

Abstract
Oxidative stress is linked to the production of reactive lipid aldehydes that non-enzymatically alkylate cysteine, histidine or lysine residues in a reaction termed protein carbonylation. Reactive lipid aldehydes and their derivatives are detoxified via a variety of phase I and phase II systems and when antioxidant defenses are compromised or oxidative conditions are increased protein carbonylation is increased. The resulting modification has been implicated as causative in a variety of metabolic states including neurodegeneration, muscle wasting, insulin resistance and aging. Although such modifications usually result in loss of protein function, protein carbonylation may be regulatory and activate signaling pathways involved in antioxidant biology and cellular homeostasis.
Keywords: 4-HNE, carbonylation, reactive lipid aldehydes, steatosis, neurodegeneration, metabolism
Carbonylation biochemistry
Aerobic respiration is invariably associated with the production of partially reduced reactive oxygen species (ROS; see Glossary). Under physiological conditions, ROS production is balanced by antioxidant detoxification of radical species. While there is strong evidence to support the role of ROS in cellular signaling [1], increased oxidative stress without parallel increases to antioxidant capacities results in damage to cellular components such as proteins, lipids and DNA [2,3]. Prolonged oxidative stress and accumulation of oxidative modification to biomolecules are linked to pathological alterations of metabolism, induction of apoptosis and cell death [4–6].
ROS are produced by radical leak from enzymes such as NADPH oxidase, xanthine oxidase, cyclooxygenases, lipoxygenases, and the mitochondrial electron transport chain system [7–9] (Figure 1). ROS production results from either radical-induced modification of molecular oxygen or incomplete consumption of molecular oxygen, resulting in formation of superoxide anion [10]. Superoxide anion is metabolized efficiently by superoxide dismutase [11] to hydrogen peroxide that in turn is transformed into water by catalase, glutathione peroxidase, or peroxiredoxin/thioredoxins [12]. If hydrogen peroxide reaches cellular regions rich in free iron (II) it can be readily converted to hydroxyl radical via Fenton chemistry [10]. Unlike superoxide anion, hydroxyl radical is not metabolized and can only be consumed via removal of hydrogen from neighboring molecules, resulting in formation of a new radical species and water.
Figure 1. Metabolism of reactive lipid aldehydes via detoxification or carbonylation.
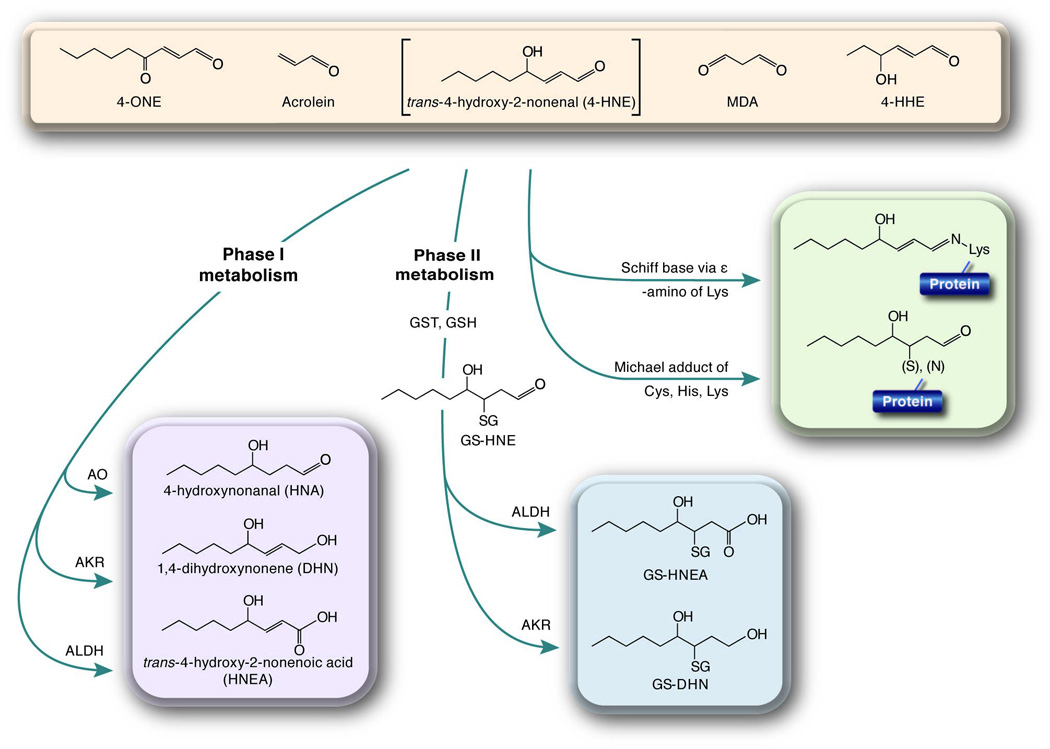
Schematic depiction of Phase I and Phase II metabolism of reactive lipid aldehydes is shown. For illustration purposes, the metabolism if 4-HNE (as shown in the bracket) is presented in detail but the reactions apply broadly to each of the indicated aldehydes. In addition, carbonylation the protein side chains (Cys, Lys or His) is shown in detail for 4-HNE but may occur for any of the other reactive lipids shown. MDA; malondialdehyde, 4-HHE; 4-hydroxy 2,3 hexenal. For other abbreviations, see text.
