

Abstract
Pulmonary fibrosis occurs in a variety of clinical settings, constitutes a major cause of morbidity and mortality, and represents an enormous unmet medical need. However, the disease is heterogeneous, and the failure to accurately discern between forms of fibrosing lung diseases leads to inaccurate treatments. Pulmonary fibrosis occurring in the context of connective tissue diseases is often characterized by a distinct pattern of tissue pathology and may be amenable to immunosuppressive therapies. In contrast, idiopathic pulmonary fibrosis (IPF) is a progressive and lethal form of fibrosing lung disease that is recalcitrant to therapies that target the immune system. Although animal models of fibrosis imperfectly recapitulate IPF, they have yielded numerous targets for therapeutic intervention. Understanding the heterogeneity of these diseases and elucidating the final common pathways of fibrogenesis are critical for the development of efficacious therapies for severe fibrosing lung diseases.
Pulmonary fibrosis: a disease spectrum
Pulmonary fibrosis can occur in a variety of clinical settings. An essential component in the evaluation of patients is the determination of whether there is evidence for systemic disease or exposure to environmental irritants (1). The importance of accurate classification of pulmonary fibrosis lies in the fact that the natural history and the potential response to treatment differ depending on the etiology (2). Connective tissue diseases such as rheumatoid arthritis and systemic sclerosis (scleroderma) commonly are accompanied by pulmonary fibrosis, and the diagnosis can often be established with reasonable confidence. However, other connective tissue diseases are less well characterized, and furthermore, the lung can be the first place in which a connective tissue disease manifests. It is important to determine the presence or absence of an underlying connective tissue disorder because some forms of connective tissue disease–related fibrosis are reversible.
Figure 1A shows a chest computed axial tomography (CAT) scan demonstrating traction bronchiectasis and chronic fibrotic remodeling. Surgical lung biopsy shows a homogenous pattern of fibrosis and chronic inflammation with a paucity of normal lung (Figure 1B). No fibroblast foci are identified. This pattern of fibrosis is referred to as fibrotic nonspecific interstitial pneumonia (NSIP). In this case, laboratory evaluation provided evidence of a connective tissue disease with elements of a mixed connective tissue disease and polymyositis (antisynthetase syndrome) (3). The patient was treated with a combination of prednisone and mycophenolate mofetil; a chest CAT scan after therapy is shown in Figure 1C. There is dramatic resolution of the fibrotic lung disease.
Figure 1. Fibrotic NSIP.

(A) Chest CAT scan from a 60-year-old woman with 6 months of cough, exertional breathlessness, and fatigue. (B) Surgical lung biopsy shows a homogenous pattern of fibrosis and chronic inflammation with a paucity of normal lung. Magnification, ×40. (C) Chest CAT after treatment with a combination of prednisone and mycophenolate mofetil.
In contrast, patients with idiopathic pulmonary fibrosis (IPF) have a different pattern of reticular opacities on chest CAT scan (Figure 2A). Imaging classically reveals traction bronchiectasis, thickened interlobular septae, and subpleural honeycombing (2). When all three are present and there is no evidence to support an associated connective tissue disease or environmental exposure, a confident diagnosis of IPF can be made (4). This means that if a surgical lung biopsy is performed, it is highly likely (>90%) that a usual interstitial pneumonia pattern (UIP) would be observed (5, 6). UIP is defined by the presence of microscopic honeycombing, fibroblastic foci, and a variegated pattern of chronic interstitial fibrosis with accentuation beneath the pleura (ref. 7 and Figure 2, B and C). When these patterns are demonstrated on chest CAT scan and lung biopsy, treatment with immunosuppressive therapy such as prednisone and azathioprine is ineffective. In fact, a recent study sponsored by the NIH evaluating the efficacy of the combination of prednisone, azathioprine, and N-acetyl cysteine together relative to placebo was halted at interim analysis for lack of efficacy. The comparison of N-acetyl cysteine with placebo continues in the study (8).
Figure 2. Classic usual interstitial pneumonia.

(A) Chest CAT scan demonstrating the cardinal radiographic features of IPF: subpleural honeycombing, traction bronchiectasis, and thickened interlobular septae. (B) Lung biopsy demonstrating the cardinal pathologic manifestations of UIP: a variegated pattern of chronic interstitial fibrosis, subpleural accentuation, and fibroblastic foci. (C) Lung biopsy demonstrating a fibroblast focus without accompanying inflammation. (B and C) Magnification, ×40.
It is important to note, however, that a UIP pattern can be seen on lung biopsies from patients that do not have idiopathic disease; specifically, hypersensitivity pneumonitis and connective tissue diseases can show a UIP pattern (9). This can make the clinical management of pulmonary fibrosis challenging. As an example, the chest CAT scan in Figure 3A shows peripheral reticular opacities with a lower lobe predominance and traction bronchiectasis. However, subpleural honeycomb changes are equivocal. Surgical lung biopsy revealed a variegated pattern of chronic interstitial pneumonia, but there were few fibroblastic foci and microscopic honeycomb changes were not evident (Figure 3, B and C). In addition, there were areas of mononuclear inflammation at a distance from established collagen deposition. Connective tissue serologies were not revealing, but immunosuppressive therapy halted disease progression while lung function remained stable and oxygen desaturation with ambulation improved. This type of pulmonary fibrosis has not been investigated rigorously, and this example emphasizes our incomplete understanding of the heterogeneity of pulmonary fibrosis. Thus, the ability to distinguish IPF and UIP from other forms of pulmonary fibrosis is of utmost importance so that clinicians can accurately identify those patients who will benefit from immune-modulating therapy. The mechanisms that lead to reversible pulmonary fibrosis are unknown, and it remains to be seen whether IPF or UIP can be reversed as novel therapeutic approaches are applied.
