

Abstract
Embryonic stem cells (ESCs) have unlimited capacity for self-renewal and can differentiate into various cell types when induced. They also have an unusual cell cycle control mechanism driven by constitutively active cyclin dependent kinases (Cdks). In mouse ESCs (mESCs). It is proposed that the rapid cell proliferation could be a necessary part of mechanisms that maintain mESC self-renewal and pluripotency, but this hypothesis is not in line with the finding in human ESCs (hESCs) that the length of the cell cycle is similar to differentiated cells. Therefore, whether rapid cell proliferation is essential for the maintenance of mESC state remains unclear. We provide insight into this uncertainty through chemical intervention of mESC cell cycle. We report here that inhibition of Cdks with olomoucine II can dramatically slow down cell proliferation of mESCs with concurrent down-regulation of cyclin A, B and E, and the activation of the Rb pathway. However, mESCs display can recover upon the removal of olomoucine II and are able to resume normal cell proliferation without losing self-renewal and pluripotency, as demonstrated by the expression of ESC markers, colony formation, embryoid body formation, and induced differentiation. We provide a mechanistic explanation for these observations by demonstrating that Oct4 and Nanog, two major transcription factors that play critical roles in the maintenance of ESC properties, are up-regulated via de novo protein synthesis when the cells are exposed to olomoucine II. Together, our data suggest that short-term inhibition of cell proliferation does not compromise the basic properties of mESCs.
Keywords: Embryonic stem cells, cell cycle, self-renewal, pluripotency
Introduction
Embryonic stem cells (ESCs) have attracted enormous attention in recent years with the expectation that ESC-derived cells will be eventually used as a source for cell-based therapy. The most distinctive features of ESCs are their unlimited capacity for self-renewal and the potential to differentiate into various cell types. These unique properties constitute the basis of embryonic development and therapeutic application in medicine. While it is exciting to recognize the potential benefits of this research in medical applications, currently we have limited knowledge of the basic physiology of ESCs.
Derived from the inner cell mass of an early stage embryo, ESCs can reproduce themselves continuously under proper culture conditions without entering senescence like differentiated cells, a property known as self-renewal. They can differentiate into all types of cells derived from the three germ layers, a property known as pluripotency [1-3]. Therefore, understanding the molecular mechanisms that control ESC differentiation, pluripotency, and self-renewal has been the center of ESC research. It is well recognized that the properties of ESCs are mainly maintained by a set of transcription factors, including Oct4, Sox2, and Nanog, which together form a master transcriptional regulatory network that maintains ESC self-renewal and pluripotency [1,4]. The unusual cell cycle control mechanism, which does not follow the conventional patterns of cell cycle regulation, is another fundamental subject that is closely related with ESC’s unique properties [5,6]. It is well established that the cell cycle is mainly controlled by cyclin-dependent kinases (Cdks) whose activity is positively regulated by cyclins and negatively regulated by cyclin-dependent kinase inhibitors (CdkIs), and modulated by several other signaling pathways, including the p53 and Rb pathways [7,8]. In differentiated cells, the activity of Cdks and CdkIs is regulated in a cell cycle-dependent manner. However, in ESCs several major check points that control cell cycle progression have little or no regulatory function. Instead, their cell cycle is driven by the Cdk2/cyclin A/E complexes that are constitutively active throughout the cell cycle [5,9,10]. Therefore, ESCs exhibit an unusual cell cycle structure, characterized by a short G1 phase and a high proportion of cells in S-phase. As ESCs differentiate, their cell cycle control changes to the mechanism that is typically seen in differentiated cells, with a significantly longer G1 phase [6,11,12]. The unique cell cycle structure and mechanism of cell cycle control indicate that the cell cycle machinery plays a key role in establishment or maintenance of the stem cell state. Mouse ESCs (mESCs) divide rapidly with a generation time of 9-11 hours and 50-60 % cells in S phase. Since cell proliferation and differentiation differ in numerous cellular activities, it is proposed that the rapid cell proliferation of mESCs could be a mechanism that prevents them from differentiation, thereby contributing to the stem cell state [1,5]. However, the doubling time of human ESCs (hESCs) and human induced pluripotent stem cells (iPSCs) is much longer (~30 h) [13,14]. Therefore, it is possible that rapid cell proliferation is important for the maintenance of stem cell state in mESCs but not in hESCs, as found in the case of leukemia inhibitory factor (LIF), which is essential for the maintenance of mESCs but not hESCs [15,16]. Alternatively, the rapid cell proliferation could be just a unique property of mECSs that is separable from self-renewal. Clarification of this issue will be important to fully understand the species difference between human and mouse ESCs.
It is interesting to note that several studies have indicated that ESCs have superior defense mechanisms against stresses to protect their self-renewal, pluripotency, and genome integrity [17,18]. In a recent study, we have demonstrated that oxidative stress caused by low level H2O2 disrupts cell cycle progression of mESCs and inhibits their proliferation. However, mESCs have remarkable recovery capacity to resume the normal cell proliferation rate without losing the capabilities of self-renewal and pluripotency, as indicated by the expression levels of pluripotency markers Oct4, Sox2, and Nanog, and by the capacity of mESCs to form embryoid bodies [19]. These observations suggest that short-term oxidative stress-induced slowdown of proliferation does not result in the loss of self-renewal and pluripotency, suggesting that cell proliferation and self-renewal may not be interdependent. However, the complex effects of oxidative stress on multiple cellular processes make it difficult to ascertain whether or not the slowdown of cell proliferation under this condition is a direct effect on the cell cycle regulatory machinery. To gain further insight into this question, in this study we directly inhibited cell proliferation of mESCs and investigated how this intervention affected their pluripotency and self-renewal capacity. We demonstrate that transient inhibition of cell proliferation does not compromise the stem cell state of mESCs.
Materials and Methods
mESC culture, cell treatment and embryoid body (EB) formation
DBA252 ESCs have been characterized as previously described [20,21]. They exhibit typical properties of mESCs and can differentiate to different cell types as described by us [22-24] and by other investigators [20,25]. D3 mESCs were obtained from (ATCC, Cat# CRL-1934). Both cells were maintained in the standard mESC Dulbecco’s modified Eagle medium (DMEM) that contains 1,000 U/ml leukemia inhibitory factor (LIF) as previously described [24]. Both DBA252 and D3 cells were routinely maintained in cell culture dishes coated with 0.1% gelatin without feeder cells at 37 °C in a humidified atmosphere at 5% CO2. Cells at 40-50% confluence were treated with olomoucine II (Olo II, Sigma) at different concentrations for the time courses as indicated. Treated cells were either analyzed or continuingly cultured in fresh medium to allow for recovery as described in individual experiments. EB formation was performed by suspending mESCs, either cultured under normal conditions as the control or recovered from Olo II treatment, in bacterial culture dishes as previously described [24].
