

Abstract
miR-122, an abundant liver-specific microRNA (miRNA), regulates cholesterol metabolism and promotes hepatitis C virus (HCV) replication. Reduced miR-122 expression in hepatocellular carcinoma (HCC) correlates with metastasis and poor prognosis. Nevertheless, the consequences of sustained loss of function of miR-122 in vivo have not been determined. Here, we demonstrate that deletion of mouse Mir122 resulted in hepatosteatosis, hepatitis, and the development of tumors resembling HCC. These pathologic manifestations were associated with hyperactivity of oncogenic pathways and hepatic infiltration of inflammatory cells that produce pro-tumorigenic cytokines, including IL-6 and TNF. Moreover, delivery of miR-122 to a MYC-driven mouse model of HCC strongly inhibited tumorigenesis, further supporting the tumor suppressor activity of this miRNA. These findings reveal critical functions for miR-122 in the maintenance of liver homeostasis and have important therapeutic implications, including the potential utility of miR-122 delivery for selected patients with HCC and the need for careful monitoring of patients receiving miR-122 inhibition therapy for HCV.
Introduction
miR-122 is a conserved liver-specific microRNA (miRNA) that constitutes 70% of the cloned hepatic miRNA in the adult mouse (1). Several key observations underscore the importance of miR-122 in liver biology and disease. First, antisense-mediated inhibition of miR-122 (also known as miR-122a) in mice leads to the induction of genes that are normally repressed in adult liver (2), suggesting that this miRNA is important for the maintenance of the terminally differentiated hepatocyte gene expression program. Furthermore, miR-122 inhibition reduces serum cholesterol by indirectly causing the downregulation of genes involved in cholesterol biosynthesis, including the rate-limiting enzyme HMG-CoA reductase (Hmgcr), thereby protecting animals from diet-induced hypercholesterolemia (3). Additionally, miR-122 plays a non-canonical role in the life cycle of HCV. Through interaction with two seed sequence binding sites located at the 5′-end of the HCV genomic RNA, miR-122 performs an incompletely understood function that is essential for replication of the virus (4). Accordingly, intravenous administration of locked nucleic acid (LNA) antisense miR-122 oligonucleotides reduces viral load in HCV-infected chimpanzees (5), a therapeutic approach that is currently under clinical investigation for HCV in humans.
Hepatocellular carcinoma (HCC) is the fifth most prevalent cancer worldwide and the third leading cause of cancer-related death (6, 7). HCC often occurs in the setting of underlying liver dysfunction, especially chronic inflammation and cirrhosis. miR-122 expression is reduced during the initiation and progression of hepatocarcinogenesis in a rat model of nonalcoholic steatohepatitis (NASH) (8), and downregulation of miR-122 is prevalent in human NASH patients (9). Moreover, reduced expression of miR-122 correlates with poor prognosis and metastasis in HCC patients, and expression of miR-122 is anti-tumorigenic in HCC cell lines (reviewed in ref. 10).
Despite these indications of a critical role for miR-122 in liver physiology and disease, and the potential for this miRNA as a therapeutic target for HCV and perhaps other disease states, the consequences of genetic loss of function of this miRNA in vivo have yet to be documented. Here we describe the generation and characterization of mice with germline knockout (KO) or liver-specific knockout (LKO) of the Mir122 locus. Both KO and LKO mice develop normally and are viable and, consistent with studies performed using antisense-mediated miR-122 inhibition, exhibit reduced serum cholesterol (2, 3, 11). In contrast to transient inhibition studies, however, Mir122-KO and -LKO animals develop microsteatosis and liver inflammation in early adult life that progresses to steatohepatitis, fibrosis, and spontaneous tumors resembling HCC. Hepatocarcinogenesis is likely initiated in these animals through the upregulation of several oncogenic pathways coupled with liver injury associated with the infiltration of inflammatory cells that produce the pro-tumorigenic cytokines IL-6 and TNF-α. Moreover, miR-122 exhibited tumor suppressor activity when delivered to livers of a non-inflammatory MYC-driven HCC mouse model. These findings provide important insight into the natural functions of this miRNA, establish its role as a tumor suppressor in vivo, and reveal potential consequences of miR-122 inhibition that may be relevant to the clinical implementation of this strategy for HCV therapy.
Results
Liver-specific (LKO) and germline (KO) Mir122 loss of function results in an altered serum lipid profile.
The mmu-Mir122 gene is located on chromosome 18 and is transcribed independently of any known gene. To determine its biological function, we generated a miR-122 conditional knockout allele (Mir122loxP) in mice using homologous recombination. Mir122loxP mice were crossed to albumin-Cre (Alb-Cre) mice to produce LKO mice (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI63539DS1). For generation of KO mice, Mir122loxP mice were initially crossed to E2a-Cre mice and then backcrossed to remove the Cre transgene. Both KO and LKO mice were born alive and fertile and were not notably different from control (floxed or wild-type) mice in terms of their body weight and growth.
Northern and Southern blotting and real-time RT-PCR were used to confirm deletion of the Mir122 gene and loss of miR-122 expression in LKO livers of 10-week-old mice. Except in rare cases, this time point was sufficient for complete Cre-mediated deletion of the floxed allele in Alb-Cre hepatocytes (ref. 12, Figure 1A, Supplemental Figure 1B, and Supplemental Figure 2A). A significant increase in the expression of known targets such as Aldoa, Slc7a1, Cs, and Ccng1 (3, 13) confirmed functional depletion of miR-122 in LKO livers (Supplemental Figure 2B). In KO mice, expression of miR-122 was abolished without influencing the levels of several other abundant liver-enriched miRNAs such as miR-148, miR-192, and miR-194 (Figure 1B). Real-time RT-PCR and Western blot analysis demonstrated increased expression of known miR-122 targets including Adam-10 (14), Pparβ/δ, Smarcd1/Baf60a (15), and Iqgap1 (ref. 2 and Supplemental Figure 2, C and D).
Figure 1. Abnormal liver structure and TG metabolism in Mir122-LKO mice in early adult life.

(A and B) Northern blot analysis of miRNA levels in liver. (C) Representative liver sections of 8-week-old control (floxed) and LKO mice after overnight fasting (n = 8–10 mice per genotype). Scale bars: top row, 200 μm; bottom row, 25 μm. (D) Oil red O– and PAS-stained liver sections from 8-week-old LKO mice after overnight fasting (n = 5 per genotype). Scale bars: top row, 100 μm; bottom row, 100 μm; insets, 25 μm. (E) Transmission electron micrographs of liver sections from 12-week-old LKO mice. Lipid droplets (L), ER, mitochondria (M), and nucleus (N) are labeled. Scale bars: top row, 2 μm; bottom row, 450 nm. (F) CK19 and A6 staining of bile duct and oval cells, respectively, in LKO livers (n = 3 mice per genotype). Scale bars: top row, 100 μm; inset, 5 μm; bottom row, 25 μm. (G) Hepatic TG and cholesterol levels in 10-week-old LKO mice. (H) De novo TG synthesis in liver as measured by 3H1-glycerol incorporation. (I) TG secretion as measured by monitoring serum TG levels after administration of Triton WR-1339. The results presented in G–I are mean ± SD. Statistical significance was calculated using a 2-tailed t test.
