

Abstract
Genome-wide association studies (GWAS) have identified a genetic variant at a locus on chromosome 1p13 that is associated with reduced risk of myocardial infarction, reduced plasma levels of LDL cholesterol (LDL-C), and markedly increased expression of the gene sortilin-1 (SORT1) in liver. Sortilin is a lysosomal sorting protein that binds ligands both in the Golgi apparatus and at the plasma membrane and traffics them to the lysosome. We previously reported that increased hepatic sortilin expression in mice reduced plasma LDL-C levels. Here we show that increased hepatic sortilin not only reduced hepatic apolipoprotein B (APOB) secretion, but also increased LDL catabolism, and that both effects were dependent on intact lysosomal targeting. Loss-of-function studies demonstrated that sortilin serves as a bona fide receptor for LDL in vivo in mice. Our data are consistent with a model in which increased hepatic sortilin binds intracellular APOB-containing particles in the Golgi apparatus as well as extracellular LDL at the plasma membrane and traffics them to the lysosome for degradation. We thus provide functional evidence that genetically increased hepatic sortilin expression both reduces hepatic APOB secretion and increases LDL catabolism, providing dual mechanisms for the very strong association between increased hepatic sortilin expression and reduced plasma LDL-C levels in humans.
Introduction
Genome-wide association studies (GWAS) have been used as an unbiased tool to identify novel genes associated with lipid traits in a population (1–4). Among the loci most strongly associated with LDL cholesterol (LDL-C) is a novel locus on chromosome 1p13 (P = 10–170) harboring several genes including SORT1. The minor allele frequency of the most strongly associated SNP is approximately 25%, and homozygosity for the minor allele is associated with a more than 10-fold increase in SORT1 expression specifically in liver, an average reduction of 16 mg/dl in plasma LDL-C, and an approximately 40% reduced risk for myocardial infarction (MI) (1). We recently reported that increasing SORT1 expression in mouse liver reduced plasma cholesterol in a variety of hyperlipidemic mouse models, consistent with the directionality predicted by the human genetics (5); however, the mechanisms by which increased hepatic SORT1 expression reduces LDL-C remain uncertain.
The protein product of the SORT1 gene, sortilin, is a multi-ligand sorting receptor originally identified by receptor-associated protein (RAP) affinity chromatography (6). Sortilin is 1 of 5 members of the mammalian VPS10 domain family and harbors an N-terminal propeptide with a furin cleavage site, an extracellular VPS10 domain for ligand binding, a single transmembrane domain, and a C-terminal cytoplasmic tail with strong homology to the cation-independent mannose-6-phosphate receptor (CI-M6PR) (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI63563DS1). The cytoplasmic tail contains 2 endolysosomal sorting motifs — a dileucine-sorting motif and a tyrosine-based sorting motif. Sortilin localizes primarily to the Golgi apparatus and has been shown to bind a variety of ligands and traffic them from the Golgi apparatus to the lysosome (6–11). Sortilin also localizes to the plasma membrane in clathrin-coated pits where it can function as an internalization receptor for progranulin (12), APOA-V (13), and LPL (ref. 14 and Supplemental Figure 1B).
Increased hepatic sortilin expression could reduce plasma LDL-C levels either by reducing the rate of LDL production, by increasing the rate of LDL catabolism, or both. The rate of LDL catabolism is an important determinant of plasma LDL-C levels. LDL is catabolized by the liver via the LDL receptor (LDLR), which binds APOB and removes LDL from circulation and traffics it to the endosomal system for degradation (15). There are LDLR-independent mechanisms of LDL clearance from plasma, but these pathways have not been fully elucidated (16). Here, we tested the hypothesis that sortilin can serve as a cell-surface receptor that binds LDL and targets it for lysosomal degradation.
LDL is generated as a product of lipolysis of VLDL with the major structural protein apolipoprotein B (APOB) remaining with the particle after conversion of VLDL to LDL. Therefore, the rate of hepatic VLDL/APOB production is also an important determinant of plasma LDL-C and APOB levels (17, 18). VLDL/APOB production is regulated mainly via posttranslational control (19). During VLDL formation in hepatocytes, APOB is lipidated initially in the ER and secondarily in the Golgi apparatus (20). When there is insufficient lipidation of nascent APOB in the ER, APOB undergoes ER-associated degradation (ERAD), which is mediated by the proteasome (21). Golgi-associated degradation of APOB is a more recently described pathway that is less well understood, but may target oxidized or misfolded APOB to the lysosome for degradation prior to secretion (22); it has been hypothesized to mediate the APOB-lowering effects of insulin and fish oil. However, the sorting receptor that traffics APOB from the Golgi apparatus to the lysosome remains to be clarified. We previously reported that increased hepatic sortilin expression in mice reduced the hepatic VLDL-triglyceride (VLDL-TG) secretion rate (5), but provided no mechanistic basis for this observation. Here, we tested the hypothesis that sortilin can act as a sorting receptor to bind intracellular APOB-containing lipoproteins and target them for lysosomal degradation. We further demonstrate that, consistent with a previously published report (23), an independently generated Sort1–/– mouse has a paradoxical reduction in APOB/VLDL secretion on a chow diet on both a wild-type and APOBEC1–/–;APOB Tg background.
Our results suggest that genetic upregulation of hepatic sortilin expression results in both reduced hepatic APOB production and increased LDL catabolism, both dependent on intact lysosomal targeting. We suggest that these dual mechanisms account for the very strong association between genetically increased hepatic sortilin expression and reduced plasma LDL-C and APOB levels in humans. In contrast, complete sortilin deficiency also leads to a reduction in VLDL secretion, possibly reflecting a parabolic relationship between hepatic sortilin expression and APOB secretion.
Results
Sortilin demonstrates high-affinity binding to APOB in a pH-dependent manner.