Cell proliferation and cell cycle analysis by flow cytometry
Cell proliferation and cell cycle analysis were performed according to the methods previously described [19]. Briefly, mESCs treated with Olo II at different concentrations were fixed with 3% paraformaldehyde for 30 min, washed with PBS and then stained with 1% toluidine blue. The wells were washed with water to remove unbound dyes. The absorbance at 630 nm was measured with a microtiter plate reader. The values, which correlate with the number of viable cells, were used as an indirect measurement of cell proliferation. Cell cycle analysis by flow cytometry was performed according our published method with an Accuri C6 flow cytometer[19]. The profiles of cell cycle progression were generated with the CFlow software.
Real-time quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using Tri-reagent (Sigma). cDNA was prepared by M-MLV reverse transcriptase. The specificity of the PCR was determined by the dissociation curve and confirmed by agarose gel electrophoresis. RT-qPCR was performed using SYBR green jumpstart Taq ready mix on a MX3000PTM Real-time PCR system (Stratagene) as previously reported [24]. Sequences of the primer sets are as follows (F, forward; R, reverse) β-actin, F: 5′-CATGTACGTAGCCATCCAGGC-3′ R: 5′-CTCTTTGATGTCACGCACGAT-3′ Cyclin B, F: 5′-CGAGAACTGCTCTTGGAGACATTG-3′ R: 5′-CCTGACACAGATACTCTTCTGCAG3′ Cyclin A, F: 5′-CAGTCACAGGACAGAGCTGG -3′ R: 5′-GGGCATGTTGTGGCGCTTTG -3′ Cyclin E, F: 5′-CCTCCAAAGTTGCACCAGTTTGC -3′ R: 5′-GACACACTTCTCTATGTCGCACC-3′ Oct4, F: 5′-AGTTGGCGTGGAGACTTTGC-3′ R: 5′-CAGGGCTTTCATGTCCTGG -3′ Sox2, F: 5′-GACAGCTACGCGCACATGA -3′ R: 5′-GGTGCATCGGTTGCATCTG -3′ Nanog, F: 5′-TTGCTTACAAGGGTCTGCTACT -3′ R: 5′-ACTGGTAGAAGAATCAGGGCT -3′ Nkx 2.5, F: 5′-GGTCTCAATGCCTATGGCTAC -3′ R: 5′-GCCAAAGTTCACGAAGTTGCT -3′ GATA4, F: 5′-CCCTACCCAGCCTACATGG -3′ R: 5′-ACATATCGAGATTGGGGTGTCT -3′ SMA, F: 5′-GGACGTACAACTGGTATTGTGC -3′ R: 5′-CGGCAGTAGTCACGAAGGAAT -3′ The mRNA level from qRT-PCR was calculated using the comparative Ct method [26]. β-actin mRNA was used as a calibrator for the calculation of relative mRNA levels of the tested genes.
Immunofluorescence analysis
Immunofluorescence analysis was performed according to our published protocols [24]. For microscopic analysis, cells were fixed with 4% paraformaldehyde and permeabilized with PBS containing 0.25% Triton X-100 for 30 min. After being blocked in 2% bovine serum albumin (BSA), the cells were incubated with antibodies against phosphorylated Rb (Cell Signaling Technology) overnight at 4 °C. The positive cells were detected by fluorescein isothiocyanate (FITC)-secondary antibodies. The nuclei were stained with 10 μM Hoescht 33258. The cells were then examined with a LSM 510 laser-scanning confocal microscope. Image analysis was performed using LSM Image Examiner software (Zeiss).
To determine the protein level by fluorescence labeling, treated cells were released by trypsin and fixed with 80% cold ethanol for 30 min at 4°C, then washed with PBS that contains 2% BSA for 20 min. Cells were incubated with primary antibodies (Oct4, Nanog, and cyclin B, Santa Cruz) at 1:100 dilution in PBS containing 2% BSA for 2 h at room temperature. The cells were then incubated with secondary antibodies conjugated with FITC or rhodamine (1:100) in PBS with 2% BSA for 1 h at room temperature. The cell suspension was examined through an Accuri C6 flow cytometer. The fluorescence intensity, which correlates with the protein level, was determined with a CFlow software.
Cell lysate preparation and Western-blot analysis
mESCs were directly lysed with 1x SDS buffer. After being heated at 95°C for 3 min, the cell lysates were passed through an insulin syringe 5 times and subjected to SDS-PAGE. Western-blot analysis was carried out as previously described [19].
siRNA transfection
siRNAs targeting Cdk2 were obtained from Santa Cruz Biotechnology. mESCs at 40-50% confluence were transfected with DharmaFECT (Thermo Scientifi) according to the manufacturer’s instructions. The cells were analyzed at 48 h and 72 h post transfection for Cdk2 knockdown efficiency and the effect on cell cycle progression.
Statistical analysis
Data are expressed as means ± SD (standard deviation). Statistical analysis was performed using a two-tailed unpaired Student t test. Differences are considered statistically significant when p<0.05.
Results
The effects of Olo II on cell proliferation, cell cycle, and cell morphology
We first designed a set of experiments to test the effects of Olo II on cell proliferation, cell cycle progression, and cell morphology within a broad range of concentrations. The number of viable cells after treatment was used as measurement of cell proliferation after 24 h treatment. As shown in Fig 1A, treatment of DBA252 cells with Olo II reduced the cell number in a dose-dependent manner with IC50 around 5 μM. The inhibitory effect nearly reached the maximal level at 10 μM. The cell cycle analysis of control cells by flow cytometry indicated that the distribution of cell populations at different phases displayed a typical profile of undifferentiated mESCs with about 60 % of the cells in the S phase. Olo II at 5-10 μM reduced the number of cells at the G1 and S phase and caused accumulation of cells at the G2/M phases. At 40 μM, Olo II caused a further pattern change characterized by dramatic cell accumulation at the G2/M phase, and to a lesser extent at the S phase (Fig. 1B), indicating multiple interferences on the cell cycle at this condition. We also tested the effects of Olo II on D3 cells, a well-characterized and commonly used mESC line by other investigators [27]. It is noted that the reduction of the G1 phase cells in D3 cells is less dramatic and the reduction in S phase populations is more obvious than that observed in DBA252 cells at the concentrations of 5 μM and 10 μM, however, the accumulation of G2 cells is similar between the two cell lines. The cell cycle profiles are nearly identical when the cells were treated with 40 μM (Fig. 1B). These results suggest that DBA252 and D3 cells showed some differences in sensitivity to Olo II, but they have an overall similar response to Olo II. At the concentrations above 10 μM, Olo II also caused some cell death (judged by the number of floating cells) which may also contribute to the reduced cell number shown in Fig.1A. Microscopic analysis of Olo II treated DBA252 cells revealed that the major effect is the reduced size of the colonies, correlating with reduced cell numbers. However, individual cells maintained the typical morphology of undifferentiated ESCs (Fig.1C). Since DBA252 and D3 cells did not show fundamental difference in responding to Olo II, further experiments were performed primarily with DBA 252 cells and the key data were confirmed in D3 cells (as specified in figure legends).
Figure 1.