Figure 3. A UIP pattern in the absence of IPF.

(A) Chest CAT scan from a 68-year-old man who presented with cough and exertional breathlessness that demonstrates subpleural reticular opacities but without definitive honeycombing. (B) Surgical lung biopsy revealed a variegated pattern of chronic interstitial fibrosis without microscopic honeycombing but with fibroblastic foci and areas of mononuclear inflammation. Magnification in B, ×40; C is a higher-magnification view of boxed area in B.
Idiopathic pulmonary fibrosis
IPF is the most common and most severe form of idiopathic interstitial pneumonia. It is often relentlessly progressive, leading to death from respiratory failure within 2–5 years of diagnosis in the majority of instances (4, 10, 11). Importantly, while the overall prognosis is poor in IPF, it is difficult to predict the rate of progression in individual patients. Over the last decade, randomized clinical trials in IPF, coupled with the increased utilization of chest CAT scans has led to the identification of patients at earlier stages of the disease process. However, predicting the clinical course in these patients is particularly challenging. The annual incidence of IPF appears to be rising and is estimated at 5–16 per 100,000 individuals; prevalence is 13–20 per 100,000 (12). IPF is more common in men, and the prevalence rises dramatically with age (7). IPF is sufficiently uncommon under the age of 50 as to mandate an exhaustive search for an underlying connective tissue disease or occult environmental exposure in young patients, particularly women. In contrast, pulmonary fibrosis in patients over the age of 70 is significantly more likely to be classified as IPF (13). Risk factors for IPF include age, male gender, and a history of cigarette smoking (14).
Genetic predisposition
Several studies have identified familial forms of pulmonary fibrosis (15). The precise contribution of genetic transmission to IPF is difficult to ascertain because of the late onset of disease presentation, but it appears to be responsible for 5% of IPF cases. Transmission is autosomal dominant, with variable and reduced penetrance. A number of different genetic mutations have been associated with the development of pulmonary fibrosis, some in genes expressed only in epithelial cells in the lung and others in more ubiquitously expressed genes. Disease-linked mutations in the surfactant protein C gene (SFTPC) were first identified in adults by Loyd and colleagues (16), and subsequently, mutations in the surfactant protein A2 (SFTPA2) gene were identified in a distinct cohort of familial pulmonary fibrosis patients (17). As both proteins are expressed by alveolar epithelial cells, these important discoveries have generated new insights into pathogenesis (discussed below) and have provided a contextual basis for the observation that fibrosis only occurs in the lungs in IPF.
The most common mutations identified in familial pulmonary fibrosis are those in the genes encoding telomerase (TERT and TERC) (18, 19). Interestingly, unlike surfactant proteins A and C, telomerase is expressed by cells outside the lung, particularly in stem cells and progenitor cells, raising intriguing questions regarding the role of these mutations in alveolar epithelial cell renewal and the development of pulmonary fibrosis that require further study (Figure 4).
Figure 4. Model of the relationship between ER stress and lung fibrosis.
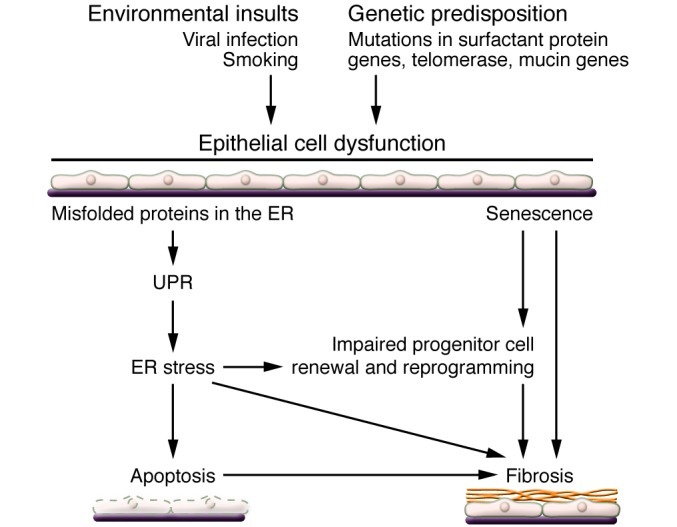
Genetic predisposition such as mutations in surfactant protein genes renders AEC2s vulnerable to environmental insults such as viral infections and cigarette smoking. These insults may result in the accumulation of misfolded proteins in the ER. Persistent injury may lead to ER stress and eventually to epithelial cell apoptosis. However, ER stress leading directly to pulmonary fibrosis in the absence of injury remains to be demonstrated.
More recently, a common polymorphism in the promoter of the MUC5B gene has been identified in both familial pulmonary fibrosis and sporadic IPF (20). This SNP was identified in 34% of subjects with familial interstitial pneumonia and in 38% of subjects with IPF (20). MUC5B is expressed in bronchiolar epithelium and not by alveolar epithelial type 2 cells (AEC2s), under physiologic conditions, and the functional consequences of this polymorphism remain to be elucidated. Nevertheless, the prevalence of the polymorphism is much greater than that of surfactant mutations, which supports an emerging theme of abnormalities in proteins uniquely expressed in the lung contributing to IPF pathogenesis.