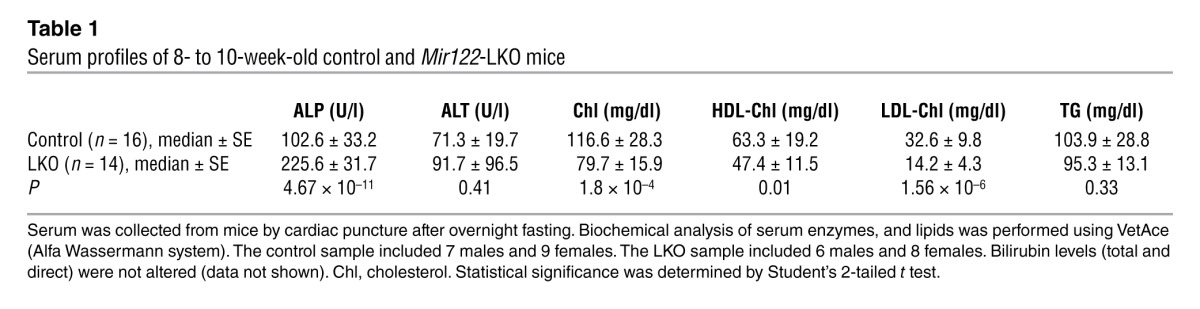
We next examined liver function in 8- to 10-week-old LKO mice by measuring the serum profile of liver enzymes and metabolites after overnight fasting (Table 1). Among the lipids, total cholesterol was reduced in the serum of LKO mice by 30% (P = 1.8 × 10–4) without significantly altering triglyceride (TG) levels, corroborating previous observations in mice (2, 3) and primates (5, 16) depleted of miR-122 by administration of antisense oligonucleotides. These mice also exhibited a pronounced decrease in LDL cholesterol (∼56.5%, P = 1.56 × 10–6) and a moderate decrease in HDL cholesterol (∼25.1%, P = 0.01). Finally, serum alkaline phosphatase (ALP) increased 2-fold (P = 4.67 × 10–11) in LKO mice. The serum profile of 5-week-old KO mice was very similar to that of the LKO mice (Supplemental Table 1).
Table 1.
Serum profiles of 8- to 10-week-old control and Mir122-LKO mice
LKO mice develop hepatic microsteatosis due to TG accumulation in early adult life.
Histopathological analysis revealed distinctive features in livers of 8- to 10-week-old LKO mice. Sinusoids in LKO mice were compressed by swollen hepatocytes containing multiple small clear vacuoles, likely representing lipid droplets (Figure 1C). Microsteatosis in LKO mice was confirmed by Oil Red O staining (Figure 1D) and transmission electron microscopy (TEM) (Figure 1E). Storage of liver glycogen was reduced in LKO mice after overnight fasting, as demonstrated by PAS staining (Figure 1D). LKO livers also exhibited proliferation of bile duct and oval cells as shown by increased CK-19– and A6-positive cells, respectively (Figure 1F). Liver histology and serology were similar in male and female LKO mice (data not shown).
Quantification of hepatic lipids revealed a 2.5-fold increase in TG levels without a change in cholesterol levels in LKO mice (Figure 1G). The accumulation of TG could be the result of altered synthesis, secretion, and/or uptake. Measurement of 3H1-glycerol incorporation into hepatic TGs showed a small (25%) but significant (P = 0.022) increase in de novo TG synthesis in LKO mice (Figure 1H). Hepatic TG secretion in LKO mice was measured by monitoring serum TG levels after injection of Triton WR-1339, an inhibitor of lipoprotein lipase. TG secretion was reduced to 43% and 46% of control levels after 1 and 3 hours, respectively (Figure 1I). Thus, increased synthesis and reduced secretion contribute to TG accumulation in the livers of LKO mice.
Genes involved in lipid metabolism, cellular proliferation, and survival are abnormally expressed in livers of LKO mice.
To investigate the mechanisms underlying the abnormalities observed in miR-122–deficient mice, we examined hepatic gene expression in 8- to 10-week-old control and LKO mice (n = 5 of each genotype) by microarray analysis. An examination of the potential enrichment of all possible hexamers, heptamers, and octamers in the 3′ UTRs of the transcripts upregulated in miR-122–deficient livers using the Sylamer algorithm (17) revealed that the only statistically significantly enriched motifs of these lengths corresponded to sites that match the miR-122 seed sequence (Figure 2A). These results indicate that the altered expression of a significant fraction of dysregulated transcripts in LKO livers is attributable to direct, canonical targeting by miR-122.
Figure 2. Altered expression of genes involved in TG metabolism and hepatocarcinogenesis in Mir122-LKO livers.

(A) Sylamer plots (17) showing the enriched hexamers (top), heptamers (middle), and octamers (bottom) in transcripts that are upregulated in LKO livers. All motifs that are statistically significantly enriched are highlighted in color on the plots and correspond to binding sites for the miR-122 seed sequence as shown on the left. (B) Expression of genes involved in TG synthesis and storage in LKO livers. For this and subsequent panels, real-time RT-PCR values represent means from triplicate measurements with multiple samples (n = 4–5). Statistical significance was calculated using a 2-tailed t test. (C) Western blot analysis of microsomal or whole liver extracts. (D) Renilla luciferase activity (LUC2) produced from wild-type or mutant (mut) Agpat1, Cidec, and Mapre1 3′ UTR reporter plasmids or empty vector (pSICHECK2) normalized to firefly luciferase activity (LUC1) produced from the same plasmid after transfection into Hepa cells together with negative control RNA (NC) or miR-122 mimic. Error bars represent SDs derived from 3 independent experiments. (E and F) Expression of transcripts (E) and proteins (F) related to hepatocarcinogenesis in LKO/KO livers.
Ingenuity pathway analysis (IPA) of molecular and cellular functions of a stringent set (threshold P ≤ 0.0001) of 194 upregulated and 121 downregulated genes in LKO livers identified 7 major networks of dysregulated genes (Supplemental Table 2). Genes involved in lipid metabolism were highly represented within these networks. Notably, among the upregulated genes were two key enzymes, Agpat1 and Mogat1, that catalyze TG biosynthesis (18). Microarray analysis also showed increased expression of several additional genes in this pathway, including Agpat3, Agpat9, Ppap2a, Ppap2c, and Dgat1, albeit at a lower significance threshold (P ≤ 0.05) (Supplemental Table 3). Real-time RT-PCR confirmed the significant upregulation of these transcripts (Figure 2B). Additionally, Cidec (Fsp27), a lipid droplet–binding protein that promotes TG accumulation in hepatocytes in vivo (19), was elevated (Figure 2B). Western blot analysis confirmed increased protein levels of Agpat1 and Ppap2a in microsomal extracts, without significant alteration of Cyp2e1, a microsomal marker. Agpat3, Agpat9, Cidec, Dgat1, and Mogat1 were elevated in whole liver extracts (Figure 2C). Collectively, the altered expression of these genes would be expected to increase TG biosynthesis and storage in the liver, as observed in LKO/KO mice.