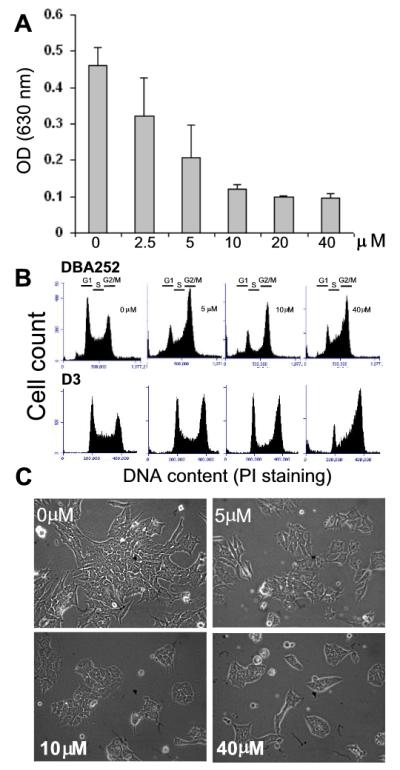
Effects of Olo II on cell proliferation, cell cycle progression, and cell morphology. DBA252 or D3 mESCs were seeded in 6-well plates (6.5 ×104/well). After incubating for 24 h, cells were treated with different concentrations of Olo II for 24 h. A, DBA252 cell proliferation was determined by the absorbance at 630 nm, which correlates with the number of cells. The results are mean ± SD from three independent experiments. Similar results were obtained with D3 cells. B, Cell cycle profile analysis by flow cytometry. C, Microscopic analysis of cell and colony morphology of DBA 252 cells under a phase contrast microscope.
The effect of Olo II on cell cycle progression
Since Cdk2 is critical for G1 to S phase transition and is considered to be a major target of Olo II, we therefore expected that Olo II would cause a reduced S phase cell population and G1 phase cell accumulation; however, the expected results were not apparent in the cells treated for 24 h (Fig. 1A). We performed a time course study that included earlier time points of treatment. As shown in Fig. 2A, the G2 phase cell accumulation started as early as 2 h after treatment and progressively increased through the entire time course, while reduction of both G1 and S phase cells was apparent at the 6 h treatment. By 10 h post treatment, the cycle profile is similar to that of 24 h treatment (Fig.1B), indicating that Olo II may affect progression of different phases of the cell cycle. However, above described experiment were unable to determine how the cell cycle progression from G1 to S phase was affected since both populations of the cells at the G1 and S phase were reduced between 6-10 h (Fig. 2A). To resolve this uncertainty, we synchronized cells with nocodazole, which arrests cells at the G2/M phase (Fig. 2B & C, Noc). The removal of nocodazole allowed the cells to reenter G1 phase in a synchronized manner (Fig. 2B & C, designated as 0 h). The cells resumed the cell cycle progression as indicated by the progressive increase of S and G2 phase cells. In the absence of Olo II, G2/M phase cells reached the maximal level at 6 h, concurrent with reduced G1 cells (Fig. 2B, 6 h). However, this process was significantly delayed by Olo II as indicated by less S and G2 cells at the same time point (Fig. 2C, 6 h) and it took about 13 h for Olo II treated cells to reach the maximal level of G2/M phase cells (Fig. 2C, 13 h vs Fig. 2B, 6 h). In the presence of Olo II, it is noted that the G1 cells decreased to the lowest level at 22 h but began to accumulate again through longer incubation (Fig. 2C, 22-45 h). This result indicates that Olo II can severely delay G2/M transition, but not cause a permanent G2 phase arrest. It is noted that apoptotic cells (sub-G1 cells) were detected in the nocodazole-synchronized cells during subsequent incubation (Fig.2B&C). Similar observation has been reported in mESCs by other investigators [28]. A likely explanation is that a certain population of cells arrested at G2/M phase by nocodazole may have some DNA damage that could trigger apoptosis during subsequent cell cycle progression after nocodazole removal.
Figure 2.
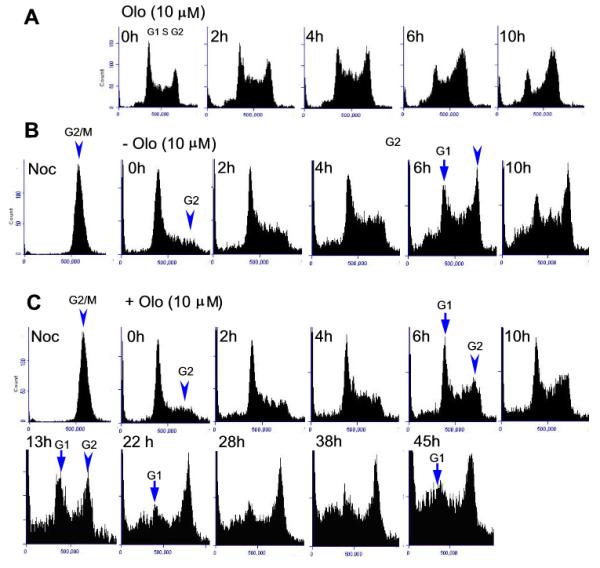
Effects of Olo II on cell cycle progression. A, The effect of Olo II on the cell cycle profiles at different time points. DBA252 cells were treated with 10 μM Olo II for the time points indicated and the cell cycle profile was analyzed by flow cytometry. B & C, DBA252 cells were synchronized by incubation with 50 ng/ml nocodazole for 16 h to arrest the cells at the G2M phases (Noc). Nocodazole was washed out with fresh medium and incubated for 1 h to allow cells to reenter the cell cycle (designated as 0 h). Afterward, the cells were cultured in the absence of (B) or in the presence of 10 μM Olo II (C) for the indicated time courses. The cells were collected at different time points and analyzed for cell cycle progression by flow cytometry.
Together, the results from the above experiments demonstrated that Olo II delayed the cell cycle progression, including G1/S and G2/M phase transition. When Cdk2 was specifically knocked down with siRNA, a cell progression profile with reduced G2 cells was observed (Fig.s1), similar to that caused by Olo II (Fig. 2C, 6 h). This result confirms that Cdk2 inhibition contributed to Olo II-induced delay in G1/S phase transition. However, the profound G2 cell accumulation caused by Olo II shown in Fig. 1B is likely a result that involves the inhibition of other Cdks.
The effects of Olo II on the expression of cell cycle regulators
In order to gain further insight into the molecular mechanisms that lead to Olo II-induced cell cycle inhibition, we examined the expression of several cell cycle regulators that are responsible for the rapid cell proliferation, cyclin A, B and E [11]. The cells were subjected to Olo II treatment under the conditions described in Fig.1, and the mRNA levels of cycle regulators were determined by RT-qPCR. As shown in Fig. 3A, the expression of cyclin A, B and E was significantly reduced by Olo II at concentrations above 10 μM. The down regulation of cyclin B was further confirmed at the protein level by two independent methods: Western blot analysis and flow cytometry of immunostained cells with antibodies against cyclin B. Both methods gave results consistent with the RT-qPCR analysis (Fig. 3B). The CdkIs including p16, p19, and p21 are expressed at very low level as expected and treatment of mESCs with Olo II did not increase their expression (data not shown). These results suggest that direct inhibition of Cdks and subsequent reduction of cyclin A, E and B could be the major mechanisms that account for Olo II-inhibited cell proliferation.
