

Abstract
Liver receptor homolog 1 (LRH-1), an established regulator of cholesterol and bile acid homeostasis, has recently emerged as a potential drug target for liver disease. Although LRH-1 activation may protect the liver against diet-induced steatosis and insulin resistance, little is known about how LRH-1 controls hepatic glucose and fatty acid metabolism under physiological conditions. We therefore assessed the role of LRH-1 in hepatic intermediary metabolism. In mice with conditional deletion of Lrh1 in liver, analysis of hepatic glucose fluxes revealed reduced glucokinase (GCK) and glycogen synthase fluxes as compared with those of wild-type littermates. These changes were attributed to direct transcriptional regulation of Gck by LRH-1. Impaired glucokinase-mediated glucose phosphorylation in LRH-1–deficient livers was also associated with reduced glycogen synthesis, glycolysis, and de novo lipogenesis in response to acute and prolonged glucose exposure. Accordingly, hepatic carbohydrate response element-binding protein activity was reduced in these animals. Cumulatively, these data identify LRH-1 as a key regulatory component of the hepatic glucose-sensing system required for proper integration of postprandial glucose and lipid metabolism.
Introduction
The liver plays a central role in metabolic homeostasis by coordinating the synthesis, storage, breakdown, and redistribution of nutrients. Adequate control of these metabolic processes is of importance to accommodate systemic fuel requirements and availability. This is achieved through regulatory complexes that modulate both the catalytic activity and the expression level of metabolic enzymes. While the first usually enables rapid changes in enzymatic activity triggered by allosteric regulation or covalent modification, the second regulatory process is slower and involves transcription factors that adjust gene expression levels. In this context, nuclear receptors and their coregulators have been shown to play a key role in the transcriptional regulation of metabolic enzyme expression in response to changes in cellular nutrient and energy status (1, 2).
Liver receptor homolog 1 (LRH-1, also known as NR5A2), a member of the NR5A superfamily of nuclear receptors, is highly expressed in the liver. Hepatic LRH-1 promotes the expression of the bile acid–synthesizing enzymes Cyp7a1 and Cyp8b1 (3–5), while it suppresses acute phase response genes (6, 7). As a consequence, bile acid metabolism is altered in liver-specific LRH-1 knockout mice (3, 4), and LRH-1 heterozygous animals show an exacerbated inflammatory response (6). Other established LRH-1 target genes in the liver are known mediators of hepatic cholesterol uptake and efflux (8, 9), HDL formation (10, 11), cholesterol exchange between lipoproteins (12), and fatty acid synthesis (13). Although these findings point to a broader role for LRH-1 in hepatic lipid metabolism and reverse cholesterol transport, their physiological impact is as yet unknown.
Independent studies have demonstrated that human LRH-1 can bind several phospholipid species, including phosphoinositides (14–17). Interestingly, dilauroyl phosphatidylcholine (DLPC), which has been identified as a ligand for both mouse and human LRH-1 in vitro, was recently shown to confer LRH-1–dependent protection against hepatic steatosis and insulin resistance in mice exposed to chronic high-fat feeding (18). While these observations suggest that hepatic LRH-1 may contribute to metabolic control, the role of LRH-1 in hepatic glucose metabolism remains largely unexplored. However, insights into the mechanisms by which LRH-1 impacts on glucose and fatty acid metabolism in the liver are required for the development of therapeutic strategies to prevent or treat hepatic steatosis.
In this study, we assessed the physiological role of LRH-1 in hepatic intermediary metabolism. We show that LRH-1 controls the first step of hepatic glucose uptake through direct transcriptional regulation of the glucokinase (Gck) gene. As a result, mice lacking LRH-1 in hepatocytes fail to adequately induce glucose-driven glycolysis, glycogen synthesis, and fatty acid synthesis in the liver. Altogether, our work identifies LRH-1 as a critical regulatory component of the glucose-sensing system that integrates hepatic postprandial glucose and lipid metabolism.
Results
Reduced glucokinase and glycogen synthase fluxes in Alb-Cre;Lrh1fl/fl mice.
To explore the putative involvement of LRH-1 in intermediary metabolism in the liver, we quantified hepatic glucose fluxes upon stable isotope infusion in mice with a somatic deletion of LRH-1 in hepatocytes (Alb-Cre;Lrh1fl/fl mice; ref. 3) and their wild-type littermates (Lrh1fl/fl mice) (Figure 1A; ref. 19). Blood glucose concentrations were similar in Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice under both normoglycemic and clamped hyperglycemic conditions (Table 1). Alb-Cre;Lrh1fl/fl mice showed significant reductions in the flux through glucokinase under both normoglycemic and hyperglycemic conditions (Figure 1B). In contrast, the glucose-6-phosphatase flux remained unaltered (Figure 1C), resulting in increased net glucose flux to the blood in Alb-Cre;Lrh1fl/fl mice (Figure 1D).
Figure 1. Reduced hepatic glucokinase and glycogen synthase fluxes in Alb-Cre;Lrh1fl/fl mice.

(A) Schematic representation of the model used for mass isotopomer distribution analysis. GP, glycogen phosphorylase; GS, glycogen synthase; G6Pase, glucose-6-phosphatase. (B–D) Glucose fluxes in Lrh1fl/fl mice (white bars) and Alb-Cre;Lrh1fl/fl mice (black bars) under normoglycemic (NG) and hyperglycemic (HG) conditions. (B) Glucokinase and (C) glucose-6-phosphatase flux and (D) glucose balance. (E–G) Glycogen fluxes in Alb-Cre;Lrh1fl/fl and Lrh1fl/fl mice under normoglycemic and hyperglycemic conditions. (E) Glycogen synthase and (F) glycogen phosphorylase flux and (G) glycogen balance. Data represent mean ± SEM for n = 5–9 per genotype. *P < 0.05 Alb-Cre;Lrh1fl/fl versus Lrh1fl/fl; #P < 0.05 hyperglycemic versus normoglycemic.
Table 1.