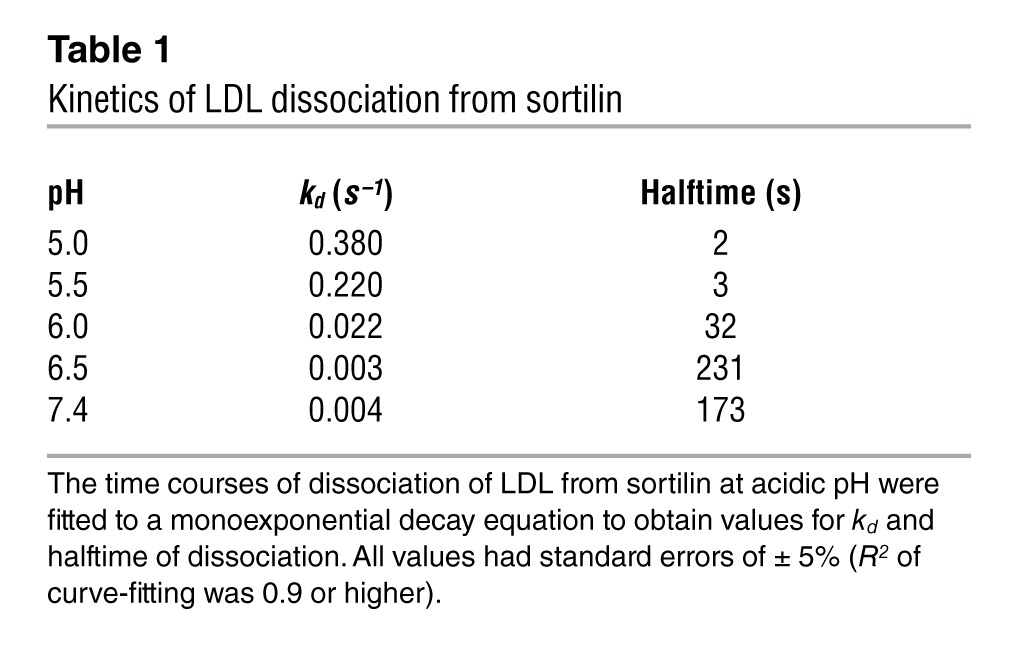
As sortilin serves as a lysosome trafficking receptor through direct binding to multiple ligands with the ability to recycle, we hypothesized that sortilin binds directly to LDL particles at neutral pH but releases them at acidic pH. Surface plasmon resonance (SPR) demonstrated a rapid association and slow dissociation phase between recombinant sortilin and LDL particles at neutral pH (Figure 1A). The kinetic behavior is characteristic of high-affinity binding, and quantitative analysis of the sensorgrams using a 2-state binding model yielded a Kd of approximately 2 nM for the LDL-sortilin interaction at pH 7.4. In contrast, human HDL and APOE 1,2-dimyristoyl-sn-glycero-e-phosphocholine (DMPC) discs did not bind to sortilin under the same conditions (Figure 1A). To determine whether sortilin might dissociate from LDL under acidic pH conditions, we measured the affinity of sortilin for LDL across a wide range of pHs using SPR. While at neutral pH, LDL was tightly bound to sortilin, showing minimal dissociation during a 4-minute dissociation phase (Figure 1A); at an acidic pH, the LDL rapidly dissociated from sortilin (Figure 1B). The change in rate constant (kd) describing this dissociation over a pH range of 5.0 to 7.4 is presented in Table 1. These data are consistent with binding of LDL to sortilin at the cell surface and Golgi apparatus and dissociation of LDL from sortilin in the endosomal compartment.
Figure 1. Sortilin demonstrates high-affinity binding to APOB.

(A) Preparations of LDL at the indicated concentrations in HBS-EP buffer (pH 7.4) were passed across the sensor chip for 240 seconds to monitor the association of LDL, HDL, and APOE DMPC discs and sortilin, and then the chip was exposed to buffer alone for an additional 240 seconds to monitor the dissociation phase. (B) 100 μg/μl of LDL was bound to immobilized human sortilin in HBS-EP buffer at pH 7.4. At the indicated time point, the buffer was exchanged with HBS-EP at lower pH values.
Table 1.
Kinetics of LDL dissociation from sortilin
Increased hepatic sortilin expression reduces APOB secretion in a lysosome-dependent manner.
We previously reported that increased hepatic sortilin expression reduced VLDL-TG secretion in mice (5). Since hepatic APOB production is a much stronger determinant of plasma LDL-C levels than VLDL-TG production (24), we tested whether increased sortilin expression reduced APOB production. Wild-type mice were injected with AAV vectors encoding the Sort1 cDNA or control AAV, and 3 weeks after AAV injection, in vivo VLDL production studies were performed. Consistent with the VLDL-TG studies, sortilin expression reduced APOB secretion by 30% (P = 0.02; Figure 2A and Supplemental Figure 1A). We repeated this study in Ldlr–/– mice and found that sortilin expression reduced APOB secretion by 50% (P = 0.01; Figure 2B and Supplemental Figure 2).
Figure 2. SORT1 expression reduces APOB secretion in a lysosome-dependent manner.

(A and B) Control and Sort1-expressing mice were fasted for 4 hours and then injected intraperitoneally with the detergent Pluronic to inhibit lipolysis, followed by radiolabeling. Mice were sacrificed 1 hour after radiolabeling, blood was harvested by retroorbital bleed, and VLDL was isolated by ultracentrifugation. Sort1 expression resulted in a 30% reduction in VLDL/APOB secretion on a wild-type background and (B) a 50% reduction in VLDL/APOB secretion on an Ldlr–/– background. (C) APOB secretion from primary mouse hepatocytes from APOBEC1–/–;APOB Tg;Ldlr+/– mice following radiolabeling for 3 hours, with or without exposure to E64d. Labeled APOB was immunoprecipitated and measured from medium and normalized to total labeled secreted protein measurements. Sort1 expression resulted in a 30% reduction in APOB secretion in the absence of E64d; the effect was reversed in the presence of E64d. (D) Control and Sort1-expressing mice were fasted for 4 hours and then injected intraperitoneally with the detergent Pluronic to inhibit lipolysis. Plasma was drawn serially 1, 2, and 4 hours after Pluronic injection to permit assessment of the VLDL-TG secretion rate. Wild-type sortilin reduced the VLDL-TG secretion rate by 30%; neither Sort.Stop nor Sort.LAYA reduced VLDL-TG secretion. (E and F) GFP-expressing (control) and sortilin-expressing HuH7 cells were metabolically labeled for 3 hours, and labeled APOB was immunoprecipitated and measured from medium and standardized to total protein measurements. Wild-type sortilin reduced secreted APOB by 40%, while Sort.Stop and Sort.LAYA did not affect secreted APOB. Albumin secretion was not affected by induction of Sort1.