Pathogenesis
The paradigms of pathogenesis of IPF have evolved as the description of the radiographic and pathologic patterns have been refined over the last two decades (21). The ability to distinguish between immune-driven pulmonary fibrosis, which is fairly obvious in the settings of rheumatoid arthritis and scleroderma but much more subtle in the context of undifferentiated connective tissue disease processes, has improved. However, diagnostic tools to distinguish between IPF and other forms of pulmonary fibrosis remain rudimentary relative to other diagnostics, such as those utilized in oncology. The conceptual transition of IPF pathogenesis from an inflammatory-driven process to a primarily fibrotic one has been well documented (22–24), largely based on the paucity of inflammatory infiltrates at the leading edge of fibrosis on lung tissue biopsy and the poor clinical response of IPF to systemic corticosteroid therapy. These observations, coupled with the identification of mutations in surfactant protein genes and evidence of alveolar epithelial cell apoptosis using histochemistry and electron microscopy analysis from patient biopsies, suggest that AEC2s are involved in the pathogenesis of fibrotic lung disease.
The vulnerable alveolar epithelium in IPF
Several lines of evidence have emerged implicating a combination of environmental, age-related, and genetic factors that coalesce to create an alveolar epithelium that is susceptible to injury from either unknown endogenous factors or exogenous insults such as viral infection or microaspiration (25–27). The mutations in surfactant proteins C and A2 that have been identified in familial pulmonary fibrosis result in abnormalities in surfactant protein folding (28). Misfolded proteins are recognized by the cell, and in order to limit their deleterious effects the cell activates the unfolded protein response (UPR) (29). The UPR is an adaptive response designed to halt protein production and augment production of chaperone proteins in order to facilitate proper protein folding. The accumulation of misfolded proteins and activation of the UPR creates a challenge to the cell, referred to as ER stress, which can also be induced by viral infection (30). If unabated, ER stress activates a program that leads to apoptosis (Figure 4).
Several groups have documented that the UPR/ER stress pathway is activated in IPF (28, 31). However, at present, the significance of this response in IPF is unknown (Figure 4). In an attempt to link misfolded proteins with pulmonary fibrosis, efforts have been made to generate transgenic mice that express the mutated surfactant protein C in AEC2 cells (28). Although these mutations do induce ER stress in murine lungs, a second insult such as low-dose bleomycin or a viral infection is still necessary to produce fibrosis (32–34). However, the threshold for bleomycin-induced fibrosis is markedly reduced, supporting the concept that activation of the UPR is a marker for a vulnerable alveolar epithelium. Further support for such a concept has been the identification of herpes virus DNA in lung tissue from IPF patients (35). Although efforts to identify an active infection in patients with progressive disease have been difficult (36, 37), previous infection could add an additional component of vulnerability to the alveolar epithelium. Future efforts need to focus on generating novel genetic models that target specific downstream mediators of the UPR/ER stress response in AEC2 cells to more precisely define the link with fibrosis.
Epithelial-mesenchymal crosstalk
Pathologic hallmarks of UIP/IPF are the destruction of basement membrane and presence of hyperplastic alveolar epithelial cells (38). This has been suggested to create an environment of “frustrated” re-epithelialization (refs. 39, 40, and Figure 5). There are a number of mediators important in regulating epithelial-mesenchymal crosstalk during alveolar development that are expressed in IPF (41, 42). Among these are the morphogenic cytokines of the TGF-β superfamily (activin, bone morphogenetic protein 4 [BMP4] and BMP7, and perhaps most importantly, TGF-β1), Wnt/β-catenin, and sonic hedgehog (Shh). In addition, several tyrosine kinase signaling pathways including those downstream of FGFs, VEGF, PDGF, epithelial growth factor, and keratinocyte growth factor mediate epithelial-mesenchymal interactions, but their precise role in IPF is unknown. Despite the fact that there is a relative paucity of data directly implicating these pathways in the pathogenesis of pulmonary fibrosis, several clinical trials studying the utility of tyrosine kinase inhibitors in the treatment of fibrotic lung disease have been undertaken. A clinical trial evaluating the efficacy of imatinib in IPF failed to demonstrate an impact on disease over the course of one year of treatment (43). Interestingly, a Phase II trial evaluating a triple tyrosine kinase inhibitor with effects on FGF, VEGF, and PDGF showed promise (44, 45), and Phase III trials are underway (46).
Figure 5. Proposed mechanisms of severe lung fibrosis.
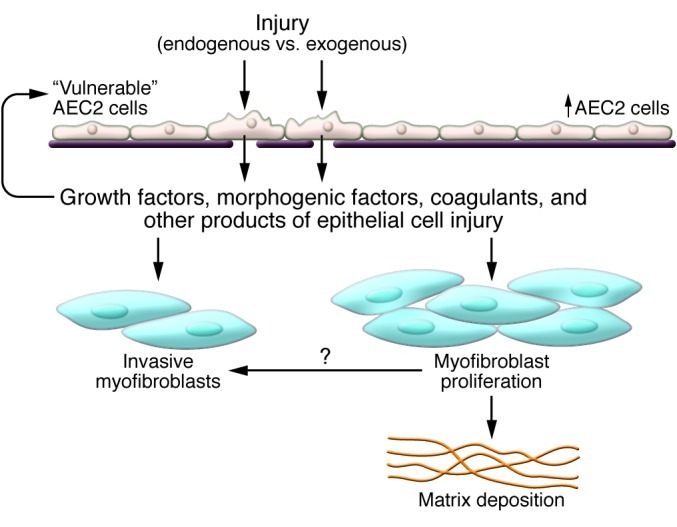
Injured AEC2s attempting to repair damage release growth factors, cytokines, coagulants, and other substances. These factors promote mesenchymal expansion and activation, leading to the accumulation of matrix-producing and invasive fibroblasts/myofibroblasts. Figure adapted with permission from the American Journal of Respiratory Cell and Molecular Biology (2).
