

Abstract
The synthesis of the marine neurotoxin azaspiracid-1 has been accomplished. The individual fragments were synthesized by catalytic enantioselective processes: A hetero-Diels-Alder reaction to afford the E- and HI-ring fragments, a carbonyl-ene reaction to furnish the CD-ring fragment, and a Mukaiyama aldol reaction to deliver the FG-ring fragment. The subsequent fragment couplings were accomplished by aldol and sulfone anion methodologies. All ketalization events to form the non-acyclic target were accomplished under equilibrating conditions utilizing the imbedded configurations of the molecule to adopt one favored conformation. A final fragment coupling of the anomeric EFGHI-sulfone anion to the ABCD-aldehyde completed the convergent synthesis of (+)-azaspiracid-1.
Introduction
Background
Azaspiracid poisoning is a recent toxic syndrome first reported in 1995. On this occasion, at least 8 people in the Netherlands became ill after consuming blue mussels (Mytilus edulis) harvested in Killary Harbor, Ireland.1 Subsequently, intoxications related to azaspiracid poisoning have been reported in several countries including the UK, Norway,2 Spain, France,3 Italy,4 as well as Morocco.5 An active search for the causative toxin provided the motivation for its isolation by the Satake group in 1998.6 Only minute amounts of the active toxin (2 mg) were obtained as an amorphous solid from 20 kg of mussel meat collected at Killary Harbor. Extensive NMR studies of this material resulted in the proposed structural assignment 1 of azaspiracid-1 (Figure 1). In this nonacyclic structure, the absolute configuration remained uncertain. In addition, the flexible methylene bridge at C27 prevented an unambiguous determination of the relative configurations between the ABCDE- and FGHI-domains. All remaining stereochemistries such as the dense acyclic C19–C21 junction between the D- and the E-rings was assigned based on an analysis of NOE interactions.6 A total of 11 azaspiracid analogues differing slightly in their methylation and hydroxylation patterns have subsequently been described, five of these analogues were isolated and characterized by NMR spectroscopy6,7 while the remaining analogues were identified by tandem mass spectrometry.8
Figure 1.

Originally proposed structure (1)6 and correct structure of (−)-azaspiracid-1 (2).22
The origin of these toxins has been proposed to be a marine dinoflagellate,4 and their occurrence has subsequently been reported in multiple shellfish species including mussels, oysters, scallops, clams, and cockles.9 Extensive studies of the azaspiracids have revealed a number of toxic effects such as cytoxicity in mammalian cell lines,10 teratogenic effects in finfish,11 pertubation of cell adhesion,12 disorganization of the actin cytoskeleton13 and inhibition of the electrical activity of neuronal networks,14 although the specific modes of all such actions of azaspiracids yet remain unclear.15 The scarcity of azaspiracids from natural sources serves as a further complication in mechanistic elucidations aiming to better understand their toxicity and ultimately prevent their hazards to human health.
The intriguing structure of this toxin has furthermore served to stimulate interest in the chemical community, in particular by the research groups of Nicolaou,16 Carter,17,18 and Forsyth,19 and others.20 The Nicolaou group reported a successful synthesis of the proposed structure 1 in 2003, only to clearly reveal its non-identity to the isolated natural product.21 Subsequent impressive efforts in the Nicolaou group using a combination of degradation studies and the synthesis of multiple diastereomers resulted in a significant structural revision of azaspiracid-1 by total synthesis. These landmark studies allowed the unambiguous structural assignment of (−)-azaspiracid-1 (2) in 2004.22 As highlighted in Figure 1, the position of the A-ring olefin (yellow) was different and the relative configurations of the CD- and FGHI-domains (green) were opposite in the correctly assigned natural product 2 compared to the originally proposed structure 1. Based on these findings, an improved synthesis of (−)-azaspiracid-1 (2) was recently reported by the Nicolaou group.23
Synthesis Plan
Conformational analysis of azaspiracid-1 suggests that each of the multiple ketals exists in their thermodynamically favored configuration on the basis of anomeric stabilization and steric effects (2, Figure 2). Accordingly, a number of thermodynamically controlled ketalization events were incorporated into the synthesis plan under the assumption that the desired configurations would be achieved under equilibrating conditions. Notably, whereas the ABC-bis(spiroketal) portion of the originally proposed structure 16 contained only one anomeric effect (not shown), the ABC-moiety of the corrected (−)-azaspiracid-1 structure (2)22 incorporates two stabilizing anomeric effects and is presumed to be in its favored configuration. Thus, a thermodynamically controlled spiroketalization should be effective in controlling both spiroketal stereocenters at C10 and C13. We also anticipated that the FG-ring bicyclic ketal might also be constructed by an acid-catalyzed ketalization. Finally, we suspected that the HI-ring spiroaminal would spontaneously cyclize to provide the desired configuration at the C36 ring junction from an acyclic precursor with an unprotected C40 amino group.
Figure 2.

3D-representation of (−)-azaspiracid-1 (2) highlighting stabilizing anomeric effects in the molecule.
In our own studies,24 we elected to pursue the synthesis of (+)-azaspiracid-1 (ent-2, Scheme 1). This target required only minimal changes in our original synthesis plan designed to pursue the initially proposed structure 1 of (−)-azaspiracid-1. Thus, the design of a new AB-ring fragment and the synthesis of the enantiomer of the E-ring fragment were required while all other regions of the molecule remain unaltered.
Scheme 1.

Retrosynthetic analysis of (+)-azaspiracid-1 (ent-2). See ref31 for abbreviations.
Our principal disconnections in the deconstruction of (+)-azaspiracid-1 (ent-2) are outlined in Scheme 1. In the final fragment coupling, the C21-anion of the anomeric EFGHI-sulfone 4 was envisioned to be a C20 electrophile such as aldehyde 3. This strategy could provide a convergent approach to (+)-azaspiracid-1 since all 9 rings are formed prior to this final sulfone anion addition. Related sulfonyl carbanion additions have substantial literature precedents, both in the original investigations from the carbohydrate field25,26 and subsequently in several advanced fragment couplings in some of our previous total synthesis projects.27–29
The plan for the deconstruction of the ABCD synthon 3 involves a thermodynamically controlled spiroketalization event to give the intermediary ketone 5. After retrosynthetic installation of a sulfone moiety at C12, an addition of this C12-sulfone anion could provide AB-ring fragment 7 and CD-ring fragment 8 of comparable structural complexity. In the assemblage of the EFGHI-sulfone 4, ketalization events under equilibrating conditions were likewise incorporated to give intermediate 9. The illustrated fragment couplings to construct the C21–C40 chain of 9 would be two aldol addition events: a boron-mediated aldol addition to form the C26–C27 bond and a chelate-controlled Mukaiyama aldol addition30 to form the C34–C35 bond. Thus, the three fragments 10, 11, and 12 of equal complexity would represent our initial targets in the synthesis of the fully elaborated EFGHI-sulfone 4.
The integration of catalytic enantioselective processes into our synthesis projects has been an ongoing objective in our research program. In this regard, the Cu-catalyzed hetero-Diels-Alder reaction to give methyl-substituted dihydropyrans (eq 1)32 was specifically developed for the synthesis of the azaspiracid E-ring fragment 10. Independently, the glyoxylate-ene reaction to afford α-hydroxy-γ,δ-unsaturated esters (eq 2)33 could be used to form the C17 stereocenter of the CD-ring fragment 8 while the Mukaiyama aldol reaction to furnish unsaturated lactones featuring 1,2-anti diol arrays (eq 3)34 could be explored to construct the C32–C33 motif of the FG-ring fragment 11.
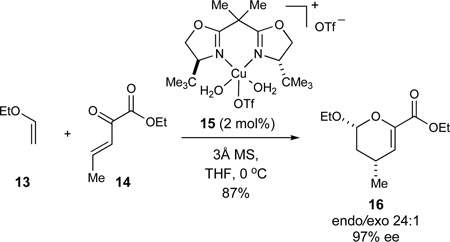 |
(1) |
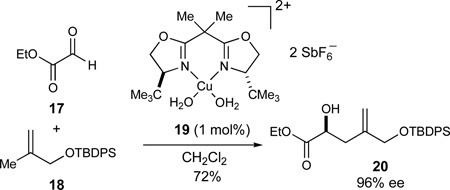 |
(2) |
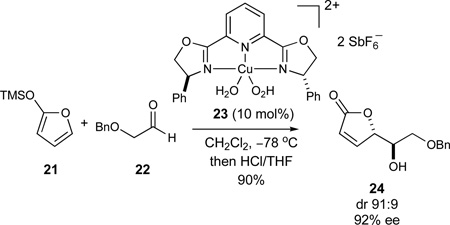 |
(3) |
Results and Discussion
Synthesis of the AB-Ring Fragment 7
In the design of the azaspiracid AB-ring fragment, the major obstacle was anticipated to be the transposed A-ring olefin in the revised azaspiracid-1 structure (2), thus creating a labile C6 divinylcarbinol stereocenter. In order to suppress the lability of this stereocenter, we selected the C10 lactol methyl ether 7, which includes the C6 divinylcarbinol in the ketal formation, as our targeted AB-ring fragment. As depicted in Figure 3, the endocyclic double bond is taken out of conjugation with the C6 C–O bond in ketal 7. This eliminates the impact of this π-bond toward hyperconjugative donation, thus influencing the stability of the C6 divinylcarbinol stereocenter in a sufficiently positive manner.
Figure 3.