Sortilin is a Golgi-to-lysosome transmembrane trafficking receptor and is known to sort a variety of ligands from the Golgi apparatus to the lysosome for degradation, including adiponectin (25), TGF-β (9), and pro–brain-derived neurotrophic factor (pro-BDNF) (8). To determine whether increased sortilin reduces APOB secretion in a lysosome-dependent manner, hypercholesterolemic mice were injected with sortilin AAV or control vector, and 10 days later, primary hepatocytes were harvested and labeled with 35S-amino acids in the presence or absence of the endolysosome inhibitor E64d. We found that increased sortilin expression reduced hepatocyte APOB secretion ex vivo by 30% (P = 0.009) and that this effect was completely reversed by E64d treatment (P = 0.0006; Figure 2C).
To further investigate the dependence of sortilin on lysosomal sorting to reduce VLDL secretion, we used site-directed mutagenesis to generate 2 sortilin mutants that cannot traffic to the lysosome because they lack both the dileucine and tyrosine lysosomal sorting motifs (Supplemental Figure 3, A and B), either by deletion of the cytoplasmic tail through introduction of a premature termination codon prior to the transmembrane domain (Sort.Stop) or by mutagenesis of key residues in the dileucine and tyrosine sorting motifs to alanine (Sort.LAYA). Others have studied similar mutations in the cytoplasmic tail of sortilin and found that they disrupt sortilin’s ability to traffic to the lysosome (6). We found that when expressed in hepatoma cells, wild-type sortilin but not Sort.Stop or Sort.LAYA colocalized with the LAMP1 lysosomal marker (Supplemental Figure 4, A–D). Sort.Stop was predominantly either secreted or in an intracellular, nonlysosomal location, while Sort.LAYA localized disproportionately to the plasma membrane (Supplemental Figure 4, A–D), consistent with previous observations (6).
AAV vectors were constructed expressing these lysosomal-sorting–defective sortilin mutants in order to test their effects on VLDL-TG secretion in vivo. Ldlr–/– mice were injected with vectors encoding wild-type sortilin, Sort.Stop, Sort.LAYA, or a control AAV. The 3 sortilin cDNAs were expressed at similar levels in mouse liver (data not shown). Three weeks after AAV injection, we performed VLDL secretion studies. Interestingly, while wild-type sortilin reduced the secretion rate by 30% (P = 0.002) as previously observed, neither of the sortilin trafficking mutants affected VLDL secretion (Figure 2D), consistent with sortilin requiring lysosomal trafficking to reduce VLDL secretion in vivo.
To further test this hypothesis and to determine the effects of the sortilin trafficking mutants on APOB secretion, we stably expressed wild-type sortilin, Sort.Stop, Sort.LAYA, or GFP (control) in the human hepatic HuH7 cell line under control of a doxycycline-inducible (dox-inducible) promoter (Supplemental Figure 4, A–D). In metabolic labeling experiments, consistent with our in vivo findings, dox-mediated induction of wild-type Sort1 led to a 40% reduction in APOB secretion, whereas induction of GFP, Sort.Stop, and Sort.LAYA had no effect on APOB secretion, consistent with sortilin requiring lysosomal targeting to reduce VLDL secretion (Figure 2, E and F).
Sortilin expression promotes LDL uptake and lysosomal catabolism.
Sortilin has been shown to act as a cell-surface receptor for multiple ligands including progranulin (12), LPL (14), and APOA-V (13), and promotes their lysosomal degradation. Given these observations, our finding that sortilin has high-affinity binding to LDL, the strong effect of increased hepatic sortilin expression on reducing plasma LDL levels in mice and humans, and a previous report suggesting that sortilin overexpression may be associated with increased cellular LDL uptake in vitro (26), we hypothesized that sortilin might bind LDL at the plasma membrane and deliver it to the lysosome for degradation. To test this hypothesis, we used our dox-inducible system expressing wild-type sortilin, Sort.Stop, and Sort.LAYA in human HuH7 hepatoma cells and assayed for LDL cell association and degradation. Both wild-type sortilin and Sort.LAYA increased LDL cell association 5-fold (P = 1.39 × 10–8 and P = 3.51 × 10–6; Figure 3A), but only wild-type sortilin increased LDL degradation (P = 0.004; Figure 3B). Sort.Stop had no significant effects on LDL cell association or degradation (Figure 3, A and B). To further confirm these findings, we performed LDL uptake studies using fluorescently labeled LDL followed by imaging studies. While wild-type sortilin increased the amount of intracellular LDL, Sort.Stop had no effect on the amount of intracellular LDL; in contrast, Sort.LAYA expression resulted in accumulation of LDL at the plasma membrane (Figure 3, C–F), consistent with an internalization defect. Wild-type sortilin expression was found to increase LDL cell association and degradation in 293T, CHO, and ldlD cells as well (Supplemental Figure 5, A–F).
Figure 3. Sortilin expression promotes LDL uptake and lysosomal catabolism in vitro.

(A and B) HuH7 cells expressing wild-type sortilin, Sort.Stop, Sort.LAYA, or GFP (control) under control of a dox-inducible promoter were assayed for (A) LDL cell association and (B) LDL degradation after incubation with 125I-LDL for 3 hours. Both wild-type sortilin and Sort.LAYA increased 125I-LDL cell association 5-fold, while only wild-type sortilin increased 125I-LDL degradation. Sort.Stop did not increase 125I-LDL cell association or degradation (C–G) Cells were incubated with fluorescently labeled LDL for 6 hours and subjected to confocal microscopy to visualize intracellular LDL. Compared with cells containing only endogenous sortilin (C), wild-type sortilin (D) increases intracellular LDL, while Sort.Stop (E) does not increase intracellular LDL. Sort.LAYA (F) resulted in profound accumulation of LDL at the cell surface without increasing intracellular LDL (G) Sortilin and LDL show strong colocalization with the lysosomal marker LAMP1. Scale bars: 15 μm.