Hydroxyl radical is responsible for removal of bisallylic hydrogen from polyunsaturated fatty acids resulting in formation of a carbon centered radical followed by addition of molecular oxygen [10]. Chemical degradation of oxidized polyunsaturated fatty acid (Hock cleavage) results in formation of a number of diffusible reactive lipophilic molecules [10]. The most widely studied class of molecules resulting from lipid peroxidation is the α,β-unsaturated aldehydes. Although a variety of lipid aldehydes are produced (e.g., acrolein, malondialdehyde), 4-hydroxy trans-2,3-nonenal (4-HNE) and 4-oxo trans-2,3-nonenal (4-ONE), products of ω6-polyunsaturated fatty acid peroxidation, have been studied in the greatest detail [2] (Figure 1). While 4-HNE and 4-ONE are known to modify DNA and RNA, the effects of lipid adduction to protein have received the most attention [13].
For a variety of reactive aldehydes (Figure 1) the carbonyl group results in an electron-poor environment centered around the 2,3 double bond. [3]. In addition, a hydroxy or keto group also withdraws electrons from the system further contributing to the reactivity of the lipid [3]. As such, carbon three of the aldehyde is highly susceptible to nucleophilic attack by the side chains of lysine, histidine, or cysteine residues [3] in a reaction termed “protein carbonylation”. This reaction produces an alkylated amino acid with a lipophilic derivative containing a free carbonyl group and specific targets of modification have been known since the early 1990’s [14]. It should be noted that acquisition of a free carbonyl group on proteins is also achieved from direct site chain oxidation. Michael addition of reactive lipids to proteins is believed to account for ~80% of all carbonyl adducts and such molecules are commonly used as a biomarker for a number of pathophysiological states and may play an active role in onset and progression of disease [13]. Reactive lipid aldehydes can also modify proteins by Schiff base formation with a primary amine (lysine side chain) resulting in dehydration of the aldehyde [3]. Schiff base adduction account for ~20% of lipid-modified proteins [3] but does not produce a free carbonyl group and as such, is not typically measured by conventional protein carbonylation reagents (e.g. aldehyde reactive probe or biotin hydrazide). In addition, lipid conjugation via Schiff base retain the electron-poor status at carbon three allowing for such molecules to modify a second amino acid side chain by Michael addition [3]. Dual modification of proteins by a single lipid aldehyde can produce either inter or intra protein crosslinks.
Damage resulting from protein modification by lipid aldehydes can be mitigated via a number of Phase I and Phase II metabolic mechanisms. Phase I metabolism decreases the reactivity of lipid aldehydes via oxidation and reduction reactions, thus preventing protein, DNA, and lipid modification (Figure 1). For illustration purposes, the metabolism of 4-HNE is shown in Figure 1 but the reactions may be applied to most of the reactive lipids shown. Oxidation of the aldehyde group of 4-HNE by aldehyde dehydrogenases (ALDH) results in formation of trans-4-hydroxy-2-nonenoic acid targeting the product for export or further metabolism by β-oxidation. 4-HNE can be reduced at the aldehyde moiety by aldo-keto reductases (AKR) resulting in 1,4-dihydroxynonene. In addition, 4-HNE can be reduced at the unsaturated 2,3 double bond by alkenal/one-oxidoreductase (AO), resulting in 4-hydroxynonanal. In each case, modification of 4-HNE results in loss of reactivity around carbon 3 decreasing its reactivity for Michael addition [3].
Phase II metabolism of reactive aldehydes is carried out primarily by enzymatic glutathionylation at carbon three [15]. The resulting glutathionylated molecule (e.g., GS-HNE) can undergo further metabolic modification by aldo-keto reductases and aldehyde dehydrogenases [16] (Figure 1). Glutathione metabolites of 4-HNE are substrates for export to the extracellular domain by RLIP76 or the multidrug resistance transporter MRP1 resulting in eventual clearance as components of urine [17].
Detection of protein carbonylation on target proteins has remained a technical challenge with a variety of blotting and mass spectrometric methods being developed to map the extent and sites of modification [11,18–20]. The majority of carbonylation target analyses involve derivatization of carbonyl groups with either hydrazide or hydrazone adducts, followed by enrichment and analysis. For simple semi-quantitative applications, the carbonyl group is conjugated with biotin-hydrazide followed by separation of proteins by traditional denaturing gel electrophoresis and blotting to a solid support. Carbonylated proteins are detected using biotin-directed probes such as avidin coupled to fluorescent reporter [20, 39] producing a carbonylation blot that may be used for general assessment of protein modification. For quantitative mass spectrometric evaluation of carbonylated proteins, complications in the analysis include oxidation of amino acid residues during the workup, the multiple types of oxidative modifications that co-exist in oxidized proteins, the variation in abundance of carbonylation proteins, and inadequate ionization of derivatized peptides during introduction into the mass spectrometer. As such, while progress is being made on quantitative analysis of protein carbonylation [21,22], a major unresolved goal of carbonylation analysis is to develop reproducible, sensitive and quantitative methods for identifying sites of modification.
Carbonylation targets and metabolic control
Protein carbonylation is associated with a variety of metabolic diseases, syndromes and conditions including aging, diabetes, heart disease, and neuropathies. Since protein carbonylation is chemically and not enzymatically driven, the targets of carbonylation are dependent upon the oxidative environment of the cell, abundance and proximity to ROS. In highly oxidative cells such as macrophages, ROS produced by the NADPH oxidase system may lead to significant amounts of lipid aldehydes that covalently modify endogenous enzymes and proteins. Moreover, activation of NADPH oxidase and the innate immune system by gram-negative bacteria may result in lipid adduction of foreign proteins and play a protective role in host defense systems. The mitochondrial respiratory chain constitutes a major source of superoxide anion production and via superoxide dismutase produces hydrogen peroxide [7–9]. Metal-catalyzed reduction of hydrogen peroxide yields hydroxyl radicals and subsequent lipid peroxidation (Figure 1). Not surprisingly, a large proportion of carbonylated proteins are localized to the mitochondria [23]. However, since reactive lipid aldehydes freely diffuse across membranes, the location of the modified proteins does not necessarily define the source of the ROS.