Depiction of C6 CO*-orbital in AB-ring fragment 7.
In order to preserve the desired oxidation state of the C1 carboxyl terminus, we incorporated the C1 carboxylic acid as its derived t-Bu ester into our synthesis plan (Scheme 2). While attempted cross metathesis35 between alkene 2536 and acrylate 2637 was low yielding with the Grubbs second-generation catalyst38 (10–35% yield), the more stable Grubbs–Hoveyda catalyst 2739 was considerably more efficient, affording Weinreb amide 30 in excellent yield (88%). The C7–C12 fragment was introduced as the racemic acetylene 29, readily prepared from acrolein by conjugate addition of thiophenol, followed by addition of the allenyl Grignard reagent (96% yield, 2 steps). The two fragments were coupled by the selective 1,2-addition of the dianion derived from acetylene 29 to the unsaturated Weinreb amide 30. After considerable experimentation, optimum conditions involved the conversion of acetylene 29 to its derived Grignard reagent by deprotonation with n-butyllithium and transmetallation with MgBr2·OEt240 followed by the addition of THF at a late stage of the reaction to facilitate solubilization. Since the product of the alkyne addition proved somewhat unstable, direct acetylation of the unpurified mixture provided the C1–C12 ene-ynone 31 in 83% overall yield for the 2 steps.
Scheme 2.

Synthesis of the C1–C12 chain of the AB-ring fragment. Reagents and conditions: a) 27 (5 mol%), CH2Cl2, 40 °C, 88%; b) PhSH, Et3N, CH2Cl2; c) allenylMgBr, Et2O, 0 °C, 96% (2 steps); d) 29, n-BuLi, Et2O, −78 °C, then MgBr2·OEt2, then 30, −78 to 0 °C, THF added, then RT; e) Ac2O, DMAP, pyr, CH2Cl2, 83% (2 steps). See ref31 for abbreviations.
Based on literature precedents for the enantioselective reduction of ene-ynones,41 the C6-stereocenter was introduced with high enantioselectivity in a supra-stoichiometric CBS-reduction42 using 2 equiv of oxazaborolidone 32 and BH3·SMe2 to afford the desired alcohol 33 (86%, 94–97% ee, Scheme 3).43 Initial attempts to use a catalytic amount of oxazaborolidone 32 were detrimental both for the yield and the observed enantioselectivity in this transformation. Semi-hydrogenation of the alkyne under Lindlar conditions proceeded in high yield, although the product was contaminated with 3–5% of the undesired C4–C5 alkane that was separated from the major product by MPLC at a later stage. Subsequent TBS-protection (92% desired product over two steps) was followed by removal of the acetate moiety (K2CO3, MeOH). In the oxidation to the β,γ-unsaturated ketone 34, Dess-Martin periodinane also oxidized the phenylsulfide to the corresponding sulfoxide.44 A Parikh-Doering oxidation45 using Et3N as the base afforded the product 34 accompanied by approximately 20% of the undesired α,β-unsaturated ketone resulting from base-induced olefin migration. This undesired side reaction was suppressed using the sterically hindered i-Pr2NEt as the base to give the desired β,γ-unsaturated ketone 34 in good yield (SO3·pyr, DMSO, i-Pr2NEt, 91%, 2 steps).
Scheme 3.

Synthesis of the AB-Ring Fragment 7. Reagents and conditions: a) 32 (2 equiv), BH3·SMe2, THF, 0 °C, 86%, 94–97% ee; b) H2 (1 atm), Pd/CaCO3/Pb (5%), pyr, benzene; c) TBSCl, DMAP, imidazole, DMF, 92%, 3–5% C4–C5 alkane (2 steps); d) K2CO3, MeOH; e) SO3·Pyr, DMSO, i-Pr2NEt, CH2Cl2, −25 °C, 91% (2 steps); f) HF·pyr, pyr, THF, 0 °C, 74%; g) PPTS, HC(OMe)3, MeOH, CH2Cl2, −10 °C, 92%; h) H2O2, (NH4)6Mo7O24, MeOH, 0 °C, 88%; i) LiAlH4, Et2O, −20 °C, 92%; j) TIPSCl, imidazole, DMF, 98%. See ref31 for abbreviations.
The stage was now set for the crucial lactol formation of the C6 alcohol. This was best effected with HF·pyr buffered with excess pyridine at 0 °C to give primarily the desired lactol (74%) accompanied by 20–25% of the undesired conjugated trienone resulting from elimination of the C6 silyl ether substituent. To our relief, this lactol was smoothly transformed into the desired lactol methyl ether in excellent yield (PPTS, MeOH, 92%). This was the first time among various substrates investigated that the C6 divinylcarbinol moiety proved stable to acidic conditions, thus validating the anticipated stability of the C6-lactol (Figure 3), and providing us with a credible lead for the anticipated spiroketalization. Finally, the sulfide moiety was oxidized to the derived sulfone (H2O2, (NH4)6Mo7O24, 88%). In a later variation on the synthesis plan, the oxidation of the carboxyl terminus needed to be altered (vide supra). Accordingly, the ester terminus of this synthon was reduced and protected as its silyl ether (LiAlH4, 92%; TIPSCl, 98%), thus concluding the synthesis of the desired AB-ring fragment 7 and its carboxylate analogue 34 in 13 linear steps from 25.

Synthesis of the CD-Ring Fragment 8
Initial synthesis via a stereoselective C14-methylation
The first of two syntheses of this fragment is illustrated in Scheme 4. In our synthesis of this synthon, we envisioned that the enantioselective Cu2+-catalyzed glyoxylate-ene reaction developed in these laboratories could provide the initial C17-stereocenter (eq 2).33 Further studies with these substrates proved that the reaction could routinely be conducted on a 20-g scale in good yields and enantioselectivities using 1 mol% of the bis(aquo)Cu2+ complex 1946 (89% yield, 98% ee, Scheme 4). In addition, the Cu2+-catalyst was sufficiently Lewis acidic that commercial ethyl glyoxalate could be used directly in its polymerized form without prior distillation to afford the monomeric aldehyde. Whereas the substrate concentration in these experiments (0.27M) was governed by the limited solubility of complex 19 in CH2Cl2, concomitant lowering of the catalyst loading and an increase of the substrate concentration (0.89M) allowed for the use of only 0.1 mol% of catalyst 19. Almost identical results were obtained (86% yield, 96% ee) although the reaction times to reach full conversion in the latter case increased considerably (12 h versus 5 days, respectively).
Scheme 4.

Initial synthesis of the CD-ring fragment. Reagents and conditions: a) NaH, BnBr, CH2Cl2, 0 °C to RT, 99%; b) DIBALH, toluene, −94 °C; c) ZnCl2, 36, 59% (2 steps), dr 93:7; d) O3, MeOH, CH2Cl2, −78 °C, then Me2S, RT, 79%; e) Et3SiH, BF3·OEt2, CH2Cl2, −78 °C, 82%, dr 90:10. See ref31 for abbreviations.
The resultant α-hydroxy ester 20 was subsequently benzylated (NaH, BnBr, 99%) and reduced to the corresponding aldehyde 35 (DIBALH, −94 °C) that was used directly without purification in the next step. The zinc homoenolate of siloxycyclopropane 3647 was then prepared in situ and added stereoselectively to aldehyde 35 according to the Kuwajima procedure.48 In accordance with presumed chelate-control exerted by the Zn2+ Lewis acid, the resulting protected syn-diol 37 was obtained in high diastereoselectivity (59%, dr 93:7, 2 steps). Ozonolysis of the olefin in the presence of methanol directly afforded the C19 lactol methyl ethers 38 in 79% yield. The C19 stereocenter was then established in a stereoselective reduction with Et3SiH mediated by BF3·OEt2 to afford the desired trisubstituted tetrahydrofuran 39 (82%, dr 90:10). The observed diastereoselectivity in this reduction is in good agreement with the stereoelectronic model 40 for nucleophilic additions to 5-membered cyclic oxocarbenium ions proposed by Woerpel.49
At this stage, our strategy for the stereoselective introduction of the C14-methyl substituent was influenced by a report by Narasaka that acyclic δ-hydroxy esters such as 41 can be alkylated with high stereoselectivity (eq 4). The presence of HMPA was proven to be critical for the observed stereoselectivity as silyl-trapping experiments afforded silyl enol ethers of opposite stereochemistry in the absence and presence of HMPA, respectively. An 8-membered internal chelate 42 in the presence of HMPA was invoked as a possibility for explaining the observed high stereoselectivity.50
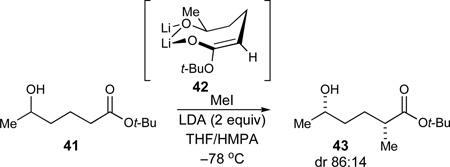 |
(4) |
Prior to the actual methylation event, ethyl ester 39 was converted into its derived amide 44 with the magnesium amide of morpholine51 (82%, Scheme 5). It was assumed that A1,3 strain in amide 44 would prevent bis(alkylation), thus allowing the use of excess external base to achieve full conversion in the methylation. The subsequent C14-methylation thus proceeded in excellent yield and moderately good diastereoselectivity to afford the alkylated product 45 (LiHMDS, 96%, dr 86:14).
Scheme 5.