There are a number of studies evaluating Wnt/β-catenin signaling in animal models of pulmonary fibrosis (47–49). Furthermore, increased expression of several components of the Wnt signaling pathway as well as downstream targets have been demonstrated in IPF (50). The challenges to sorting out the role of Wnt/β-catenin signaling in pulmonary fibrosis derive from the protean expression of the pathway and the lack of AEC2-specific targeting approaches to interrogate the functional significance following lung injury.
In addition to mediators of lung development that may be implicated in fibrogenesis, studies investigating the pathogenesis of non-infectious lung injury have identified other salient pathways. Following lung injury, a constellation of events is set into place as the lung attempts to limit the extent of damage and restore functional integrity. Epithelial cells and innate immune cells (particularly macrophages) are the first responders charged with assessing the severity of the insult and mounting a host response. Diffuse alveolar damage occurs at focal sites of injury, with hyaline membrane formation and a fibronectin-rich provisional matrix as the coagulation cascade is locally activated in the context of increased alveolar permeability. The coagulation cascade has been suggested as a therapeutic target in IPF (51, 52), but a recent trial evaluating Coumadin (warfarin) was terminated for lack of efficacy (25, 53). However, more directed approaches such as targeting factor Xa, which has been shown to be expressed in IPF tissue and may contribute to TGF-β activation through interaction with the PAR-1 receptor, could be more fruitful (52).
The interplay between epithelial cells and macrophages in orchestrating the recruitment of fibroblasts is an area of active investigation. Both populations are capable of releasing a variety of mediators that have chemotactic properties for fibroblasts (54–56), but the relative contributions of these cell types has been difficult to discern. Recently, lysophosphatidic acid (LPA) has been shown to be present following lung injury, and its cognate receptor, LPAR1, is necessary for the recruitment of fibroblasts to the lung and the development of fibrosis following lung injury (55). Based on these data, an antagonist of LPAR1 is in early clinical development for IPF (57).
As noted above, activation of TGF-β is undoubtedly a critical ingredient for fibrogenesis. Several studies have suggested that fibrosis is abrogated in the absence of TGF-β signaling in the epithelium (58, 59). The integrin αvβ6 has been shown to be upregulated on epithelium following lung injury and to be necessary for activation of latent TGF-β (60). Anti-β6 antibodies are being studied in a Phase II clinical trial for IPF (52, 61). The precise and fundamental signals generated by epithelium that promote mesenchymal expansion have not been elucidated, but a recent study in acute kidney injury suggested that epithelial cell cycle arrest may generate signals that promote fibrosis (62). Interestingly, this process appeared to be mediated by JNK, and fibrosis was abated with a JNK inhibitor. Of note, a JNK inhibitor is in clinical development for IPF (63, 64).
Mesenchymal expansion
Patients suffering from IPF succumb to respiratory failure due to the inexorable accumulation of gas-exchanging regions of the lung that are obliterated by extracellular matrix. Analysis of lung tissue shows advancing regions of fibroblast accrual and destruction of the normal interstices of alveolar tissue. The source of mesenchymal expansion in IPF and experimental models of lung fibrosis have been an area of active investigation. In addition, the source of the pathologic hallmark of IPF — the emergence of α-SMA–staining myofibroblasts juxtaposed to vulnerable alveolar epithelium — remains unexplained.
The long-standing belief that the source of mesenchymal expansion and myofibroblast accumulation was derived from resident lung fibroblasts has been challenged over the last several years by data suggesting that either a bone marrow–derived cell or alveolar epithelium could be the source of mesenchymal expansion. Several studies have suggested that fibrocytes — circulating mesenchymal cells — can be recruited to the lung following injury and potentially contribute to the burden of fibroblast accumulation and tissue remodeling (65–67). In addition, fibrocytes have been identified in the blood of patients with IPF (68, 69) and might be useful as a clinical marker for disease progression (70). Difficulties have arisen in the interpretation of these studies because of challenges with identifying and following the course of fibrocyte trafficking, due to the lack of specific markers and alterations in cell surface expression of fibrocyte markers in lung tissue.
Recently, a great deal of interest has been generated by the hypothesis that AEC2 cells may be a major source of fibroblast accumulation in experimental models of lung fibrosis and in IPF through epithelial-to-mesenchymal transition (EMT). EMT is an accepted biological process in cancer progression and is an appealing concept in IPF because of the unique features of fibroblastic foci. In addition, EMT has been suggested to be an important contributor to kidney fibrosis, although it remains an area of controversy (41, 71). Studies suggested that stimulating isolated AEC2 cells in vitro with TGF-β would lead to the expression of fibroblast markers and loss of AEC2 cell markers (72). In addition, immunohistochemistry suggested that there might be co-localization of AEC2 and fibroblast markers in IPF (72), but these observations have not been confirmed in all studies (73). To test the hypothesis that EMT contributes to experimental pulmonary fibrosis in vivo, two groups independently performed AEC2 cell lineage tracing experiments using a fragment of the human surfactant protein C (SFTPC) promoter driving β-galactosidase as a lineage label (74, 75). Either repeated doses of bleomycin or adenoviral delivery of active TGF-β were used to cause pulmonary fibrosis (74, 75). Both studies suggested that AEC2 cells are a significant source of mesenchymal cells in experimental pulmonary fibrosis.
An alternative approach of in vivo genetic lineage tracing was designed to explore the putative role of EMT in mesenchymal expansion and myofibroblast accumulation following lung injury (76). An Sftpc-CreERT2 knock-in allele was generated to induce the heritable expression of a fluorescent lineage tag in mature AEC2 cells. Lineage tracing of adult Sftpc-positive AEC2s revealed no evidence to suggest that they give rise to any type of stromal cells after bleomycin administration (76). Interestingly, this study did suggest that pericytes, a population of cells associated with blood vessels that has been implicated as a source of myofibroblasts in other organs (77, 78), are a source of stromal cell expansion following lung injury (76).