Metabolic parameters during stable isotope infusion in Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice
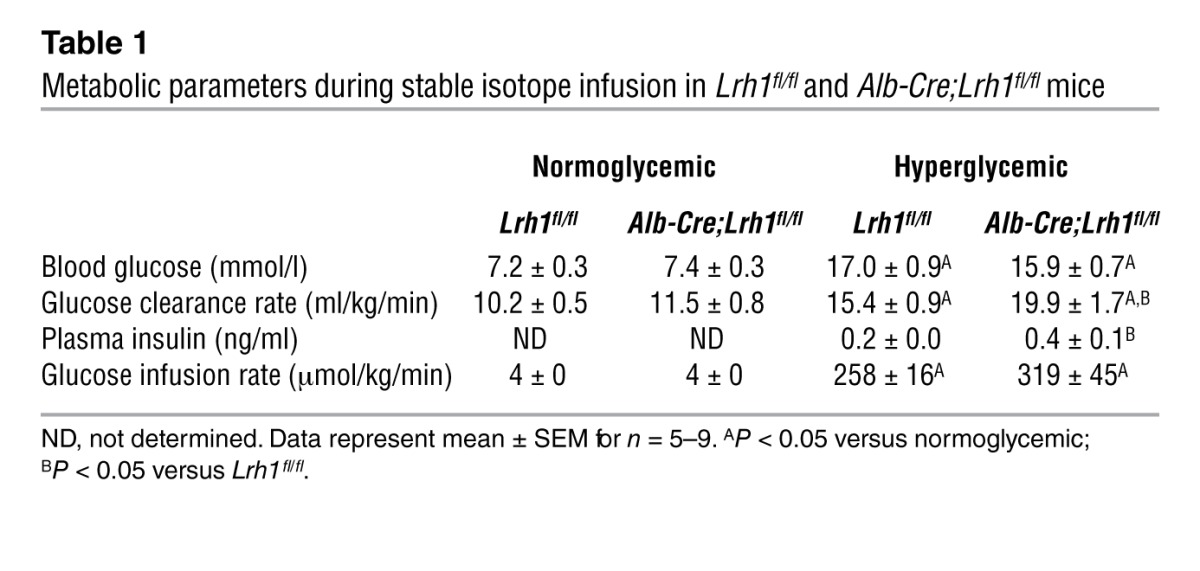
Hepatic LRH-1 deficiency also affected the conversion of glucose-6-phosphate (G6P) into glycogen. Normoglycemic and hyperglycemic glycogen synthase fluxes were lowered in Alb-Cre;Lrh1fl/fl mice (Figure 1E), while glycogen phosphorylase fluxes remained unchanged (Figure 1F). As a consequence, hepatic glycogen balances were markedly reduced in Alb-Cre;Lrh1fl/fl mice under both conditions (Figure 1G). Overall, hepatic ablation of LRH-1 reduced glucose phosphorylation via glucokinase and impaired the capacity of the liver to convert G6P into glycogen. Of interest, the whole-body glucose clearance rate was increased in Alb-Cre;Lrh1fl/fl mice under hyperglycemic conditions, presumably as a consequence of elevated insulin levels (Table 1). Alb-Cre;Lrh1fl/fl mice therefore required higher glucose infusion rates to maintain hyperglycemic states similar to those of their wild-type littermates (Table 1). However, these changes did not impact systemic energy metabolism. Alb-Cre;Lrh1fl/fl mice showed normal lean and fat masses, and food and water intake was unchanged compared with that of Lrh1fl/fl mice (Supplemental Figure 1, A and B; supplemental material available online with this article; doi: 10.1172/JCI62368DS1). Energy expenditure and substrate utilization were also similar to those of wild-type controls (Supplemental Figure 1, C and D).
Glucokinase is a transcriptional target of LRH-1.
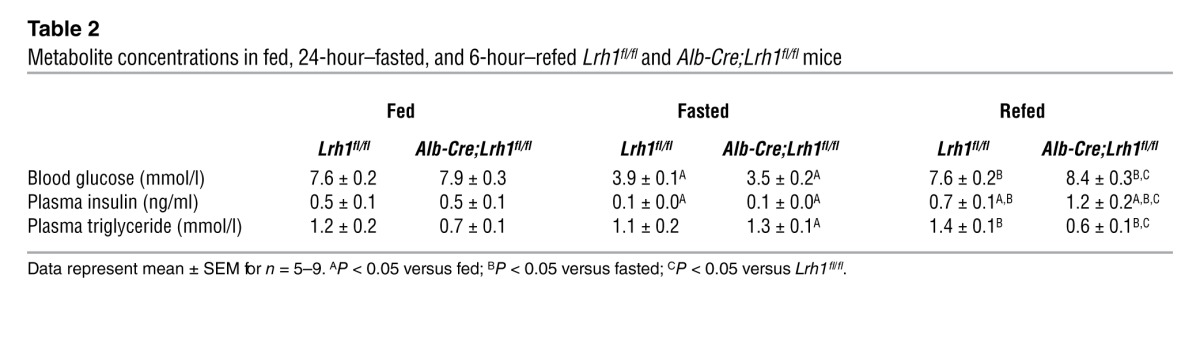
To establish the basis of the reduced glucose fluxes, we evaluated the acute hepatic response to a glucose bolus in Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice. Glucose and insulin levels showed the expected increases (Figure 2, A and B) and were not affected in Alb-Cre;Lrh1fl/fl mice. The expression of Lrh1 and its established target and corepressor SHP (20) was undetectable in Alb-Cre;Lrh1fl/fl mice (Figure 2C). Interestingly, the mRNA level of Gck, but not Hk1, was consistently reduced in the livers of Alb-Cre;Lrh1fl/fl mice, and its glucose-mediated increase was markedly blunted as compared with that in Lrh1fl/fl mice (Figure 2D). To evaluate whether the impaired induction of Gck in response to acute glucose availability also occurred upon prolonged glucose exposure, Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice were analyzed under fed, fasted, and refed conditions. Circulating glucose and insulin levels were similar between genotypes in the fed and fasted states but significantly increased in refed Alb-Cre;Lrh1fl/fl mice (Table 2). This was associated with lower pancreatic insulin content (Supplemental Figure 2), suggesting compensatory pancreatic insulin release as a result of impaired GCK-mediated hepatic glucose uptake in refed Alb-Cre;Lrh1fl/fl mice. As with Lrh1 and its targets Shp and Cyp8b1 (Figure 2E), the hepatic mRNA levels of Gck were robustly reduced in Alb-Cre;Lrh1fl/fl mice in all nutritional states (Figure 2F). Hk1 expression remained unaffected by genotype or nutritional status (Figure 2F). Transient transfection assays revealed that LRH-1 contributes to basal Gck transcription irrespective of glucose availability, since activation of a heterologous and an endogenous LRH-1 reporter construct by LRH-1 was similar under low (5 mM) and high (25 mM) levels of glucose (Figure 2G).
Figure 2. Reduced glucokinase expression in Alb-Cre;Lrh1fl/fl mice.

(A) Blood glucose and (B) plasma insulin concentrations (n = 10 per genotype) and (C and D) hepatic mRNA levels of Lrh1, Shp, Gck, and Hk1 during an oral glucose tolerance test (n = 4–8 per genotype) in Lrh1fl/fl mice (white boxes) and Alb-Cre;Lrh1fl/fl mice (black boxes). (E and F) Hepatic mRNA levels in fed, 24-hour–fasted or 6-hour–refed Lrh1fl/fl mice (white bars) and Alb-Cre;Lrh1fl/fl mice (black bars) (n = 7–9 per genotype). (G) LRH-1 transcriptional activity in HeLa cells transfected with a heterologous (4x LRH-1 response element–Luc [4xLRE-Luc]) or an endogenous LRH-1 reporter driven by the Shp promoter (SHP-Luc). Luciferase activity was determined in the absence (empty; white bars) or presence (LRH-1; black bars) of LRH-1 after exposure to 5 or 25 mM glucose for 24 hours. Data are expressed as relative light units (RLUs) compared with empty reporter (pGL3). Data represent mean ± SEM. *P < 0.05 versus Lrh1fl/fl or versus empty vector (pCMX).