To determine whether increased sortilin expression promotes LDL catabolism in vivo, wild-type mice were injected with 2 different doses of sortilin AAV or null AAV (control), and 3 weeks later, in vivo LDL clearance studies were performed. Compared with mice injected with null AAV, mice injected with low and high doses of sortilin AAV had an increase of 70% (P = 8 × 10–5) and 200% (P = 4 × 10–5) in fractional catabolic rate (FCR), respectively (Figure 4, A and B).
Figure 4. Sortilin expression promotes LDL uptake and lysosomal catabolism in vivo.

(A–D) Control mice and mice receiving AAVs encoding wild-type or mutant Sort1 were injected with 2 × 106 counts of 125I-LDL via tail vein. Blood was drawn serially 2 minutes and 1, 3, 6, 9, and 24 hours after injection. All counts were normalized to the 2-minute count. Clearance curves (A) and FCRs (B) demonstrated a dose-dependent increase in LDL clearance in wild-type mice expressing Sort1. Clearance curves (C) and FCRs (D) demonstrated that wild-type sortilin and Sort.LAYA increased LDL clearance, while Sort.Stop did not increase plasma LDL clearance (E and F) Clearance and FCRs demonstrating a 40% reduction in FCR in Sort1–/– mice compared with wild-type mice and a 50% reduction in FCR in Ldlr–/–;Sort1–/– mice compared with Ldlr–/– mice.
To study the effects of sortilin on LDL turnover in a hyperlipidemic mouse model and to study the interdependence of sortilin and the LDLR, we repeated the turnover study in Ldlr–/– mice. Sortilin expression in these mice increased the FCR 3-fold (P = 2 × 10–5), indicating that increased hepatic sortilin expression promotes LDL catabolism in an LDLR-independent manner (Figure 4, C and D).
To determine whether intact lysosomal targeting is required for the effect of sortilin expression on LDL catabolism in vivo, we expressed the 2 sortilin trafficking mutants Sort.Stop and Sort.LAYA in Ldlr–/– mice and determined the effect on plasma lipids and LDL clearance compared with wild-type sortilin. Sort.Stop was well expressed, but had no effect on plasma lipid levels or LDL catabolism. In contrast, Sort.LAYA reduced plasma total and LDL-C (Supplemental Figure 6, A–D) and increased LDL clearance from plasma (Figure 4, C and D). However, in distinct contrast to wild-type sortilin, the liver-associated 125I-LDL counts were 6-fold higher in mice expressing Sort.LAYA (P = 0.001, Supplemental Figure 6E), indicating lack of effective degradation of the radiolabeled LDL. This is consistent with the in vitro LDL imaging studies suggesting that Sort.LAYA acts as a cell-surface reservoir for LDL binding, but fails to target the LDL to the lysosome for degradation.
While the above experiments confirm that increased hepatic sortilin expression promotes LDL catabolism in vivo, they leave open the question of whether endogenous sortilin plays a role in LDL catabolism. To test this hypothesis, we obtained Sort1–/– mice that have previously been reported to be completely lacking in sortilin protein (7). Compared with wild-type mice, Sort1–/– mice displayed a 40% reduction in FCR (P = 0.002; Figure 4, E and F). To determine whether sortilin deficiency also reduces LDL clearance in the absence of the LDLR, we assessed LDL clearance in Sort1–/–;Ldlr–/– mice. Ldlr–/–;Sort1–/– mice had a 50% reduction in FCR (P = 0.02) compared with Ldlr–/– mice, consistent with sortilin and the LDLR acting separately to facilitate LDL uptake (Figure 4, E and F).
Remarkably, despite the substantially reduced catabolism of LDL in Sort1-deficient mice, we noted that they were not hypercholesterolemic, either on the Ldlr–/– background or on an APOBEC1–/–;APOB TG background (Figure 5, A–E). We therefore tested the effect of sortilin deficiency on VLDL/APOB secretion and found that sortilin deficiency in the hypercholesterolemic APOBEC1–/–;APOB TG background is associated with a 60% reduction in APOB secretion (P = 0.04; Figure 5F and Supplemental Figure 1B), offsetting the reduced LDL catabolism and resulting in the unremarkable plasma lipid profile. This is consistent with a previous report of reduced VLDL production in a different sortilin-knockout mouse on a wild-type background (23). Thus, while increased hepatic expression of sortilin results in both increased LDL catabolism and reduced APOB secretion with additive effects in reducing LDL-C levels, complete deficiency of sortilin results in delayed LDL catabolism and reduced APOB secretion with offsetting effects on LDL-C levels.
Figure 5. Sortilin deficiency is associated with reduced or normal plasma cholesterol and reduced APOB/VLDL secretion.

(A–D) Mice (8- to 10-week-old females) were fasted for 4 hours, and blood was drawn by retroorbital bleed; plasma total cholesterol was measured by autoanalyzer and FPLC. (A and B) Sort1 deficiency on an Ldlr–/– background was associated with a 20% reduction in plasma cholesterol. (C–E) Sort1 deficiency on an APOBEC1–/–;APOB transgenic background did not affect plasma cholesterol or APOB levels. (F) Mice (8- to 10-week-old females) were fasted for 4 hours and injected intraperitoneally with the detergent Pluronic to inhibit lipolysis followed by radiolabeling. Mice were sacrificed 1 hour after radiolabeling, plasma was collected, and VLDL was isolated by ultracentrifugation. Sort1 deficiency was associated with a 60% reduction in VLDL/APOB secretion on an APOBEC1–/–;APOB transgenic background.
Discussion
In humans, a common genetic variant leading to a greater than 10-fold increase in sortilin expression specifically in the liver is associated with substantially reduced plasma levels of LDL-C and APOB and with substantial reduction in coronary heart disease. Here, we provide evidence in mice and hepatocytes that increased sortilin expression reduces plasma LDL-C both by reducing the hepatic production of APOB and by increasing the catabolism of LDL (Supplemental Figure 7). We demonstrate that sortilin binds LDL with high affinity at neutral pH and with much less affinity at acidic pH, consistent with release in the endosome. We further demonstrate that both consequences of increased hepatic sortilin expression — reduced APOB production and increased LDL catabolism — require intact endolysosomal trafficking.