C14-Methylation. Reagents and conditions: a) i-PrMgCl, morpholine, THF, −78 to −20 °C, 82%; b) LiHMDS, MeI, THF, −78 °C, 96%, dr 86:14. See ref31 for abbreviations.
Improved synthesis of the CD-ring fragment 8 via a direct introduction of a C14-methylated fragment
While the above findings afforded us a workable route to the CD-ring fragment, we decided to investigate a modified approach to the orthogonally protected CD-fragment 8 (Scheme 1) which would involve the direct introduction of a fragment incorporating the C14 methyl group (Scheme 6). In this regard, the hydroxy ester 20 derived from the carbonyl ene reaction (Scheme 4) was converted into the corresponding Weinreb amide52 (MeNH(OMe)·HCl, Me3Al, 98%). The C17 hydroxy group was then protected as its derived PMB ether 46 under standard basic conditions with no epimerization of the lone stereocenter (PMBBr, NaH, DMF, 95%), presumably prevented by A1,3 strain of the Weinreb amide.
Scheme 6.

Second synthesis of the CD-ring fragment 8. Reagents and conditions: a) MeNH(OMe)·HCl, Me3Al, THF, 98%; b) PMBBr, NaH, DMF, 0 °C, 95%; c) 47,53 t-BuLi, Et2O, −78 °C, then 46, 90%; d) L-Selectride, THF, −78 °C, 96%, dr > 95:5; e) i) O3, pyridine, CH2Cl2/MeOH (5:1 v/v), −78 °C, then Me2S, −78 °C to RT; ii) PPTS, MeOH, 90%; f) Et3SiH, BF3·OEt2, CH2Cl2, −78 °C, 84%, dr >95:5; g) DDQ, CH2Cl2/pH 7 buffer (9:1 v/v), 0 °C, 94%; h) TESCl, imidazole, DMF, 97%; i) LiDBB, THF, −78 °C, 96%; j) SO3·Pyr, DMSO, i-Pr2NEt, CH2Cl2, −30 °C, 97%. See ref31 for abbreviations.
Metallation of the readily available iodide 4753 with t-BuLi54 followed by addition of Weinreb amide 46 then introduced the remaining 4-carbon subunit (−78 °C, 90%). Notably, optimal results were obtained when 1.5 equiv of t-BuLi relative to the iodide 47 were used, while 2 equiv of t-BuLi afforded substantial amounts of the undesired t-Bu-ketone. The resulting α-alkoxyketone was then reduced with L-Selectride to afford alcohol 48 as the only detectable diastereomer (96%, dr >95:5). This stereoselective reduction, presumably the result of Felkin control, was confirmed by advanced Mosher's ester analysis.55 Careful ozonolysis of the 1,1-disubstituted olefin in the presence of Sudan III dye as an indicator56 allowed for the selective oxidative cleavage of the olefin (O3, −78 °C, then Me2S) without any accompanying oxidation of the benzylic ether protecting groups. Subsequent ketalization (PPTS, MeOH) afforded the methyl ketal 49 (90%, 2 steps). In analogy to the reduction to give 39 described in Scheme 4, stereoselective reduction with Et3SiH mediated by BF3·OEt2 then provided the desired trisubstituted tetrahydrofuran 50 (84%, dr > 95:5) with the primary by-product being derived from elimination to the corresponding undesired furan.
Whereas the observed diastereoselectivity in the 49→50 oxocarbenium ion reduction (dr > 95:5) is in agreement with the stereoelectronic model 40 proposed by Woerpel,49 it is noteworthy that the analogous reduction of the O17-TBS protected methyl ketal 51 proceeded with no diastereoselectivity to give tetrahydrofuran 52 (dr 1:1, eq 5). This result suggests that steric factors also play a significant role for these sterically encumbered substrates.
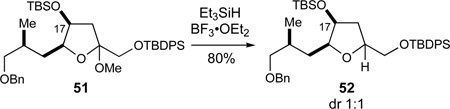 |
(5) |
Since a labile C17-silyl ether was required for the projected ketalization events, the PMB group was exchanged for a TES ether by oxidative cleavage with DDQ (94%) and re-protection under standard conditions (TESCl, 97%, Scheme 6). As this TES ether proved to be labile to standard palladium-catalyzed hydrogenolysis conditions, the terminal benzyl ether was removed under radical anion conditions (LiDBB,57 96%). Finally, a Parikh–Doering oxidation45 afforded aldehyde 8 in 97% yield. Altogether, this second-generation sequence obviates the need for any diastereomer separation and efficiently provided the desired CD-ring fragment 8.
Union of the AB- and CD-ring fragments
With the AB- and CD-ring fragments in hand, we proceeded to investigate their coupling and further elaboration into the ABCD-bis(spiroketal). In the initial synthesis plan, we anticipated that a selective kinetic deprotonation of the AB-ring fragment might be feasible α to the C12-sulfone under the appropriate experimental conditions in the presence of a C1 t-Bu ester, although the α-protons of esters and sulfones are of comparable thermodynamic acidity. This expectation was born out by initial investigations using the sulfone 53, prepared by essentially the same sequence as previously described for AB-ring fragment 7, for which the C6 divinylcarbinol moiety was masked only as its acyclic silyl ether. Thus, selective addition of sulfone 53 to the C17-TBS-protected aldehyde 54 proceeded to give a mixture of diastereomeric hydroxysulfones 55 (74% yield, eq 6).
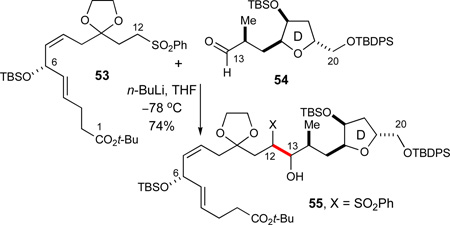 |
(6) |
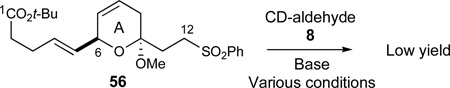 |
(7) |
Unfortunately, as eluded to earlier in our discussion of Figure 3, adducts 55 incorporating acyclic C6-divinylcarbinol moieties proved to be too unstable to successfully participate in the subsequent derivatization into the ABCD-bis(spiroketal) under a variety of conditions and were abandoned for further studies. However, much to our surprise, the corresponding addition of the superficially similar sulfone 56 to C17-TES-protected aldehyde 8 was nonselective for C12 deprotonation under a variety of experimental conditions (eq 7). These findings suggest that although α-deprotonation of the C12 sulfone moiety may be kinetically favored over enolization of the C1 ester, subtle steric differences may have a negative impact on the kinetics in such desired α-deprotonations.
Assembly of the ABCD-aldehyde 3
A simple solution to the above obstacle was realized using the AB-ring fragment 7 with the C1 terminus protected as its silyl ether (Scheme 7). With this pattern of orthogonal functionalities, sulfone deprotonation could readily be effected with LDA as also reported for structurally similar sulfone anion additions,18 a base which in previous experiments had displayed inferior kinetic selectivity for the sulfone moiety (eqs 6–7). The addition of the sulfonyl carbanion derived from 7 (1.6 equiv) to aldehyde 8 proceeded to give an initial mixture of four C12–C13 hydroxysulfones mixed with excess sulfone 7. Subsequent Dess-Martin oxidation59 afforded the two C12-diastereomeric ketosulfones 6 in 92% yield for the 2 steps. Subsequent recovery of unreacted sulfone 7 was achieved in 76% yield. The yield in this step is based on the limiting aldehyde reaction component. Sodium amalgam excision of the sulfone moiety under mild conditions (Na/Hg, NaH2PO4)60 then provided ketone 5 as a single diastereomer in 92% yield
Scheme 7.