Carbonylation muscle pathophysiology and aging
Increased levels of carbonylated proteins have been detected in skeletal muscle under several pathological conditions including sepsis, ischemia-reperfusion, diabetes, and chronic obstructive pulmonary disease [24,25]. The affected proteins include those that are part of contractile machinery of the muscle (actin, myosin light and heavy chains, desmin, tropomysin). However, it is unclear whether carbonylation leads to dysfunction of these proteins or skeletal muscle or is simply associated with such conditions.
Protein carbonylation in aging muscle mitochondria increases with regard to both the number of targets and extent of carbonylation [23]. Using the Fischer 344 rat system, skeletal muscle aging is associated with increased carbonylation in both, fast-twitch, and slow-twitch muscle, respectively. The proteins that exhibited the largest carbonylation increase with aging were the α polypeptide of electron-transferring flavoprotein (detected in both muscle types), malate dehydrogenase and the long-chain specific acyl-CoA dehydrogenase (detected in fast-twitch muscle), and the S36 mitochondrial ribosomal protein detected in the slow-twitch muscle. The degree of carbonylation also varies between muscle fiber types. Fast-twitch, glycolytic fibers have decreased superoxide-scavenging capacity relative to slow-twitch, oxidative fibers leading to a 2-fold increase of mitochondrial protein carbonylation [23]. Because of the predominance of carbonylation in rat fast-twitch muscle proteins, this muscle type may bear dysfunction as a result of carbonylation. The pathways that may be affected in fast-twitch glycolytic fibers included oxidative phosphorylation, the citric acid cycle, and fatty acid metabolism (Figure 2).
Figure 2. Protein carbonylation targets in metabolic disease.
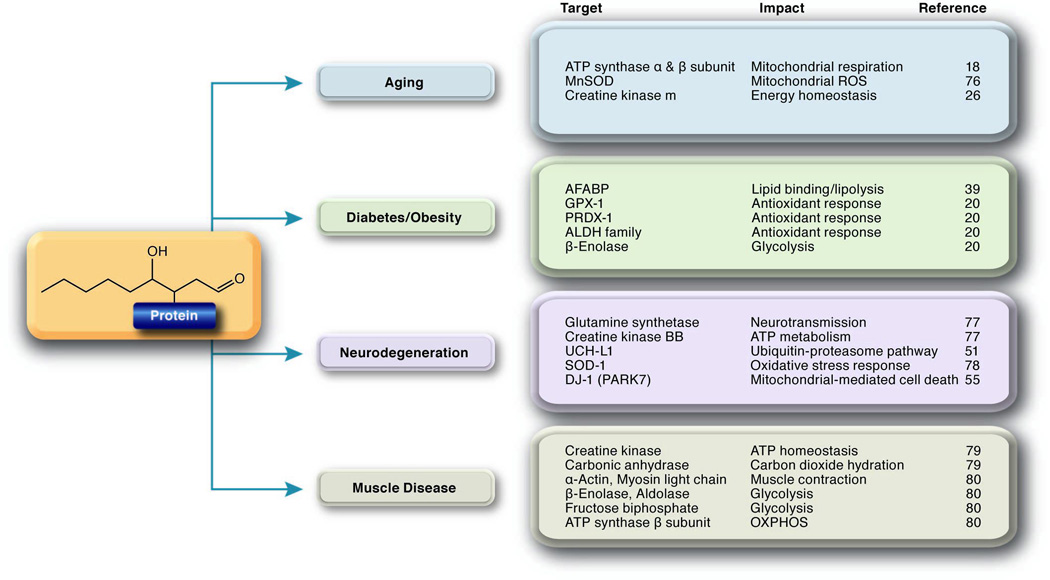
Protein carbonylation targets in a variety of metabolic diseases and their impact on cellular biology are listed. The list is not meant to be inclusive but exemplary of the breadth of biological targets affected by protein carbonylation.
Muscle creatine kinase is a target of oxidative modification [26]. Carbonylation of creatine kinase in mouse quadriceps increased 2.5 fold in the old and middle age relative to the young mice. Similarly, carbonylated creatine kinase was less active and found in insoluble aggregates and high molecular weight oligomers in the old and middle-aged relative to the young mice. Because muscle creatine kinase is critical to homeostasis of creatine phosphate and creatine during periods of high- and low-energy requirements, these correlative studies suggest that carbonylation-induced structural instability may contribute to loss of protein function and collectively translate to loss of muscle function with age.
Carbonylation in liver and steatosis
Age-associated increases of ROS and protein carbonylation have also been detected in the liver [27], However, the majority of studies identifying carbonylated proteins fail to provide functional impact and conversely studies that show functional impact of oxidative stress do not directly identify targets of carbonylation. Elevated carbonylation of endoplasmic reticulum resident proteins such as the endoplasmic reticulum chaperone protein GRP78 and calreticulin occurs in the liver with age, suggesting that carbonylation may interfere with protein folding and quality control [28]. Additionally, a study comparing various animal models of longevity found that longer-lived species exhibited reduced nuclear accumulation of carbonylated proteins relative to shorter-lived species [29]. Additional liver pathologies associated with increased protein carbonylation include alcoholic liver disease, non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Histopathological studies using anti-HNE antibodies found significantly increased levels of protein modification in patients with these types of liver disease [30]. The degree of protein modification correlated with the severity of steatosis, but not fibrosis, likely due to the abundance of lipids associated with steatosis. Combined with an inflammatory environment, fatty liver provides ideal conditions for the generation of reactive lipid species and subsequent protein modification.