Two independent mouse lines, NG2-Cre-ER and forkhead box J1–CreER (FoxJ1-CreER), were used to induce expression of a heritable fluorescent tag in pericyte-like cells in the lung and to follow their fate after bleomycin treatment (76). Pericyte-like cells were found to proliferate and expand in areas of fibrosis, but they did not express high levels of α-SMA, suggesting that a yet-unidentified stromal cell type might be the source of the expanding population of myofibroblasts following injury. This study significantly questions the relevance of in vitro studies examining AEC2 expression of fibroblast markers to in vivo fibrosis. Immunohistochemistry and confocal microscopy using a panel of stromal markers in fresh biopsy samples from patients with IPF recapitulated the heterogeneity of stromal markers described in the mouse and failed to demonstrate co-localization of NG2 or α-SMA in fibroblastic foci (76). Further studies are needed to delineate the significance of the variety of fibroblast populations and the source of myofibroblast expansion in both the bleomycin mouse model and in human IPF.
Emergence of pathologic mesenchymal phenotypes
In addition to the challenges in identifying the sources of fibroblast expansion, there have also been difficulties in elucidating the properties of fibroblasts that promote unrelenting tissue remodeling rather than normal repair. One hypothesis to explain the failure of fibrosis to resolve in the setting of IPF is that IPF fibroblasts may be resistant to apoptosis (79, 80). This hypothesis is appealing, but in vivo evidence supporting an apoptosis-resistant phenotype driving severe fibrosis has been elusive. The unrelenting nature of progressive fibrosis in IPF could be considered analogous to tumor cell growth, although fibroblasts isolated from IPF patients are not monoclonal or transformed (81–83). The pathologic hallmarks of IPF include matrix deposition, basement membrane destruction, and expansion of the mesenchyme. These properties suggest that fibroblasts acquire functions that overlap with tumorigenesis, such as invasive potential and insensitivity to growth inhibitory signals.
Several studies have demonstrated an abnormal proliferative capacity of IPF fibroblasts and have focused on the interactions with extracellular matrix (83–86). Polymerized collagen is naturally found in tissues and acts as a negative regulator of fibroblast proliferation (86). In contrast, monomeric collagen that is present during tissue injury supports fibroblast proliferation (86, 87). This physiologic process that accompanies normal wound repair is aberrant in IPF fibroblasts and is mediated by pathological integrin signaling (86). On normal fibroblasts, β1-integrin interacts with polymerized collagen to activate the negative growth regulator tumor suppressor phosphatase and tensin homolog. In IPF fibroblasts, this negative feedback mechanism is defective, facilitating the circumvention of the negative feedback signals to inhibit proliferation (86). This is accompanied by aberrant activation of the PI3K/Akt/S6K1 signaling pathway in IPF fibroblasts (86, 88). There is also evidence of genome-wide derangements in translational control in IPF fibroblasts, detectable after nine generations in culture and resulting in ribosomal recruitment of proteins that promote tissue remodeling (89).
Additional evidence of a pathological mesenchymal phenotype driving unrelenting pulmonary fibrosis derives from the examination of the invasive capacity of IPF fibroblasts (90, 91). One of the hallmarks of IPF is the destruction of basement membrane in alveolar tissue leading to collapse of alveoli (92). IPF fibroblasts/myofibroblasts have been shown to spontaneously invade tissue in in vitro models (93). The mechanisms that regulate myofibroblast functions in vivo remain poorly understood, although recent evidence suggests that NADPH oxidase 4 is essential for TGF-β–induced differentiation of fibroblasts to myofibroblasts in vitro and for bleomycin-induced pulmonary fibrosis in vivo (94). When hyaluronan synthase 2 (HAS2) was transgenically overexpressed by myofibroblasts in vivo, a severe fibrotic phenotype followed bleomycin-induced lung injury (95). The development of this fibrosis was dependent on the hyaluronan receptor CD44 (95). Hyaluronan is a glycosaminoglycan produced in great abundance during noninfectious lung injury and regulates the inflammatory response (95, 96). Mice with deletion of HAS2 in mesenchymal cells fail to develop the same degree of fibrosis as wild-type mice (95). Knockdown of HAS2 or treatment with anti-CD44 antibodies inhibits the invasive capacity of IPF fibroblasts (95).
Mice deficient in β-arrestins are protected from bleomycin-induced pulmonary fibrosis (97). β-Arrestins regulate G-protein–coupled receptor signaling, and a deficiency in fibroblasts thwarts the invasive capacity of both murine and IPF fibroblasts (97). Lysyl oxidases modify extracellular matrix and have been suggested to contribute to tumor metastasis (98). Inhibition of lysyl oxidase-like 2 has been shown to blunt the development of lung and liver fibrosis (99), and this approach is in early clinical development for IPF therapy (100, 101). Collectively, these studies address components of fibroblast/myofibroblast biology and suggest that the unrelenting nature of severe pulmonary fibrosis may relate to processes that promote tumor-like qualities in the expanding mesenchyme (Figure 5). Elucidating the key regulators of these properties, such as microRNAs (102–105) or epigenetic processes (106–108), are important areas of future investigation.
Summary
Severe pulmonary fibrosis is a complex process involving the interplay of a variety of cell types contributing to architectural distortion and loss of gas exchange function. Recognizing the complexities and heterogeneity of fibrosis in clinical settings is necessary for optimal clinical care and clinical trial enrollment. An understanding of how communication between the alveolar epithelium and mesenchyme leads to pathological phenotypes may unlock the mysteries of progressive pulmonary fibrosis and lead to the development of efficacious therapies for IPF.
Acknowledgments
This work was supported by NIH grants R01-HL060539, R01-AI052201, R01-HL077291, and P50-HL084917.