To assess whether miR-122 can indeed regulate the expression of the aforementioned factors involved in TG metabolism, we measured their mRNA levels in a mouse hepatoma cell line (Hepa) after transient overexpression (∼40-fold increase) or depletion (∼40% decrease) of miR-122 (Supplemental Figure 3A). Ectopic miR-122 reduced Agpat1, Agpat3, Agpat9, Dgat1, Cidec, Ppap2a, and Ppap2c expression, whereas depletion of miR-122 upregulated these transcripts (Supplemental Figure 3B). Mogat1 was not detectable in these cells (data not shown). Furthermore, reporter plasmids were constructed with a Renilla luciferase open reading frame followed by the 3′ UTRs of Agpat1 or Cidec (harboring 3 and 1 miR-122 binding sites, respectively, as predicted by TargetScan; ref. 20). miR-122 strongly repressed luciferase expression from the Agpat1 reporter and, to a lesser extent, from the Cidec reporter (Figure 2D). Mutations in the putative miR-122 binding sites abrogated reporter repression, consistent with the direct targeting of these transcripts by this miRNA. Finally, knockdown of Agpat1 in hepatocytes isolated from LKO/KO mice reduced TG synthesis, suggesting that Agpat1 plays a key role in TG accumulation in LKO/KO livers (Supplemental Figure 3, C and D).
In addition to genes that regulate lipid metabolism, IPA highlighted the abnormal expression of many genes involved in development, cellular proliferation and death, and cancer (Supplemental Table 2). Many of these dysregulated genes are known to exhibit altered expression in HCC and, in some cases, functionally contribute to hepatocarcinogenesis. Such genes include components of insulin-like growth factor 2 (Igf2), Ras, and β-catenin (Ctnnb1) signaling pathways (Supplemental Figure 4A), as well as other genes known to play a role in HCC such as Epcam (21), c-Myc (22), Mapre1 (23), and Rhoa (24). Real-time RT-PCR validated the upregulation of several of these transcripts in LKO livers (Figure 2E and Supplemental Table 4), and Western blotting documented the increased expression of the majority of these proteins in KO livers (Figure 2F). In addition, miR-122 overexpression or inhibition in Hepa cells resulted in concordant changes in expression of selected key genes, including H19, Igf2, Ctnnb1, Epcam, Mapre1, Mapkapk2, and c-Myc (Supplemental Figure 4B). Among these, Mapre1 was validated as a target of miR-122 using reporter assays (Figure 2D). In contrast, Igf2 and H19 do not appear to be directly regulated by miR-122, since comparable upregulation of both unspliced heterogeneous nuclear RNA (hnRNA) and fully spliced mRNA was observed for these transcripts (Supplemental Figure 5A). Although Igf2 and H19 appear to be transcriptionally upregulated in LKO/KO livers, bisulfite sequencing did not reveal significant changes in the methylation status of the differentially methylated region (DMR) located between these two genes (Supplemental Figure 5B). The observed upregulation of these genes in early adult life suggested that these mice might be predisposed to liver cancer as they age.
Recruitment of monocytes and neutrophils to livers of KO mice leads to inflammation and production of pro-tumorigenic cytokines.
Since many genes involved in cell proliferation and survival were significantly upregulated in LKO/KO livers, we aged these animals to determine whether they are tumor prone or exhibit any other adult-onset pathology. Importantly, aged LKO mice did not exhibit significant repopulation of the liver with hepatocytes that escape Cre-mediated deletion, as has been observed in mice with liver-specific deletion of Dicer (ref. 25 and Supplemental Figure 6). At 6 months of age, LKO/KO mice developed steatohepatitis (Figure 3, A–D), with visible foci of altered hepatocytes (inset, Figure 3A). Trichrome staining revealed bridging fibrosis. Like younger LKO mice, 6-month-old KO mice exhibited increased hepatic TG levels without a significant change in hepatic cholesterol after overnight fasting (Figure 3E). A significant increase in serum ALP and γ-glutamyl transpeptidase (GGT) levels, consistent with hepatobiliary disease, was observed (Supplemental Table 5).
Figure 3. Mir122-LKO and -KO mice develop hepatitis and fibrosis with age.

(A and C) Portal inflammation, steatosis, and fibrosis in 6-month-old LKO (A) and KO (C) mice. The inset in A depicts foci of altered hepatocytes as observed in some livers. Eight to 10 mice of each genotype were analyzed after overnight fasting. Scale bars for A: top row, 200 μm; second and third rows and inset, 25 μm; fourth row, 200 μm. Scale bars for C: top row, 200 μm; middle row, 25 μm; bottom row, 100 μm. (B and D) Inflammation, steatosis, and fibrosis scores generated through blinded evaluation of histopathology. (E) Hepatic TG and cholesterol levels in 6-month-old mice. The results shown in B–D are mean ± SD. Statistical significance was calculated using a 2-tailed t test.
Inflammation is a major factor contributing to malignant transformation in HCC and other tumor types (26). In particular, the inflammatory cytokines IL-6 and TNF-α have been shown to promote HCC development (27). We therefore characterized the immune cells that infiltrate hepatic parenchyma in Mir122-KO mice and the cytokines they produce. Consistent with the histologic appearance of LKO/KO livers (Figure 3), direct quantification of infiltrating inflammatory cells revealed a 2-fold increase in the livers of KO mice (Figure 4A). Flow cytometry documented that the hepatic population of CD11bhiGr1+ cells, previously classified as monocytes and neutrophils (28), was greater than 3-fold higher in 10-month-old KO mice than in controls (Figure 4, B and C). Intracellular flow cytometry demonstrated that CD11bhiGr1+ cells from KO livers produced a high level of IL-6 (Figure 4D) and TNF-α (Supplemental Figure 7A).
Figure 4. Infiltration of IL-6–producing CD11bhiGr1+ cells in livers of Mir122-KO mice.

(A) Immune cells from the liver of 10-week-old male KO and control mice were quantified by trypan blue exclusion. Statistical significance was calculated using a 2-tailed t test. (B and C) The percentage of CD11bhiGr1+ cells is significantly increased in 10-month-old non-tumor-bearing LKO/KO mice. Flow cytometric data for 1 representative pair of mice (B) and summary data for 3 mice (C) are shown. (D) Intracellular flow cytometric analysis indicates that CD11bhiGr1+ cells but not lymphocytes from the liver express IL-6. (E) Real-time RT-PCR analysis of Ccl2 expression in LKO (10-week-old) and KO (5-week-old) livers compared with age-matched controls. (F and G) Ccl2 expression (F) is reduced in LKO/KO hepatocytes (isolated from 2 LKO mice and 1 KO mouse) upon overexpression of miR-122 (G). NC-S, scrambled negative control. (H and I) Ccl2 (H) or Mir122 (I) expression in Hepa cells transfected with miR-122 mimic versus control (NC-S) or anti–miR-122 (miR-122-AS) versus control (NC-AS). (J) Induction of spliced Ccl2 mRNA and unspliced Ccl2 hnRNA in KO livers (paired t test). (K) Predicted miR-122 binding site in the 3′ UTR of Ccl2 and corresponding mutant site. (L) Luciferase reporter assays as described in Figure 2D. Results are mean ± SD.