Figure 3.
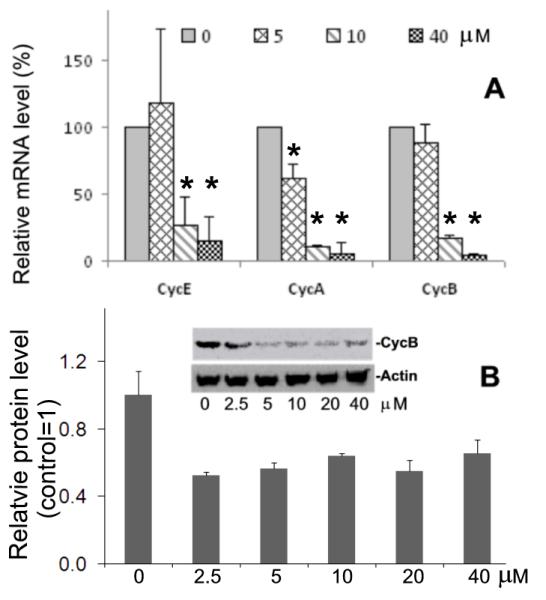
Effects of Olo II on the expression of cycle regulators. DBA 252 cells were treated under the conditions described in Fig.1. A, The mRNA levels of cyclins E, A, and B were determined by RT-qPCR. The mRNA level of tested gene in control cells (0 μM) is designated as 100%. The results are means ± SD of three independent experiments. Statistical analysis was performed with Student t test. The difference was considered statistically significant when p<0.05 (*). B, Graph, the protein levels of cyclin B determined by flow cytometry. The protein levels were determined by fluorescence intensity from a flow cytometer. The value determined from control cells (0 μM, control) is designated as 1. Inset, the protein levels of cyclin B was analyzed by Western blot. β-actin were used as a loading control.
The members of the Rb protein family are negative regulators of cell proliferation. In differentiated cells, hypo-phosphorylated Rb is active and acts as a tumor suppressor by blocking cell cycle progression through inhibition of E2F transcription [7]. Phosphorylation inactivates Rb and allows cell cycle progression to proceed [29]. In mESCs, it is thought that Rb is locked in its inactive form (phosphorylated), which may contribute to the accelerated proliferation of mESCs [6,12,30]. We examined Rb phosphorylation using three antibodies that specifically recognize the phosphorylated Rb (pRb) at S780, S795, and T807/811. Microscopic analysis by immunostaining revealed that all antibodies gave positive results, with the antibody against pRB-S780 showing the strongest signal. The pRB-S780 detected a subpopulation with hyperphosphorylated Rb (pRb). It is noted that all of these cells were at the mitotic phase as identified by their condensed chromosomes with extremely high intensity of antibody staining (Fig. 4A, indicated by arrows). When analyzed by flow cytometry, this population was identified as a shoulder peak with the most intense fluorescence labeling (Fig. 4B, indicated by arrow). Upon Olo II treatment, the fluorescence signal was reduced in a dose-dependent manner and completely disappeared at 10μM Olo II as analyzed by flow cytometry (Fig. 4C) and by microscopy (data not shown). Additional flow cytometry that combines cell cycle progression analysis and Rb phosphorylation analysis was performed using two additional antibodies against pRB-S780 and pRB-T807/811. In both cases, nearly all cells with high levels of fluorescence intensity (phosphorylation level) were at the G2/M phases (Fig. 4D, boxed areas), confirming their mitotic status revealed by microscopic analysis (Fig. 4 A, B, respectively). These results indicate that Olo II reduced Rb phosphorylation level and could activate cell cycle repressor function of Rb, thereby contributing to Olo II-induced inhibition of cell proliferation.
Figure 4.
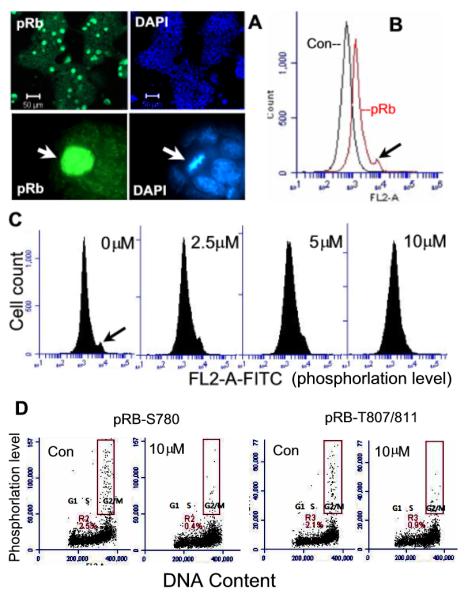
Rb phosphorylation and the effect of Olo II. A, Detection of hyperphosphorylated Rb (pRb-S780) by fluorescence microscopic analysis of DBA 252 cells labeled with anti-pRb antibodies and DAPI. The arrow indicates a single cell with intensive fluorescence staining of pRb, which is at the mitotic phase identified by its condensed mitotic chromosomes (DAPI). B, Flow cytometry analysis of pRb. Control (Con) represents the cells without first antibody incubation. pRb-S780 is detected as a shoulder peak (the cell population with the strongest fluorescence intensity, indicated by arrow). C, Olo II-induced dephosphorylation of pRb. Cells were treated with Olo II at different concentrations for 24 h. Dephosphorylation was indicated by the disappearance of the shoulder peak in the flow cytometry profiles. D, Dual analysis of Rb phosphorylation and cell cycle by flow cytometry. DBA 252 cells treated with Olo II for 24 h or control cells were doubly labeled with anti-Rb-S780 or anti-Rb-T807/811 antibodies to detect pRb and PI to detect DNA. Nearly all pRb positive cells are in G2/M phases and shown in the boxed areas.
The effects of Olo II on the expression of pluripotency and differentiation markers
To test how pluripotency and self-renewal of mESCs are affected by the interrupted cycle progression, we first determined the mRNA levels of Oct4, Nanog, and Sox-2 in cells that were treated for 24 h with different concentrations of Olo II, the same conditions as described for cell proliferation analysis. As shown in Fig. 5A, the transcript levels of all three genes were down regulated. The mRNA of Oct4, the most abundantly expressed pluripotency marker, was moderately reduced even at 40 μM Olo II while the mRNAs of Sox2 and Nanog were more sensitive and were reduced more than 50% by 10 μM Olo II. However, when the expression of Oct4 at the protein level was analyzed by Westernblot, it was surprising to find that Oct4 was actually increased by Olo II at concentrations above 10 μM (Fig. 5B, inset). To confirm this finding, the protein level of Oct4 was further analyzed by flow cytometry after immunostaining. As illustrated in Fig.5B, the level of Oct4 parallels the result of Western blot analysis. The Nanog protein was increased in a dose-dependent manner by Olo II treatment, similar to Oct4, as measured by flow cytometry (Fig.5C). We also tested the effect of Olo on the expression of several differentiation markers. Up-regulation of two differentiation markers, GATA-4 and smooth muscle actin (SMA) was noted in the cells treated with 5 μM Olo II, but this was not the case in the cells treated with 10 μM and 40 μM Olo II, where Olo II either had no effect on SMA or reduced basal expression of GATA4 and Nestin (Fig.s 2).