Carbonylation in adipose tissue and obesity
Numerous reports using various rodent models of obesity describe a chronic low-grade inflammatory state in adipose tissue [36,37]. Characteristic of this inflammation is an accumulation of macrophages, T-cells, dendritic cells and other inflammatory cells within adipose depots. Activated adipose tissue macrophages release a variety of cytokines, most notably tumor necrosis factor-alpha (TNF-α) that acts on adipocytes to increase ROS production, lipid peroxidation and protein carbonylation, which collectively leads to mitochondrial dysfunction [38]. Indeed, elevated levels of protein carbonylation have been reported in the adipose tissue of diet induced obese mice [20], as well as in obese human subjects [39]. One such example is that of fatty acid binding protein 4 (FABP4) protein, that is carbonylated on Cys117 resulting in markedly decreased affinity for fatty acids [20]. The resulting loss of fatty acid binding by FABPs in adipose and other tissues may lead to aberrant fatty acid trafficking and lipotoxicity. Acute fatty acid treatment has been shown to induce insulin resistance in both muscle and liver [40]. Also of interest is that glutathione S-transferase A4 (GSTA4), a central Phase II antioxidant enzyme responsible for conjugating glutathione to reactive lipid aldehydes, [41,42] (Figure 1), is also a carbonylation target [20]. GSTA4 was found to be down regulated in mouse models of obesity as well as in insulin resistant obese humans, suggesting that cellular oxidative protection capacity may be reduced in these conditions [43]. Indeed, treatment with the pro-inflammatory cytokine TNF-α decreases GSTA4 expression and increases protein carbonylation in 3T3-L1 adipocytes similar to obese states [43,44]. These results suggest that in adipocytes, cytokine-induced mitochondrial dysfunction is mechanistically linked to protein carbonylation and may underlie insulin resistance (Figure 2).
Carbonylation adducts in neurodegenerative diseases
A number of carbonylated proteins have also been reported as elevated in the hippocampus and inferior parietal lobule of aged humans with mild cognitive impairment, a diagnosis that typically precedes Alzheimer’s disease [31]. These proteins include several important enzymes in glucose metabolism such as lactate dehydrogenase B, phosphoglycerate kinase, pyruvate kinase, α-enolase and the α-subunit of the ATP synthase. The specific activity of lactate dehydrogenase, ATP synthase and pyruvate kinase are all decreased in brains of humans with mild cognitive impairment suggesting a correlation between increased covalent modification, the loss of enzyme function and disease pathology [32].
In the brain, the formation of lipid adducts impairs protein function and leads to neuronal death, both contributing factors to diminished cognition. Accumulation of carbonylated proteins with age is partly due to an age-associated decline of proteasomal activity [33]. Carbonylated proteins are primarily degraded by the 20S core proteasome in a ubiquitin-independent manner where the oxidative modifications often cause structural rearrangements and expose hydrophobic residues on the protein surface [34]. These exposed hydrophobic residues serve as a recognition motif for the 20S core proteasome [33]. Indeed, interventions that extend lifespan, such as caloric restriction, also attenuate aging-induced loss of proteasomal activity and accumulation of protein carbonyls [33]. The accumulation of oxidative protein and DNA modifications over a lifetime is thought to be negatively associated with longevity. Reducing caloric intake confers weight loss, enhances insulin sensitivity, and decreases inflammation, mitochondrial superoxide production, and accumulation of protein carbonyls [35].
Inflammation, oxidative stress and protein carbonylation have been implicated in a number of neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis and multiple sclerosis. Indeed, the central nervous system is enriched in polyunsaturated fatty acids, thus making it particularly vulnerable to membrane-associated oxidative stress [45]. Alzheimer’s disease is the most common form of dementia and pathologic changes include accumulation of β-amyloid plaques and neurofibril tangles, decreased neurotransmission and eventually gross atrophy of specific brain regions [46]. Specifically, 4-HNE modification of tau, a microtubule-associated protein, restricts tau dephosphorylation and results in stabilization of neurofibrillary tangles, a hallmark of the disease [47]. Strikingly, increased carbonylated proteins are reported in affected brain regions at each stage of disease progression [32,48,49]. Targets of lipid modification in Alzheimer’s disease include several metabolic proteins as well as protein involved in proteasomal degradation [50,51]. For instance, the abundant de-ubiquitylating enzyme ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1) is highly susceptible to oxidative modification [51]. Investigators have shown that 4-HNE modification of UCH-L1 causes a conformational change, a loss in solubility and defective interactions with tubulin leading to protein aggregation [52] (Figure 2). Another critical carbonylation target in Alzheimer’s patients is neuropolypeptide h3 that is an important modulator of neurotransmission through control of acetylcholine levels. The characteristic cholinergic deficiency found in Alzheimer’s disease and the use of acetyl cholinesterase inhibitors to treat symptoms in affected patients highlight the significance of the oxidative modification of neuropolypeptide h3. [31,53].
Parkinson’s disease presents with progressive loss of dopaminergic neurons in the substantia nigra and a variety of studies have demonstrated increased oxidative stress in this region from Parkinson’s patients. Hallmarks of this oxidative stress are reduced glutathione levels, reduced manganese superoxide dismutase (mnSOD), as well as increased oxidative modifications to lipids, proteins and DNA [54,55]. Both acute administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and systemic treatment with rotenone, both of which are used to mimic Parkinson’s disease pathology in rodent models, inhibit Complex I of the electron transport system. The resulting mitochondrial dysfunction leads to in increased protein carbonylation in the substantia nigra similar to that which is observed in Parkinson’s patients [56]. Indeed, the majority of experimental in vivo models of Parkinson’s disease employ induction of oxidative stress in dopaminergic neurons in the substantia nigra.