In settings of chronic liver injury, the myeloid chemoattractant Ccl2 is induced in hepatocytes and other liver-resident cells (29) and is an important driver of hepatic inflammation (30). In LKO/KO mice, the fraction of intrahepatic CD11bhiGr1+ cells expressing Ccr2, the Ccl2 receptor, was much greater than the fraction of CD11bhiGr1+ cells expressing Ccr2 in peripheral blood (Supplemental Figure 7B). We therefore hypothesized that activation of the Ccl2/Ccr2 axis in LKO/KO mice leads to recruitment of CD11bhiGr1+ cells, causing hepatic inflammation and injury. Indeed, microarray analysis of mRNA from 5-week-old KO livers showed a greater than 3-fold (P = 0.0003) increase in expression of Ccl2 (data not shown), which was confirmed by real-time RT-PCR in LKO/KO livers (Figure 4E) and cultured hepatocytes (Supplemental Figure 7C). Ectopic miR-122 expression downregulated Ccl2 in LKO/KO hepatocytes (Figure 4, F and G) and in Hepa cells (Figure 4, H and I), whereas depletion of miR-122 increased Ccl2 levels (Figure 4, H and I). Moreover, in KO livers, Ccl2 mRNA levels increased more than unspliced hnRNA levels (Figure 4J), consistent with both transcriptional and post-transcriptional mechanisms contributing to Ccl2 upregulation. The RNA22 algorithm (31) identified a potential miR-122 binding site in the 3′ UTR of Ccl2 (Figure 4K). Upon inhibition of miR-122 in Hepa cells, a Ccl2 3′ UTR reporter construct produced a moderate but significant increase in luciferase activity, whereas a reporter with a mutation in the miR-122 binding site was not affected by miR-122 inhibition (Figure 4L). These data support a model whereby Ccl2 is induced in miR-122–deficient livers both directly through the targeting of this transcript by miR-122 and indirectly as a response to the underlying hepatocyte injury present in these animals. The resulting recruitment of CD11bhiGr1+ cells leads to the production of proinflammatory and tumor-promoting cytokines including IL-6 and TNF-α, initiating a well-described pathogenic sequence that would be expected to predispose to HCC (27).
LKO and KO mice develop HCC with age.
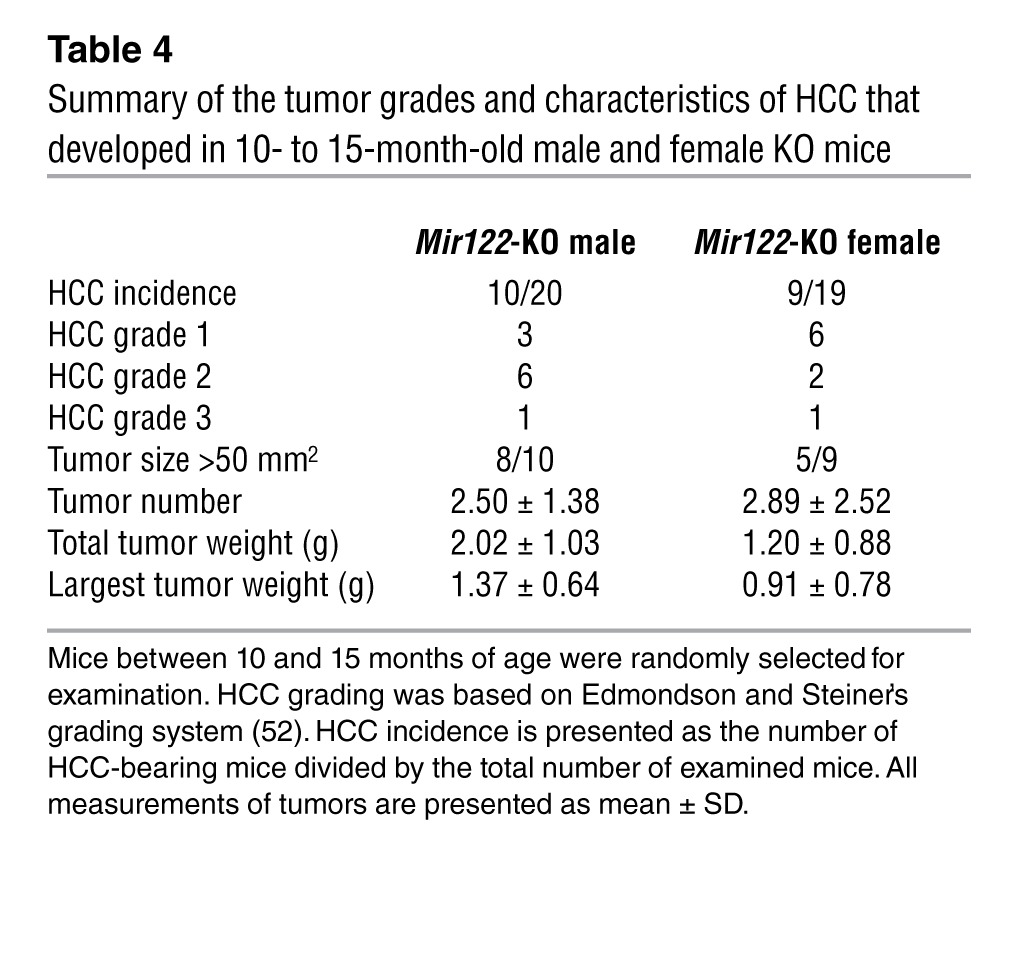
Consistent with the upregulation of oncogenic pathways (Figure 2) and the infiltration of liver with inflammatory cells that produce proinflammatory cytokines (Figure 4), both LKO and KO mice developed moderately to poorly differentiated Afp-positive HCCs with age (Figure 5A). Thirteen of 26 male LKO mice developed spontaneous liver tumors (1–12 macroscopic tumors observed per animal), whereas 2 of 20 female LKO mice developed HCC in 12–17 months (Table 2). In contrast, both male (10 of 20) and female (9 of 19) KO mice developed HCCs with approximately equal penetrance after 11–15 months (Table 3). However, the average tumor weight and grade were significantly higher in male compared with female KO mice (Table 4). Two of 6 heterozygous LKO and 1 of 20 heterozygous KO mice also developed HCC at 20 and 5 months of age, respectively (data not shown). Among the tumor-bearing mice, 3 were also found to have lung metastases (Figure 5A). Tumorigenesis in these animals was associated with liver damage, as demonstrated by a dramatic increase in serum ALT, ALP, bile acid, and GGT levels (Figure 5B). Serum IL-6 was significantly increased in tumor-bearing male but not female LKO/KO mice (Figure 5C), which correlated with greater tumor incidence in male LKO mice and higher tumor burden and tumor grade in the male KO mice.
Figure 5. Mir122-LKO/KO mice develop spontaneous HCC with age.

(A) Representative photographs of liver and lung tumors that developed in LKO/KO mice. Left lower panel shows a representative section stained with H&E and Afp (inset). Inset in lower right panel shows an H&E stain identifying the lung tumor as metastatic HCC. Scale bars: lower left panel, 25 μm; left inset, 35 μm; right inset, 25 μm. (B) Analysis of serum markers of liver function in control and tumor-bearing LKO/KO mice represented as mean ± SEM. P values were calculated using the Welch’s test after log transformation ALT, alanine aminotransferase. (C) Serum IL-6 levels in control and tumor-bearing LKO/KO mice represented as mean ± SEM. P values were calculated using 2-tailed t test. (D) Heat map and dendrogram showing that the expression levels of genes that are dysregulated in tumors from LKO/KO mice are sufficient to classify human HCCs into high– and low–miR-122–expressing subsets. Significance of this classification was assessed by using a bootstrap method.
Table 2.