Table 2.
Metabolite concentrations in fed, 24-hour–fasted, and 6-hour–refed Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice
We then evaluated whether glucokinase is subject to direct transcriptional control by LRH-1. Since Lrh1 and Shp expression was abolished in the livers of Alb-Cre;Lrh1fl/fl mice (Figure 2, C and E), we first assessed whether changes in Gck mRNA levels arise from altered LRH-1 or SHP activity by means of adeno-associated viral vector-serotype 8–mediated (AAV8-mediated) SHP reconstitution in Alb-Cre;Lrh1fl/fl mice. Consistent with the lower expression of LRH-1 targets Cyp8b1 and Srb1, SHP reconstitution also did not normalize the expression of Gck (Figure 3A). Reduced Gck mRNA levels in Alb-Cre;Lrh1fl/fl mice translated into lower GCK protein expression (Figure 3B). The LRH-1–mediated control of Gck expression was also confirmed in vitro. In line with reduced Gck mRNA levels in the absence of LRH-1, adenoviral overexpression of LRH-1 in mouse primary hepatocytes markedly induced Lrh1, Shp, Cyp8b1, and Gck expression (Figure 3C). Moreover, Shp and Gck mRNA levels were robustly elevated in mouse hepatoma cells that overexpressed LRH-1 (Figure 3D), resulting in enhanced glucose uptake and phosphorylation (Figure 3E). Computational analysis revealed 6 putative LRH-1 binding sites in the mouse Gck promoter (Figure 3F), and ChIP analysis of hepatic DNA from Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice identified specific recruitment of LRH-1 to sites 5 and 6, which are most proximal to the transcription initiation site in the hepatic Gck promoter (ref. 21; Figure 3, F and G). Site-directed mutagenesis of these sites identified site 6 downstream from the transcription initiation site as the major functional site that confers LRH-1 responsiveness (Figure 3H).
Figure 3. Gck is a direct transcriptional target of LRH-1.

(A) Hepatic mRNA levels in Alb-Cre;Lrh1fl/fl mice 5 weeks after in vivo transduction of the liver using AAV8-SHP virus (gray bars) in comparison with those in Lrh1fl/fl mice (white bars) and Alb-Cre;Lrh1fl/fl mice (black bars) (n = 4–5 per group). (B) Hepatic GCK protein expression in refed Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice. (C and D) Expression levels of LRH-1 and its targets in (C) wild-type primary hepatocytes and (D) Hepa 1.6 mouse hepatoma cells transduced with AdGFP (white bars) or AdLRH-1 (black bars) viruses (n = 3 per condition). (E) 2-deoxyglucose (2-DG) uptake and 2-deoxyglucose-6-phosphate (2-DG6P) production in Hepa 1.6 cells transduced with AdGFP (white bars) or AdLRH-1 (black bars) viruses (n = 6 per condition). (F) Schematic presentation of the 6 putative LRH-1 response elements in the mouse Gck promoter. (G) Assessment of LRH-1 recruitment to these sites, as depicted in F, determined by ChIP analysis using genomic DNA from livers of Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice. (H) Luciferase activities in HeLa cells transfected with empty luciferase reporter (pGL3; white bar) or long and short Gck promoter constructs (black bars). Data are expressed as fold induction in luciferase activity upon LRH-1 cotransfection. Data represent mean ± SEM. *P < 0.05 versus Lrh1fl/fl, versus GFP, or versus empty reporter (pGL3); #P < 0.05 versus Alb-Cre;Lrh1fl/fl.
Reduced glycogen synthesis, glycolysis, de novo lipogenesis, and Chrebp activity in Alb-Cre;Lrh1fl/fl mice.
Perturbed glucokinase activity impacts on hepatic glucose and lipid metabolism (22, 23). We hypothesized that hepatic ablation of LRH-1 would limit the availability of G6P for glycogen synthesis, glycolysis, and de novo lipogenesis. We therefore analyzed these biochemical pathways downstream of glucokinase in Alb-Cre;Lrh1fl/fl mice. In accordance with the reduction in the flux to glycogen (Figure 1, E and G), we observed that the accumulation of hepatic glycogen, which usually occurs within 30 minutes after an oral glucose bolus in Lrh1fl/fl mice, was markedly delayed in Alb-Cre;Lrh1fl/fl mice, in which glycogen levels were maximally increased after only 1 hour (Figure 4, A and B). In addition to this acute effect, glycogen content was also significantly lower in the livers of fed and refed Alb-Cre;Lrh1fl/fl mice (Figure 4C). In parallel to the reduced glucokinase flux in Alb-Cre;Lrh1fl/fl mice, we observed that primary hepatocytes from Alb-Cre;Lrh1fl/fl mice exhibited lower extracellular acidification rates when exposed to glucose, which is indicative of reduced glycolysis (Figure 4D). Besides its role in ATP generation, glycolysis also provides substrates for de novo fatty acid synthesis. Correspondingly, we found that gene expression of lipogenic enzymes was reduced in Alb-Cre;Lrh1fl/fl mice (Figure 5A). The induction of Acaca expression upon refeeding was attenuated in Alb-Cre;Lrh1fl/fl mice, while Fasn and Scd1 mRNA levels were lower in all conditions studied (Figure 5A). Acacb expression was also reduced in refed Alb-Cre;Lrh1fl/fl mice (Figure 5A). To unequivocally establish the physiological relevance of these findings, we used 13C-acetate to enrich the acetyl-CoA pool and quantified fractional de novo palmitate, stearate, and oleate synthesis rates (Figure 5B). Consistent with the reduction in glycolysis, de novo lipogenesis was reduced in fed and refed Alb-Cre;Lrh1fl/fl mice as compared with that in wild-type littermates (Figure 5B). Accordingly, plasma triglyceride concentrations were decreased in fed and refed Alb-Cre;Lrh1fl/fl mice compared with those in Lrh1fl/fl mice (Table 2).
Figure 4. Delayed glycogen synthesis and reduced glycolysis in Alb-Cre;Lrh1fl/fl mice.

(A) Representative glycogen stainings during oral glucose tolerance test (n = 3–8 per genotype). Scale bar: 200 μm. (B) Quantification of hepatic glycogen content in Lrh1fl/fl mice (white boxes) and Alb-Cre;Lrh1fl/fl mice (black boxes) during oral glucose tolerance test. (C) Hepatic glycogen content in fed, 24-hour–fasted, or 6-hour–refed Lrh1fl/fl mice (white bars) and Alb-Cre;Lrh1fl/fl mice (black bars) (n = 7 per genotype). (D) Extracellular acidification rates (ECARs) in Lrh1fl/fl (white boxes) and Alb-Cre;Lrh1fl/fl (black boxes) primary hepatocytes (n = 5–6 per genotype). Data represent mean ± SEM. *P < 0.05 versus Lrh1fl/fl.
Figure 5. Reduced de novo lipogenesis in Alb-Cre;Lrh1fl/fl mice.