The classic pathway of LDL uptake and degradation is via the hepatic LDLR, which binds LDL in circulation, mediates its uptake by clathrin-dependent endocytosis, and delivers it to the lysosome for degradation while recycling back to the plasma membrane (15). Mutations in the LDLR that reduce or eliminate protein expression or impair its ability to deliver LDL to the lysosome for degradation cause familial hypercholesterolemia (FH) (27). Although LDL catabolism is severely impaired in humans and mice deficient in the LDLR, LDLR-deficient livers still clear a large amount of LDL from the plasma through pathways that are not fully explained by existing mechanisms (28). To date, several additional receptors have been reported to facilitate clearance of TG-rich lipoproteins, including heparan-sulfate proteoglycans (HSPGs) (29), LDLR-related protein-1 (LRP1) (30), and syndecan-1 (31). However, additional pathways of LDL clearance are poorly understood. Sortilin is known to localize to clathrin-coated pits on the plasma membrane and mediate the uptake and lysosomal degradation of several ligands. Here, we show that increased hepatic expression of sortilin mediates accelerated clearance of LDL from blood and, conversely, that sortilin deficiency is associated with markedly reduced LDL clearance. These effects were independent of the LDLR itself. A mutant of sortilin containing the ligand-binding domain but lacking the transmembrane domain and cytoplasmic tail did not affect LDL catabolism. Moreover, a different mutant of sortilin containing the ligand binding and transmembrane domains but with mutations in the cytoplasmic tail that preclude lysosomal targeting was capable of binding LDL and mediating its clearance from blood, but incapable of delivering it to the lysosome for degradation. The identification of sortilin as an LDLR-independent pathway for LDL uptake helps explain the strong association between increased hepatic sortilin expression and reduced LDL-C levels. It may also account for the variability of residual LDL clearance in individuals with FH.
The rate of hepatic APOB production is an important determinant of plasma LDL-C and APOB levels in humans (17, 18). Familial combined hyperlipidemia is an inherited condition characterized by hepatic overproduction of APOB and, as a consequence, elevated levels of LDL-C and APOB in plasma (32–34). APOB production is posttranslationally regulated through presecretory degradation (19), either by the classic pathway of proteasome degradation (ERAD) (21) or by a more recently described post-ER lysosomal pathway (22). Presecretory lysosomal degradation of APOB-containing lipoproteins is believed to play a role in the clinical efficacy of fish oils as well as in the physiological effects of insulin on VLDL-TG secretion (22). Sortilin is a known Golgi-to-lysosome transporter for other ligands in other cell types (8, 9, 11, 25). We show here that increased expression of sortilin in the hepatocyte results in reduced hepatic production of APOB. Our results are consistent with and complementary to recent results from Ai et al. in mice, indicating that pharmacological or genetic disruption in liver sortilin expression both result in increased APOB/VLDL secretion, while restoration of sortilin expression in mouse liver using AAVs and mouse models with increased sortilin expression have reduced VLDL/APOB secretion (35). Our data further indicate that the sortilin-mediated reductions in APOB/VLDL secretion are dependent on intact lysosomal targeting. Sortilin mutants defective in their ability to traffic to the lysosome had no effect on hepatic APOB secretion. Thus, sortilin appears to be a key protein responsible for the previously described post-ER shunting of APOB from the Golgi to the lysosome for degradation.
While our studies are focused on explaining the molecular basis of the association between genetically increased hepatic sortilin expression and substantially reduced plasma LDL-C levels in humans, the physiology of sortilin and lipoprotein metabolism is clearly complex, as evidenced by the phenotype of the whole-body sortilin-knockout mouse. It was previously reported that a sortilin-knockout mouse that had some residual truncated sortilin expression (12), upon feeding with a Western-type diet, had reduced VLDL production (23). Here, we confirm and extend this finding. Using a different sortilin-knockout mouse proven to be devoid of any sortilin protein (7), we found that on a chow diet, sortilin deficiency was associated with reduced APOB production (data not shown). Furthermore, when crossed onto the APOBEC1–/–;APOB transgenic background, sortilin deficiency still resulted in reduced APOB production. Thus, both increased hepatic sortilin expression and total-body sortilin deficiency are associated with reduced VLDL/APOB production. The basis for this observation remains unknown. Sortilin is widely expressed, and while the causal SNP in humans and our in vivo sortilin expression manipulations were liver specific, the knockout mouse was deficient of sortilin in all tissues. It is possible that the reduction in APOB secretion in the whole-body knockout mouse could be related to sortilin deficiency in extrahepatic tissues. Indeed, sortilin is strongly expressed in brain, adipose, and skeletal muscle, all tissues have that been shown to influence hepatic VLDL production (36, 37). It is possible that selective sortilin deficiency in liver only would have a different phenotype related to APOB secretion. Alternatively, the deficiency of sortilin from early embryogenesis could have led to compensatory changes that resulted in reduced APOB production. Finally, it is possible that hepatic sortilin expression may have a parabolic relationship with regard to APOB secretion, with low levels of sortilin facilitating APOB secretion and substantially higher levels of sortilin — such as those seen in homozygotes for the minor allele associated with reduced LDL-C — serving to traffic some APOB from the Golgi to the lysosome, thus reducing its secretion. Interestingly, a recent study suggested that neuronal sortilin can serve as either a chaperone or an intracellular degrader of pro-BDNF, depending on its ADAM10 cleavage status (8). This suggests a model by which sortilin could serve as either a chaperone or an intracellular degrader of APOB, depending on its proteolytic processing or intracellular abundance.
In summary, we present evidence that increased hepatic expression of sortilin, as seen in humans carrying a common variant at the SORT1 locus, is associated with both reduced hepatic APOB production and increased LDL catabolism. Furthermore, we show that both effects are dependent on lysosomal targeting, consistent with sortilin’s role as a lysosomal trafficking protein. This dual effect on both production and catabolism may explain the very strong association of the SORT1 locus with reduced plasma levels of LDL-C and APOB as well as with substantial protection from coronary heart disease.
Methods
Antibodies.
Rabbit anti-sortilin (ab16640), rabbit anti-APOB (ab20737), and mouse anti-actin (ab8226) were purchased from Abcam. Mouse anti-sortilin (612101) was purchased from BD Biosciences.