Synthesis of the ABCD-aldehyde 3. Reagents and conditions: a) 7 (1.6 equiv), LDA, THF, −78 °C; then 8 (1.0 equiv); b) Dess-Martin periodinane, pyr, CH2Cl2, 92% (2 steps); c) Na/Hg (5%), NaH2PO4, THF/MeOH, −10 °C, 92%; d) TBAF, THF, −20 °C (workup); e) PPTS, CH2Cl2, 0 °C (83%, 2 steps); f) TBAF, AcOH, DMF, 86%; g) SO3·Pyr, DMSO, i-Pr2NEt, CH2Cl2, −30 °C, 91%. See ref31 for abbreviations.
In the spirocyclization event, the TES ether was selectively removed with 1 equiv of TBAF at low temperature to form the intermediary lactol 57. This step was followed by treatment of the unpurified mixture with PPTS in a non-hydroxylic solvent such as CH2Cl2. Under these conditions, the desired bis(spiroketal) isomer 58 was obtained in 83% yield (2 steps).61 The inclusion of a hydroxylic solvent such as MeOH led to the undesired formation of significant amounts of dimethylketals under otherwise identical spirocyclization conditions. The desired, thermodynamically favored bis(spiroketal) configuration in 58 was established by an NOE interaction between H6 and the methyl group at C14 (see 3D-depiction of 58) as also previously reported for analogous azaspiracid-derived bis(spiroketal) structures.6,22 Finally, the C20 terminus was elaborated by the selective removal of the TBDPS ether62 (TBAF, AcOH, 86%) followed by a Parikh-Doering oxidation45 (SO3·pyr, DMSO, 91%) to furnish the desired aldehyde 3. In summary, the above convergent sequence afforded the fully elaborated ABCD-aldehyde.
Enantioselective hetero-Diels-Alder approach to the syn-1,3-dimethyl syn-thons of azaspiracid-1
With a viable route to the ABCD-portion of azaspiracid-1 established, we turned our attention to the remaining EFGHI-region of the molecule. Initially, we focused on synthesizing the E-ring fragment to investigate potential strategies to form the central C20–C21 bond as will be discussed in the following sections. By inspection, two syn 1,3-dimethyl synthons of the same configuration are embedded in the E- and the I-rings of azaspiracid-1 (Scheme 8). We were particularly attracted to the possibility of constructing both of these subunits from the common precursor 59. Tetrahydropyran 59 could potentially be accessible from dihydropyran 60, which could be prepared using the hetero-Diels-Alder reaction previously developed in our laboratories (eq 1)32 for this particular target structure.
Scheme 8.

Syn-1,3-dimethyl synthons embedded in azaspiracid-1.
The two components of the hetero-Diels-Alder reaction, 6263 and 64,64 were readily prepared on multi-gram scales using slight modifications of reported literature procedures (Scheme 9). The subsequent cycloaddition proceeded readily using low loadings of the bench-stable hydrated copper complex 15 (2 mol%), provided it was dehydrated with molecular sieves prior to use. In contrast to the related cycloadditions reported previously for which THF was identified as the optimal solvent,32 these conditions gave rise to a mixture of cis and trans diastereomers of dihydropyran 60. Diethyl ether proved to be the optimal solvent for this particular hetero-Diels-Alder reaction (97% ee, dr 94:6), and the desired product 60 could be isolated in 84% yield as a single isomer on a 10–20 g scale. Since neither propenyl ether 62 nor dihydropyran 60 were observed to isomerize in control experiments, we believe the less than perfect diastereomeric ratios in the cycloaddition could potentially be explained by a stepwise reaction pathway under these Lewis acid catalyzed conditions. In the subsequent hydrogenation of the tetrasubstituted olefin, EtOAc proved to be superior to alcoholic solvents, giving rise to tetrahydropyran 59 in excellent yield and diastereoselectivity (H2, Pd/C, EtOAc, dr 98:2, 95% of 59).
Scheme 9.

Synthesis of the tetrahydropyran 59. Reagents and conditions: a) KOt-Bu, DMSO, 70 °C, 70%; b) 2-propenylMgBr, THF/Et2O, −78 °C, 80%; c) 15 (2 mol%), 3Å MS, Et2O, −40 °C, dr 94:6, 97% ee, 84% 60; d) H2 (1 atm), Pd/C (5 mol%), EtOAc, dr 98:2, 95% 59. See ref31 for abbreviations.
Chelate-controlled vinylmetal addition to form the central C20–C21 bond
With the syn 1,3-dimethyl motif of the E-ring thus in hand, we turned our attention to the elaboration of tetrahydroparan 59 into an appropriate E-ring intermediate that would allow us to establish the crucial C20–C21 bond. These studies would identify in which order the remaining EFGHI-region of the molecule should be assembled. One possible option we investigated as a viable C20–C21 fragment coupling was a chelate-controlled vinyllithium addition65,66 to a D-ring aldehyde synthon 65 (eq 8). As facile elaboration of the C21-1,1-disubstituted olefin was thought to be incompatible with the two olefins contained in the AB-ring fragment 7, this C20–C21 bond construction to join the CD- and the E-ring fragments was investigated prior to the introduction of the AB-ring fragment.
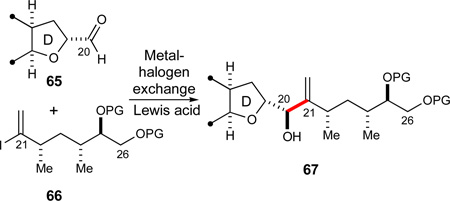 |
(8) |
Synthesis of the E-ring vinyl iodide 71
In order to arrive at an appropriately protected vinyl iodide fragment 66, the ester substituent of tetrahydropyran 59 was epimerized to its equatorial diastereomer (t-BuOK, THF, −50 °C, Scheme 10) prior to pyran ring opening to give the dithiane 68 (1,3-propanedithiol, BF3·OEt2, 95%, 2 steps). Subsequent ester reduction (LiBH4) and acetonide protection proceeded uneventfully in excellent yields (Me2C(OMe)2, PPTS, 98%, 2 steps). In the dithiane methylation,67 it was found that a 5-min deprotonation time of the intermediary dithiane at −78 °C was optimal. This afforded dithiane 69 in good yield (MeI, t-BuLi, HMPA/THF, 92%). Longer deprotonation times or higher temperatures afforded substantial amounts of addition of the reactive dithiane anion to HMPA as the electrophile. Oxidative removal of the dithiane moiety under mild conditions revealed the methyl ketone (Hg(ClO4)2, CaCO3,68 88%). Finally, the desired vinyl iodide was conveniently installed in a Shapiro reaction69 via an intermediate trisylhydrazone.70 Thus, the methyl ketone was converted into the intermediate trisylhydrazone 70 (trisylhydrazine, 93%) which proved slightly unstable in solution but could be stored neat in the freezer for weeks without any detectable decomposition. Metallation with t-BuLi followed by warming to 0 °C generated the intermediate vinyl anion before an iodine quench at −78 °C furnished vinyl iodide 71 in 86% overall yield. In summary, this sequence provided the desired E-ring vinyl iodide 71 in 10 linear steps from the hetero-Diels-Alder precursors 62/64.
Scheme 10.

Synthesis of the E-ring vinyl iodide 71. Reagents and conditions: a) t-BuOK, THF, −50 °C, quant.; b) 1,3-propanedithiol, BF3·OEt2, CH2Cl2, 95% (2 steps); c) LiBH4, THF, quant; d) 2,2-dimethoxypropane, PPTS, CH2Cl2, 98%; e) t-BuLi, HMPA, THF, −78 °C, then MeI, 92%; f) Hg(ClO4)2, CaCO3, H2O, THF, 88%; g) trisylhydrazine, THF, 93%; h) t-BuLi, THF, −78 to 0 °C, then I2, −78 °C, 86%. See ref31 for abbreviations.
Chelate-controlled vinylmetal addition and elaboration into the ABCDE-region 77
In preparation for the subsequent chelate-controlled vinyl iodide addition to stereoselectively form the C20–C21-bond, the CD-ring tetrahydrofuran 50 was desilylated (TBAF, AcOH, 97%) and converted in a Parikh-Doering oxidation45 to the corresponding D-ring aldehyde 72 (SO3·pyr, DMSO, 94%, Scheme 11). Metal-halogen exchange of the vinyl iodide fragment 71 was then effected with t-BuLi in Et2O at −78 °C followed by cannulation of this vinyllithium solution into a solution of the aldehyde 72 pre-complexed to MgBr2·OEt2 (prepared in an Et2O/toluene solution40) in CH2Cl2. The observed diastereoselectivity was excellent at temperatures ranging from −78 to 0 °C while the best yields were obtained at −20 °C (80%, dr > 99:1). With 2 equiv of vinyl iodide 71 relative to the aldehyde 72, on the order of 0.6–0.7 equiv of vinyl iodide 71 could additionally be recovered by a final I2-quench, thus emphasizing the stability of the vinylmetal species in CH2Cl2.
Scheme 11.