Summary of the incidence and characteristics of the tumors that developed in 12- to 17-month-old LKO mice
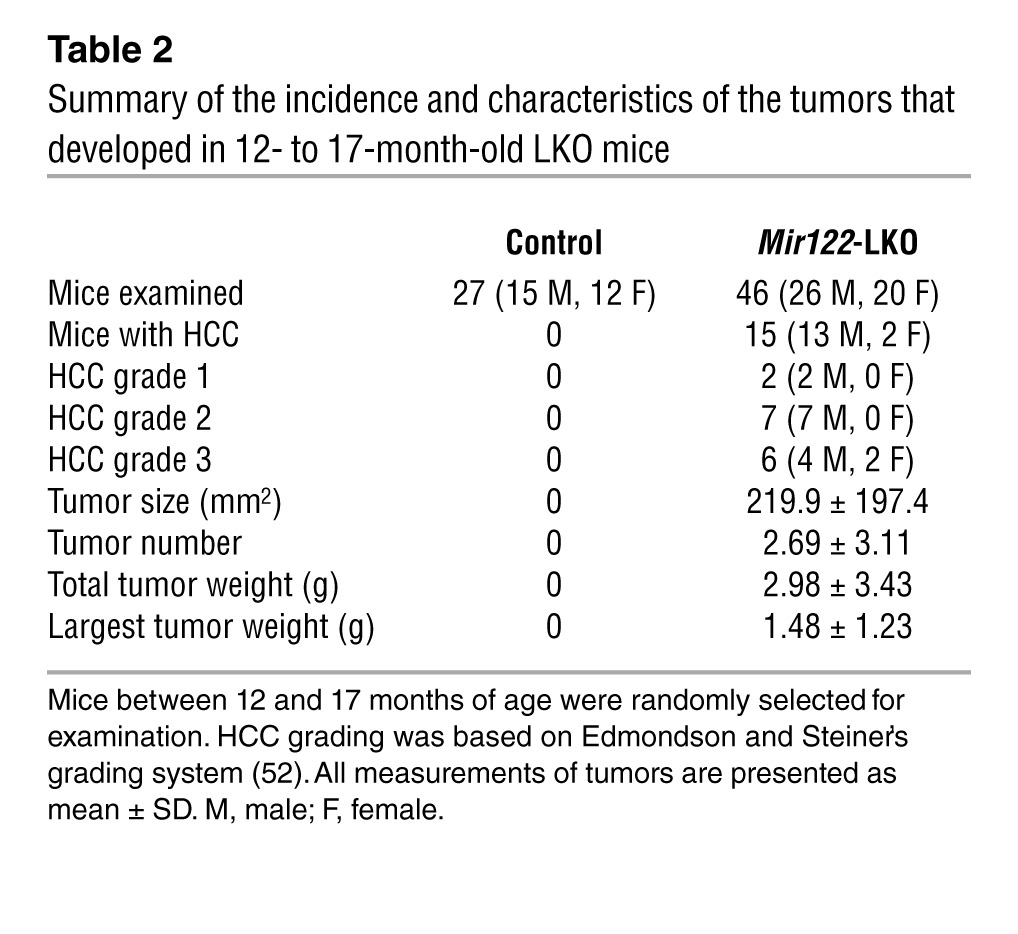
Table 3.
Summary of the incidence and characteristics of the tumors that developed in 10- to 15-month-old KO mice

Table 4.
Summary of the tumor grades and characteristics of HCC that developed in 10- to 15-month-old male and female KO mice
We next analyzed gene expression in the tumors that arose in Mir122-LKO/KO mice. As we observed in normal liver, motifs complementary to the miR-122 seed sequence were significantly enriched in the 3′ UTRs of upregulated transcripts (Supplemental Figure 8). Additionally, we compared gene expression in LKO/KO tumors with a previously reported analysis of gene expression in human HCCs with high and low miR-122 expression (32). A moderated t test was used to obtain a list of 29 upregulated genes and 51 downregulated genes in LKO/KO tumors, which was sufficient to stratify the human tumor samples into high– and low–miR-122–expressing clusters (P = 0.046) (Figure 5D). Thus, loss of miR-122 results in similar effects on the expression of these genes in human and mouse tumors.
AAV-mediated delivery of miR-122 suppresses tumorigenesis in tet-o-MYC;LAP-tTA mice.
In order to determine whether miR-122 performs a tumor suppressor function that is independent of its role in preventing liver injury and inflammation, we directly tested the anti-tumorigenic activity of miR-122 in a non-inflammatory mouse model of HCC. Bi-transgenic mice harboring a tetracycline-repressible (tet-repressible) MYC transgene (tet-o-MYC) and a transgene that produces the tet transactivator protein (tTA) driven by the liver activator promoter (LAP) develop tumors resembling HCC with complete penetrance without preceding liver damage or inflammation (33). Notably, miR-122 levels were dramatically reduced in tumors from tet-o-MYC;LAP-tTA mice compared with normal liver from tet-o-MYC littermates (which lack the LAP-tTA transgene and therefore exogenous MYC expression) (Figure 6A). miR-122 levels were not reduced in normal liver tissue in tet-o-MYC;LAP-tTA mice, where Myc expression is only minimally increased (34).
Figure 6. AAV-mediated delivery of miR-122 impedes liver tumor growth in tet-o-MYC;LAP-tTA mice.

(A) Northern blot showing miR-122 expression in normal liver (N) or tumor (T) from mice of the indicated genotypes. The normalized miR-122 level is presented below each lane. (B) Time line of miR-122 delivery experiment. Dox, doxycycline; vg, vector genomes. (C) Gross tumor burden of livers from control-treated and miR-122–treated animals, as determined by quantification of tumor area using the ImageJ software package ( http://rsbweb.nih.gov/ij/). Each box represents the range of tumor burden observed. The ends of the boxes represent the 25th and 75th percentiles, the bars indicate the 10th and 90th percentiles, and horizontal lines within the boxes represent the median. (D) Representative images of livers from control-treated or miR-122–treated animals.
In order to assess whether miR-122 downregulation is necessary for tumor progression in tet-o-MYC;LAP-tTA mice, we utilized a previously described self-complementary adeno-associated virus (scAAV) vector system (35) to deliver this miRNA to livers of these animals (scAAV.miR122.eGFP; Supplemental Figure 9A). Transfection studies established that the vector produces robust miR-122 expression (Supplemental Figure 9B). For in vivo delivery, vectors were packaged with the AAV8 serotype, and 1 × 1012 vector genomes were administered per animal, a dose that we have shown previously to transduce greater than 90% of hepatocytes (35). At this dose, scAAV8.miR122.eGFP produces a level of miR-122 that is very similar to endogenous levels, as revealed by administration of the virus to Mir122-KO mice (Supplemental Figure 9C). As negative control, a vector lacking a miRNA (scAAV.eGFP) or a vector expressing a synthetic miRNA targeting firefly luciferase expressed from a miR-30 backbone as previously described (36) (scAAV.shLuc.eGFP) was utilized. For experiments in tet-o-MYC;LAP-tTA mice, MYC expression was induced at 4 weeks of age, and virus was administered at 11 weeks of age, a time point at which multiple macroscopic tumors are typically present in each animal (35). At 14 weeks of age, animals were sacrificed, and tumor burden was assessed (Figure 6, B–D). As expected, mice that received the control scAAV8.eGFP or scAAV8.shLuc.eGFP vectors were characterized by extensive tumor progression (scAAV8.eGFP median tumor burden, 40.0%; scAAV8.shLuc.eGFP median tumor burden, 53.5%). In contrast, tumor burden was strongly reduced in mice that received scAAV.miR122.eGFP (median tumor burden, 7.7%). These results support a tumor suppressor function for miR-122 that is independent of its role in preventing hepatic injury and inflammation.