In amyotrophic lateral sclerosis (ALS), a loss in upper and lower motor neurons leads to a signature muscle atrophy and is usually fatal within 3–5 years of diagnosis [45]. Approximately 90% of cases are sporadic while ~10% or the cases are familial. Among the familial amyotrophic lateral sclerosis cases, many map to the copper/zinc superoxide dismutase SOD1 locus [45], which encodes the cytosolic antioxidant enzyme. While the exact mechanism of how mutations in SOD1 lead to ALS pathology is unclear, recent studies show that mutant SOD1 can lead to misfolding and protein aggregation specifically in motor neurons of affect tissue [ 57]. To further understand the role of SOD1 mutants in ALS, a transgenic mouse model was created to mimic ALS pathology. The G93A-SOD-1 mouse has an SOD1 mutation that results in production of hydroxyl radical independent of SOD1 superoxide reduction, resulting in massive amounts of radical production and oxidative stress. Motor neurons in the spinal cords of ALS patients as well as G93A-SOD-1 transgenic mice show elevated protein carbonylation, which increases with disease progression [45,58]. Using a mass spectroscopy approach Perluigi and colleagues identified axonal outgrowth protein dihydropyrimidinase-related protein 2, chaperone heat shock protein 70, and glycolytic α-enolase as proteins with increased carbonyl modification in the G93A-SOD-1 transgenic model of ALS [45].
Multiple sclerosis is an autoimmune neurodegenerative disease of the central nervous system, characterized by inflammation, demyelination and axonal degeneration. Post mortem analyses reveal that multiple sclerosis patients have elevated protein carbonyls in white and gray matter [59]. Likewise, in a mouse model of multiple sclerosis, experimental autoimmune encephalitis, a marked increase in protein carbonylation in cerebellar astrocytes was reported. Major targets of protein carbonylation throughout disease progression were identified as the structural proteins β-actin, β-tubulin and glial fibrillary acidic protein as well as the protein folding chaperone heat shock cognate 71 [60].
Activation of cell signaling via protein carbonylation
Whereas many carbonylation targets are inactivated resulting in decreased function and associated metabolic activity, carbonylation also serves to activate several signaling systems. Recent studies have focused on the Nuclear factor (erythroid-derived-2)-like 2 (NRF2) – Kelch-like ECH-associated protein 1 (KEAP1) pathway that controls Phase II antioxidant enzyme expression [61] and the Thioredoxin (TRX) – Apoptosis signal regulating kinase 1 (ASK1) that controls the c-Jun N-terminal kinase (JNK) [62,63].
Cytoplasmic KEAP1 binds to NRF2 sequestering it from the nucleus [64] and targeting it for proteasomal degradation. Upon carbonylation of critical thiols in KEAP1 (Cys273, Cys288) NRF2 is released and translocates to the nucleus where it dimerizes with a variety of nuclear factors including MAF and NRF1. This complex binds to the antioxidant response element (ARE) activating expression of a cassette of genes linked to the antioxidant response (Figure 3 left). Carbonylated KEAP1 is degraded cytoplasmically by the proteasome [65,66]. Treatment of COS-7 cells with HNE induced NRF2 nuclear translocation resulting in active ARE signaling [67] while RNAi-mediated silencing of NRF2 in a variety of cancer cell lines ablated HNE-dependent activation of downstream targets [68]. These studies illustrate a negative feedback loop where lipid peroxidation-induced protein carbonylation of KEAP1 activates the antioxidant response, resulting in increased Phase II metabolic enzymes and decreased oxidative stress. Decreased oxidative stress in turn leads to decreased KEPA1 carbonylation.
Figure 3. Activation of cell signaling by protein carbonylation.

Carbonylation of proteins results not only in enzyme inactivation, but also activation of several signaling pathways. (L) 4-HNE modification of KEAP1 at critical cysteine residues releases NRF2 that translocates to the nucleus. Activated NRF2 heterodimerizes with other factors and activates the transcription of a variety of antioxidant gene targets. (R) 4-HNE modification of thioredoxin releases ASK1 kinase that becomes phosphorylated and initiates a signaling cascade via SEK and JNK. Activation/phosphorylation of JNK leads to phosphorylation of IκBα and translocation/activation of p50/p65 to the nucleus and expression of pro-inflammatory gene targets.
Similar to the NRF2/KEAP1 system, reduced thioredoxin complexes with and negatively regulates the activity of the apoptosis signal-regulating kinase (ASK) [69] (Figure 3 right). Under basal conditions, thioredoxin forms a complex with ASK1 inhibiting its autophosphorylation and activation. Lipid peroxidation products such as HNE modify structurally vicinyl thiol groups of thioredoxin (Cys32, Cys35), releasing ASK1 from the complex, facilitating its autophosphorylation (Thr813, Thr838, Thr842) and subsequent activation [70]. Thioredoxin can also be carbonylated at Cys73, a residue distal to the catalytic site that also results in thioredoxin inactivation, but not ASK1 release. Activation of ASK1, through carboylation of thioredoxin, leads to a cascade of phosphorylation events linked to the phosphorylation of SEK and JNK culminating in NF-κB activation [71] contributing to the development of insulin resistance [69,72].
Concluding Remarks
Protein carbonylation has been appreciated for decades; however the absence of identified carbonylation targets with mapped sites of modification has made it difficult to assess the functional relevance of this modification. The possible existence of enzymatic decarbonylation may further complicate such assessment [73]. Moreover, since the extent of carbonylation is almost always sub-stoichiometric, the implications of partial loss of activity have remained enigmatic. However, when carbonylation leads to gain of function, small changes in carbonylation may result in significant changes in cellular function. For example, Velez et al. reported that 4-HNE adduction to proteins activates Bcl-2 phosphorylation and triggers autophagy [74]. Similarly, carbonylation of critical active site thiols on PTEN (phosphatase and tensin homolog) inactivates it leading to potentiated growth factor signaling [75]. Such examples point towards the ability of small changes in carbonylation to affect major pathways, particularly if such modification is followed by signal amplification.