(A) Hepatic lipogenic gene expression and (B) de novo synthesis of hepatic palmitate, stearate, and oleate in fed, fasted, and refed Lrh1fl/fl mice (white bars) and Alb-Cre;Lrh1fl/fl mice (black bars) (n = 7–9 per genotype). Data represent mean ± SEM. *P < 0.05 versus Lrh1fl/fl.
Previous work has shown that hepatic GCK ablation reduces carbohydrate response element-binding protein (Chrebp) and pyruvate kinase (Pklr) expression (23). In accordance with this, and consistent with the observed reduction in glycolysis (Figure 4D) and de novo lipogenesis (Figure 5), we observed that the expression of Chrebp and Pklr was lower in fed and refed Alb-Cre;Lrh1fl/fl mice (Figure 6A). On the other hand, the mRNA levels of sterol regulatory element binding protein 1c (Srebp-1c) and its target gene glucose-6-phosphate dehydrogenase (G6pd1) were not changed in Alb-Cre;Lrh1fl/fl mice (Figure 6A). Nuclear ChREBP protein was also reduced in refed Alb-Cre;Lrh1fl/fl mice (Figure 6B), consistent with lower hepatic G6P content under these conditions (Figure 6C; ref. 24). This was not attributed to the concomitant loss of hepatic SHP in the Alb-Cre;Lrh1fl/fl mice, since Shp reconstitution did not restore Chrebp and Pklr mRNA levels (Figure 6D). To establish whether the reduction in glycolytic and lipogenic gene expression in the livers of Alb-Cre;Lrh1fl/fl mice results from direct regulation of Chrebp transcription by LHR-1, we evaluated the effect of LRH-1 on Chrebp promoter activation. Unlike the Gck promoter (Figure 3H), LRH-1 did not directly activate the mouse Chrebp promoter, while a robust transactivation of the Shp promoter was observed in response to LRH-1 (Figure 6E), indicating that LRH-1 impacts ChREBP in an indirect manner.
Figure 6. Impaired GCK activity in Alb-Cre;Lrh1fl/fl mice reduces ChREBP expression and activity.

(A) Hepatic Chrebp, Pklr, Srebp-1c, and G6pd1 expression in fed, 24-hour–fasted, or 6-hour–refed Lrh1fl/fl mice (white bars) and Alb-Cre;Lrh1fl/fl mice (black bars) (n = 7–9 per genotype). (B) Nuclear ChREBP protein expression in 24-hour–fasted and 6-hour–refed Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice. (C) Hepatic G6P content in fasted and refed Lrh1fl/fl mice (white bars) and Alb-Cre;Lrh1fl/fl mice (black bars) (n = 6–7 per genotype). (D) Hepatic Chrebp and Pklr expression in Alb-Cre;Lrh1fl/fl mice 5 weeks after in vivo transduction of the liver using AAV8-SHP virus (gray bars) in comparison with that in Lrh1fl/fl mice (white bars) and Alb-Cre;Lrh1fl/fl mice (black bars) (n = 4–5 per group). (E) Luciferase activities in HeLa cells transfected with Chrebp (ChREBP-Luc) or SHP (SHP-Luc) promoter (black bars) constructs in the absence (empty; white bars) or presence (LRH-1; black bars) of LRH-1. Data are expressed as relative light units and normalized to empty reporter (pGL3). (F) Hepatic mRNA levels in Alb-Cre;Lrh1fl/fl mice 5 weeks after in vivo transduction of the liver using 2 different titers of AAV8-GCK virus (low, 1011 particles per mouse [light gray bars], and high, 1012 particles per mouse [dark gray bars]) in comparison with those in Lrh1fl/fl mice (white bars) and Alb-Cre;Lrh1fl/fl mice (black bars) (n = 4–5 per group). Data represent mean ± SEM. *P < 0.05 versus Lrh1fl/fl or versus empty vector (pCMX); #P < 0.05 versus Alb-Cre;Lrh1fl/fl.
Based on previous studies showing that GCK is required for optimal ChREBP expression and activity (23–25), we postulated that LRH-1 indirectly modulates ChREBP via GCK. To support this hypothesis, we rescued hepatic Gck expression in Alb-Cre;Lrh1fl/fl mice by AAV8-mediated gene transfer (Figure 6F). Consistent with reduced nuclear ChREBP expression and hepatic G6P content (Figure 6, B and C), Pklr and Acaca mRNA levels were normalized in Alb-Cre;Lrh1fl/fl mice upon Gck reconstitution, indicating that the lower expression levels of these ChREBP target genes occurs secondary to a reduced flux through GCK in Alb-Cre;Lrh1fl/fl mice. Hepatic Fasn expression, on the other hand, remained low upon GCK reconstitution in Alb-Cre;Lrh1fl/fl mice, consistent with a previous study that identified this gene as a direct target of LRH-1 (13). Scd1 mRNA levels showed a dose-dependent response to Gck rescue in Alb-Cre;Lrh1fl/fl mice (Figure 6F). Combined, these data indicate that LRH-1 acts as an upstream regulator of GCK and as such indirectly impacts glycolytic and, at least in part, lipogenic gene expression.
Discussion
Hepatic LRH-1 is an established transcriptional regulator of cholesterol and bile acid homeostasis (3–5, 14, 26). Here, we report that LRH-1 directly regulates intrahepatic glucose and fatty acid metabolism during the postprandial phase. Our data show that both loss and gain of LRH-1 function control glucokinase expression and impact hepatic glucose uptake in liver and hepatocytes. The LRH-1–driven flux through glucokinase in turn critically determines the partitioning of G6P toward glycogen storage, glycolysis, and de novo lipogenesis. Hence, we believe that this study confirms the unique and essential ability of hepatic glucokinase to properly accommodate glucose availability to its oxidation and storage (22, 23, 27) and reveals a novel upstream regulator of its activity that determines the capacity for hepatic glucose uptake upon glucose exposure.
Glucokinase mediates hepatic glucose uptake and, as such, represents the first step of glucose metabolism in the liver. Its product, G6P, is a central metabolite for intrahepatic glucose and lipid homeostasis. This metabolite serves as the substrate for the oxidation of glucose as well as its storage as glycogen and fat. Hepatic glucokinase expression depends on the nutritional state and is subject to reciprocal regulation by insulin and glucagon (28). Different transcription factors, including liver X receptor (LXR), PPARγ, and SREBP-1c, are known to interact to control glucokinase expression (29). The induction of Gck in response to refeeding is, however, not significantly impaired in LXR- or Srebp-1c–deficient mice (30–32). In this study, we observed a profound and consistent reduction in Gck mRNA levels in fed, fasted, and refed Alb-Cre;Lrh1fl/fl mice, confirming previous observations in insulin-resistant mice (18). Together, these data suggest that LRH-1 is a major regulator of hepatic glucokinase expression. Metabolic alterations in Alb-Cre;Lrh1fl/fl mice may, however, also be related to lower Shp expression. This is of importance given that SHP acts as a transrepressor on LXRα- or PPARγ-mediated Gck induction (29). However, a recent report showed that SHP deficiency does not impact hepatic Gck expression in chow-fed mice (33). This observation is consistent with the results obtained in this study. By means of ChIP analysis, site-directed mutagenesis, and SHP reconstitution experiments, we have firmly established that LRH-1 directly controls Gck transcription.