To investigate the cellular mechanisms of miR-122–mediated tumor suppression, we examined tumors 5 days after virus administration. Tumor cell proliferation was reduced, as indicated by Ki-67 staining in scAAV8.miR122.eGFP-treated animals compared with scAAV8.eGFP-treated animals, whereas no difference in tumor cell apoptosis was observed (Supplemental Figure 9, D–F). Since large tumors in this model are relatively resistant to AAV-mediated transduction, it is likely that the phenotypic effects of AAV-delivered miR-122 are even more pronounced in microscopic tumor foci, which are not assayable at this time point, yet ultimately contribute to terminal tumor burden.
Discussion
In this study, we investigated the biologic function of miR-122 through the generation and characterization of mice with liver-specific (LKO) and germline (KO) deletion of this miRNA locus. Analysis of these mice revealed that loss of miR-122 does not lead to overt defects in liver development. This finding suggests that a previously described positive feedback loop involving hepatocyte nuclear factor 6 (HNF6) and miR-122 that has been proposed to regulate hepatocyte differentiation (37) is not required for grossly normal liver development. Furthermore, HNF6 expression was unaltered at the RNA or protein level in adult LKO and KO mice (data not shown). While this suggests that endogenous miR-122 does not regulate HNF6 expression in adults, it remains possible that HNF6 is controlled by this miRNA during embryogenesis and HNF6–miR-122 reciprocal regulation influences the kinetics of hepatocyte differentiation in a manner that is no longer evident at birth.
Transient inhibition of miR-122 using antisense oligonucleotides is well tolerated in adult mice and results in reduced serum cholesterol (2). While genetic deletion of miR-122 also lowers serum cholesterol, our study revealed that prolonged loss of function of this miRNA leads to accumulation of hepatic TGs in young mice. This abnormality is associated with the upregulation of several gene products that catalyze TG biosynthesis and storage, including the newly identified direct miR-122 targets Agpat1 and Cidec. As Mir122-LKO/KO mice aged, hepatic inflammation ensued, preceding the progressive onset of fibrosis and, eventually, tumors resembling HCC. Although a tumor suppressor role for miR-122 has previously been proposed based on in vitro studies and expression analyses of human HCC samples, these findings provide the first in vivo evidence to our knowledge that loss of this miRNA is sufficient to initiate highly penetrant HCC development. It is well established that liver damage and inflammation potently promote the development of HCC, and, in humans, this tumor type nearly always arises in the setting of underlying liver injury (26). Thus, chronic steatohepatitis in Mir122-LKO/KO mice is an important component of the pathology that leads to HCC in these animals (38). In addition, upregulation of the chemokine Ccl2, both directly through loss of targeting by miR-122 and indirectly as a consequence of liver injury, resulted in the intrahepatic recruitment of CD11bhiGr1+ inflammatory cells that locally produce pro-tumorigenic cytokines including IL-6 and TNF-α.
The importance of underlying inflammation in HCC development in these mice is further highlighted by the greater tumor incidence and higher tumor burden and grade in male LKO and KO mice, respectively. In humans, HCC exhibits a similar sex bias, with 2- to 4-fold greater incidence in males (7). This bias is believed to be partially attributable to the greater susceptibility of males to injury-induced hepatic inflammation and hepatocyte proliferation mediated by IL-6, whose production is suppressed by estrogens in females (39). Indeed, circulating IL-6 is increased in tumor-bearing male, but not female, LKO/KO mice, likely contributing to the increased severity of hepatocarcinogenesis in male mice in these animals. Nevertheless, it will be important for future studies to examine the contribution of additional factors that are known to play a role in sexual dimorphism of HCC, such as Foxa1/a2 (40), in the phenotype of miR-122–deficient animals.
The ability of miR-122 to suppress tumorigenesis when delivered to a non-inflammatory MYC-driven HCC model establishes that this miRNA performs a tumor suppressor function that is independent of its role in reducing inflammation and maintaining hepatocyte integrity. This activity is likely mediated by the ability of miR-122 to directly and indirectly control a broad program of gene expression, which includes key factors that influence HCC pathogenesis such as Igf2, Ctnnb1, Ccnd1, c-Myc, Epcam, Mapre1, Iggap1, and Rhoa. miR-122 also appears to be essential for maintenance of the mature hepatocyte gene expression program as manifested by the reactivation of fetal genes including Afp, H19, and Igf2 in LKO/KO mice. In the setting of tonically increased signaling through canonical oncogenic pathways, the expansion of immature hepatocytes in an inflammatory microenvironment likely results in a state of highly increased susceptibility to cellular transformation and tumorigenesis. It is also noteworthy that the cancer phenotype is more severe in germline Mir122-KO mice compared with those with liver-specific deletion of the miRNA. While this may simply reflect the earlier and more uniform Mir122 deletion in KO mice, another intriguing possibility is that a non-hepatocyte function of miR-122 might contribute to the more severe phenotype. Indeed, low-level expression of miR-122 in fibroblasts has been demonstrated (41).
Beyond contributing to our understanding of the physiologic functions of miR-122, the findings reported herein have important therapeutic implications. First, our results demonstrate that delivery of miR-122 using AAV is sufficient to suppress tumor progression in a highly aggressive HCC model. We have previously reported similar results using miR-26a, another miRNA that is frequently downregulated in human HCC (35). Both of these miRNAs, therefore, represent promising candidates for miRNA replacement therapy in patients with this tumor type. Additionally, since miR-122 is downregulated in NASH (9), the efficacy of miR-122 delivery for reducing progression to cirrhosis and HCC in relevant animal models of this disease is worthy of investigation. However, given the essential role of miR-122 in HCV replication (4) the treatment of HCC or other liver diseases arising in the context of HCV infection would not be an appropriate setting for miR-122 delivery.
Finally, miR-122 inhibition therapy using an LNA-modified antisense oligonucleotide, SPC3639, is currently in phase II clinical trials for the treatment of HCV infection (5). Treatment of HCV-infected chimpanzees with SPC3639 reduced viremia and hepatitis without causing any adverse effects during the 12-week period of the study (5). However, our examination of Mir122-LKO and -KO mice has demonstrated that the chronic loss of miR-122 causes steatohepatitis and altered liver function that ultimately leads to liver cancer. Multiple explanations may account for the apparent discrepancy between the phenotypes observed after transient inhibition versus genetic deletion of miR-122. First, the more severe phenotype in LKO and KO mice might arise due to a developmental defect resulting from the absence of miR-122 throughout gestation, or it may result from the effects of complete deletion of the miRNA as opposed to the partial loss of function achieved with chemical inhibitors. Alternatively, the development of liver damage and the resulting sequelae might simply require a longer period of miR-122 depletion beyond that which has been examined using injected inhibitors. Distinguishing between these possibilities will be a priority for future research. Nevertheless, the insights gained through the study of mice lacking miR-122 should aid the design of safe therapeutic strategies based on miR-122 inhibition.
Methods
Generation of liver-specific (LKO) and germline (KO) Mir122 knockout mice.