Figure 5.
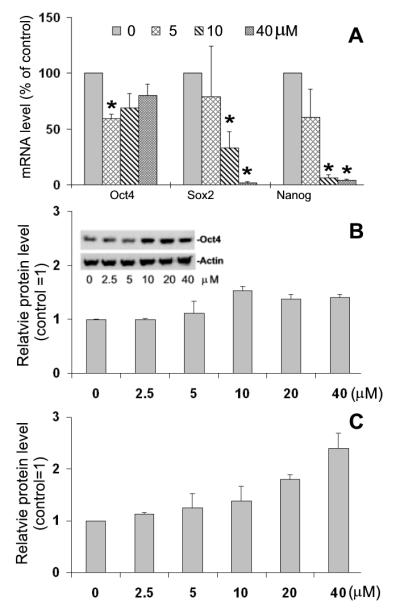
Effect of Olo II on the expression of pluripotency markers. A, DBA 252 cells were treated with Olo II at different concentrations for 24 h. The mRNA levels of Oct4, Sox 2 and Nanog were determined by RT-qPCR. The results are means ± SD of three independent experiments. The mRNA level of tested gene in control cells (0 μM) is designated as 100%. Statistical analysis was performed with Student t test. The p<0.05 (*) was considered statistically significant. B&C, Determination of protein levels of Oct4 (B) and Nanog (C) were determined by flow cytometry. Oct4 was confirmed by Westernblot (B, inset. β-actin was used as a loading control). The fluorescence intensity determined from control cells (0 μM, control) is designated as 1.
mESCs can recover from Olo II-induced cell cycle interruption without losing ESC properties
To determine the long-term effects of Olo II on mESC properties, cells treated with Olo II for 24 h were cultured in fresh medium to allow them recovery. The cells treated with 2.5-5 μM Olo II could resume the normal growth rate within 48 h post treatment, whereas it took 5 days and 10 days for cells treated with 10 μM and 20-40 μM Olo II, respectively. It is noted that during the course of recovery, significant numbers of cells died in the samples that had been treated with Olo II at concentrations above 10 μM. However, the cells that survived the treatment were able to resume normal cell proliferation and showed similar morphology of individual cells and colonies to the control cells (Fig. 6A), and their cell proliferation profiles were also similar to undifferentiated mESCs (Fig. 6C). The expression of cyclins and stem cells markers in recovered cells was returned to normal levels of untreated mESCs (data not shown). Furthermore, recovered cells were able to form embryoid bodies (EBs) with size and shape similar to those formed from the control cells (Fig. 6B). Using RT-qPCR analysis, cell differentiation in EBs was quantitatively determined by the expression of Nkx2.5, GATA-4, and SMA. In EBs derived from DBA 252 cells, the expression levels of all three genes were significantly induced in comparison with undifferentiated ESCs, but differences between the EBs formed from control cells and cells recovered from Olo II treatment are not significant (Fig. 6D). The relatively large variations (standard deviations) are attributed to the nature of long-term EB differentiation and the heterogeneity of environment within individual EBs [24]. When the same experiments were performed in D3 cells, comparable results were obtained for Nkx2.5 and GATA-4, but SMA was not induced in both control EBs and Olo II treated EBs (Fig. 6E). These results suggest that there may be slight difference in sensitivity of EB-induced differentiation between the two mESC lines, but Olo II treatment did not compromise the differentiation potential of both mESCs.
Figure 6.
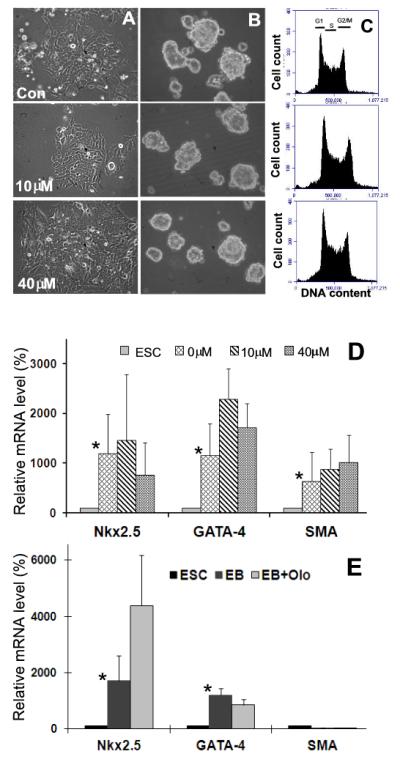
Recovery of mESCs from Olo II treatment. A, DBA 252 cells were treated with 10 μM or 40 μM Olo II. The cells were then cultured in the fresh medium for 5 and 10 days, respectively, and photographed under a phase contrast microscope. B, Five day old EBs formed from control cells and cells recovered from Olo II treatment as shown in A. C, The cell cycle profiles of the cells shown in A were determined by flow cytometry. D, The differentiation capacities of EBs shown in B were determined by the expression of three differentiation markers. The mRNA level of each gene was determined by qRT-PCR and compared with its mRNA level in undifferentiated ESCs (designated as 100%). The results are means ± SD of three independent experiments. E, The same experiments were performed with D3 cells. EB and EB+Olo are EBs formed from control cells and cells recovered from Olo II treatment (10 μM), respectively. In both D and E, the differences in the expression of tested genes between ESCs and EBs are statistically significant (* p<0.05, with the exception of SMA in D), but the differences between control EBs and Olo II treated EBs are not.
In a separate set of experiments, we cultured mESCs in the presence of 5 μM Olo II continuously for up to 3 days and the cell cycle profiles were analyzed at each day. At the third day, the cells were split to prevent overgrowth and passed to a new dish at the same density as in the initial experiment and cultured again in the presence of 5 μM Olo II for an additional two days. Although a moderate accumulation of cells at the G2/M phase was observed in Olo II treated cells, no difference in the protein level of Oct4 was detected between the control cells and the treated cells under this condition (Fig.s3).
Olo II increases the de novo protein synthesis of Oct4 and Nanog without significant alteration of protein stability
It is intriguing to note that expression of Oct4 and Nanog at the mRNA level was down regulated whereas their protein level was increased. The reduced mRNA can be explained by the fact that Olo II at higher concentrations can inhibit RNA polymerase II, thus causing a general transcription inhibition [31]. Therefore, we investigated the mechanisms that caused the increase of Oct4 and Nanog at the protein level. It is conceivable that this can be either due to increased protein synthesis rate or the stability of existing proteins, or both. We first used the protein synthesis inhibitor cycloheximide (Chx) to block de novo protein synthesis. As shown in Fig. 7A, Chx by itself caused moderate decrease of Oct4 and Nanog (Chx, top panels), reflecting the degradation of existing proteins. Treatment of mESCs with Olo II significantly increased the levels of Oct4 and Nanog (Olo, middle panels), which was completely blocked by Chx (Olo + Chx vs Chx, bottom panels). These results clearly demonstrate that the de novo protein synthesis contributed to Olo II-induced increase of Oct4 and Nanog.