Synthesis of the ABCDE-region 77. Reagents and conditions: a) TBAF, AcOH, THF 97%; b) SO3·Pyr, DMSO, i-Pr2NEt, CH2Cl2, −30 °C, 94%; c) 71 (2 equiv), t-BuLi, Et2O, −78 °C, then 72, MgBr2·OEt2,40 CH2Cl2, −20 °C, 80%, dr > 99:1; d) Ac2O, DMAP, Et3N, CH2Cl2, 96%; e) i) O3, MeOH, CH2Cl2, −78 °C, then Me2S; ii) MeOH, HC(OMe)3, PPTS, 75% (2 steps); f) TBDPSCl, imidazole, CH2Cl2; g) DDQ, CH2Cl2/pH 7 buffer (9:1 v/v), 0 °C; 78% (2 steps); h) TESCl, imidazole, CH2Cl2, 90%; i) H2, RaNi (W-2),71 EtOH, 93%; j) SO3·Pyr, DMSO, i-Pr2NEt, CH2Cl2, −30 °C, 75%. See ref31 for abbreviations.
Subsequently, the coupled product was acetylated under standard conditions to give 73 (Ac2O, 96%). Careful ozonolysis of the 1,1-disubstituted olefin in the presence of Sudan III as a color indicator56 allowed for an almost completely selective cleavage of the olefin accompanied by only trace amounts of undesired oxidation of the benzyl ether protecting groups. Subsequent ketalization in the presence of a mild acid allowed for the formation of lactol methyl ether 74 (PPTS, MeOH, 75% of 74, 2 steps). Intermediates observed during investigations of this ketalization procedure suggested that the product was partially dehydrated to give the E-ring C21-dihydropyran which in the presence of strong acids such as p-TsOH could be more rapidly ketalyzed to give the desired C21 methyl lactol ether, albeit in only moderate overall yields upon scale-up. A subsequent series of standard protecting group manipulations involved TBDPS-protection (TBDPSCl, imidazole), PMB-removal (DDQ, 78%, 2 steps), and C17 TES installation (TESCl, imidazole, 90%). The final debenzylation in the presence of the TES silyl ether proceeded readily using W-2 RaNi71 (H2, RaNi, EtOH, 93%) followed by a Parikh-Doering oxidation45 to give the C13 aldehyde 75 (SO3·pyr, DMSO, 75%). In preliminary experiments, aldehyde 75 could be readily elaborated in a five-step sequence analogous to the one already discussed in Scheme 7 to give access to the ABCDE-region 77 of azaspiracid-1.
The C20–C21 bond via an anomeric sulfone anion addition
Although we had thus established a viable route to form the C20–C21 bond of azaspiracid-1 as our initial fragment coupling, we remained attracted to the possibility of performing this crucial C20–C21 bond formation last. This would constitute a more convergent approach to azaspiracid-1 since all 9 rings of the final target would be formed prior to such an ultimate fragment coupling as briefly outlined in Scheme 1.
Synthesis of cyclic E-ring fragments containing a C21 sulfide moiety
We began these investigations by synthesizing several E-ring fragments containing a C21-sulfide substituent and different electrophiles at C26 (Scheme 12). Treatment of tetrahydropyran 59 with one equivalent of thiophenol and a slight excess of BF3·OEt2 provided thioglycoside 78 (dr 96:4, 83% of 78) accompanied by minor amounts of the acyclic dithioacetal as a byproduct. We believe the diastereoselectivity observed in this reaction is the result of initial formation of the axial thioglycoside, followed by Lewis acid induced isomerization to the more stable equatorial isomer 78. In support of this hypothesis, the observed diastereoselectivity at C21 was inferior when substoichiometric amounts of BF3·OEt2 were employed. Subsequent epimerization of the ester substituent to give the tetrahydropyran 79 with all four substituents occupying equatorial positions proceeded readily with catalytic amounts of t-BuOK in THF at low temperature (dr 96:4, 88% of 79). The only major byproduct in this reaction was the corresponding t-Bu ester and its formation was minimized by using only 5 mol% of t-BuOK. Ester 79 could then alternatively be converted to Weinreb amide 80 (Me(OMe)NH·HCl, i-PrMgCl, 93%), or reduced directly to aldehyde 10 (DIBALH, 93%). In summary, this sequence provided the E-ring fragment 10 in 5 steps (54% yield) from the hetero-Diels-Alder precursors 62/64.
Scheme 12.

Synthesis of cyclic E-ring fragments. Reagents and conditions: a) PhSH, BF3·OEt2, CH2Cl2, −20 °C, dr 96:4, 83% 78; b) t-BuOK, THF, −50 °C, dr 96:4, 88% 79; c) Me(OMe)NH·HCl, i-PrMgCl,51 THF, −78 to −20 °C, 93%; d) 79, DIBALH, toluene, −94 to −78 °C, 93%. See ref31 for abbreviations.
Model experiments to form the C20–C21 bond by an anomeric sulfone anion addition
At this stage, we decided to test the viability of the final anomeric sulfone anion addition to form the C20–C21 bond of azaspiracid-1 using less elaborate model systems. To the best of our knowledge, the most related literature precedent for such a fragment coupling can be found in our altohyrtin synthesis where an anomeric sulfone anion was added to an N-acylbenzotriazole as the electrophile.27,28 In a simple azaspiracid model system, the anion of sulfone 81 could likewise be added to a host of simple activated esters and amides as exemplified by the N-acylpyrazole 82 (Scheme 13). Although the yields of these additions to give the slightly unstable C21-β ketosulfone 83 were only moderate at this stage, we were more concerned with the identification of sufficiently mild conditions for the subsequent anomeric sulfone excision to arrive at the desired C21-lactol moiety. Very little is reported about such anomeric sulfone derivatizations apart from the single altohyrtin precedent that required forcing conditions for converting one of the anomeric sulfone anomers,27,28 conditions which were judged to be too harsh in the present azaspiracid case.
Scheme 13.

Model studies of final C20–C21 sulfone fragment coupling and product elaboration. Reagents and conditions: a) EtMgBr, THF, −78 °C, 99%; b) TMSCH2Li, THF, −78 °C, 70%; c) KH, THF, 99%; d) H2O2, Na2WO4·2H2O,72 MeOH, 91%; e) 81 (1 equiv), LDA (1.2 equiv), THF, then 82 (1.5 equiv), −78 °C, dr(C21): 6:1, 51% 83; f) PhSLi, Et2O, −78 °C, 99%; g) KHBEt3, THF, −78 to −20 °C, dr 5–6:1, 56% major isomer; h) HgCl2, pH 7 buffer, MeCN/THF, 0 °C, 86%; i) KHBEt3, CH2Cl2, −78 to −20 °C, dr 10:1, 91% major isomer. See ref31 for abbreviations.
Initially, it was found that the transformation of ketosulfone 83 into ketosulfide 84 could be effected with PhSH in the presence of MgBr2·OEt2 and i-Pr2NEt in relatively nonpolar solvents (CH2Cl2 or Et2O, −20 °C, >80% yield). Subsequently, we discovered that the same transformation could be effected in quantitative yield under very mild conditions (PhSLi, Et2O, −78 °C, 99%) in an interesting transformation that proceeds with complete inversion at the C21 stereocenter. The resulting ketosulfide 84 could be stereoselectively reduced to afford either C20 alcohol diastereomer utilizing a simple change of solvent (KHBEt3 in THF or CH2Cl2, dr 5–6:1 or 1:10, respectively). Finally, the phenylsulfide moieties could be readily converted with aqueous HgCl2 into the lactols 85 and 86 for which the C21 hydroxyl spontaneously occupied an axial position. This mild reaction sequence was judged to be viable for our more functionalized azaspiracid target.
As may be noted, the above investigations were performed with the enantiomer of the Weinreb amide E-ring fragment (ent-80) since they were part of our early studies targeting the originally proposed structure of azaspiracid-1 (1). By similar approaches as depicted in Scheme 13, we additionally synthesized the C19-epimers of lactols 85 and 86, thus providing access to all four diastereomers of the C19–C20 junction. Comparison of 1H NMR data for these four C19–C20 diastereomeric lactols with the published 1H NMR spectrum of azaspiracid-16 clearly suggested that the acyclic C19–C20 junction between the ABCD- and the E-rings was one likely region to have been originally miss-assigned. This assumption was confirmed by Nicolaou in 2004 with the structural revision of azaspiracid-1 (2).22
Unfortunately, more elaborate model systems in the correct enantiomeric series of the D-ring that comprised activated esters and amides derived either from CD-ring aldehyde 72 (Scheme 11) or ABCD-aldehyde 3 (Scheme 7) and the addition of the anion of E-ring sulfone 87 (Scheme 14) proved significantly less promising under a variety of conditions, also in the subsequent transformation with PhSLi (not shown). As an alternative, a significantly more rapid entry to the C21-lactol region of the azaspiracid skeleton would be to utilize aldehyde 3 directly, a strategy with some promising precedent in the literature.29 The well-known competing side reaction in such a transformation is the sulfinate elimination to the corresponding C21-dihydropyran.25,28 Nevertheless, when this reaction was carried out with aldehyde 3 and the E-ring model sulfone 87 followed by a quench with slightly acidic buffer at −78 °C, the desired lactols 88 were preferentially formed over dihydropyrans 89 (3:1 ratio, 65% combined yield). The desired lactols 88 could subsequently be interconverted by an oxidation/reduction sequence of the C20 moiety to give either C20 alcohol in high stereoselectivty. A limited number of subsequent studies with several bases (nBuLi, LDA), additives (HMPA, MgBr2·OEt2), solvents (THF, Et2O, and CH2Cl2 for pre-complexation of the aldehyde), and different quenching buffers all failed to provide a full conversion of aldehyde 3, a goal yet to be achieved, and did not improve the ratio of desired lactols 88 to dihydropyrans 89. Although by no means optimal, this coupling experiment depicted in Scheme 15 still provided a credible lead for the final C20–C21 coupling experiments and established that the C21 lactols 88 could indeed be accessed directly from ABCD-aldehyde 3 upon appropriate work up conditions. With this result in hand, a decision was made to embark on the synthesis of the fully elaborated EFGHI-sulfone 4 (Scheme 1) as will be discussed in the following sections.
Scheme 14.

Model studies of final C20–C21 sulfone fragment coupling via the C20-aldehyde 3. Reagents and conditions: a) LiAlH4, Et2O, −20 °C; b) TBDPSCl, imidazole, DMF, 99% (2 steps); c) m-CPBA, NaHCO3, CH2Cl2, 94%; d) 87 (2.3 equiv), n-BuLi, THF, −78 °C, then 3 (1.0 equiv), then sat. aq. NH4Cl, −78 °C to RT, 65%, 3:1 88:89. See ref31 for abbreviations.
Scheme 15.