Footnotes
Conflict of interest: P.W. Noble serves or has served as a consultant for Intermune, Boehringer-Ingelheim, Bristol-Meyers-Squibb, Celgene, Anthera, Stromedix, GlaxoSmithKline, Actelion, Gilead, Regeneron, Moerae Matrix, and Takeda in the development of therapeutics for idiopathic pulmonary fibrosis.
Citation for this article: J Clin Invest. 2012;122(8):2756–2762. doi:10.1172/JCI60323.
References
- 1.Interstitial Lung Disease. Schwarz MI, King TE Jr, eds. 5th ed. Shelton, Connecticut, USA: People’s Medical Publishing House; 2010. [Google Scholar]
- 2.Noble PW, Homer RJ. Back to the future: historical perspective on the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2005;33(2):113–120. doi: 10.1165/rcmb.F301. [DOI] [PubMed] [Google Scholar]
- 3.Fischer A, et al. Anti-synthetase syndrome in ANA and anti-Jo-1 negative patients presenting with idiopathic interstitial pneumonia. Respir Med. 2009;103(11):1719–1724. doi: 10.1016/j.rmed.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 2008;3:8. doi: 10.1186/1750-1172-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunninghake GW, et al. Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;164(2):193–196. doi: 10.1164/ajrccm.164.2.2101090. [DOI] [PubMed] [Google Scholar]
- 6.Raghu G, Mageto YN, Lockhart D, Schmidt RA, Wood DE, Godwin JD. The accuracy of the clinical diagnosis of new-onset idiopathic pulmonary fibrosis and other interstitial lung disease: A prospective study. Chest. 1999;116(5):1168–1174. doi: 10.1378/chest.116.5.1168. [DOI] [PubMed] [Google Scholar]
- 7.[No authors listed] American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 pt 1):646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 8. National Heart, Lung, and Blood Institute (NHLBI). Evaluating the Effectiveness of Prednisone, Azathioprine, and N-acetylcysteine in People With Idiopathic Pulmonary Fibrosis (PANTHER-IPF). NIH Web site. http://clinicaltrials.gov/ct2/show/NCT00650091 . Updated January 23, 2012. Accessed April 2, 2012.
- 9.Churg A, Muller NL, Flint J, Wright JL. Chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2006;30(2):201–208. doi: 10.1097/01.pas.0000184806.38037.3c. [DOI] [PubMed] [Google Scholar]
- 10.Noble PW. Idiopathic pulmonary fibrosis: natural history and prognosis. Clin Chest Med. 2006;27(1 suppl 1):S11–S16. doi: 10.1016/j.ccm.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Martinez FJ, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142(12 pt 1):963–967. doi: 10.1001/archinte.1982.00340180121022. [DOI] [PubMed] [Google Scholar]
- 12.Demedts M, et al. Interstitial lung diseases: an epidemiological overview. Eur Respir J Suppl. 2001;32:2s–16s. [PubMed] [Google Scholar]
- 13.Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150(4):967–972. doi: 10.1164/ajrccm.150.4.7921471. [DOI] [PubMed] [Google Scholar]
- 14.Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155(1):242–248. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 15.Steele MP, et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005;172(9):1146–1152. doi: 10.1164/rccm.200408-1104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas AQ, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in 1 kindred. Am J Respir Crit Care Med. 2002;165(9):1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armanios MY, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 19.Tsakiri KD, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104(18):7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seibold MA, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaherty KR, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164(9):1722–1727. doi: 10.1164/ajrccm.164.9.2103074. [DOI] [PubMed] [Google Scholar]
- 22.Haddad R, Massaro D. Idiopathic diffuse interstitial pulmonary fibrosis (fibrosing alveolitis), atypical epithelial proliferation and lung cancer. Am J Med. 1968;45(2):211–219. doi: 10.1016/0002-9343(68)90039-9. [DOI] [PubMed] [Google Scholar]
- 23.Turner-Warwick M, Lebowitz M, Burrows B, Johnson A. Cryptogenic fibrosing alveolitis and lung cancer. Thorax. 1980;35(7):496–499. doi: 10.1136/thx.35.7.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med. 2000;161(1):5–8. doi: 10.1164/ajrccm.161.1.9906062. [DOI] [PubMed] [Google Scholar]
- 25.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 26.Naik PK, Moore BB. Viral infection and aging as cofactors for the development of pulmonary fibrosis. Expert Rev Respir Med. 2010;4(6):759–771. doi: 10.1586/ers.10.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134(2):136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 28.Lawson WE, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A. 2011;108(26):10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 30.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 31.Korfei M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degryse AL, et al. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L442–L452. doi: 10.1152/ajplung.00026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawson WE, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 34.Kropski JA, Lawson WE, Blackwell TS. Right place, right time: the evolving role of herpesvirus infection as a “second hit” in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302(5):L441–L444. doi: 10.1152/ajplung.00335.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lasithiotaki I, et al. Detection of herpes simplex virus type-1 in patients with fibrotic lung diseases. PLoS One. 2011;6(12):e27800. doi: 10.1371/journal.pone.0027800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zamo A, et al. HHV-8 and EBV are not commonly found in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22(2):123–128. [PubMed] [Google Scholar]