LRH-1 specifically acts on glucokinase transcription, since no significant differences in hexokinase expression were observed between Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mice. This is of particular interest, because it has been shown that only G6P generated through glucokinase determines the metabolic fate of intrahepatic glucose (34, 35). Liver-specific glucokinase knockout mice show a reduction in glycogen synthesis under hyperglycemic conditions (22). In this study, we also observed reduced glycogen storage and a lower glycogen synthase flux in the livers of Alb-Cre;Lrh1fl/fl mice. Glucokinase is furthermore required to increase Chrebp mRNA levels in response to refeeding and is critical for the ChREBP-mediated induction of glycolytic and lipogenic genes in response to glucose (23). Importantly, recent work has identified G6P as the actual metabolic signal that drives ChREBP-mediated control of glycolysis and lipogenesis (24, 25). In line with this, we observed that refed Alb-Cre;Lrh1fl/fl mice showed reduced hepatic G6P content along with a suboptimal induction of glycolytic and lipogenic genes. The reconstitution of hepatic SHP furthermore revealed that, similar to those of Gck, the lower expression levels of Chrebp and its target Pklr in Alb-Cre;Lrh1fl/fl mice are also caused by the absence of LRH-1, and not SHP, in the liver. Reduced mRNA levels of Chrebp and its targets in Alb-Cre;Lrh1fl/fl mice were associated with lowered glycolytic and lipogenic fluxes as well as reduced plasma triglyceride levels. The reduction in glycolysis in Alb-Cre;Lrh1fl/fl mice results from impaired GCK activity, as Pklr expression was completely normalized upon hepatic Gck rescue. Gck reconstitution also restored the mRNA levels of the ChREBP target Acaca, but not the expression of Fasn, which is a direct target of both ChREBP and LRH-1 (13, 23). The reduced de novo lipogenesis in Alb-Cre;Lrh1fl/fl mice therefore most likely results from a concerted impairment of both ChREBP-dependent and LRH-1–dependent lipogenesis. Thus, although the flux through glucokinase is only partially inhibited in Alb-Cre;Lrh1fl/fl mice, these animals present a striking phenotypic resemblance to liver-specific glucokinase knockout mice with regard to glycogen storage and ChREBP-mediated glycolysis and lipogenesis (22, 23).
Genetic models of obesity and type 2 diabetes, including ob/ob and db/db mice, have been reported to display increased hepatic glucokinase expression and flux (36–38), along with ChREBP hyperactivity and hepatic steatosis, which is partially reversible by ChREBP inhibition (39). Importantly, glucokinase expression is also associated with lipogenic activity and fatty liver in humans (40). This implies that a reduction in LRH-1 activity would have a potential protective effect on steatosis development via reduced glucokinase-mediated ChREBP activation when hepatic glucose availability is high, e.g., in obese diabetic states. Although Alb-Cre;Lrh1fl/fl mice are not protected against high-fat diet–induced glucose intolerance and insulin resistance (Supplemental Figure 3 and ref. 18), it was recently shown that DLPC administration improves insulin sensitivity and liver steatosis in an LRH-1–dependent manner (18). It was proposed that DLPC-mediated LRH-1 activation interferes with the insulin/SREBP-1c signaling cascade that drives lipogenesis in the liver. In this study, we observed that the hepatic mRNA levels of Srebp-1c and its target G6pd1 were unaltered in insulin-sensitive Alb-Cre;Lrh1fl/fl mice. This is in agreement with a report showing that Srebp-1c and G6pd1 expression levels are not altered upon glucokinase overexpression, while those of the ChREBP targets Pklr and Acaca/Acacb are induced (41). Combined, current and previous findings (18) indicate that LRH-1 impacts on hepatic lipogenesis via alternate routes under different nutritional and pathophysiological conditions.
In conclusion, these data indicate that LRH-1 is a major and essential transcriptional regulator of glucokinase, the gatekeeper of hepatic glucose metabolism. As a consequence, LRH-1 not only controls the initial step of glucose uptake but also determines the intrahepatic fate of its product G6P. Our data identifies LRH-1 as a critical component of the hepatic glucose-sensing system that integrates glucose and lipid homeostasis in the postprandial phase.
Methods
For more details, see the Supplemental Methods. Unless otherwise indicated, all chemical reagents were obtained from Sigma-Aldrich.
Generation of viral vectors.
AAV8 encoding the mouse SHP or GCK cDNAs (AAV8-SHP or AAV8-GCK), driven by the hybrid cytomegalovirus enhancer/chicken β-actin constitutive promoter or the liver-specific α1-antitrypsin promoter (hAAT; ref. 42), respectively, were generated and titrated as described previously to enable their use in vivo (43, 44). Plasmids used to generate viral particles and the hAAT promoter were provided by K.A. High, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA. For in vitro experiments, cells were transduced with an adenoviral vector encoding the mouse Lrh1 gene (AdLRH-1; derived from pCMX-mLRH-1; ref. 8) using the Adeno-X Expression System (Clontech).
Animal studies.
LRH-1 floxed mice (3) were crossed with serum albumin-Cre mice (The Jackson Laboratory) and then further intercrossed to generate hepatocyte-specific LRH-1 knockout (Alb-Cre;Lrh1fl/fl) and wild-type (Lrh1fl/fl) mice on a pure C57BL/6J background. Animals had ad libitum access to regular chow (Teklad no. 2016, Harlan) and drinking water and were kept under a 12-hour-dark/12-hour-light cycle (lights on 7 AM to 7 PM). At the end of the experiments, mice were sacrificed by cardiac puncture under isoflurane anesthesia, and livers were quickly snap frozen.
For quantification of carbohydrate fluxes, mice were equipped with a permanent jugular vein catheter, after which they were allowed a recovery period of at least 3 days (45). Prior to the infusion experiment, mice were fasted overnight. They were infused with a solution containing [U-13C]glucose, [2-13C]glycerol, [1-2H]galactose, and paracetamol at an infusion rate of 0.6 ml per hour, as previously described, for 210 minutes (19). Then, the infusion of a second solution containing glucose (1,067 mM) and [U-13C]glucose (50 mM) was initiated, while the infusion of the first solution was continued. The infusion rate of the second solution was adjusted according to the blood glucose concentration in order to maintain comparable hyperglycemia in both genotypes. These infusions were prolonged for another 210 minutes. During the entire experiment, blood glucose concentrations were measured and blood and urine samples were collected on filter paper at regular time intervals (46). After 420 minutes, mice were sacrificed.
For oral glucose tolerance tests, mice were fasted overnight and subsequently challenged with a glucose bolus (2 g/kg) by gavage. At regular time points, blood glucose concentrations were measured, and blood samples were collected to determine insulin levels. Separate groups of mice were sacrificed after 0, 30, and 60 minutes.