Synthesis of the FG-ring fragment 11. Reagents and conditions: a) O3, MeOH, CH2Cl2, −78 °C, then Me2S, 61%; then vacuum sublimation, 73%; b) 10 mol% 93, CH2Cl2, −78 °C, 94% conversion, dr > 40:1, 97% ee (67% after recrystallization, >99% ee); c) H2 (1 atm), 2 mol% [(COD)Ir(PCy3)(py)]PF6, CH2Cl2, 98%, dr > 95:5; d) PMBBr, NaH, DMF, −40 to −20 °C, 67%; e) KHBEt3, THF, 0 °C to RT, 94%; f) TBSCl, imidazole, DMF, 99%; g) HF·pyr, THF, −10 °C, 87%; h) TsCl, DMAP, pyridine, 98%; i) NaCN, DMSO, 55 °C, 99%; j) Boc2O, DMAP, CH3CN, 99%;kj) LiBH4, H2O, THF, 0 °C to RT, 99%; l) TMSCl, imidazole, CH2Cl2, 99%; m) MeLi, Et2O, 0 °C, 84%; n) SO3·Pyr; DMSO, i-Pr2NEt, CH2Cl2, −30 °C, 99%. See ref31 for abbreviations.

Synthesis of the FG-ring fragment 11
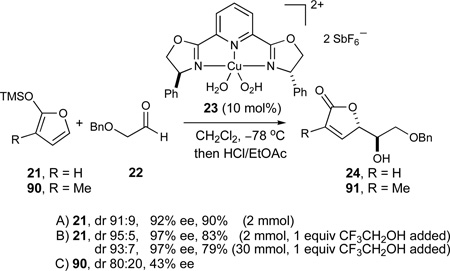
In the synthesis of the FG-ring fragment 11, we envisioned that the C32–C33 anti diol array could be efficiently obtained by an extension of our enantioselective catalytic Mukaiyama aldol reaction of siloxyfuran 2173 and (benzyloxy)acetaldehyde 2274 (eq 9, entry A).34 In contrast to the original studies where hydrated complex 23 was formed in situ with CuCl2·2H2O,34 further investigations revealed that bis(aquo) complex 23 could conveniently be isolated and stored as a bench-stable solid prior to use. The inclusion of CF3CH2OH as a silicon-scavenging reagent75 further increased the observed stereoselectivities and was readily amenable to reliable large-scale preparations of the desired product 24 (entry B, 79%, dr 93:7, 97% ee), thus suggesting the suppression of a racemic silicon-catalyzed background reaction. However, much inferior stereoselectivities were obtained in preliminary experiments with the corresponding methyl-substituted siloxyfuran 9078 (entry C, dr 80:20, 43% ee), thus prompting us to investigate alternative conditions for this particular transformation.
 |
(9) |
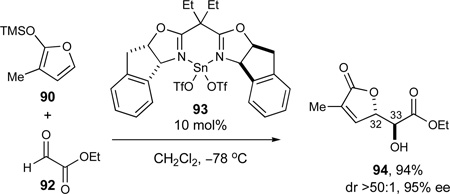
Further studies revealed that the corresponding Sn(2+)-catalyzed aldol reaction79 with Sn(2+)-complex 93 alternatively afforded optimal results in the desired transformation. Thus, methyl-substituted siloxyfuran 90 underwent stereoselective aldol addition with ethyl glyoxalate (92) to give the desired lactone 94 in excellent yield and selectivities (94%, 95% ee, dr >50:1, eq 10). During these investigations, it was found that the ethyl substituents on the chiral ligand were important for obtaining high stereoselectivities, for reasons that remain unclear.
 |
(10) |
Another practical variant of this addition process using N-phenyl glyoxamide 96,80 readily prepared by ozonolysis of olefin 95 and dehydration of the intermediary glyoxamide hydrate by vacuum sublimation, is depicted in Scheme 15 and afforded adduct 97 (94% conversion, dr > 40:1, 97% ee) as a highly crystalline solid. In this transformation, it was found that the highest stereoselectivities were observed using a sequential addition of the reagents in 0.1 equiv increments rather than a slow addition of either reagent. The resulting lactone 97 could readily be recrystallized to enantiopurity prior to the subsequent transformations (99% ee, 67%). Another benefit of the N-phenyl amide 97 was its propensity to undergo highly diastereoselective directed hydrogenations to install the methyl-bearing stereocenter, a goal that was not successfully achieved with the corresponding ethyl ester 94 under the same range of hydrogenation conditions. The directed hydrogenation of 97 proved to be optimal with Crabtree’s catalyst81 that afforded the resulting lactone as a single isomer (H2, [(COD)Ir(PCy3)(py)]PF6, 98%, dr > 95:5). Subsequently, PMB-protection of the secondary alcohol to give 98 was sufficiently chemoselective under basic conditions (PMBBr, NaH, 67%) whereas a variety of acidic conditions afforded both decomposition and epimerization of the newly set methyl stereocenter.
The lactone 98 was next reduced to the diol (KHBEt3, 94%), persilylated (TBSCl, 99%), and converted into the corresponding primary alcohol by selective desilylation (HF·pyr, 87%). We elected to introduce C28 as a cyano function at this stage of the synthesis to provide maximal flexibility in subsequent fragment couplings since the nitrile could be either converted into an electrophile (an aldehyde) or a nucleophile (a methyl ketone). This was accomplished in high yield by tosylation of the primary alcohol and displacement with sodium cyanide to give nitrile 99 (97% yield, 2 steps). At the other terminus of the molecule, the N-phenyl amide functionality was reduced in a two-step sequence82 involving Boc-activation and reduction with lithium borohydride to give alcohol 100 in 98% yield for the two steps. Preliminary investigations of our various fragment coupling strategies revealed that a C27-methyl ketone was the most desirable for coupling the FG-ring fragment to the E-ring fragment 10. The methyl ketone was therefore accessed by a methyllithium addition to the nitrile in a step that required transient protection of the primary alcohol to effect full conversion (TMSCl, 99%). Thus, the nitrile was converted into the C27 methyl ketone with methyllithium with concomitant TMS ether hydrolysis during the workup procedure (84%). Finally, a Parikh-Doering oxidation45 (SO3·Pyr, DMSO, 97%) concluded the synthesis of the FG-ring ketoaldehyde 11.
Synthesis of the HI-ring fragment 12
Chiral auxiliary approach via known intermediate 104
In our initial efforts aimed at gaining access to the HI-ring fragment 12 for further studies, we utilized the known hydroxy ester 104 (Scheme 16). This intermediate is readily available in 8 steps83 based on a double stereodifferentiating directed hydrogenation84 of the homoallylic olefin 103 synthesized via a chiral auxiliary-based diastereoselective aldol reaction.85 The remaining four transformations comprised ester reduction, selective tosylation of the primary alcohol to give 105, azide substitution and finally a Swern oxidation86 (80%, 4 steps) to deliver the desired keto azide 12.
Scheme 16.

Auxiliary-based synthesis of the HI-ring fragment 12. Reagents and conditions: a) 104,83 LiAlH4, THF, 0 °C, 95%; b) TsCl, Et3N, CH2Cl2, 0 °C to RT, 95%; c) NaN3, TBAI, DMF, 60 °C, 93%; d) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C to RT, 95%. See ref31 for abbreviations.
Catalytic enantioselective approach via Hetero-Diels-Alder intermediate 59
Alternatively, we envisioned that our 1,3-syn-dimethyl substituted tetrahydropyran 59 generated in the enantioselective hetero-Diels-Alder reaction could likewise be utilized to synthesize the acyclic HI-ring fragment 12 by incorporation of a ring fragmentation. Thus, ester 59 was converted to alcohol 106 by sequential acetal and ester reduction (Et3SiH, BF3•OEt2; LiAlH4, 81% yield), followed by iodine installation (Scheme 17). Whereas initial investigations of the latter transformation were frustrated by the lack of reactivity of intermediate sulfonate esters derived from the primary alcohol 106, the iodide 107 was smoothly obtained by heating of the intermediate phosphonium salt derived from alcohol 106 to 60 °C (I2, PPh3, imidazole,87 94%). Although the fragmentation of iodide 107 could be accomplished under a number of conditions (Zn, MeOH or n-BuLi, Et2O), the resultant primary alcohol was too volatile for convenient manipulation. As a solution, the intermediary lithium alkoxide was formed in THF and trapped in situ with TsCl to give 108 in 95% yield. The terminal olefin was smoothly oxidized to the methyl ketone under standard Wacker conditions (O2, PdCl2, CuCl, DMF, 89%), with the remainder of the material consisting of olefin isomers. Finally, an azide introduction (NaN3, DMSO, 60 °C, 97%) concluded this catalytic asymmetric synthesis of the HI-ring fragment 12.
Scheme 17.