Figure 7.
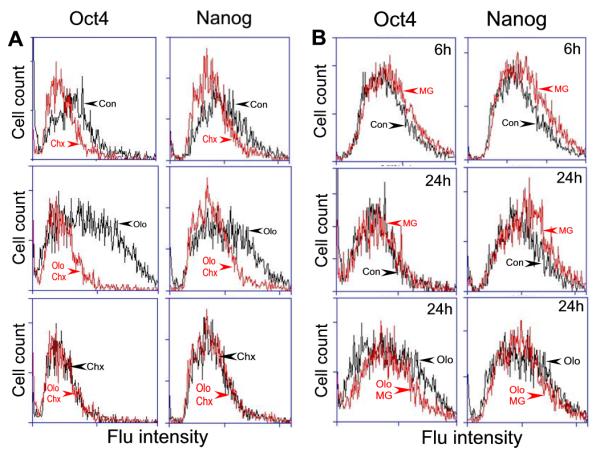
A, cycloheximide (Chx) inhibits Olo II-induced de novo synthesis of Oct4 and Nanog. DBA 252 cells were treated with 20 μM Olo II and 20 μg/ml Chx alone or in combination as indicated for 24 h. B, Effect of MG-132 on Olo II-induced Oct4 and Nanog protein synthesis. DBA 252 cells were treated with 20 μM Olo II (Olo) and 3 μM MG-132 (MG) alone or in combination for the times indicated. Con represents the control experiments. The protein levels of Oct4 and Nanog were indicated by fluorescence (Flu) intensity via flow cytometry analysis.
Since the half-life of cellular proteins is mainly regulated by the activity of proteosomes [32], there are at least two additional possibilities that may contribute to Olo II -induced increase of Oct4 and Nanog; first, the proteosomes that target Oct4 and Nanog could be inactivated; second, Oct4 and Nanog could be modified by post-translational mechanisms thereby becoming resistant to proteosome degradation. We tested these possibilities by using a potent inhibitor of proteosomes, MG-132 [33]. As shown in Fig.7B, cells treated with MG-132 alone slightly increased the level of both Oct4 and Nanog at 6 h in the absence of Olo II treatment (top panels), indicating that MG-132 slowed down the degradation of existing proteins by proteosomes. However, this effect is no longer obvious at 24 h treatment (middle panels), which may represent a steady state. The presence of MG-132 did not significantly alter Olo II-induced Oct4 or Nanog expression at 24 h (bottom panels) or at 6 h treatment (data not shown). Together, these results suggest that the stability of Oct4 and Nanog is subjected to the regulation by proteasome activity, but it is not significantly altered by Olo II treatment. Therefore, de novo synthesis, not enhanced stability of preexisting proteins, is the major contributor to the increased protein level of Oct4 and Nanog caused by Olo II.
Discussion
The relationship between rapid cell proliferation of mESCs and self-renewal is an important subject that has been under intensive investigation, but whether or not the accelerated cell division is essential for the maintenance of the stem cell state is still controversial. Based on the finding that the constitutively active Cdks, especially Cdk2, are the driving force behind the rapid mESC proliferation, it is rational to assume that inhibition of Cdk activity, and thereby cell proliferation, will provide an opportunity to analyze the effects of such intervention on mESC self-renewal and pluripotency. A previous study [11] demonstrated that inhibition of Cdk2 with Ro09-3033, although slowed down cell cycle progression, did not change the characteristics of mESCs, indicating that rapid cell division is not essential to maintain mESC state. On the contrary, a recent study suggested that Olo II delayed G1 to S phase transition, decreased the rate of DNA replication, and resulted in a somatic cell-like cell cycle in mESCs [34], suggesting that rapid cell proliferation is important for the establishment of mESC state.
In order to get further insight into this unresolved issue, we investigated in parallel the effects of Olo II on cell proliferation, self-renewal, and pluripotency in mESCs. Olo II is a potent and relatively specific inhibitor for Cdk2 and Cdk9 (IC50 = 0.06-01 μM), but at higher concentrations it also inhibits Cdk1 (IC50>5 μM) or other members of kinase families (IC50> 100 μM) [31,34]. We recognize that the non-specific effects of chemical kinase inhibitors are problematic in interpreting their physiological effects. However, this may be less of a concern for the purpose of this study since our intention is not to identify the specific functions of individual Cdks, but rather to utilize the collective inhibition of Cdks on cell proliferation so that the relationship between the mESC cell cycle and self-renewal/pluripotency can be analyzed. Therefore, we used a wide range of concentrations of Olo II (2.5-40 μM) and demonstrated that it inhibited cell proliferation and disrupted cell cycle progression in a dose-dependent manner. Olo II also reduced expression levels of cyclin A, B, and E, three major cyclins that account for the rapid cell proliferation in ESCs [11]. This is likely due to the fact that the expression of these cyclins is under the control of E2F transcription factors, which depend on the activity of Cdk2 and Cdk1 [35-37]. Therefore, inhibition of these two kinases by Olo II in turn resulted in the reduced expression of cyclins. Alternatively, this can be explained by the inhibition of Cdk9 of Olo II, which can lead to a general inhibition of transcription [31].
Our data indicated that Olo II may interrupt cell cycle of mESCs at multiple phases. In unsynchronized cells, the most notable effect of Olo II is causing G2 cell accumulation. Although this effect is less prominent in D3 cells than in DBA252 cells at lower concentration of Olo II (5 μM), but at the concentrations above 10 μM, there is no fundamental difference between the two cells lines. By using nocodazole-synchronized cells in a time course study, we can demonstrate that Olo II can caused G1 cell accumulation, reduced S phase cells, and delayed G1 to S phase transition, but these effects seemed to be masked by G2 cell accumulation resulted from severely delayed G2/M transition in unsynchronized cells. Although the precise mechanisms that caused the delayed G2/M transition remain to be determined, it is conceivable that inhibition of Cdk1, which is required for G2/M transition, could be inhibited by Olo II, whereas inhibition of Cdk9 may interfere with the transcription of multiple cell cycle regulators. Therefore, the G2 cell accumulation caused by Olo II could be a collective result of cell cycle interruption at multiple phases. Interestingly, a previous study reported that Olo II mainly caused delayed G1 cell accumulation in mESCs although delayed G1/S and G2/M phase transitions were also noted in [34]. The discrepancy between the two studies is not clear, but could be due to sensitivity of the different cell lines to Olo II and different experimental conditions.
Our results also provide an important insight into the role of the Rb pathway in the regulation of ESC proliferation. In differentiated cells, it is well established that the activation of Rb is critical in repressing cell proliferation by shutting down the transcription of E2F target genes, especially under stress conditions [38], but is generally believed that the Rb pathway is not operational in mESCs [6,30]. We indeed detected highly phosphorylated Rb in mitotic cells confirming the inactive status of Rb in these cells. However, we demonstrated that Olo II treatment reduced Rb phosphorylation level, indicating that the Rb pathway could be activated in mESCs, thereby contributing to Olo II-inhibited cell proliferation.