- 37.Konishi K, et al. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;180(2):167–175. doi: 10.1164/rccm.200810-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hyde DM, et al. Idiopathic pulmonary fibrosis. Quantitative assessment of lung pathology. Comparison of a semiquantitative and a morphometric histopathologic scoring system. Am Rev Respir Dis. 1992;146(4):1042–1047. doi: 10.1164/ajrccm/146.4.1042. [DOI] [PubMed] [Google Scholar]
- 39.Noble PW. Idiopathic pulmonary fibrosis. New insights into classification and pathogenesis usher in a new era therapeutic approaches. Am J Respir Cell Mol Biol. 2003;29(3 suppl):S27–S31. [PubMed] [Google Scholar]
- 40.Brass DM, Wise AL, Schwartz DA. Host-environment interactions in exposure-related diffuse lung diseases. Semin Respir Crit Care Med. 2008;29(6):603–609. doi: 10.1055/s-0028-1101270. [DOI] [PubMed] [Google Scholar]
- 41.Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol. 2011;73:413–435. doi: 10.1146/annurev-physiol-012110-142225. [DOI] [PubMed] [Google Scholar]
- 42.Horowitz JC, Thannickal VJ. Epithelial-mesenchymal interactions in pulmonary fibrosis. Semin Respir Crit Care Med. 2006;27(6):600–612. doi: 10.1055/s-2006-957332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daniels CE, et al. Imatinib treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial Results. Am J Respir Crit Care Med. 2010;181(6):604–610. doi: 10.1164/rccm.200906-0964OC. [DOI] [PubMed] [Google Scholar]
- 44.Antoniu SA, Kolb MR. Intedanib, a triple kinase inhibitor of VEGFR, FGFR and PDGFR for the treatment of cancer and idiopathic pulmonary fibrosis. IDrugs. 2010;13(5):332–345. [PubMed] [Google Scholar]
- 45.Richeldi L, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 46. Boehringer Ingelheim Pharmaceuticals. Roll Over Study From 1199.30 BIBF 1120 in Idiopathic Pulmonary Fibrosis (IPF). NIH website. http://clinicaltrials.gov/ct2/show/NCT01170065 . Updated March 7, 2012. Accessed April 2, 2012.
- 47.Morrisey EE. Wnt signaling and pulmonary fibrosis. Am J Pathol. 2003;162(5):1393–1397. doi: 10.1016/S0002-9440(10)64271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konigshoff M, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119(4):772–787. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henderson WR, Jr, et al. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A. 2010;107(32):14309–14314. doi: 10.1073/pnas.1001520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chilosi M, et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol. 2003;162(5):1495–1502. doi: 10.1016/S0002-9440(10)64282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chambers RC. Procoagulant signalling mechanisms in lung inflammation and fibrosis: novel opportunities for pharmacological intervention? Br J Pharmacol. 2008;153(suppl 1):S367–S378. doi: 10.1038/sj.bjp.0707603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scotton CJ, et al. Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. J Clin Invest. 2009;119(9):2550–2563. doi: 10.1172/JCI33288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. National Heart, Lung, and Blood Institute (NHLBI). AntiCoagulant Effectiveness in Idiopathic Pulmonary Fibrosis (ACE-IPF). NIH Web site. http://clinicaltrials.gov/ct2/show/NCT00957242 . Updated December 9, 2011. Accessed April 2, 2012.
- 54.Tager AM, et al. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol. 2004;31(4):395–404. doi: 10.1165/rcmb.2004-0175OC. [DOI] [PubMed] [Google Scholar]
- 55.Tager AM, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14(1):45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 56.Jiang D, et al. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J Clin Invest. 2010;120(6):2049–2057. doi: 10.1172/JCI38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Datta A, Scotton CJ, Chambers RC. Novel therapeutic approaches for pulmonary fibrosis. Br J Pharmacol. 2011;163(1):141–172. doi: 10.1111/j.1476-5381.2011.01247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li M, et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest. 2011;121(1):277–287. doi: 10.1172/JCI42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Degryse AL, et al. TGFbeta signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol. 2011;300(6):L887–L897. doi: 10.1152/ajplung.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Munger JS, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–328. doi: 10.1016/S0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 61. Stromedix, Inc. STX-100 in Patients With Idiopathic Pulmonary Fibrosis (IPF). NIH website. http://clinicaltrials.gov/ct2/show/NCT01371305 . Updated December 13, 2011. Accessed April 2, 2012.
- 62.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–543. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plantevin Krenitsky V, et al. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorg Med Chem Lett. 2012;22(3):1433–1438. doi: 10.1016/j.bmcl.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 64. Celgene Corporation. A Study to Characterize the Safety, PK and Biological Activity of CC-930 in Idiopathic Pulmonary Fibrosis (IPF). NIH website. http://clinicaltrials.gov/ct2/show/NCT01203943 . Updated January 31, 2011. Accessed April 2, 2012.