For fasting-refeeding experiments, fed and 24-hour–fasted mice were sacrificed at 7 AM. Refed mice were fasted for 24 hours, starting at 7 AM, and were subsequently refed with normal chow diet for 6 hours. To quantify fatty acid synthesis rates, all mice received sodium[1-13C] acetate via their drinking water (2%) 24 hours prior to sacrifice.
For SHP reconstitution experiments, Alb-Cre;Lrh1fl/fl mice received an injection of SHP-AAV8 vector (1012 particles per mouse) into the jugular vein under isoflurane anesthesia. Five weeks after vector administration, the mice were sacrificed after a 6-hour fast. For GCK reconstitution experiments, Alb-Cre;Lrh1fl/fl mice received an injection of GCK-AAV8 vector (1011 or 1012 particles per mouse) into the jugular vein under isoflurane anesthesia. Five weeks after vector administration, the mice were sacrificed after 6 hours of refeeding. Hepatic gene expression levels were compared with those of Lrh1fl/fl and Alb-Cre;Lrh1fl/fl mouse AAV8 vector particles carrying a noncoding genome (AAV8-null).
Biochemical analysis.
The extraction of glucose from blood and urine spots, the derivatization of the extracted compounds, the GC-MS analysis of these derivatives, and the calculation of hepatic carbohydrate fluxes were performed as described previously (46, 47). Blood glucose concentrations were measured using a Maxi Kit glucometer (Bayer Diagnostics). Plasma insulin and triglyceride concentrations were determined using commercially available ELISA (Crystal Chem Inc.) and enzymatic kits (Roche), respectively. Hepatic triglyceride content was quantified after lipid extraction (48) using an enzymatic assay (Roche). Fatty acids derived from these hepatic lipid extracts were liberated, derivatized, and subjected to GC-MS analysis in order to calculate fractional synthesis rates from the incorporation of 13C-acetate as described previously (49). Hepatic glycogen and G6P content were determined using enzymatic assays (50, 51).
Histological analysis.
In order to visualize hepatic glycogen disposition, hematoxylin and eosin and periodic acid-Schiff stainings were performed on 4-μm-thick liver sections.
Cell culture.
Primary hepatocytes were isolated using collagenase perfusion as described previously with minor modifications (23). Hepatocytes from Lrh1fl/fl mice were plated in DMEM 4.5 g/l glucose with 10% FBS. Four hours after plating, cells were transduced with AdLRH-1 or AdGFP. Hepatocytes were then incubated in DMEM 4.5 g/l glucose without serum for 36 hours and lysed for RNA isolation.
Primary hepatocytes from Alb-Cre;Lrh1fl/fl and Lrh1fl/fl mice were seeded in Seahorse XF24 plates (Seahorse Bioscience) containing glucose-free medium without serum for 24 hours. They were subsequently exposed to 25 mM glucose, and the extracellular acidification rate (a measure of glycolysis) was measured at 5-minute intervals by the Seahorse XF24 system (Seahorse Bioscience).
Hepa 1.6 mouse hepatoma cells were cultured in DMEM 4.5 g/l glucose with 10% FBS, 1% NEAA, and 0.1% gentamicin. These cells were transduced with AdLRH-1 or AdGFP. After 12 hours, media were changed and incubation was continued for another 30 hours. Then, cells were rinsed with PBS 3 times and either lysed in Tri Reagent (Invitrogen) for RNA isolation or exposed to 2-[14C]deoxyglucose (DOG; 0.1 mCi) for 10 minutes to quantify DOG incorporation as described previously (52). In short, cells were rinsed with 0.1 mM phloretin to further inhibit sugar efflux and lysed in 1 M NaOH at 60°C for 10 minutes. The lysates were neutralized in 1 M HCl and centrifuged. 0.3 M ZnSO4 and 0.3 M Ba(OH)2 were added to the supernatant, and the mixture was centrifuged. Then, 6% perchloric acid was added to the supernatant. After the centrifugation, the radioactivity of the supernatant was measured by a liquid scintillation counter. Radioactive counts were normalized to DNA content to determine DOG uptake and G6P production.
Cloning and site-directed mutagenesis.
A 0.6-kb fragment (–601 to +10) of the murine Shp promoter, as well as a long (~1.2 kb; –1,063 to +141) and short (~0.6 kb; –514 to +141) fragment of the murine hepatic Gck promoter (21) (GCK long and short), were amplified from mouse liver genomic DNA. These fragments were inserted into the multiple cloning site of the pGL3-basic reporter (Promega Life Science). The heterologous LRH-1 reporter vector was generated by subcloning 4 repeats of an established LRH-1 response element in the human Shp promoter (–79 to –71) into the multiple cloning site of the TK-pGL3 reporter (Promega Life Science).
Mutations in putative LRH-1 responsive elements 5 and 6 (Figure 3F) in the GCK short reporter were introduced using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Additional mutagenesis was performed to obtain GCK short mutant elements 5 and 6 (mt5-6), from the GCK short mt5. The sequences of all reporter constructs were analyzed and confirmed.
Transient transfection and cell reporter assays.
All transfections were performed using Lipofectamin 2000 (Invitrogen). HeLa cells were transfected under low (5 mM) glucose conditions with reporter constructs driven by a heterologous promoter, consisting of 4 consensus LRH-1 response elements, or driven by the endogenous Shp promoter in the presence of either pCMX-mLRH-1 or pCMX-empty (8). After 6 hours, media were changed to either high (25 mM) or low (5 mM) glucose. Luciferase activities were determined 24 hours later and normalized for β-galactosidase activities. Luciferase activities in HeLa cells transfected with Chrebp promoter reporter (53), the Shp promoter reporter, or the different Gck promoter reporters in normal culture conditions (25 mM glucose) in the presence of either pCMX-mLRH-1 or pCMX-empty were determined 30 hours after transfection and normalized for β-galactosidase activities. All experiments were performed 3 times, and data represent average values of a representative experiment, which was performed in quadruplicate and normalized for empty luciferase reporter (pGL3) activity.
Quantitative RT-PCR and ChIP.
RNA was isolated from liver samples and cells using Tri Reagent, and quantitative RT-PCR was performed as described previously (3). Primer sequences are listed in Supplemental Table 1. Gene expression levels were normalized for β2 microglobulin.
ChIP analysis was performed as described previously with minor adaptations (54). DNA was purified using the PCR clean-up Gel extraction Kit (Macherey-Nagel), after which qPCR was performed as described previously (3). Data were normalized for GAPDH promoter binding and expressed relative to IgG. ChIP primer sequences are listed in Supplemental Table 1.
Western blot analysis.
Total liver and nuclear protein extracts were prepared, and lysates were subjected to Western blotting as described previously (3). Blots were probed with GCK (55), ChREBP (Novus), LRH-1 (56), Parp-1 (Santa Cruz Biotechnology Inc.), and HSP90 (BD Transduction Laboratories).
Statistics.
Data are presented as mean ± SEM. Statistical significance (P < 0.05) was assessed using the Kruskal-Wallis test followed by the Conover test for post-hoc analysis.
Study approval.