An additional consideration when addressing protein carbonylation is the effect of lipid adduction on protein localization. That is, in a number of in vivo and model systems, covalent attachment of lipophilic groups to proteins affects their localization and assembly on membranes (e.g., palmitoylation, prenylation). Studies to date have not assessed carbonylation of proteins as a regulatory process potentially affecting membrane association and/or protein-protein interactions. When additional characterized carbonylation targets are identified such studies should be forthcoming and may represent a new theme linking protein carbonylation to cellular physiology and metabolism.
Acknowledgements
We would like to thank members of the Bernlohr and Arriaga groups for comments on this review and to Anthony Hertzel for artwork. Supported by NIH F32 DK091004 to EKL and NIH RO1 DK084669 to DAB.
Glossary
- Protein carbonylation
As it relates to lipid peroxidation, the covalent adduction of a lipid aldehyde to cysteine, lysine, and histidine side chains resulting in the introduction of a free carbonyl group to a target protein. See also Michael addition.
- Reactive oxygen species
A generic term used to denote one or more partially reduced, highly reactive oxygen species including superoxide anion, hydroxyl radical, hydrogen peroxide.
- Phase I metabolism
Metabolism of biomolecules by oxidation/reduction reactions resulting in alterations in functional groups of molecules such as α,β-unsaturated aldehydes.
- Phase II metabolism
Metabolism of biomolecules by conjugation to metabolic cofactors such as glutathione or glucuronic acid, typically targeting the metabolite for export or further metabolism.
- Lipid peroxidation
Free radical-mediated addition of molecular oxygen to polyunsaturated fatty acids resulting in hydroperoxy-fatty acids subject to spontaneous degradation to small, diffusible, carbonyl-containing lipids.
- Hock cleavage
The mechanism by which lipid hydroperoxides degrade to small carbonyl-containing molecules such as trans-4-hydroxy-2-nonenal.
- Michael addition
Conjugation of α,β-unsaturated aldehydes to cysteine, lysine, and histidine residues of proteins and peptides by nucleophilic attack on carbon three of the α,β-unsaturated aldehyde.
- Schiff Base
Conjugation of α,β-unsaturated aldehydes to lysine residues of proteins and peptides by nucleophilic attack by the primary amino group of lysine on the aldehydic carbon one of α,β-unsaturated aldehydes resulting in dehydration and adduct formation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esterbauer H, et al. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 3.Schaur RJ. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Aspects Med. 2003;24:149–159. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 4.Furukawa S, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houstis N, et al. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 6.Hou N, et al. Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology. 2008;149:1654–1665. doi: 10.1210/en.2007-0988. [DOI] [PubMed] [Google Scholar]
- 7.Drechsel DA, Patel M. Differential contribution of the mitochondrial respiratory chain complexes to reactive oxygen species production by redox cycling agents implicated in parkinsonism. Toxicological Sciences. 2009;112:427–434. doi: 10.1093/toxsci/kfp223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kou B, et al. Xanthine oxidase interaction with vascular endothelial growth factor in human endothelial cell angiogenesis. Microcirculation. 2008;15:251–267. doi: 10.1080/10739680701651495. [DOI] [PubMed] [Google Scholar]
- 9.Zuo L, et al. Lipoxygenase-dependent superoxide release in skeletal muscle. J. Appl. Physiol. 2004;97:661–668. doi: 10.1152/japplphysiol.00096.2004. [DOI] [PubMed] [Google Scholar]
- 10.Gardner HW. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic. Biol. Med. 1989;7:65–86. doi: 10.1016/0891-5849(89)90102-0. [DOI] [PubMed] [Google Scholar]
- 11.Møller IM, et al. Protein carbonylation and metal-catalyzed protein oxidation in a cellular perspective. J Proteomics. 2011;74:2228–2242. doi: 10.1016/j.jprot.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Codoñer-Franch P, et al. Oxidant mechanisms in childhood obesity: the link between inflammation and oxidative stress. Transl Res. 2011;158:369–384. doi: 10.1016/j.trsl.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Poli G, et al. 4-Hydroxynonenal-protein adducts: A reliable biomarker of lipid oxidation in liver diseases. Mol. Aspects Med. 2008;29:67–71. doi: 10.1016/j.mam.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 14.Uchida K, Stadtman ER. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4544–4548. doi: 10.1073/pnas.89.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long EK, et al. Ethanol withdrawal increases glutathione adducts of 4-hydroxy-2-hexenal but not 4-hydroxyl-2-nonenal in the rat cerebral cortex. Free Radic. Biol. Med. 2010;48:384–390. doi: 10.1016/j.freeradbiomed.2009.10.048. [DOI] [PubMed] [Google Scholar]
- 16.Zhong L, et al. Aldo-keto reductase family 1 B10 protein detoxifies dietary and lipid-derived alpha, beta-unsaturated carbonyls at physiological levels. Biochem. Biophys. Res. Commun. 2009;387:245–250. doi: 10.1016/j.bbrc.2009.06.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singhal SS, et al. RLIP76: A novel glutathione-conjugate and multi-drug transporter. Biochem. Pharmacol. 2009;77:761–769. doi: 10.1016/j.bcp.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meany DL, et al. Identification of carbonylated proteins from enriched rat skeletal muscle mitochondria using affinity chromatography-stable isotope labeling and tandem mass spectrometry. Proteomics. 2007;7:1150–1163. doi: 10.1002/pmic.200600450. [DOI] [PubMed] [Google Scholar]
- 19.Roe MR, et al. Proteomic mapping of 4-hydroxynonenal protein modification sites by solid-phase hydrazide chemistry and mass spectrometry. Anal. Chem. 2007;79:3747–3756. doi: 10.1021/ac0617971. [DOI] [PubMed] [Google Scholar]
