Curie Institute, Paris, France) with mice in which the floxed MAPK14 gene (encoding the p38 protein) had been tagged for recombination (acquired via generous donation from Huiping Jiang, Boehringer Ingelheim Pharmaceuticals, Inc.; Ridgefield, CT). Littermates lacking Cre were used as wild type (WT) controls. The gene knockout was induced by giving intraperitoneal (IP) tamoxifen injections (0.5mg/day) for three consecutive days (Days 1–3, Figure 1). We have shown that this system reliably and efficiently deleted the gene of interest (Figure 2).
Figure 1. Tumorigenesis Protocol.

Inducible, enterocyte-specific p38 deletion was induced by intraperitoneal (IP) tamoxifen (TAM) injection (0.5mg/day) on days 1–3. Azoxymethane (AOM, 10mg/kg) was injected IP on day 8. On day 13 2.5% dextran sodium sulfate (DSS) was administered to mice in their drinking water for seven days. On day 20 mice were given water for 14 days to recover. Mice underwent three cycles of DSS treatment followed by water. Control mice were given water for the entire protocol instead of DSS. Animals were sacrificed on day 77.
Figure 2. Efficiency of p38 deletion.
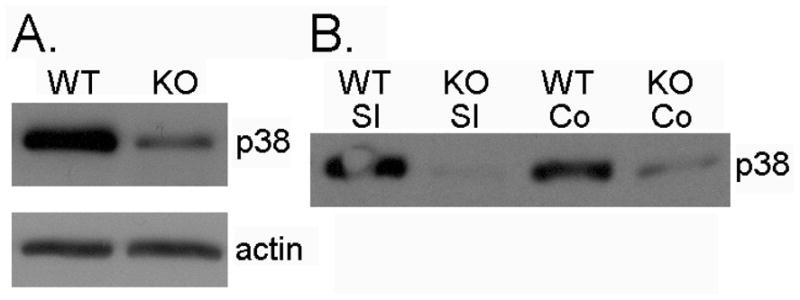
Western blots for total p38 MAPK (p38) and actin from the intestine mucosa of wild type (WT) and inducible, intestine epithelial-deleted p38 mice (KO). A. Small intestine epithelium collected 77 days after initial tamoxifen injection. B. Small intestine (SI) and colon (Co) epithelium harvested 7 days after tamoxifen injection.
Genotyping/Phenotyping
Polymerase chain reaction was used to determine mouse genotype from tail clippings in the standard fashion using previously published protocols for villin-Cre25 and MAPK14(f/f)23. In addition, we confirmed the phenotype (presence or absence of p38 protein) of every mouse that survived to the end of the experiment by Western immunoblotting. Antibodies against p38-alpha and total p38 MAPK were obtained from Cell Signalling (Danvers, MA).
Experimental design
For these experiments we used four groups of mice: WT mice given water control (WT H2O; n=10), WT mice given DSS (WT DSS; n=17), ed-p38 mice given water control (KO H2O; n=11), and ed-p38 mice given DSS (KO DSS; n=12). The protocol lasted a total of 77 days (Figure 1). On the first three days, all mice were given an IP injection of 0.5mg of tamoxifen. On day 8, mice were given an IP injection of AOM at a dose of 10mg/kg. On day 13, some mice were given 2.5% DSS dissolved in their drinking water for seven days. Control mice were given regular drinking water for the entirety of the experiment. After seven days of DSS treatment, the mice were given water for 14 days to recover. Mice underwent three cycles of DSS treatment followed by water. Animals were sacrificed on day 77.
Data Collection
Throughout the experiment animal weight and feces character were documented three times per week. For example, following day 13 the mice were weighed and their feces were characterized on days 16, 18, and 20 (afterwards this 3, 2, 2 cycle continued until the end of the protocol). Feces from individual mice were collected by isolating them in individual containers and waiting for each mouse to defecate to ensure a fresh sample. A scale was then used to allow for statistical analyses: normal=1, loose=2, bloody=3, loose and bloody=4. In this way, a greater number reflects more severe colitis.