All animal studies were performed in accordance with Swiss animal protection law and were approved by the Cantonal Veterinary Office (Vaud, Switzerland) and the local committee for animal experimentation (license no. 2331; Ecole Polytechnique Fédérale de Lausanne).
Supplementary Material
Acknowledgments
The authors thank Thibaud Clerc, Jiujiu Yu, Amandine Signorino-Gelo, Dongryeol Ryu, Pablo Fernandez-Marcos, Sabrina Bichet, Aycha Bleeker, Rick Havinga, and Niels Kloosterhuis for technical assistance and discussion. The ChREBP reporter was provided by K. Gauthier (Institut de Génomique Fonctionnelle de Lyon, Université de Lyon, Lyon, France). This work was supported by the Ecole Polytechnique Fédérale de Lausanne, the Swiss National Science Foundation, the Swiss Cancer League, and the FP7 programme EUGENE2. M.H. Oosterveer is supported by a fellowship from the Royal Netherlands Academy of Arts and Sciences. J. Auwerx is the Nestlé Chair in Energy Metabolism.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(8):2817–2826. doi:10.1172/JCI62368.
References
- 1.Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007;17(6):292–301. doi: 10.1016/j.tcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 2.McKenna NJ, et al. Minireview: evolution of NURSA, the nuclear receptor signaling atlas. Mol Endocrinol. 2009;23(6):740–746. doi: 10.1210/me.2009-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mataki C, et al. Compromised intestinal lipid absorption in mice with a liver-specific deficiency of liver receptor homolog 1. Mol Cell Biol. 2007;27(23):8330–8339. doi: 10.1128/MCB.00852-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Out C, et al. Liver receptor homolog-1 is critical for adequate up-regulation of Cyp7a1 gene transcription and bile salt synthesis during bile salt sequestration. Hepatology. 2011;53(6):2075–2085. doi: 10.1002/hep.24286. [DOI] [PubMed] [Google Scholar]
- 5.Lee YK, et al. Liver receptor homolog-1 regulates bile acid homeostasis but is not essential for feedback regulation of bile acid synthesis. Mol Endocrinol. 2008;22(6):1345–1356. doi: 10.1210/me.2007-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venteclef N, Smith JC, Goodwin B, Delerive P. Liver receptor homolog 1 is a negative regulator of the hepatic acute-phase response. Mol Cell Biol. 2006;26(18):6799–6807. doi: 10.1128/MCB.00579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venteclef N, Delerive P. Interleukin-1 receptor antagonist induction as an additional mechanism for liver receptor homolog-1 to negatively regulate the hepatic acute phase response. J Biol Chem. 2007;282(7):4393–4399. doi: 10.1074/jbc.M608993200. [DOI] [PubMed] [Google Scholar]
- 8.Schoonjans K, et al. Liver receptor homolog 1 controls the expression of the scavenger receptor class B type I. EMBO Rep. 2002;3(12):1181–1187. doi: 10.1093/embo-reports/kvf238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freeman LA, et al. The orphan nuclear receptor LRH-1 activates the ABCG5/ABCG8 intergenic promoter. J Lipid Res. 2004;45(7):1197–1206. doi: 10.1194/jlr.C400002-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Delerive P, Galardi CM, Bisi JE, Nicodeme E, Goodwin B. Identification of liver receptor homolog-1 as a novel regulator of apolipoprotein AI gene transcription. Mol Endocrinol. 2004;18(10):2378–2387. doi: 10.1210/me.2004-0132. [DOI] [PubMed] [Google Scholar]
- 11.Venteclef N, Haroniti A, Tousaint JJ, Talianidis I, Delerive P. Regulation of anti-atherogenic apolipoprotein M gene expression by the orphan nuclear receptor LRH-1. J Biol Chem. 2008;283(7):3694–3701. doi: 10.1074/jbc.M706382200. [DOI] [PubMed] [Google Scholar]
- 12.Luo Y, Liang CP, Tall AR. The orphan nuclear receptor LRH-1 potentiates the sterol-mediated induction of the human CETP gene by liver X receptor. J Biol Chem. 2001;276(27):24767–24773. doi: 10.1074/jbc.M100912200. [DOI] [PubMed] [Google Scholar]
- 13.Matsukuma KE, Wang L, Bennett MK, Osborne TF. A key role for orphan nuclear receptor liver receptor homologue-1 in activation of fatty acid synthase promoter by liver X receptor. J Biol Chem. 2007;282(28):20164–20171. doi: 10.1074/jbc.M702895200. [DOI] [PubMed] [Google Scholar]
- 14.Lee YK, Moore DD. Liver receptor homolog-1, an emerging metabolic modulator. Front Biosci. 2008;13:5950–5958. doi: 10.2741/3128. [DOI] [PubMed] [Google Scholar]
- 15.Ingraham HA, Redinbo MR. Orphan nuclear receptors adopted by crystallography. Curr Opin Struct Biol. 2005;15(6):708–715. doi: 10.1016/j.sbi.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 16.Mullaney BC, et al. Regulation of C. elegans fat uptake and storage by acyl-CoA synthase-3 is dependent on NR5A family nuclear hormone receptor nhr-25. Cell Metab. 2010;12(4):398–410. doi: 10.1016/j.cmet.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musille PM, Pathak MC, Lauer JL, Hudson WH, Griffin PR, Ortlund EA. Antidiabetic phospholipid-nuclear receptor complex reveals the mechanism for phospholipid-driven gene regulation. Nat Struct Mol Biol. 2012;19(5):532–537. doi: 10.1038/nsmb.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JM, et al. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature. 2011;474(7352):506–510. doi: 10.1038/nature10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derks TG, et al. Inhibition of mitochondrial fatty acid oxidation in vivo only slightly suppresses gluconeogenesis but enhances clearance of glucose in mice. Hepatology. 2008;47(3):1032–1042. doi: 10.1002/hep.22101. [DOI] [PubMed] [Google Scholar]
- 20.Lee YK, Moore DD. Dual mechanisms for repression of the monomeric orphan receptor liver receptor homologous protein-1 by the orphan small heterodimer partner. J Biol Chem. 2002;277(4):2463–2467. doi: 10.1074/jbc.M105161200. [DOI] [PubMed] [Google Scholar]
- 21.Postic C, et al. Cloning and characterization of the mouse glucokinase gene locus and identification of distal liver-specific DNase I hypersensitive sites. Genomics. 1995;29(3):740–750. doi: 10.1006/geno.1995.9943. [DOI] [PubMed] [Google Scholar]
- 22.Postic C, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274(1):305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 23.Dentin R, et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem. 2004;279(19):20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- 24.Dentin R, et al. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J Hepatol. 2012;56(1):199–209. doi: 10.1016/j.jhep.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 25.Li MV, et al. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem Biophys Res Commun. 2010;395(3):395–400. doi: 10.1016/j.bbrc.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fayard E, Auwerx J, Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004;14(5):250–260. doi: 10.1016/j.tcb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Niswender KD, Shiota M, Postic C, Cherrington AD, Magnuson MA. Effects of increased glucokinase gene copy number on glucose homeostasis and hepatic glucose metabolism. J Biol Chem. 1997;272(36):22570–22575. doi: 10.1074/jbc.272.36.22570. [DOI] [PubMed] [Google Scholar]