Creation of adenoassociated viruses for gene overexpression studies in mouse liver.
The murine Sort1 cDNA (MR210834; Origene) was subcloned into a specialized AAV8 vector provided by the University of Pennsylvania Vector Core. Site-directed mutagenesis was performed using the Stratagene kit to generate both trafficking mutants. To make the truncated mutant, the following primers were used: GCAACTTCTTGAACCCCACAAAGTAGAATTCCAAGTCAAATTCTG (forward); CAGAATTTGACTTGGAATTCTACTTTGTGGGGTTCAAGAAGTTGC (reverse). This created a C2251T transition mutation, which created a Q751X nonsense mutation. To make the double-sortilin trafficking mutant, 2 sequential PCR reactions were performed. The first reaction converted the dileucine sorting motif to a dialanine (L826A/L827A) by introducing the following base pair changes: CT2476-77GC and CT2479-80GC using the primers CACGACGACTCAGATGAGGACGCCGCGGAATAGTCTAGAAAAGATATCC (forward) and GGATATCTTTTCTAGACTATTCCGCGGCGTCCTCATCTGAGTCGTCGCGTG (reverse). The second reaction converted the tyrosine sorting motif to an alanine (Y793A) by introducing the base pair changes TA2377-2378GC using the primers GAAGGTTCCTGGTGCACCGGGCCTCGGTGCTACAGCAG (forward) and CTGCTGTAGCACCGAGGCCCGGTGCACCAGGAACCTTC (reverse). Viruses were produced with a chimeric packaging construct in which the AAV2 rep gene was fused with the cap gene of AAV serotype 8. Empty AAV8 viral particles were also provided by the University of Pennsylvania Vector Core. Mice were injected intraperitoneally with either 1 × 1011 or 1 × 1012 viral particles of AAV in PBS.
Measurement of mouse plasma lipids and lipoproteins.
Plasma samples were drawn prior to and 2 weeks after AAV injection. Animals were anesthetized by isoflurane inhalation, and blood was collected by retroorbital bleed followed by centrifugation to isolate plasma. Plasma lipids were analyzed individually by analytical chemistry (Cobas Mira Autoanalyzer; Roche Diagnostic Systems) and pooled (150 μl total) by fast protein liquid chromatography (FPLC). Cholesterol plate assays were performed on FPLC fractions using Infinity Cholesterol Reagents.
VLDL secretion studies.
To measure the VLDL-TG secretion rate, mice were fasted for 4 hours, prebled by retroorbital bleeding, and injected intraperitoneally with 400 μl of 1 mg/g Pluronic P407 Detergent resuspended in PBS to inhibit lipolysis. Plasma samples were drawn serially 1, 2, and 4 hours after Pluronic injection. Plasma TG from each time point was measured by plate assay using the Infinity TG Reagent.
APOB secretion studies.
Mice were fasted for 4 hours and then injected intraperitoneally with 1 mg/g of the detergent P407 to inhibit peripheral lipolysis. Fifteen minutes after detergent injection, mice were injected via tail vein with 0.5 mCi of 35S-methionine/cysteine. Blood was drawn by retroorbital bleed at 2 minutes for normalization purposes. One hour after 35S-methionine/cysteine injection, a terminal bleed was performed and plasma was isolated. VLDL was harvested by ultracentrifugation and run on an acrylamide gel. Bands were cut and counted and normalized to the 2-minute plasma count.
LDL turnover studies.
Mice were injected via tail vein with 2 × 106 counts of 125I-LDL. Blood was drawn by retroorbital bleed 2 minutes after LDL injection for normalization purposes and 1, 3, 6, 9, and 24 hours after LDL injection for measurement of the LDL clearance rate.
Generation of stable HuH7 cell lines.
The GC-rich 5′ third of the human SORT1 gene was synthesized (Genewiz), and the remainder of the gene was amplified from cDNA, with mutations introduced by PCR engineering. The assembled wild-type or mutant genes were subcloned into the LV-tetO lentiviral vector (38). The SORT1 lentiviral vectors as well as FUΔGW-rtTA encoding the reverse tetracycline transactivator (Addgene) were used to generate viral particles and cellular transduction, as previously described (39). Huh7 cells were transduced with lentiviral supernatant 24 hours after passaging at about 40% confluency. Six-well dishes were transduced using 1,000 μl lenti-rtTA virus and 1,000 μl lenti-SORT1 or lenti-SORT1 mutants or lenti-GFP virus per well. Cells were incubated at 37°C overnight, viral supernatant was removed, and cells were further cultured in growth medium.
Immunofluorescence.
HuH7 cells were plated on glass coverslips in a 24-well plate and allowed to adhere for 24 hours. Dox (700 ng/ml) was added for 72 hours to induce gene expression. For LDL uptake, cells were serum starved overnight and treated with LDL-DyLight 549 (10011125; Cayman) for 6 hours at 37°C. Cells were then washed extensively to remove unbound LDL before immunohistochemistry. For immunohistochemistry, cells were permeabilized with 0.1% Triton X-100 for 10 minutes and blocked for 1 hour in 3% BSA in PBS. The cells were then incubated with sortilin antibody (which is able to recognize wild-type sortilin as well as Sort.Stop and Sort.LAYA) and APOB antibody (1:1000 dilution) at 4°C overnight, washed with PBS, and incubated for 1 hour at room temperature with different secondary antibodies (1:500 dilution) (Invitrogen). Hoechst (1:5,000) was used for nuclear staining. Cells were then mounted with an antifade mounting medium (Vector Laboratories), and immunofluorescence was done using a confocal microscope (LSM 710; Zeiss).
In vitro LDL studies.
Cells were plated in 12-well plates, transfected where indicated, and incubated overnight in 2 mg/ml BSA in DMEM to upregulate the LDLR. Cells were then incubated for 3 hours at 37°C in DMEM + 5 μg/ml 125I-LDL. To assay for LDL degradation, medium was harvested, unreacted iodine was TCA-precipitated out of the medium, and the remaining 125I (representing the lysosomal LDL degradation product) was counted. To assay for total LDL uptake, cells were treated with 100 U/ml of heparin to release LDL bound to the LDLR and HSPGS; cells were dissolved in 0.2 M NaOH, and cell lysates were counted. Values were standardized by well protein content, which was measured by standard bicinchoninic acid (BCA) assay (Thermo) on the NaOH-dissolved samples.