Mouse Sacrifice and Tissue Collection
Mice were sacrificed by cervical dislocation under inhaled isoflourane anesthesia. The intestine was removed from the pylorus to the anus and immediately flushed with ice-cold phosphate-buffered saline containing protease and phosphatase inhibitors (0.2 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 1 mM benzamidine, 1 mM sodium orthovanadate, 2 μM cantharidin). The distal 3cm of small intestine and entire large intestine were incised longitudinally and fixed in 10% formalin. After three hours the intestine was placed in 70% ethanol for storage. A 10 cm portion of the mid-small bowel was saved fresh for enterocyte isolation26. Briefly, the small intestine was opened longitudinally and placed in isolation buffer containing ice-cold balanced salt solution (1.5 mM KCl, 96 mM NaCl, 27 mM sodium citrate, 8 mM KH2PO4, 5.6 mM Na2HPO4), 15 mM EDTA, 1 mM dithiothreitol, and protease and phosphatase inhibitors (as above). The samples were then subjected to mechanical vortexing for about 20 minutes. We have found that this causes the crypts and villi to dissociate from the underlying basement membrane. The remnant intestine was then discarded and the crypts and villi collected for future analysis. This results in a specimen of intestinal mucosa with little to no contamination from the submucosal or muscular layers26.
Colon Analysis
The pinned segments of intestine were photographed at 7X magnification using an Olympus microscope and DP controller software. Tumor measurements were obtained using image analysis software (Metamorph, UIC, Downingtown, PA). Colon area, individual tumor area, and number of tumors were measured. In this way, total tumor area could be derived and tumor ratio (total tumor area/total colon area) was calculated. Inflammatory lesions, such as ulcers and regions with thickened mucosa, were also measured.
Histology
Formalin fixed specimens of colons were embedded in paraffin and five-micrometer-thick longitudinal sections were mounted on glass slides and used for morphologic analyses. Hematoxylin- and eosin–stained sections were used to assess tumor morphology qualitatively for overall structure and degree of atypia.
Statistics
Statistical analyses were performed by an experienced statistician (Liu) as follows: Fisher’s exact test was used to test the association between tumor development and groups. Differences between groups with regard to number of tumors, total tumor area, tumor ratio, and inflammation were determined by Kruskal-Wallis tests. By necessity, these tests were only performed for mice that survived to sacrifice. Weight and feces character were evaluated by repeated measures method analysis for all subjects alive on the day of collection. Means and standard error are reported. Two-sided p values less than 0.05 were considered significant. SAS Version 9.1 (Cary, NC) was used to perform all statistical analysis.
Results
Efficiency of p38 deletion
Attenuated expression of p38 protein after tamoxifen injection was confirmed in the small intestine epithelium of all mice by Western immunoblotting after 77 days (Figure 2A). Earlier time points after tamoxifen (7 days) revealed more efficient knockout of p38 protein expression in both intestinal epithelial cells and colon (Figure 2B).
Severity of chronic DSS-induced colitis is not altered by p38 deletion
Weight change
As expected, both WT and ed-p38 mice lost weight compared to control mice given water during treatment with DSS (p<0.05). However, the change in weight during treatment interval did not differ between WT DSS and KO DSS groups (p=0.69). Mouse weight was not statistically different between WT DSS and KO DSS groups at any day of the treatment protocol (Figure 3). Of note, 3 of 17 mice in the WT DSS group and 1 of 12 mice in the KO DSS group died during treatment. No mice died in the water control groups during the treatment protocol.
Figure 3. Mouse weight.

Mean animal weights (with standard error) of WT mice given DSS (WT DSS, n=17) and KO mice administered DSS (KO DSS, n=12). Three mice in the WT DSS group and one animal in the KO DSS group died during the treatment protocol. Values for these mice were included until death. Mouse weight did not differ between WT DSS and KO DSS groups overall or at any day of the treatment protocol.
Stool character
Both WT and ed-p38 mice developed clinically evident colitis, manifested by loose and bloody stools after DSS treatment compared to the water controls (p<0.0001). Over the entire protocol, no significant difference was demonstrated for stool character over time between the WT DSS and KO DSS groups (p=0.82; Figure 4).
Figure 4. Feces character.

Mouse feces were collected on the protocols days shown and scored to quantify the severity of colitis as follows: normal feces = 1, loose feces = 2, bloody feces = 3, loose and bloody feces = 4. No significant difference was found for feces score over time between the WT DSS and KO DSS groups.
Colon Inflammation
Both groups of mice exposed to DSS had a significant percentage of the colon affected by inflammatory lesions compared to mice that were given water control. WT H2O mice had 0.25 +/− 0.10%, while WT DSS mice had 4.30 +/− 0.97% of the colon with inflammatory lesions (p<0.0001). The KO H2O group had 0.47 +/− 0.17% and the KO DSS group had 3.79 +/− 1.20% (p=0.0005). WT DSS and KO DSS mice had similar total inflamed area and percentage of inflamed colon (p=0.87 and 0.83 respectively; Figure 5).