- 28.Iynedjian PB. Mammalian glucokinase and its gene. Biochem J. 1993;293(pt 1):1–13. doi: 10.1042/bj2930001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim TH, et al. Interrelationship between liver X receptor alpha, sterol regulatory element-binding protein-1c, peroxisome proliferator-activated receptor gamma, and small heterodimer partner in the transcriptional regulation of glucokinase gene expression in liver. J Biol Chem. 2009;284(22):15071–15083. doi: 10.1074/jbc.M109.006742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oosterveer MH, et al. Lxralpha deficiency hampers the hepatic adaptive response to fasting in mice. J Biol Chem. 2008;283(37):25437–25445. doi: 10.1074/jbc.M801922200. [DOI] [PubMed] [Google Scholar]
- 31.Denechaud PD, et al. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J Clin Invest. 2008;118(3):956–964. doi: 10.1172/JCI34314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. 2002;277(11):9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- 33.Park YJ, et al. Dissociation of diabetes and obesity in mice lacking orphan nuclear receptor small heterodimer partner. J Lipid Res. 2011;52(12):2234–2244. doi: 10.1194/jlr.M016048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seoane J, Gomez-Foix AM, O’Doherty RM, Gomez-Ara C, Newgard CB, Guinovart JJ. Glucose 6-phosphate produced by glucokinase, but not hexokinase I, promotes the activation of hepatic glycogen synthase. J Biol Chem. 1996;271(39):23756–23760. doi: 10.1074/jbc.271.39.23756. [DOI] [PubMed] [Google Scholar]
- 35.O’Doherty RM, Lehman DL, Seoane J, Gomez-Foix AM, Guinovart JJ, Newgard CB. Differential metabolic effects of adenovirus-mediated glucokinase and hexokinase I overexpression in rat primary hepatocytes. J Biol Chem. 1996;271(34):20524–20530. doi: 10.1074/jbc.271.34.20524. [DOI] [PubMed] [Google Scholar]
- 36.Bandsma RH, et al. Enhanced glucose cycling and suppressed de novo synthesis of glucose-6-phosphate result in a net unchanged hepatic glucose output in ob/ob mice. Diabetologia. 2004;47(11):2022–2031. doi: 10.1007/s00125-004-1571-8. [DOI] [PubMed] [Google Scholar]
- 37.Hron WT, Sobocinski KA, Menahan LA. Enzyme activities of hepatic glucose utilization in the fed and fasting genetically obese mouse at 4-5 months of age. Horm Metab Res. 1984;16(suppl 1):32–36. doi: 10.1055/s-2007-1014687. [DOI] [PubMed] [Google Scholar]
- 38.Yen TT, Stamm NB. Constitutive hepatic glucokinase activity in db/db and ob/ob mice. Biochim Biophys Acta. 1981;657(1):195–202. doi: 10.1016/0005-2744(81)90143-1. [DOI] [PubMed] [Google Scholar]
- 39.Dentin R, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55(8):2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 40.Peter A, et al. Hepatic glucokinase expression is associated with lipogenesis and fatty liver in humans. J Clin Endocrinol Metab. 2011;96(7):E1126–E1130. doi: 10.1210/jc.2010-2017. [DOI] [PubMed] [Google Scholar]
- 41.Scott DK, et al. A modest glucokinase overexpression in the liver promotes fed expression levels of glycolytic and lipogenic enzyme genes in the fasted state without altering SREBP-1c expression. Mol Cell Biochem. 2003;254(1–2):327–337. doi: 10.1023/A:1027306122336. [DOI] [PubMed] [Google Scholar]
- 42.Manno CS, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 43.Ayuso E, et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17(4):503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- 44.Lock M, et al. Characterization of a recombinant adeno-associated virus type 2 Reference Standard Material. Hum Gene Ther. 2010;21(10):1273–1285. doi: 10.1089/hum.2009.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuipers F, Havinga R, Bosschieter H, Toorop GP, Hindriks FR, Vonk RJ. Enterohepatic circulation in the rat. Gastroenterology. 1985;88(2):403–411. doi: 10.1016/0016-5085(85)90499-8. [DOI] [PubMed] [Google Scholar]
- 46.Van Dijk TH, Boer TS, Havinga R, Stellaard F, Kuipers F, Reijngoud DJ. Quantification of hepatic carbohydrate metabolism in conscious mice using serial blood and urine spots. Anal Biochem. 2003;322(1):1–13. doi: 10.1016/j.ab.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 47.Lee WN, Byerley LO, Bergner EA, Edmond J. Mass isotopomer analysis: theoretical and practical considerations. Biol Mass Spectrom. 1991;20(8):451–458. doi: 10.1002/bms.1200200804. [DOI] [PubMed] [Google Scholar]
- 48.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 49.Oosterveer MH, et al. High fat feeding induces hepatic fatty acid elongation in mice. PLoS One. 2009;4(6):e6066. doi: 10.1371/journal.pone.0006066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hohorst HJ. Methoden der Enzymatischen Analyse. D–Glucose–6–phosphat und D–fructose–6–phosphat. In: Bergmeyer HU, ed. pp. 1200–1204. Weinheim, Germany; Verlag Chemie: 1970: [Google Scholar]
- 51.Keppler D, Decker K. Methoden der Enzymatischen Analyse. Glykogen. Bestimmung mit amyloglucosidase. In: Bergmeyer HU, ed. pp. 1089–1094. Weinheim, Germany; Verlag Chemie: 1970: [Google Scholar]
- 52.Mullin JM, McGinn MT, Snock KV, Kofeldt LM. Na+-independent sugar transport by cultured renal (LLC-PK1) epithelial cells. Am J Physiol. 1989;257(1 pt 2):F11–F17. doi: 10.1152/ajprenal.1989.257.1.F11. [DOI] [PubMed] [Google Scholar]
- 53.Gauthier K, et al. Thyroid hormone receptor beta (TRbeta) and liver X receptor (LXR) regulate carbohydrate-response element-binding protein (ChREBP) expression in a tissue-selective manner. J Biol Chem. 2010;285(36):28156–28163. doi: 10.1074/jbc.M110.146241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duggavathi R, et al. Liver receptor homolog 1 is essential for ovulation. Genes Dev. 2008;22(14):1871–1876. doi: 10.1101/gad.472008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang Y, Jetton TL, Zimmerman EC, Najafi H, Matschinsky FM, Magnuson MA. Effects of alternate RNA splicing on glucokinase isoform activities in the pancreatic islet, liver, and pituitary. J Biol Chem. 1991;266(11):6999–7007. [PubMed] [Google Scholar]
- 56.Coste A, et al. LRH-1-mediated glucocorticoid synthesis in enterocytes protects against inflammatory bowel disease. Proc Natl Acad Sci U S A. 2007;104(32):13098–13103. doi: 10.1073/pnas.0702440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.