Mir122 conditional knockout (Mir122loxP/loxP or floxed) mice were generated as depicted in Supplemental Figure 1A. Mir122loxP/loxP littermates served as controls in all studies. Animals were housed in a Helicobacter-free facility and were handled and euthanized following institutional guidelines.
Serological, histological, and immunohistochemical analyses.
Serum was isolated from mice by cardiac puncture after CO2 asphyxiation and cervical dislocation following overnight fasting. Biochemical analysis of enzymes and lipids in the sera was performed at the Ohio State University Comparative Pathology and Mouse Phenotyping Shared Resource using VetAce (Alfa Wassermann system).
For histology, tissues were fixed in 4% paraformadehyde and frozen in O.C.T. or embedded in paraffin. H&E, Oil Red O, PAS, and Masson’s trichrome staining of liver sections were performed as described previously (25, 42). For immunohistochemical analysis, the slides were dewaxed and subjected to antigen retrieval at 95°C for 30 minutes, followed by incubation with the antibodies and color development by the DAB method. CK-19 (43) and A6 (44) antibodies were provided by Joshua Friedman (University of Pennsylvania School of Medicine, Children’s Hospital of Philadelphia Research Institute, Philadelphia, Pennsylvania, USA) and Valentina Factor (Center for Cancer Research, National Cancer Institute, NIH, Bethesda, Maryland, USA), respectively.
H&E-stained sections (×100 magnification) were analyzed for inflammation and steatosis according to the following scoring system: (a) for inflammation: 0, no inflammation; 1, mild lymphocytic infiltration in the portal triad; 2, severe lymphocytic infiltration in portal triad; 3, extended infiltration of lymphocytes throughout liver; (b) for steatosis: 0, no steatosis; 1, microsteatosis; 2, microsteatosis and mild macrosteatosis; 3, severe macrosteatosis. All scoring was performed by two pathologists blinded to the experimental protocol.
TEM.
For TEM, animals were perfused through the portal vein with 2.5% glutaraldehyde. The liver was postfixed in 2% OsO4, embedded in resin, and sectioned. After locating the periportal area in thick (1 μm) toluidine blue (EMS), thin (<90 nm) sections were cut on an ultramicrotome (Leica UC6) and were post-stained with uranyl acetate. Electron micrographs were taken using an FEI Philips Tecnai T-12.
Liver lipid analysis.
Total hepatic lipid was purified and analyzed as described previously (45).
Measurement of hepatic TG synthesis.
The in vivo TG synthesis rate was determined by measuring 3H1-glycerol incorporation into hepatic TGs following a published protocol (46, 47).
Hepatic TG secretion.
After overnight fasting, mice were injected via the tail vein with Triton WR-1339 (Sigma-Aldrich) (48), a lipoprotein lipase inhibitor. Blood was collected from the tail vein, and the serum TG levels were measured at 0, 1, and 3 hours after injection.
Microarray analysis of liver RNA.
Total RNA from the livers of male mice was isolated using TRIzol (Invitrogen) and purified using RNeasy Mini columns (QIAGEN), and the integrity and quantity of the RNA were assessed using an Agilent Bioanalyzer and Nanodrop RNA 6000, respectively. Total RNA was labeled using the Affymetrix Whole Transcript Sense Target Labeling kit and hybridized to the Affymetrix Mouse Exon 1.0 ST array following the manufacturer’s protocol at the Microarray Shared Resource Facility, Ohio State University Comprehensive Cancer Center. The microarray data were deposited in the GEO database (GSE20610). Microarray data analysis methodology is described in Supplemental Methods.
Real-time RT-PCR analysis.
Real-time RT-PCR analysis of mRNAs was performed using SYBR Green chemistry. Relative expression was calculated using the ΔΔCT method (49). The primer sequences are provided in Supplemental Methods.
Western blot analysis.
Mouse liver microsomes were purified according to a published protocol (50). Whole tissue or microsomal extracts were prepared as described previously (49) and subjected to Western blot analysis with specific antibodies using the supplier’s protocol. The signal was detected using an ECL Western blotting reagent (Pierce). Details regarding the antibodies used are provided in Supplemental Materials.
Microarray analysis of the tumor RNA.
Agilent 4X44 platform was used to assess gene expression in liver tumors from 4 LKO and 4 KO mice and in normal liver from age-matched control mice according to the manufacturer’s protocol. Further details are provided in Supplemental Methods. The microarray data have been deposited in the GEO database (GSE31731).
Flow cytometric analysis.
Flow cytometric analysis of liver immune cells was performed as described previously (51). The following antibodies reactive with murine cells were obtained from BD: Gr-1 (RB6-8c5), CD11b (M1/70), CD3 (145-2C11), CD19 (1D3), NK1.1 (PK136), IL-6 (MP5-20F3), and TNF-α (MP6-XT22). CCR2 mAb (clone 475301) was purchased from R&D Systems.
Plasmid construction and AAV packaging.
The scAAV.miR122.eGFP vector was constructed by amplifying Mir122 and approximately 200 bp of flanking sequence from human genomic DNA (primers provided in Supplemental Methods) and ligating this fragment into the FseI site of the previously described scAAV.EF1α.eGFP backbone (35). An shRNA targeting firefly luciferase in a miR-30 backbone (sequence provided in Supplemental Methods) was synthesized by GenScript and cloned into the FseI site of scAAV.EF1α.eGFP to produce scAAV.shLuc.eGFP. Packaging with the AAV8 serotype was performed as described previously (35).
Statistics.
The significances of all results were determined using Student’s unpaired, 2-tailed t test except the analysis of serum marker of liver functions in tumor-bearing mice (Figure 5B), for which Welch’s test after log transformation of the data was used. All results are presented as mean (horizontal lines or rectangular bars) and SD, except the measurements of serum marker of liver functions (Figure 5B) and IL-6 (Figure 5C) in tumor-bearing mice. P values less than 0.05 were considered significant.
Study approval.
All animal studies were reviewed and approved by the Ohio State University Institutional Laboratory Animal Care and Use Committee.
Supplementary Material
Acknowledgments
We thank the Ohio State University Comprehensive Cancer Center Microarray, Microscopy, Nucleic Acids, Comparative Pathology and Mouse Phenotyping, and Nationwide Children’s Hospital Microarray facilities for technical assistance and the University of Michigan Transgenic Animal Model Core for generating miR-122 chimeric mice. The Center for Gene Therapy and Vector Manufacturing Facility at Nationwide Children’s Hospital supplied viral vector for gene transfer. We thank Wei Huang, Xiaocheng Dong, and Tsonwin Hai for valuable discussions; Corie Klepper, Julia Shreve, Jian Liu, and Nuzhat Khan for technical assistance; and Jennifer Chao and Kim M. Shontz for mouse genotyping. This work was supported, in part, by NIH grants CA122694 (to K. Ghoshal), DK088076 (to K. Ghoshal), CA086978 (to K. Ghoshal and S.T. Jacob), Pelotonia Idea Grant (to J. Yu and K. Ghoshal), CA120185 (to J.T. Mendell), CA134292 (to J.T. Mendell), and the Cancer Prevention and Research Institute of Texas (to J.T. Mendell). Bo Wang is supported by a Pelotonia graduate fellowship.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(8):2871–2883. doi:10.1172/JCI63539.
Shoumei Bai’s present address is: Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
See the related Commentary beginning on page 2773.