Catalytic enantioselective synthesis of the HI-ring fragment 12. Reagents and conditions: a) Et3SiH, BF3·OEt2, CH2Cl2, 0 °C, 91%; b) LiAlH4, Et2O, 0 °C, 91%; c) I2, PPh3, imidazole, CH3CN/benzene, RT to 60 °C, 94%; d) t-BuLi, THF, −78 °C, then TsCl, −78 to 0 °C, 95%; e) O2, PdCl2, CuCl, H2O, DMF, 89%; f) NaN3, DMSO, 60 °C, 97%. See ref31 for abbreviations.

Synthesis of the EFGHI-sulfone 4
Assembly of the EFGHI carbon skeleton and formation of the FG bicyclic ketal 115
With the required E-, FG-, and HI-ring fragments in hand as described above, we proceeded to investigate their assembly to the pentacyclic EFGHI-sulfone 4. We first formed the C34–C35 bond to unite the FG- and the HI-ring fragments in a chelate-controlled Mukaiyama aldol reaction.30 The required enolsilane 109 was readily formed from methyl ketone 12 under standard conditions (LiHMDS, TMSCl, Et3N, 89%, Scheme 18). A careful evaluation of a range of Lewis acids to effect this transformation established that freshly prepared solid MgBr2·OEt268,89 was superior to other Lewis acids such as TiCl2(Oi-Pr)2, TiCl4, SnCl4, MgI2, MgBr2, Mg(OTf)2, and commercial MgBr2·OEt2. In addition, a high concentration (0.4M) was crucial for obtaining a complete conversion with this mild and selective Lewis acid. Consequently, the desired chelate-controlled addition of enolsilane 109 to aldehyde 11 proceeded in excellent yield to afford the desired aldol adduct 110 as a single diastereomer (93%, dr > 95:5). Subsequent model studies on adduct 110 revealed that treatment with aqueous HF in acetonitrile88 smoothly effected the formation of the desired FG bicyclic ketal 112 in 92% yield.
Scheme 18.

Chelate-controlled Mukaiyama aldol reaction and initial investigations of FG-ketalization. Reagents and conditions: a) LiHMDS, TMSCl, Et3N, THF, −78 °C, 89%; b) MgBr2·OEt2, CH2Cl2, 0 °C, 93%, dr > 95:5; c) HF, H2O, CH3CN, 0 °C, 92%; d) TBSCl, imidazole, DMF, 99%. See ref31 for abbreviations.
The aldol addition to form the C26–C27 bond, was implemented with Cy2BCl (2.4 equiv, i-Pr2NEt) due to its excellent regioselectivity in the enolization of methyl ketones (Scheme 19).90 The excess Cy2BCl is presumed to function as a transient protecting group for the C34–C36 hydroxyketone moiety, and prevent any retroaldol reactions from taking place as depicted in the proposed intermediate 113. When treated with a slight excess of E-ring aldehyde 10, the desired aldol adduct 9 was formed in near quantitative yield (dr 60:40). When adduct 9 was subsequently exposed to aqueous HF in acetonitrile, the lone TBS ether was cleaved and the molecule spontaneously cyclized to form the desired FG bicyclic ketal 114 in excellent yield (92%, 2 steps). It is noteworthy that the analogous cyclization of the O34-TBS-protected aldol adduct 111 afforded a multitude of products.
Scheme 19.

Assembly of the EFGHI carbon skeleton and formation of the FG bicyclic ketal 115. Reagents and conditions: a) Cy2BCl (2.4 equiv), i-Pr2NEt, CH2Cl2, −78 °C, then 10 (1.4 equiv), −78 to 0 °C; then H2O2, MeOH, pH 7 buffer, 0 °C to RT, dr 60:40; b) HF, H2O, CH3CN, 0 °C, 92% (2 steps); c) Dess-Martin periodinane, pyr, CH2Cl2, 85%. See ref31 for abbreviations.
The lack of diastereoselectively in the boron aldol addition was inconsequential since the C26 secondary alcohols 114 were subsequently oxidized to the corresponding ketone 115 with Dess-Martin periodinane59 (85%). Pyridine was crucial to buffer this latter oxidation step in order to suppress any over-oxidation and subsequent loss of the C21-phenylsulfide moiety, a prominent side reaction observed in initial unbuffered experiments.
Completion of the EFGHI-sulfone 4
Subsequently, the PMB ether in ketone 115 was oxidatively removed to afford an equilibrium mixture of lactols 116 and open-chain tautomers (DDQ, pH 7 buffer, Scheme 20). Decomposition of this intermediate upon exposure to silica gel was readily circumvented by buffering of the silica gel with Et3N. With two of the three functional groups of the HI spiroaminal unmasked, the terminal azide was reduced to the primary amine. Whereas Staudinger conditions provided a mixture of isomers, hydrogenation under standard conditions induced the spontaneous formation of the thermodynamically favored HI-spiroaminal 117 as a single diastereomer (H2, Pd/C, 77%, dr > 95:5, 2 steps). The desired configuration of the spiroaminal 117 was verified by the key NOE interactions depicted in Figure 4.
Scheme 20.

Completion of the EFGHI-sulfone 4. Reagents and conditions: a) DDQ, pH 7 buffer, CH2Cl2, 0 °C; b) H2 (1 atm), Pd/C, THF, dr >95:5, 77% (2 steps); c) TeocCl,91 i-Pr2NEt, THF, 0 °C, 89%, dr > 97:3; d) Tebbe reagent,92 pyr, toluene, −40 °C, 55%;93 (41% recovered ketone); e) H2O2, pyr, (NH4)6Mo7O24, t-BuOH, 96%. See ref31 for abbreviations.
Figure 4.

Key NOEs for spiroaminal 117 (C6D6, 500 MHz).
To mask all reactive functionalities in the molecule, we then elected to protect the spiroaminal nitrogen of 117a as its Teoc derivative. This transformation proceeded in excellent selectivity when conducted in THF at high concentrations (TeocCl, 89%, >97:3 mixture of isomers). Somewhat surprisingly, all Teoc-protected intermediates proved to be slightly unstable to acidic conditions while their purification was readily executed using silica gel buffered with organic bases. The stage was now set for the olefination of the C26 ketone. We had deliberately postponed this transformation until after the closure of the FG bicyclic ketal in our synthesis plan to avoid any intermediates containing an β,γ-unsaturated ketone prone to isomerization. After initial experiments using relatively non-basic methylenedianion equivalents such as the Takai-Nozaki reagent,94 the Tebbe reagent95 buffered with pyridine to avoid any C36-epimerization proved superior in this transformation. The Tebbe reagent could conveniently be prepared in situ from Cp2TiCl2 and AlMe392 and afforded the desired olefinated product in 58% yield along with 41% of the recovered ketone.93 The yield for this step could be elevated to 78% after two recycles. The key NOE interactions of the spiroaminal portion remained unchanged from those depicted in Figure 4, thus verifying that the C36 spiroaminal had not epimerized during these latter transformations. Finally, the oxidation of the C21-phenylsulfide moiety to sulfone 4 proceeded readily (H2O2, (NH4)6Mo7O24, 96%) In analogy to the Dess-Martin oxidation described above, buffering of the reaction mixture with pyridine was crucial to suppress concomitant loss of the C21-sulfur substituent as an undesired side reaction. In summary, the above sequence allowed the efficient synthesis of the EFGHI-sulfone 4.
Sulfone coupling and completion
With the desired ABCD-aldehyde 3 and EFGHI-sulfone 4 in hand, we proceeded to the final fragment coupling to form the C20–C21 bond of azaspiracid utilizing the reaction conditions developed during our model studies (Scheme 14). Thus, the sulfone anion derived from 4 was generated with nBuLi and added to aldehyde 3 followed by a quench at −78 °C with pH 5 buffer. Gratifyingly, this procedure afforded a 50% overall yield of the readily separable lactol diastereomers 118 and 119 in nearly equal amounts (Scheme 21).96 Interestingly, the undesired alcohol 118 was obtained as an inseparable mixture of the C21-lactol and the corresponding hydroxyketone, thus suggesting a lower configurational stability of this C20 diastereomer. Next, the undesired lactol 118 was converted into the desired lactol 119 by an oxidation/reduction sequence. Oxidation to the corresponding C20 ketone was best effected under Swern conditions (oxalyl chloride, DMSO, Et3N, 63%), a transformation somewhat complicated by the equilibrium between the C21 lactol and the hydroxyketone, since the latter is oxidized to the corresponding undesired diketone during the course of the reaction. A variety of reduction conditions were then evaluated in the diastereoselective reduction of the C20 ketone to test both traditional chelation and Felkin control, but unfortunately most reducing agents selectively provided the undesired lactol 118. However, LiBH4 in CH2Cl2 at −40 °C exclusively afforded the desired C20 diastereomer 119, although in moderate yield (56%, dr > 20:1).
Scheme 21.