SPR studies.
Studies of the binding of LDL (APOB) to sortilin were performed with a Biacore 3000 SPR instrument (Biacore) using a CM3 or CM5 sensor chip. The soluble fragment (residues 45–725) of human sortilin was attached to the chip using amine coupling chemistry according to the manufacturer’s instructions. In brief, the chip surface was prepared by exposing the carboxylated dextran matrix to an aqueous solution containing 0.4 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and 0.1 M N-hydroxysuccinimide (10 μl/min for 7 minutes), and then sortilin (50 μg/ml in 10 mM sodium acetate buffer, pH 5.0) was flowed across the chip surface at the same rate for 7 minutes, followed by 1 M ethanolamine-HCl (pH 8.5) at 19 μl/min for 7 minutes to deactivate excess reactive groups and remove any noncovalently bound sortilin. This procedure led to approximately 2,500 response units (RU) of sortilin immobilized (corresponding to a surface coverage of ∼3 ng sortilin/mm2). To monitor LDL association with sortilin, LDL (isolated by sequential ultracentrifugation from human plasma as described before; ref. 40) solutions in HBS-EP buffer (Biacore) (0.01 M Hepes, 0.15 M NaCl, 3 mM EDTA, 0.005% surfactant P20, pH 7.4) in the concentration range of 10 to 300 μg/ml were flowed across the chip at 20 μl/min for 4 minutes at room temperature. The dissociation of LDL was then monitored by washing the surface for 4 minutes with buffer alone over a pH range of 5.0 to 7.4. A chip freshly coated with sortilin was used to obtain each LDL sensorgram. LDL was flowed across an activated but uncoated CM3 chip under the same conditions as control for nonspecific binding, and this was found to be negligible. The changes in RU giving the LDL association and dissociation time courses were analyzed using BIA evaluation software, version 4.1 (Biacore). The response curves were fitted to a 2-state binding model, as described before (41); this analysis yielded the association and dissociation rate constants and, thereby, the Kd. A similar protocol was used to monitor HDL association with sortilin.
Metabolic labeling studies.
For labeling experiments, cells were switched to cystine/methionine-free DMEM with 1% FBS, 1% antibiotics/antimycotics, and 0.4 mM oleic acid for 1 hour, followed by addition of 200 μCi/well of 35S-methionine/cysteine. In some cases, 10 μM E64d was added prior to the labeling. After 3 hours, medium from the cells was harvested, and APOB was immunoprecipitated with the antibody ab20737 (Abcam). The immunoprecipitate was subjected to SDS-PAGE, and the gel was exposed to film at –80°C overnight. Relative secreted APOB levels were determined by quantitation of appropriately sized bands by densitometry and by cutting and counting bands from the gel.
To determine relative total secreted protein levels for normalization, 50 μl of 2 mg/ml BSA and 25 μl of 50% trichloroacetic acid (TCA) were added to 50 μl of harvested medium, followed by incubation on ice for 20 minutes. The samples were centrifuged for 15 minutes, and the pellets were washed with 1 ml of 50% TCA and resuspended by boiling in 1 ml of 0.2 M NaOH. 200 μl of the NaOH suspension was analyzed in a scintillation counter for 35S counts. Values were normalized to total cellular protein in each sample, which was determined using a classic BCA Assay (Thermo Scientific).
Statistics.
Statistical analyses were done using 2-tailed Student’s t test. VLDL secretion rates were determined using a linear regression model and testing for statistical significance was performed using GraphPad Prism software. Error bars reflect SD. P < 0.05 was considered significant.
Study approval.
All animal protocols were reviewed and approved by the University of Pennsylvania IACUC.
Supplementary Material
Acknowledgments
We acknowledge Hui Li, Aisha Wilson, Edwige Eduoard, and Mao-Sen Sun for their help with the animal studies; Deborah Cromley for her help with lipid measurements; and Jeffrey Billheimer, William Lagor, Ilia Fuki, Robert Brown, and Jane Glick for scientific discussions and mentorship. This work was supported by 10PRE3010061 from the American Heart Association (AHA) (to A. Strong), RC2HL101864 and R01HL109489 from the National Heart, Lung and Blood Institute (NHLBI) (to D.J. Rader), and K99 HL098364 from the NHLBI (to K. Musunuru). We would also like to acknowledge the NHLBI Gene Therapy Resource Program for providing support for the vector production as well as the Vector Core Laboratory of the University of Pennsylvania for producing the vectors.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(8):2807–2816. doi:10.1172/JCI63563.