- 20.Grimsrud PA, et al. Carbonylation of adipose proteins in obesity and insulin resistance: identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol. Cell Proteomics. 2007;6:624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Madian AG, Regnier FE. Proteomic identification of carbonylated proteins and their oxidation sites. J. Proteome Res. 2010;9:3766–3780. doi: 10.1021/pr1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madian AG, et al. Determining the effects of antioxidants on oxidative stress induced carbonylation of proteins. Anal. Chem. 2011;83:9328–9336. doi: 10.1021/ac201856g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng J, et al. Quantitative proteomic profiling of muscle type-dependent and age-dependent protein carbonylation in rat skeletal muscle mitochondria. J Gerontol A Biol Sci Med Sci. 2008;63:1137–1152. doi: 10.1093/gerona/63.11.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson EJ, Neufer PD. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H(2)O(2) generation. Am. J. Physiol., Cell Physiol. 2006;290:C844–C851. doi: 10.1152/ajpcell.00402.2005. [DOI] [PubMed] [Google Scholar]
- 25.Barreiro E, Hussain SNA. Protein carbonylation in skeletal muscles: impact on function. Antioxid. Redox Signal. 2010;12:417–429. doi: 10.1089/ars.2009.2808. [DOI] [PubMed] [Google Scholar]
- 26.Nuss JE, et al. Oxidative modification and aggregation of creatine kinase from aged mouse skeletal muscle. Aging (Albany NY) 2009;1:557–572. doi: 10.18632/aging.100055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaudhuri AR, et al. Detection of protein carbonyls in aging liver tissue: A fluorescence-based proteomic approach. Mech. Ageing Dev. 2006;127:849–861. doi: 10.1016/j.mad.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 28.Rabek JP, et al. Carbonylation of ER chaperone proteins in aged mouse liver. Biochem. Biophys. Res. Commun. 2003;305:566–572. doi: 10.1016/s0006-291x(03)00826-x. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharya A, et al. Attenuation of liver insoluble protein carbonyls: indicator of a longevity determinant? Aging Cell. 2011;10:720–723. doi: 10.1111/j.1474-9726.2011.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seki S, et al. Pathological significance of oxidative cellular damage in human alcoholic liver disease. Histopathology. 2003;42:365–371. doi: 10.1046/j.1365-2559.2003.01599.x. [DOI] [PubMed] [Google Scholar]
- 31.Reed T, et al. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer's disease. Neurobiol. Dis. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 32.Reed TT, et al. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Research. 2009;1274:66–76. doi: 10.1016/j.brainres.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 33.da Cunha FM, et al. Aging and calorie restriction modulate yeast redox state, oxidized protein removal, and the ubiquitin-proteasome system. Free Radic. Biol. Med. 2011;51:664–670. doi: 10.1016/j.freeradbiomed.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 34.Jung T, Grune T. The proteasome and its role in the degradation of oxidized proteins. IUBMB Life. 2008;60:743–752. doi: 10.1002/iub.114. [DOI] [PubMed] [Google Scholar]
- 35.Hunt ND, et al. Bioenergetics of aging and calorie restriction. Ageing Res. Rev. 2006;5:125–143. doi: 10.1016/j.arr.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 36.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 37.Unger RH, et al. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta. 2010;1801:209–214. doi: 10.1016/j.bbalip.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Chen XH, et al. TNF-alpha induces mitochondrial dysfunction in 3T3-L1 adipocytes. Molecular and Cellular Endocrinology. 2010;328:63–69. doi: 10.1016/j.mce.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Frohnert BI, et al. Increased adipose protein carbonylation in human obesity. Obesity (Silver Spring) 2011;19:1735–1741. doi: 10.1038/oby.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samuel VT, Shulman GI. Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Awasthi YC, et al. Regulation of 4-hydroxynonenal mediated signaling by glutathione S-transferases. Meth. Enzymol. 2005;401:379–407. doi: 10.1016/S0076-6879(05)01024-4. [DOI] [PubMed] [Google Scholar]
- 42.Cheng JZ, et al. Transfection of mGSTA4 in HL-60 cells protects against 4-hydroxynonenal-induced apoptosis by inhibiting JNK-mediated signaling. Arch. Biochem. Biophys. 2001;392:197–207. doi: 10.1006/abbi.2001.2452. [DOI] [PubMed] [Google Scholar]
- 43.Curtis JM, et al. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes. 2010;59:1132–1142. doi: 10.2337/db09-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valerio A. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. Journal of Clinical Investigation. 2006;116:2791–2798. doi: 10.1172/JCI28570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perluigi M, et al. Proteomic analysis of 4-hydroxy-2-nonenal-modified proteins in G93A-SOD1 transgenic mice--a model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2005;38:960–968. doi: 10.1016/j.freeradbiomed.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 46.Wenk GL. Neuropathologic changes in Alzheimer's disease. J Clin Psychiatry. 2003;64:7–10. [PubMed] [Google Scholar]
- 47.Liu Q, et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic. Biol. Med. 2005;38:746–754. doi: 10.1016/j.freeradbiomed.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Butterfield DA, et al. Oxidative stress in Alzheimer's disease brain: new insights from redox proteomics. Eur. J. Pharmacol. 2006;545:39–50. doi: 10.1016/j.ejphar.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 49.Cenini G, et al. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer's disease. J. Cell. Mol. Med. 2008;12:987–994. doi: 10.1111/j.1582-4934.2008.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sultana R, et al. Oxidatively modified proteins in Alzheimer's disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Acta Neuropathol. 2009;118:131–150. doi: 10.1007/s00401-009-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi J, et al. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson“s and Alzheimer”s diseases. J. Biol. Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 52.Kabuta T, et al. Aberrant molecular properties shared by familial Parkinson's disease-associated mutant UCH-L1 and carbonyl-modified UCH-L1. Hum. Mol. Genet. 2008;17:1482–1496. doi: 10.1093/hmg/ddn037. [DOI] [PubMed] [Google Scholar]