Figure 5. Colon Inflammation.

Inflammatory lesions such as ulcers and regions with thickened mucosa were measured in resected colons. Shown are plots for A. total inflamed colon area and B. colon inflammation ratio (inflamed area/total colon area). No significant differences were found between WT DSS (n=14) and KO DSS groups (n=11).
Epithelial-deleted p38 mice develop more colon tumors following AOM and DSS
Tumor incidence
As expected, both WT and ed-p38 mice developed tumors after treatment with AOM and DSS compared to their littermate controls given AOM treatment alone (Figure 6). Zero of 10 WT H2O mice developed tumors, while 7 of 14 WT DSS mice had tumors at sacrifice (p=0.02). Zero of 11 KO H2O mice had colon tumors, while 9 of 11 KO DSS mice grew tumors after treatment (p<0.0001). While 82% of KO DSS mice developed tumors compared to only 50% of WT DSS mice, this difference was not statistically significant (p=0.21).
Figure 6. Mouse colons after the tumor induction protocol.

Mouse colons were removed at the time of sacrifice, pinned, fixed, and photographed at 7X magnification. The arrowheads indicate tumors.
Tumor morphology
Tumors were typical adenomas that have been previously described after AOM and DSS treatment of mice5–7. Qualitatively, there were no apparent morphologic differences between the tumors that grew in the DSS treated WT and ed-p38 mice [viewed by an experienced pathologist (Stappenbeck), Figure 7)].
Figure 7. Colon tumor morphology.

Shown are representative photomicrographs (4X) of mouse rectums after the treatment protocol. Mouse colons were fixed, sectioned, and stained with hematoxylin and eosin. Tumor morphology was assessed for degree of atypia by an experience pathologist (TSS). Tumors that developed after AOM and DSS treatment in the WT and KO groups were typical adenomas with no apparent morphologic differences between groups.
Number of tumors
As expected, mice treated with DSS developed more tumors than mice given water control. WT H2O mice had zero tumors and WT DSS mice had 1.07 +/− 0.38 tumors per mouse (p=0.01). KO H2O mice had zero tumors and KO DSS mice had 3.73 +/− 0.85 tumors per mouse (p=0.0003). The ed-p38 mice developed significantly more tumors than their WT littermates after AOM and DSS treatment (p=0.008; Figure 8A).
Figure 8. Tumor Burden.

Mouse colons were removed at the time of sacrifice and photographed. Colon area, individual tumor area, and number of tumors were measured. In this way total tumor area and tumor percentage was calculated. KO DSS (n=11) mice developed statistically more tumors than WT DSS (n=14). A. Shown is the mean number of colon tumors per mouse. B. Depicted is the total tumor area per mouse. C. The ratio of tumor area to colon area is shown. * indicates p<.05, ** indicates p<.01.
Total tumor area
Both genotypes given DSS had a greater tumor burden than mice given water. WT DSS mice had 4.84 +/− 1.84 mm2 of colon tumor involvement versus KO DSS mice had 17.39 +/− 5.57 mm2 of colon tumor burden (p=0.03; Figure 8B).
Percentage of colon affected by tumors
Mice treated with DSS had a greater percentage of their colons affected by tumors than mice given water control. WT H2O mice had no tumors, while WT DSS mice had 0.84 +/− 0.34 % of their colons impacted by tumors (p=0.01). KO H2O mice had zero tumors, while KO DSS mice had 2.50 +/− 0.78% of the colon affected by tumors (p=0.0003). Ed-p38 mice had a significantly larger percentage of tumors compared to WT mice (p=0.04; Figure 8C).
Discussion
In the present study, we show that p38 deletion increases colon tumorigenesis following exposure to a potent carcinogen in the setting of a chronic inflammatory state. Mice with inducible, enterocyte-specific deletion of p38 MAPK developed more tumors and had a greater tumor burden than wild type controls. These data suggest that under normal physiologic conditions, p38 MAPK serves as an important cell cycle inhibitor. Overall, our findings support the hypothesis that p38 MAPK may be an important contributor to the development of colon cancer, particularly in the context of inflammatory diseases of the colon.