Our results clearly demonstrated that Olo II induced delayed cell cycle progression and reduced cell proliferation rate. Under these conditions, we observed that at mRNA level the expression of the pluripotency markers was transiently repressed, concurrent with the up-regulation of two differentiation markers, GATA-4 and smooth muscle actin at 5 μM Olo II. However, these disturbances did not result in notable differentiating phenotype. Treated cells could resume normal proliferation rate upon removal of Olo II and retained basic mESC properties including expression of ESC markers, cell and colony morphology, the ability to form EBs that show similar differentiation potential to control cells. It is noted that the cells treated with Olo II at the concentrations above 10 μM resulted in apoptosis of many cells during recovery. While the differentiation state of apoptotic cells could not be assessed, recovered cells have normal cell proliferation rate and display stem cell properties similar to control cells even after a relatively long period of recovery (5-10 days). It is generally believed that perturbation of the expression of pluripotency markers causes ESCs to differentiate. We were first surprised by the finding that the mRNA of Oct4, Nanog and Sox-2 were down regulated by Olo II, but the stem cell state of mESCs was maintained. However, the this phenomenon could be explained by the results that although the mRNA of Oct4, Nanog, and Sox2 level was transiently decreased by OIo II, presumably by the inhibition of Cdk9 [31], their protein levels (as demonstrated in the case of Oct4 and Nanog) were actually increased via increased protein synthesis, which could effectively maintain the stem cell. state of ESCs during short period of treatment. Oct4 is expressed at a high and relatively constant level in mESCs. This steady-state is in a dynamic balance between de novo synthesis and degradation [39-41]. It has been shown that Oct4 stability can be regulated at multiple levels including fine-tuning by ubiquitination and sumoylation [40,42]. Our data suggest that the increased level of Oct4 by Olo II is mainly through elevated protein synthesis, rather than increased stability against proteosome activity. It is interesting to note that a recent study demonstrated that stresses, such as hypoxia and genotoxic agents, can alter Oct4 translation efficiency [43], which may provide a potential mechanistic explanation for our observation.
In summary, our data suggest that Olo II can dramatically slow down cell proliferation of mESCs. However, this intervention does not compromise their capacity for self-renewal. Our findings that mESCs can maintain or even increase protein synthesis of Oct4 under the conditions where cell proliferation is inhibited by oxidative stress [19] and by Olo II (this study) support the hypothesis that Oct4 is a stress responsive gene that can preserve self-renewal capacity under certain stress conditions [44,45].
Supplementary Material
Highlights.
Inhibition of Cdks slows down mESCs proliferation
mESCs display remarkable recovery capacity from short-term cell cycle interruption
Short-term cell cycle interruption does not compromise mESC self-renewal
Oct4 and Nanog are up-regulated via de novo synthesis by cell cycle interruption.
Acknowledgements
This work was supported in part by NIH grant HL082731 (Y-L.G.) and by Mississippi INBRE funded by the National Institute of General Medical Sciences (8 P20 GM103476-11). We thank Baobin Kang for microscopy analysis and Mississippi Functional Genomics Network for the use of the facility.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Niwa H. How is pluripotency determined and maintained? Development. 2007;134:635–646. doi: 10.1242/dev.02787. [DOI] [PubMed] [Google Scholar]
- [2].Wobus AM, Boheler KR. Embryonic stem cells: prospects for developmental biology and cell therapy. Physiol.Rev. 2005;85:635–678. doi: 10.1152/physrev.00054.2003. [DOI] [PubMed] [Google Scholar]
- [3].Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- [4].Boheler KR. Stem cell pluripotency: a cellular trait that depends on transcription factors, chromatin state and a checkpoint deficient cell cycle. J. Cell Physiol. 2009;221:10–17. doi: 10.1002/jcp.21866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Takumi M, Mattson M, Rao MS. Cellular lifespan and senescence signaling in embryonic stem cells. Aging Cell. 2008;3:333–343. doi: 10.1111/j.1474-9728.2004.00134.x. [DOI] [PubMed] [Google Scholar]
- [6].Burdon T, Smith A, Savatier P. Signalling, cell cycle and pluripotency in embryonic stem cells. Trend. Cell Biol. 2002;12:432–438. doi: 10.1016/s0962-8924(02)02352-8. [DOI] [PubMed] [Google Scholar]
- [7].Macaluso M, Montanari M, Giordano A. Rb family proteins as modulators of gene expression and new aspects regarding the interaction with chromatin remodeling enzymes. Oncogene. 2006;25:5263–5267. doi: 10.1038/sj.onc.1209680. [DOI] [PubMed] [Google Scholar]
- [8].Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- [9].Singh AM, Dalton S. The cell cycle and myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell. 2009;5:141–149. doi: 10.1016/j.stem.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Burdon T, Chambers I, Stracey C, Niwa H, Smith A. Signaling mechanisms regulating self-renewal and differentiation of pluripotent embryonic stem cells. Cells Tissues Organs. 1999;165:131–143. doi: 10.1159/000016693. [DOI] [PubMed] [Google Scholar]
- [11].Stead E, White J, Faast R, Conn S, Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D, Dalton S. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene. 2002;21:8320–8333. doi: 10.1038/sj.onc.1206015. [DOI] [PubMed] [Google Scholar]
- [12].White J, Stead E, Faast R, Conn S, Cartwright P, Dalton S. Developmental activation of the Rb-E2F pathway and establishment of cell cycle-regulated cyclin-dependent kinase activity during embryonic stem cell differentiation. Mol.Biol.Cell. 2005;16:2018–2027. doi: 10.1091/mbc.E04-12-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dalton S. Exposing hidden dimensions of embryonic stem cell cycle control. Cell Stem Cell. 2009;4:9–10. doi: 10.1016/j.stem.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Singh AM, Dalton S. The cell cycle and myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell. 2009;5:141–149. doi: 10.1016/j.stem.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Daheron L, Opitz SL, Zaehres H, Lensch WM, Andrews PW, Itskovitz-Eldor J, Daley GQ. LIF/STAT3 signaling fails to maintain self-renewal of human embryonic stem cells. Stem Cells. 2004;22:770–778. doi: 10.1634/stemcells.22-5-770. [DOI] [PubMed] [Google Scholar]