- 65.Moore BB, et al. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166(3):675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Phillips RJ, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114(3):438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113(2):243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353(1):104–108. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- 69.Andersson-Sjoland A, et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40(10):2129–2140. doi: 10.1016/j.biocel.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 70.Moeller A, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179(7):588–594. doi: 10.1164/rccm.200810-1534OC. [DOI] [PubMed] [Google Scholar]
- 71.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Willis BC, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166(5):1321–1332. doi: 10.1016/S0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamada M, et al. Dual-immunohistochemistry provides little evidence for epithelial-mesenchymal transition in pulmonary fibrosis. Histochem Cell Biol. 2008;129(4):453–462. doi: 10.1007/s00418-008-0388-9. [DOI] [PubMed] [Google Scholar]
- 74.Kim KK, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119(1):213–224. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tanjore H, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180(7):657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rock JR, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108(52):E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Humphreys BD, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Göritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisén J. A pericyte origin of spinal cord scar tissue. Science. 2011;333(6039):238–242. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- 79.Moodley YP, et al. Comparison of the morphological and biochemical changes in normal human lung fibroblasts and fibroblasts derived from lungs of patients with idiopathic pulmonary fibrosis during FasL-induced apoptosis. J Pathol. 2004;202(4):486–495. doi: 10.1002/path.1531. [DOI] [PubMed] [Google Scholar]
- 80.Vuga LJ, et al. WNT5A is a regulator of fibroblast proliferation and resistance to apoptosis. Am J Respir Cell Mol Biol. 2009;41(5):583–589. doi: 10.1165/rcmb.2008-0201OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jordana M, et al. Heterogeneous proliferative characteristics of human adult lung fibroblast lines and clonally derived fibroblasts from control and fibrotic tissue. Am Rev Respir Dis. 1988;137(3):579–584. doi: 10.1164/ajrccm/137.3.579. [DOI] [PubMed] [Google Scholar]
- 82.Westergren-Thorsson G, Sime P, Jordana M, Gauldie J, Särnstrand B, Malmström A. Lung fibroblast clones from normal and fibrotic subjects differ in hyaluronan and decorin production and rate of proliferation. Int J Biochem Cell Biol. 2004;36(8):1573–1584. doi: 10.1016/j.biocel.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 83.Mio T, Nagai S, Kitaichi M, Kawatani A, Izumi T. Proliferative characteristics of fibroblast lines derived from open lung biopsy specimens of patients with IPF (UIP). Chest. 1992;102(3):832–837. doi: 10.1378/chest.102.3.832. [DOI] [PubMed] [Google Scholar]
- 84.Torry DJ, Richards CD, Podor TJ, Gauldie J. Anchorage-independent colony growth of pulmonary fibroblasts derived from fibrotic human lung tissue. J Clin Invest. 1994;93(4):1525–1532. doi: 10.1172/JCI117131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramos C, et al. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol. 2001;24(5):591–598. doi: 10.1165/ajrcmb.24.5.4333. [DOI] [PubMed] [Google Scholar]
- 86.Xia H, et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med. 2008;205(7):1659–1672. doi: 10.1084/jem.20080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schor SL. Cell proliferation and migration on collagen substrata in vitro. J Cell Sci. 1980;41:159–175. doi: 10.1242/jcs.41.1.159. [DOI] [PubMed] [Google Scholar]
- 88.Tian B, Lessan K, Kahm J, Kleidon J, Henke C. beta 1 integrin regulates fibroblast viability during collagen matrix contraction through a phosphatidylinositol 3-kinase/Akt/protein kinase B signaling pathway. J Biol Chem. 2002;277(27):24667–24675. doi: 10.1074/jbc.M203565200. [DOI] [PubMed] [Google Scholar]
- 89.Larsson O, et al. Fibrotic myofibroblasts manifest genome-wide derangements of translational control. PLoS One. 2008;3(9):e3220. doi: 10.1371/journal.pone.0003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.White ES, Lazar MH, Thannickal VJ. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J Pathol. 2003;201(3):343–354. doi: 10.1002/path.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thannickal VJ, et al. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278(14):12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 92.Fukuda Y, Ferrans VJ, Schoenberger CI, Rennard SI, Crystal RG. Patterns of pulmonary structural remodeling after experimental paraquat toxicity. The morphogenesis of intraalveolar fibrosis. Am J Pathol. 1985;118(3):452–475. [PMC free article] [PubMed] [Google Scholar]
- 93.White ES, et al. Integrin alpha4beta1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. Am J Respir Crit Care Med. 2003;168(4):436–442. doi: 10.1164/rccm.200301-041OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hecker L, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15(9):1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li Y, et al. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med. 2011;208(7):1459–1471. doi: 10.1084/jem.20102510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Teder P, Heldin P. Mechanism of impaired local hyaluronan turnover in bleomycin-induced lung injury in rat. Am J Respir Cell Mol Biol. 1997;17(3):376–385. doi: 10.1165/ajrcmb.17.3.2698. [DOI] [PubMed] [Google Scholar]
- 97.Lovgren AK, et al. beta-arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix. Sci Transl Med. 2011;3(74):74ra23. doi: 10.1126/scitranslmed.3001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440(7088):1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 99.Barry-Hamilton V, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16(9):1009–1017. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- 100.Kolb MR, Gauldie J. Idiopathic pulmonary fibrosis: the matrix is the message. Am J Respir Crit Care Med. 2011;184(6):627–629. doi: 10.1164/rccm.201107-1282ED. [DOI] [PubMed] [Google Scholar]
- 101. Arresto Biosciences, Inc. Safety and Pharmacokinetics Study With AB0024 in Patients With Idiopathic Pulmonary Fibrosis. NIH website. http://clinicaltrials.gov/ct2/show/NCT01242189 . Updated November 15, 2010. Accessed April 2, 2012.
- 102.Liu G, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207(8):1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pandit KV, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182(2):220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xie T, Liang J, Guo R, Liu N, Noble PW, Jiang D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol Genomics. 2011;43(9):479–487. doi: 10.1152/physiolgenomics.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xie T, et al. MicroRNA-127 inhibits lung inflammation by targeting IgG Fcγ receptor I. J Immunol. 2012;188(5):2437–2444. doi: 10.4049/jimmunol.1101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bechtel W, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16(5):544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hu B, Gharaee-Kermani M, Wu Z, Phan SH. Essential role of MeCP2 in the regulation of myofibroblast differentiation during pulmonary fibrosis. Am J Pathol. 2011;178(4):1500–1508. doi: 10.1016/j.ajpath.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sanders YY, Tollefsbol TO, Varisco BM, Hagood JS. Epigenetic regulation of thy-1 by histone deacetylase inhibitor in rat lung fibroblasts. Am J Respir Cell Mol Biol. 2011;45(1):16–23. doi: 10.1165/rcmb.2010-0154OC. [DOI] [PMC free article] [PubMed] [Google Scholar]