In our studies we concluded that p38 deletion did not alter the clinical severity of colitis after DSS treatment since the change in animal weight and feces character over time were similar between the WT and ed-p38 mice. These findings are in contrast to those recently reported by Otsuka et al who found that constitutive deletion of p38 in the intestine mucosa worsened DSS-induced colitis24. In their study, the ed-p38 mice lost more weight and had more severe scores of epithelial injury after six days of DSS treatment than wild type controls. One explanation for this discrepancy could be the different time course of exposure to DSS. Our mice were given DSS over three-one week cycles in order to induce chronic inflammation instead of the short course of exposure required to cause acute colitis. In addition, mice in our study were allowed two weeks to heal after their last treatment before sacrifice, while Otsuksa’s group waited only three days. Nevertheless, we believe that p38 deletion did not cause more severe colitis. This fact further strengthens the claim for p38’s role as a suppressor of colon cancer in that an augmented inflammatory response did not drive tumor progression; rather, the loss of p38’s inhibitory effects on the cell cycle allowed more tumors to form.
The present study does have several methodological points to address. The time course of our experiment was relatively short. We sacrificed mice about 10 weeks after starting treatment. Other investigators have waited 20 weeks before assessing for tumors5. Allowing tumors more time to develop would likely result in more tumors at sacrifice and, potentially, a more consistent number of tumors in each mouse. However, longer treatment times would likely result in more tumor development in the WT mice. We chose the shorter time course to accentuate the difference between the ed-p38 and WT mice. Currently we found a trend toward the ed-p38 mice being more likely to have developed colonic tumors. However, given the difference in tumor incidence found (82% vs 50%), a larger number of mice in each group would be needed to achieve statistical significance.
Although levels of p38 were significantly reduced in our knockout group, some p38 protein was still detectable at later time points by Western blotting. A partial explanation for this could be that we used an antibody against total p38 MAPK, which would detect all four isoforms of p38 (alpha, beta, gamma, and delta). The gene that was floxed, thus, tagging it for deletion in this study was MAPK14. Therefore, the signal could be generated from the other three isoforms not deleted by our knockout model. However, we have found that p38-alpha is the primary isoform expressed in the intestine, so this, likely, does not account for the entire signal found22. Another explanation is that a small minority of cells which did not undergo genetic recombination by Cre (and, thus, still express p38) may repopulate the epithelium, creating a mosaic pattern of p38 expression. This phenomenon has been shown to occur for c-myc deletion in the intestine mucosa by an identical inducible Villin-Cre recombinase system 21 days after tamoxifen injection27. Furthermore, our lab has previously found a profound (>95%) reduction in p38 protein levels in bowel harvested 7 days after initiating p38 deletion with only two tamoxifen injections22. Therefore, we believe that the p38 was robustly deleted in this study, but unperturbed cells do repopulate the epithelium if given enough time.
It will be interesting in future studies to determine the impact of p38 deletion in the context of other models for CRC development. Our current model uses a setting of chronic inflammation analogous to inflammatory bowel disease to drive tumor formation. To determine if p38 MAPK plays a role in sporadic CRC, the APCMin model would be useful28–29. Alternatively, it will be important to determine the effects of p38 overexpression on tumor development in the context of both models.
Acknowledgments
Support: This work was supported by National Institutes of Health Grants T32 CA009621 (Wakeman), R01 DK059288 (Wandu, Erwin, Guo, Warner), R01 AI084887 (Stappenbeck), P30 CA91842 (Liu) and P30 DK52574 - Morphology and Murine Models Cores of the Digestive Diseases Research Core Center of the Washington University School of Medicine. Additional support was obtained from the Summer Undergraduate Research Fellowship (SURF) Program at Washington University (Schneider) and the St. Louis Children’s Hospital Foundation - Children’s Surgical Sciences Institute at Washington University School of Medicine.
The authors wish to acknowledge Susan Shi, and Nicole Malvin for their contributions to this research.
Abbreviations
- CRC
colorectal cancer
- AOM
azoxymethane
- DSS
dextran sodium sulfate
- p38 MAPK, p38
p38-alpha mitogen-activated protein kinase
- ed-p38 mice
epithelial-deleted p38-alpha mice
- WT
wild type
- IP
intraperitoneal
- WT H2O
WT mice given water control
- WT DSS
WT mice given DSS
- KO H2O
ed-p38 mice given water control
- KO DSS
ed-p38 mice administered DSS
- TAM
tamoxifen
Footnotes
Disclosures: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Centers for Disease Control and Prevention. www.cdc.gov.
- 2.SEER Database. http://seer.cancer.gov/statfacts/html/colorect.html#incidence-mortality.