References
- 1.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12(9):735–739. doi: 10.1016/S0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 2.Krutzfeldt J, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 3.Esau C, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3(2):87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309(5740):1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 5.Lanford RE, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327(5962):198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27(9):1485–1491. doi: 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 8.Kutay H, et al. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem. 2006;99(3):671–678. doi: 10.1002/jcb.20982. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Cheung O, et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology. 2008;48(6):1810–1820. doi: 10.1002/hep.22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filipowicz W, Grosshans H. The liver-specific microRNA miR-122: biology and therapeutic potential. Prog Drug Res. 2011;67:221–238. [PubMed] [Google Scholar]
- 11.Kojima K, et al. MicroRNA122 is a key regulator of alpha-fetoprotein expression and influences the aggressiveness of hepatocellular carcinoma. Nat Commun. 2011;2:338. doi: 10.1038/ncomms1345. [DOI] [PubMed] [Google Scholar]
- 12.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26(2):149–150. doi: 10.1002/(SICI)1526-968X(200002)26:2<149::AID-GENE16>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 13.Chang J, et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1(2):106–113. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- 14.Bai S, et al. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem. 2009;284(46):32015–32027. doi: 10.1074/jbc.M109.016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatfield D, et al. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev. 2009;23(11):1313–1326. doi: 10.1101/gad.1781009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elmen J, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452(7189):896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 17.van Dongen S, Abreu-Goodger C, Enright AJ. Detecting microRNA binding and siRNA off-target effects from expression data. Nat Methods. 2008;5(12):1023–1025. doi: 10.1038/nmeth.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43(2):134–176. doi: 10.1016/S0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 19.Matsusue K, et al. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab. 2008;7(4):302–311. doi: 10.1016/j.cmet.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita T, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136(3):1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thorgeirsson SS, Lee JS, Grisham JW. Molecular prognostication of liver cancer: end of the beginning. J Hepatol. 2006;44(4):798–805. doi: 10.1016/j.jhep.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 23.Orimo T, et al. Proteomic profiling reveals the prognostic value of adenomatous polyposis coli-end-binding protein 1 in hepatocellular carcinoma. Hepatology. 2008;48(6):1851–1863. doi: 10.1002/hep.22552. [DOI] [PubMed] [Google Scholar]
- 24.Man JH, et al. Gankyrin plays an essential role in Ras-induced tumorigenesis through regulation of the RhoA/ROCK pathway in mammalian cells. J Clin Invest. 2010;120(8):2829–2841. doi: 10.1172/JCI42542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sekine S, et al. Disruption of Dicer1 induces dysregulated fetal gene expression and promotes hepatocarcinogenesis. Gastroenterology. 2009;136(7):2304–2315. doi: 10.1053/j.gastro.2009.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuraishy A, Karin M, Grivennikov SI. Tumor promotion via injury- and death-induced inflammation. Immunity. 2011;35(4):467–477. doi: 10.1016/j.immuni.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011;70(suppl 1):i104–i108. doi: 10.1136/ard.2010.140145. [DOI] [PubMed] [Google Scholar]
- 28.Lagasse E, Weissman IL. Flow cytometric identification of murine neutrophils and monocytes. J Immunol Methods. 1996;197(1–2):139–150. doi: 10.1016/0022-1759(96)00138-X. [DOI] [PubMed] [Google Scholar]
- 29.Karlmark KR, Wasmuth HE, Trautwein C, Tacke F. Chemokine-directed immune cell infiltration in acute and chronic liver disease. Expert Rev Gastroenterol Hepatol. 2008;2(2):233–242. doi: 10.1586/17474124.2.2.233. [DOI] [PubMed] [Google Scholar]
- 30.Baeck C, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61(3):416–426. doi: 10.1136/gutjnl-2011-300304. [DOI] [PubMed] [Google Scholar]
- 31.Miranda KC, et al. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126(6):1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 32.Lee JS, Thorgeirsson SS. Comparative and integrative functional genomics of HCC. Oncogene. 2006;25(27):3801–3809. doi: 10.1038/sj.onc.1209561. [DOI] [PubMed] [Google Scholar]
- 33.Beer S, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol. 2004;2(11):e332. doi: 10.1371/journal.pbio.0020332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shachaf CM, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431(7012):1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 35.Kota J, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137(6):1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silva JM, et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet. 2005;37(11):1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- 37.Laudadio I, et al. A feedback loop between the liver-enriched transcription factor network and miR-122 controls hepatocyte differentiation. Gastroenterology. 2012;142(1):119–129. doi: 10.1053/j.gastro.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Anderson N, Borlak J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol Rev. 2008;60(3):311–357. doi: 10.1124/pr.108.00001. [DOI] [PubMed] [Google Scholar]
- 39.Naugler WE, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 40.Li Z, Tuteja G, Schug J, Kaestner KH. Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell. 2012;148(1–2):72–83. doi: 10.1016/j.cell.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burns DM, D’Ambrogio A, Nottrott S, Richter JD. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature. 2011;473(7345):105–108. doi: 10.1038/nature09908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang B, Hsu SH, Frankel W, Ghoshal K, Jacob ST. Hepatology. Stat3-mediated activation of miR-23a suppresses gluconeogenesis in hepatocellular carcinoma by downregulating G6PC and PGC-1alpha [published online ahead of print February 9, 2012]. doi: 10.1002/hep.25632 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hand NJ, Master ZR, Le Lay J, Friedman JR. Hepatic function is preserved in the absence of mature microRNAs. Hepatology. 2009;49(2):618–626. doi: 10.1002/hep.22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishikawa T, et al. Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology. 2012;55(4):1215–1226. doi: 10.1002/hep.24796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang W, Bansode R, Mehta M, Mehta KD. Loss of protein kinase Cbeta function protects mice against diet-induced obesity and development of hepatic steatosis and insulin resistance. Hepatology. 2009;49(5):1525–1536. doi: 10.1002/hep.22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams GM, Norton SJ. Lipid metabolism. I. Effects of pressure and gas composition on acetate-C14 incorporation into liver lipids. Aerosp Med. 1971;42(2):146–148. [PubMed] [Google Scholar]
- 47.Chang BH, et al. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol. 2006;26(3):1063–1076. doi: 10.1128/MCB.26.3.1063-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uchiyama S, Shimizu T, Shirasawa T. CuZn-SOD deficiency causes ApoB degradation and induces hepatic lipid accumulation by impaired lipoprotein secretion in mice. J Biol Chem. 2006;281(42):31713–31719. doi: 10.1074/jbc.M603422200. [DOI] [PubMed] [Google Scholar]
- 49.Wang B, et al. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009;50(4):1152–1161. doi: 10.1002/hep.23100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hill JR. In vitro drug metabolism using liver microsomes. Curr Protoc Pharmacol. 2004;Chapter 7:Unit7.8. doi: 10.1002/0471141755.ph0708s23. [DOI] [PubMed] [Google Scholar]
- 51.Yu J, et al. NKp46 identifies an NKT cell subset susceptible to leukemic transformation in mouse and human. J Clin Invest. 2011;121(4):1456–1470. doi: 10.1172/JCI43242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edmondson HA, Steiner PE. Primary carcinoma of the liver: a study of 100 cases among 48,900 necropsies. Cancer. 1954;7(3):462–503. doi: 10.1002/1097-0142(195405)7:3<462::AID-CNCR2820070308>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.