- 53.Mehta M, et al. New acetylcholinesterase inhibitors for Alzheimer's disease. Int J Alzheimers Dis. 2012;2012:728983. doi: 10.1155/2012/728983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi D-H, et al. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson's disease. Antioxid. Redox Signal. 2012;16:1033–1045. doi: 10.1089/ars.2011.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ren H, et al. Oxidized DJ-1 interacts with the mitochondrial protein BCL-XL. J. Biol. Chem. 2011;286:35308–35317. doi: 10.1074/jbc.M110.207134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang P, et al. Neuroprotective effect of gypenosides against oxidative injury in the substantia nigra of a mouse model of Parkinson's disease. J. Int. Med. Res. 2010;38:1084–1092. doi: 10.1177/147323001003800336. [DOI] [PubMed] [Google Scholar]
- 57.Brotherton TE, et al. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc. Natl. Acad. Sci. U.S.A. 2012;109:5505–5510. doi: 10.1073/pnas.1115009109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaw PJ, et al. Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann. Neurol. 1995;38:691–695. doi: 10.1002/ana.410380424. [DOI] [PubMed] [Google Scholar]
- 59.Bizzozero OA, et al. Elevated protein carbonylation in the brain white matter and gray matter of patients with multiple sclerosis. J. Neurosci. Res. 2005;81:687–695. doi: 10.1002/jnr.20587. [DOI] [PubMed] [Google Scholar]
- 60.Zheng J, Bizzozero OA. Accumulation of protein carbonyls within cerebellar astrocytes in murine experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2010;88:3376–3385. doi: 10.1002/jnr.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu H, et al. Antioxidants and Phase 2 Enzymes in Macrophages: Regulation by Nrf2 Signaling and Protection Against Oxidative and Electrophilic Stress. Experimental Biology and Medicine. 2008;233:463–474. doi: 10.3181/0711-RM-304. [DOI] [PubMed] [Google Scholar]
- 62.Blüher M, et al. Activated Ask1-MKK4-p38MAPK/JNK stress signaling pathway in human omental fat tissue may link macrophage infiltration to whole-body Insulin sensitivity. J. Clin. Endocrinol. Metab. 2009;94:2507–2515. doi: 10.1210/jc.2009-0002. [DOI] [PubMed] [Google Scholar]
- 63.Yu Y, Richardson DR. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. Journal of Biological Chemistry. 2011;286:15413–15427. doi: 10.1074/jbc.M111.225946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McMahon M, et al. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 65.Levonen A-L, et al. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McMahon M, et al. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Numazawa S, et al. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am. J. Physiol., Cell Physiol. 2003;285:C334–C342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- 68.Mahaffey CM, et al. Multidrug-resistant protein-3 gene regulation by the transcription factor Nrf2 in human bronchial epithelial and non-small-cell lung carcinoma. Free Radic. Biol. Med. 2009;46:1650–1657. doi: 10.1016/j.freeradbiomed.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fujino G, et al. Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol. Cell. Biol. 2007;27:8152–8163. doi: 10.1128/MCB.00227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fang J, Holmgren A. Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo. J. Am. Chem. Soc. 2006;128:1879–1885. doi: 10.1021/ja057358l. [DOI] [PubMed] [Google Scholar]
- 71.Sharma R, et al. 4-Hydroxynonenal self-limits fas-mediated DISC-independent apoptosis by promoting export of Daxx from the nucleus to the cytosol and its binding to Fas. Biochemistry. 2008;47:143–156. doi: 10.1021/bi701559f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang L, et al. Depletion of cytosolic or mitochondrial thioredoxin increases CYP2E1-induced oxidative stress via an ASK-1-JNK1 pathway in HepG2 cells. Free Radic. Biol. Med. 2011;51:185–196. doi: 10.1016/j.freeradbiomed.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong CM, et al. Cell signaling by protein carbonylation and decarbonylation. Antioxid. Redox Signal. 2010;12:393–404. doi: 10.1089/ars.2009.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Velez JM, et al. p53 Regulates oxidative stress-mediated retrograde signaling: a novel mechanism for chemotherapy-induced cardiac injury. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0018005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shearn CT, et al. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibition by 4-hydroxynonenal leads to increased Akt activation in hepatocytes. Mol. Pharmacol. 2011;79:941–952. doi: 10.1124/mol.110.069534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Padrão AI, et al. Effect of lifestyle on age-related mitochondrial protein oxidation in mice cardiac muscle. Eur. J. Appl. Physiol. 2012;112:1467–1474. doi: 10.1007/s00421-011-2100-3. [DOI] [PubMed] [Google Scholar]
- 77.Castegna A, et al. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 78.Choi J, et al. Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 2005;280:11648–11655. doi: 10.1074/jbc.M414327200. [DOI] [PubMed] [Google Scholar]
- 79.Barreiro E, et al. Expression and carbonylation of creatine kinase in the quadriceps femoris muscles of patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2005;33:636–642. doi: 10.1165/rcmb.2005-0114OC. [DOI] [PubMed] [Google Scholar]
- 80.Marin-Corral J, et al. Redox balance and carbonylated proteins in limb and heart muscles of cachectic rats. Antioxid. Redox Signal. 2010;12:365–380. doi: 10.1089/ars.2009.2818. [DOI] [PubMed] [Google Scholar]