References
- 1.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40(2):189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willer CJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41(1):25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kathiresan S, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41(1):56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466(7307):714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nielsen MS, et al. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. Embo J. 2001;20(9):2180–2190. doi: 10.1093/emboj/20.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng J, Racicott J, Morales CR. The inactivation of the sortilin gene leads to a partial disruption of prosaposin trafficking to the lysosomes. Exp Cell Res. 2009;315(18):3112–3124. doi: 10.1016/j.yexcr.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 8.Evans SF, et al. Neuronal brain-derived neurotrophic factor is synthesized in excess, with levels regulated by sortilin-mediated trafficking and lysosomal degradation. J Biol Chem. 2011;286(34):29556–29567. doi: 10.1074/jbc.M111.219675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwon S, Christian JL. Sortilin associates with transforming growth factor-beta family proteins to enhance lysosome-mediated degradation. J Biol Chem. 2011;286(24):21876–21885. doi: 10.1074/jbc.M111.228262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefrancois S, Zeng J, Hassan AJ, Canuel M, Morales CR. The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. Embo J. 2003;22(24):6430–6437. doi: 10.1093/emboj/cdg629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ni X, Morales CR. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic. 2006;7(7):889–902. doi: 10.1111/j.1600-0854.2006.00429.x. [DOI] [PubMed] [Google Scholar]
- 12.Hu F, et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68(4):654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nilsson SK, Christensen S, Raarup MK, Ryan RO, Nielsen MS, Olivecrona G. Endocytosis of apolipoprotein A-V by members of the low density lipoprotein receptor and the VPS10p domain receptor families. J Biol Chem. 2008;283(38):25920–25927. doi: 10.1074/jbc.M802721200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nielsen MS, Jacobsen C, Olivecrona G, Gliemann J, Petersen CM. Sortilin/neurotensin receptor-3 binds and mediates degradation of lipoprotein lipase. J Biol Chem. 1999;274(13):8832–8836. doi: 10.1074/jbc.274.13.8832. [DOI] [PubMed] [Google Scholar]
- 15.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232(4746):34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 16.Ye ZJ, Go GW, Singh R, Liu W, Keramati AR, Mani A. LRP6 protein regulates low density lipoprotein (LDL) receptor-mediated LDL uptake. J Biol Chem. 2012;287(2):1335–1344. doi: 10.1074/jbc.M111.295287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turley SD. Cholesterol metabolism and therapeutic targets: rationale for targeting multiple metabolic pathways. Clin Cardiol. 2004;27(6 suppl 3):III16–III21. doi: 10.1002/clc.4960271506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab. 2011;22(9):353–363. doi: 10.1016/j.tem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ginsberg HN, Fisher EA. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res. 2009;50(suppl):S162–S166. doi: 10.1194/jlr.R800090-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sundaram M, Yao Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr Metab (Lond). 2010;7:35. doi: 10.1186/1743-7075-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou M, Fisher EA, Ginsberg HN. Regulated Co-translational ubiquitination of apolipoprotein B100. A new paradigm for proteasomal degradation of a secretory protein. J Biol Chem. 1998;273(38):24649–24653. doi: 10.1074/jbc.273.38.24649. [DOI] [PubMed] [Google Scholar]
- 22.Pan M, et al. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-B: a pathway for late-stage quality control. Proc Natl Acad Sci U S A. 2008;105(15):5862–5867. doi: 10.1073/pnas.0707460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kjolby M, et al. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010;12(3):213–223. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Taghibiglou C, et al. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J Biol Chem. 2000;275(12):8416–8425. doi: 10.1074/jbc.275.12.8416. [DOI] [PubMed] [Google Scholar]
- 25.Karki S, Chakrabarti P, Huang G, Wang H, Farmer SR, Kandror KV. The multi-level action of Fatty acids on adiponectin production by fat cells. PLoS One. 2011;6(11):e28146. doi: 10.1371/journal.pone.0028146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linsel-Nitschke P, et al. Genetic variation at chromosome 1p13.3 affects sortilin mRNA expression, cellular LDL-uptake and serum LDL levels which translates to the risk of coronary artery disease. Atherosclerosis. 2010;208(1):183–189. doi: 10.1016/j.atherosclerosis.2009.06.034. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cain WJ, et al. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a]. J Lipid Res. 2005;46(12):2681–2691. doi: 10.1194/jlr.M500249-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Bishop JR, Stanford KI, Esko JD. Heparan sulfate proteoglycans and triglyceride-rich lipoprotein metabolism. Curr Opin Lipidol. 2008;19(3):307–313. doi: 10.1097/MOL.0b013e3282feec2d. [DOI] [PubMed] [Google Scholar]
- 30.Rohlmann A, Gotthardt M, Hammer RE, Herz J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. 1998;101(3):689–695. doi: 10.1172/JCI1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanford KI, et al. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J Clin Invest. 2009;119(11):3236–3245. doi: 10.1172/JCI38251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kissebah AH, Alfarsi S, Adams PW. Integrated regulation of very low density lipoprotein triglyceride and apolipoprotein-B kinetics in man: normolipemic subjects, familial hypertriglyceridemia and familial combined hyperlipidemia. Metabolism. 1981;30(9):856–868. doi: 10.1016/0026-0495(81)90064-0. [DOI] [PubMed] [Google Scholar]
- 33.Janus ED, Nicoll A, Wootton R, Turner PR, Magill PJ, Lewis B. Quantitative studies of very low density lipoprotein: conversion to low density lipoprotein in normal controls and primary hyperlipidaemic states and the role of direct secretion of low density lipoprotein in heterozygous familial hypercholesterolaemia. Eur J Clin Invest. 1980;10(2 pt 1):149–159. doi: 10.1111/j.1365-2362.1980.tb02075.x. [DOI] [PubMed] [Google Scholar]
- 34.Venkatesan S, Cullen P, Pacy P, Halliday D, Scott J. Stable isotopes show a direct relation between VLDL apoB overproduction and serum triglyceride levels and indicate a metabolically and biochemically coherent basis for familial combined hyperlipidemia. Arterioscler Thromb. 1993;13(7):1110–1118. doi: 10.1161/01.ATV.13.7.1110. [DOI] [PubMed] [Google Scholar]
- 35.Ai D, et al. The regulation of hepatic sortilin-1 in obese mice by activation of mTORC1 and ER stress. J Clin Invest. 2012;122(5):1677–1687. doi: 10.1172/JCI61248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grundy SM, Barnett JP. Metabolic and health complications of obesity. Dis Mon. 1990;36(12):641–731. [PubMed] [Google Scholar]
- 37.Costandi J, Melone M, Zhao A, Rashid S. Human resistin stimulates hepatic overproduction of atherogenic ApoB-containing lipoprotein particles by enhancing ApoB stability and impairing intracellular insulin signaling. Circ Res. 2011;108(6):727–742. doi: 10.1161/CIRCRESAHA.110.238949. [DOI] [PubMed] [Google Scholar]
- 38.Stadtfeld M, Brennand K, Hochedlinger K. Reprogramming of pancreatic beta cells into induced pluripotent stem cells. Curr Biol. 2008;18(12):890–894. doi: 10.1016/j.cub.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahfeldt T, et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat Cell Biol. 2012;14(2):209–219. doi: 10.1038/ncb2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lund-Katz S, Phillips MC. Packing of cholesterol molecules in human low-density lipoprotein. Biochemistry. 1986;25(7):1562–1568. doi: 10.1021/bi00355a016. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S. Molecular mechanism of apolipoprotein E binding to lipoprotein particles. Biochemistry. 2009;48(13):3025–3032. doi: 10.1021/bi9000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.