- [17].Saretzki G, Armstrong L, Leake A, Lako M, von ZT. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells. 2004;22:962–971. doi: 10.1634/stemcells.22-6-962. [DOI] [PubMed] [Google Scholar]
- [18].Saretzki G, Walter T, Atkinson S, Passos JF, Bareth B, Keith WN, Stewart R, Hoare S, Stojkovic M, Armstrong L, von Zglinicki T, Lako M. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells. 2008;26:455–464. doi: 10.1634/stemcells.2007-0628. [DOI] [PubMed] [Google Scholar]
- [19].Guo YL, Chakraborty S, Rajan S, Wang R, Huang F. Effects of oxidative stress on mouse embryonic stem cell proliferation, apoptosis, senescence, and self-Renewal. Stem Cells Dev. 2010;19:1321–1331. doi: 10.1089/scd.2009.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Allen M, Svensson L, Roach M, Hambor J, McNeish J, Gabel CA. Deficiency of the stress kinase p38{alpha} results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J.Exp.Med. 2000;191:859–870. doi: 10.1084/jem.191.5.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Roach ML, Stock JL, Byrum R, Koller BH, McNeish JD. A new embryonic stem cell line from DBA/1lacJ mice allows genetic modification in a murine model of human inflammation. Exp.Cell Res. 1995;221:520–525. doi: 10.1006/excr.1995.1403. [DOI] [PubMed] [Google Scholar]
- [22].Chakraborty S, Kang B, Huang F, Guo YL. Mouse embryonic stem cells lacking p38[alpha] and p38[delta] can differentiate to endothelial cells, smooth muscle cells, and epithelial cells. Differentiation. 2009;78:143–150. doi: 10.1016/j.diff.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guo YL, Yang B. Altered cell adhesion and cell viability in a p38alpha mitogen-activated protein kinase-deficient mouse embryonic stem cell line. Stem Cells Dev. 2006;15:655–664. doi: 10.1089/scd.2006.15.655. [DOI] [PubMed] [Google Scholar]
- [24].Guo YL, Ye J, Huang F. p38alpha MAP kinase-deficient mouse embryonic stem cells can differentiate to endothelial cells, smooth muscle cells, and neurons. Dev.Dyn. 2007;236:3383–3392. doi: 10.1002/dvdy.21374. [DOI] [PubMed] [Google Scholar]
- [25].Kim JM, White JM, Shaw AS, Sleckman BP. MAPK p38{alpha} is dispensable for lymphocyte development and proliferation. J Immunol. 2005;174:1239–1244. doi: 10.4049/jimmunol.174.3.1239. [DOI] [PubMed] [Google Scholar]
- [26].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucl.Acids.Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Toumadje A, Kusumoto KI, Parton A, Mericko P, Dowell L, Ma G, Chen L, Barnes D, Denry Sato J. Pluripotent differentiation in vitro of murine ES-D3 embryonic stem cells. In Vitro Cell.Dev. Biol. - Animal. 2003;39:449–453. doi: 10.1290/1543-706X(2003)039<0449:PDIVOM>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- [28].Chuykin IA, Lianguzova MS, Pospelova TV, Pospelov VA. Activation of DNA damage response signaling in mouse embryonic stem cells. 2008;7:2922–2928. doi: 10.4161/cc.7.18.6699. [DOI] [PubMed] [Google Scholar]
- [29].Buchkovich K, Duffy LA, Harlow E. The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell. 1989;58:1097–1105. doi: 10.1016/0092-8674(89)90508-4. [DOI] [PubMed] [Google Scholar]
- [30].Savatier P, Huang S, Szekely L, Wiman KG, Samarut J. Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene. 1994;9:809–818. [PubMed] [Google Scholar]
- [31].Krystof V IW, McNae MD, Walkinshaw PM, Fischer P, Muller B, Vojitesek M, Orsag L, Havl!eek M. Strnad, Antiproliferative activity of olomoucine II, a novel 2,6,9-trisubstituted purine cyclin-dependent kinase inhibitor. Cell.Mol.Life Sci. 2005;62:1763–1771. doi: 10.1007/s00018-005-5185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Konstantinova IM, Tsimokha AS, Mittenberg AG. Role of proteasomes in cellular regulation. Intl. Rev. Cell Mol. Biol. 2008;28:59–124. doi: 10.1016/S1937-6448(08)00602-3. [DOI] [PubMed] [Google Scholar]
- [33].Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trendi Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- [34].Koledova Z, Kafkova LR, Calabkova L, Krystof V, Dolezel P, Divoky V. Cdk2 inhibition prolongs G1 phase progression in mouse embryonic stem cells. Stem Cells Dev. 2009;19:181–194. doi: 10.1089/scd.2009.0065. [DOI] [PubMed] [Google Scholar]
- [35].Schulze A, Zerfass K, Spitkovsky D, Middendorp S, Berg+?s J, Helin K, Jansen-Durr P, Henglein B. Cell cycle regulation of the cyclin A gene promoter is mediated by a variant E2F site. Proc. Natl. Acad. Sci. USA. 1995;92:11264–11268. doi: 10.1073/pnas.92.24.11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Botz J, Zerfass-Thome K, Spitkovsky D, Delius H, Vogt B, Eilers M, Hatzigeorgiou A, Jansen-Durr P. Cell cycle regulation of the murine cyclin E gene depends on an E2F binding site in the promoter. Mol.Cell.Biol. 1996;16:3401–3409. doi: 10.1128/mcb.16.7.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trend Biochem. Sci. 2004;29:409–417. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- [38].Ben Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Intl. J BiochemCell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- [39].Zhang Z, Liao B, Xu M, Jin Y. Post-translational modification of POU domain transcription factor Oct-4 by SUMO-1. FASEB J. 2007;21:3042–3051. doi: 10.1096/fj.06-6914com. [DOI] [PubMed] [Google Scholar]
- [40].Wei F, Scholer HR, Atchison ML. Sumoylation of Oct4 enhances its stability, DNA binding, and transactivation. J Biol. Chem. 2007;282:21551–21560. doi: 10.1074/jbc.M611041200. [DOI] [PubMed] [Google Scholar]
- [41].Xu HM, Liao B, Zhang QJ, Wang BB, Li H, Zhong XM, Sheng HZ, Zhao YX, Zhao YM, Jin Y. Wwp2, an E3 ubiquitin ligase that targets transcription factor oct-4 for ubiquitination. J. Biol. Chem. 2004;279:23495–23503. doi: 10.1074/jbc.M400516200. [DOI] [PubMed] [Google Scholar]
- [42].Naujokat C, Saric T. Concise review: role and function of the ubiquitin-proteasome system in mammalian stem and progenitor cells. Stem Cells. 2007;25:2408–2418. doi: 10.1634/stemcells.2007-0255. [DOI] [PubMed] [Google Scholar]
- [43].Wang X, Zhao Y, Xiao Z, Chen B, Wei Z, Wang B, Zhang J, Han J, Gao Y, Li L, Zhao H, Zhao W, Lin H, Dai J. Alternative translation of OCT4 by an internal ribosome entry site and its novel function in stress response. Stem Cells. 2009;27:1265–1275. doi: 10.1002/stem.58. [DOI] [PubMed] [Google Scholar]
- [44].Kang J, Shakya A, Tantin D. Stem cells, stress, metabolism and cancer: a drama in two Octs. Trend Biochem Sci. 2009;34:491–499. doi: 10.1016/j.tibs.2009.06.003. [DOI] [PubMed] [Google Scholar]
- [45].Kang J, Gemberling M, Nakamura M, Whitby FG, Handa H, Fairbrother WG, Tantin D. A general mechanism for transcription regulation by Oct1 and Oct4 in response to genotoxic and oxidative stress. Genes Dev. 2009;23:208–222. doi: 10.1101/gad.1750709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.