- 3.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–35. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Hogezand RA, Eichhorn RF, Choudry A, Veenendaal RA, Lamers BHW. Malignancies in inflammatory bowel disease: fact or fiction? Scand J Gastroenterol Suppl. 2002;235:48–53. doi: 10.1080/003655202320621454. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94(11):965–73. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaker A, Swietlicki EA, Wang L, Jiang S, Onal B, Bala S, et al. Epimorphin deletion protects mice from inflammation-induced colon carcinogenesis and alters stem cell niche myofibroblast secretion. J Clin Invest. 2010;120(6):2081–93. doi: 10.1172/JCI40676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007;2(8):1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- 8.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–49. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 9.Rincon M, Davis RJ. Regulation of the immune response by stress-activated protein kinases. Immunol Rev. 2009;228:212–24. doi: 10.1111/j.1600-065X.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- 10.Bradham C, McClay DR. p38 MAPK in development and cancer. Cell Cycle. 2006;5(8):824–8. doi: 10.4161/cc.5.8.2685. [DOI] [PubMed] [Google Scholar]
- 11.Ambrosino C, Nebreda AR. Cell cycle regulation by p38 MAP kinases. Biol Cell. 2001;93:47–51. doi: 10.1016/s0248-4900(01)01124-8. [DOI] [PubMed] [Google Scholar]
- 12.Thornton TM, Rincon M. Non-classical p38 MAP kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. 2009;5:44–51. doi: 10.7150/ijbs.5.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deacon K, Mistry P, Chernoff J, Blank JL, Patel R. p38 Mitogen-activated protein kinase mediates cell death and p21-activated kinase mediates cell survival during chemotherapeutic drug-induced mitotic arrest. Mol Biol Cell. 2003;14:2071–87. doi: 10.1091/mbc.E02-10-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Losa JH, Parada Cobo C, Viniegra JG, Sánchez-Arevalo Lobo VJ, Ramón y Cajal S, Sánchez-Prieto R. Role of the p38 MAPK pathway in cisplatin-based therapy. Oncogene. 2003;22:3998–4006. doi: 10.1038/sj.onc.1206608. [DOI] [PubMed] [Google Scholar]
- 15.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11(2):191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 16.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nature Genet. 2002;31:210–5. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 17.Yu W, Imoto I, Inoue J, Onda M, Emi M, Inazawa J. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene. 2007;26:1178–87. doi: 10.1038/sj.onc.1209899. [DOI] [PubMed] [Google Scholar]
- 18.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iyoda K, Sasaki Y, Horimoto M, Toyama T, Yakushijin T, Sakakibara M, et al. Involvement of the p38 mitogen-activated protein kinase cascade in hepatocellular carcinoma. Cancer. 2003;97:3017–26. doi: 10.1002/cncr.11425. [DOI] [PubMed] [Google Scholar]
- 20.Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, et al. p38α suppresses normal and cancer cell proliferation by antagonizing the JNK–c-Jun pathway. Nature Genet. 2007;39:741–9. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- 21.Ventura JJ, Tenbaum S, Perdiguero E, Huth M, Guerra C, Barbacid M, et al. p38α MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nature Genet. 2007;39:750–8. doi: 10.1038/ng2037. [DOI] [PubMed] [Google Scholar]
- 22.Wakeman D, Guo J, Santos JA, Wandu WS, Schneider JE, McMellen ME, Leinicke JA, et al. p38 MAPK Regulates Bax Activity and Apoptosis in Enterocytes at Baseline and after Intestinal Resection. Am J Physiol Gastrointest Liver Physiol. 2012 Mar 1; doi: 10.1152/ajpgi.00485.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, et al. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004;24(24):10611–20. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otsuka M, Kang YJ, Ren J, Jiang H, Wang Y, Omata M, et al. Distinct effects of p38alpha deletion in myeloid lineage and gut epithelia in mouse models of inflammatory bowel disease. Gastroenterology. 2010;138(4):1255–65. doi: 10.1053/j.gastro.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39(3):186–93. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 26.Guo J, Longshore S, Nair R, Warner BW. Retinoblastoma protein (pRb), but not p107 or p130, is required for maintenance of enterocyte quiescence and differentiation in small intestine. J Biol Chem. 2009;284(1):134–40. doi: 10.1074/jbc.M806133200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bettess MD, Dubois N, Murphy MJ, Dubey C, Roger C, Robine S, et al. c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol Cell Biol. 2005;25(17):7868–78. doi: 10.1128/MCB.25.17.7868-7878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–4. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 29.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, et al. Suppression of intestinal polyposis in Apc_716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–9. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]