Final fragment coupling and completion of the synthesis. Reagents and conditions: a) 4 (2.2 equiv), n-BuLi, −78 °C, then 3 (1.0 equiv), then NaOAc/AcOH buffer, −78 °C to RT, 50% (27% 118, 23% 119); b) (COCl)2, DMSO, Et3N, CH2Cl2, −78 to −20 °C, 63%; c) LiBH4(THF), CH2Cl2, −40 °C, dr > 20:1, 56%; d) TBAF, THF, 0 °C, 93%; e) Dess-Martin periodinane, CH2Cl2, 0 °C; f) NaClO2, NaH2PO4·H2O, 2-methyl-2-butene, t-BuOH, 90% (2 steps). See ref31 for abbreviations
With the desired lactol 119 thus prepared, only a few steps remained to complete the synthesis. Thus, the two silyl protecting groups were readily removed with TBAF (93% yield). Next, the C1 terminus was oxidized in a mild two-step sequence since initial experiments using TEMPO-type oxidations proved too harsh for the remainder of the molecule. Interestingly, a Dess-Martin oxidation at 0 °C chemoselectively afforded the desired C1 aldehyde even though a large excess of Dess-Martin periodinane was required to reach a full conversion into the aldehyde. In contrast, the C20 secondary alcohol of lactol 119 could rapidly be oxidized with Dess-Martin periodinane at room temperature. Finally, the resulting C1-aldehyde was cleanly oxidized with NaClO2 to the corresponding carboxylic acid97 in the presence of 2-methyl-2-butene as a chlorine scavenger98 (90%, 2 steps). This sequence concluded the total synthesis of (+)-azaspiracid-1 (ent-2) and has allowed us to synthesize 75 mg of the target molecule. The spectroscopic data for (+)-azaspiracid-1 corresponded with those reported for (−)-azaspiracid (2) apart from the optical rotation which was of equal magnitude but of opposite sign ((+)-azaspiracid-1 (ent-2), [α]24D +21.7 (c = 1.00, MeOH), natural (−)-azaspiracid-1 (2), [α]20D −21 (c = 0.1, MeOH),6 synthetic (−)-azaspiracid-1 (2), [α]33D −19.0 (c = 0.07, MeOH)22).
Conclusions
In projects of this level of complexity, the casual reader is rarely informed of the intricacies associated with developing a viable synthesis. For structures such as azaspiracid, assembled from a number of complex subunits, once the viability of the successful route has been established, we routinely re-analyze the individual synthons as a collection of “new targets”. If more aggressive approaches to the syntheses of these individual subunits can be perceived, the “first-generation” route to that subunit is re-evaluated as far as the final publication is concerned. In this article, we have attempted to reveal some of the hidden complexity associated with the reported synthesis of azaspiracid.
Supplementary Material
Acknowledgement
Financial support has been provided by the National Institutes of Health (GM-33328-22), the National Science Foundation (CHE-0608664), the Merck Research Laboratories, Amgen, and Eli Lilly. Fellowships were provided to L.K. by the Villum Kann Rasmussen and the Carlsberg Foundations (Denmark), to T.B.D. by the National Science Foundation, to A.B. by the NSERC (Canada), to J.A.M. by the National Institutes of Health, and to M.J. by the Technical University of Denmark.
Footnotes
Supporting Information Available: Experimental details and analytical data including copies of 1H and 13C NMR spectra for all new compounds (154 pages) (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.McMahon T, Silke J. Harmful Algae News. 1996;14:2. [Google Scholar]
- 2.James KJ, Furey A, Lehane M, Ramstad H, Aune T, Hovgaard P, Morris S, Higman W, Satake M, Yasumoto T. Toxicon. 2002;40:909–915. doi: 10.1016/s0041-0101(02)00082-x. [DOI] [PubMed] [Google Scholar]
- 3.Magdalena AB, Lehane M, Krys S, Fernandez ML, Furey A, James KJ. Toxicon. 2003;42:105–108. doi: 10.1016/s0041-0101(03)00105-3. [DOI] [PubMed] [Google Scholar]
- 4.James KJ, Moroney C, Roden C, Satake M, Yasumoto T, Lehane M, Furey A. Toxicon. 2003;41:145–151. doi: 10.1016/s0041-0101(02)00244-1. [DOI] [PubMed] [Google Scholar]
- 5.Taleb H, Vale P, Amanhir R, Benhadouch A, Sagou R, Chafik A. J. Shellfish Res. 2006;25:1067–1070. [Google Scholar]
- 6.Satake M, Ofuji K, Naoki H, James KJ, Furey A, McMahon T, Silke J, Yasumoto T. J. Am. Chem. Soc. 1998;120:9967–9968. [Google Scholar]
- 7.(a) Ofuji K, Satake M, McMahon T, Silke J, James KJ, Naoki H, Oshima Y, Yasumoto T. Nat. Toxins. 1999;7:99–102. doi: 10.1002/(sici)1522-7189(199905/06)7:3<99::aid-nt46>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]; (b) Ofuji K, Satake M, McMahon T, James KJ, Naoki H, Oshima Y, Yasumoto T. Biosci. Biotechnol. Biochem. 2001;65:740–742. doi: 10.1271/bbb.65.740. [DOI] [PubMed] [Google Scholar]
- 8.James KJ, Sierra MD, Lehane M, Magdalena AB, Furey A. Toxicon. 2003;41:277–283. doi: 10.1016/s0041-0101(02)00288-x. [DOI] [PubMed] [Google Scholar]
- 9.Furey A, Moroney C, Magdalena AB, Saez MJF, Lehane M, James KJ. Environ. Sci. Technol. 2003;37:3078–3084. doi: 10.1021/es020246z. [DOI] [PubMed] [Google Scholar]
- 10.Twiner MJ, Hess P, Dechraoui MYB, McMahon T, Samons MS, Satake M, Yasumoto T, Ramsdell JS, Doucette GJ. Toxicon. 2005;45:891–900. doi: 10.1016/j.toxicon.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 11.Colman JR, Twiner MJ, Hess P, McMahon T, Satake M, Yasumoto T, Doucette GJ, Ramsdell JS. Toxicon. 2005;45:881–890. doi: 10.1016/j.toxicon.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Ronzitti G, Hess P, Rehmann N, Rossini GP. Toxicol. Sci. 2007;95:427–435. doi: 10.1093/toxsci/kfl167. [DOI] [PubMed] [Google Scholar]
- 13.Vilarino N, Nicolaou KC, Frederick MO, Cagide E, Ares IR, Louzao MC, Vieytes MR, Botana LM. Chem. Res. Toxicol. 2006;19:1459–1466. doi: 10.1021/tx060131z. [DOI] [PubMed] [Google Scholar]
- 14.Kulagina NV, Twiner MJ, Hess P, McMahon T, Satake M, Yasumoto T, Ramsdell JS, Doucette GJ, Ma W, O'Shaughnessy TJ. Toxicon. 2006;47:766–773. doi: 10.1016/j.toxicon.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 15.(a) Alfonso A, Vieytes MR, Ofuji K, Satake M, Nicolaou KC, Frederick MO, Botana LM. Biochem. Biophys. Res. Commun. 2006;346:1091–1099. doi: 10.1016/j.bbrc.2006.06.019. [DOI] [PubMed] [Google Scholar]; (b) Vale C, Nicolaou KC, Frederick MO, Gomez-Limia B, Alfonso A, Vieytes MR, Botana LM. J. Med. Chem. 2007;50:356–363. doi: 10.1021/jm061063g. [DOI] [PubMed] [Google Scholar]
- 16.(a) Nicolaou KC, Pihko PM, Diedrichs N, Zou N, Bernal F. Angew. Chem. Int. Ed. 2001;40:1262–1265. [PubMed] [Google Scholar]; (b) Nicolaou KC, Qian WY, Bernal F, Uesaka N, Pihko PM, Hinrichs J. Angew. Chem. Int. Ed. 2001;40:4068–4071. [PubMed] [Google Scholar]
- 17.(a) Carter RG, Weldon DJ. Org. Lett. 2000;2:3913–3916. doi: 10.1021/ol006674w. [DOI] [PubMed] [Google Scholar]; (b) Carter RG, Graves DE. Tetrahedron Lett. 2001;42:6035–6039. [Google Scholar]; (c) Carter RG, Bourland TC, Graves DE. Org. Lett. 2002;4:2177–2179. doi: 10.1021/ol026033w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Carter RG, Graves DE, Gronemeyer MA, Tschumper GS. Org. Lett. 2002;4:2181–2184. doi: 10.1021/ol026034o. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhou XT, Carter RG. Chem. Commun. 2004:2138–2140. doi: 10.1039/b410092a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhou XT, Lu L, Furkert DP, Wells CE, Carter RG. Angew. Chem. Int. Ed. 2006;45:7622–7626. doi: 10.1002/anie.200603353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Carter RG, Bourland TC, Zhou XT, Gronemeyer MA. Tetrahedron. 2003;59:8963–8974. [Google Scholar]; (b) Zhou XT, Carter RG. Angew. Chem. Int. Ed. 2006;45:1787–1790. doi: 10.1002/anie.200503733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Dounay AB, Forsyth CJ. Org. Lett. 2001;3:975–978. [PubMed] [Google Scholar]; (b) Aiguade J, Hao JL, Forsyth CJ. Org. Lett. 2001;3:979–982. [PubMed] [Google Scholar]; (c) Aiguade J, Hao JL, Forsyth CJ. Tetrahedron Lett. 2001;42:817–820. [Google Scholar]; (d) Hao JL, Aiguade J, Forsyth CJ. Tetrahedron Lett. 2001;42:821–824. [Google Scholar]; (e) Forsyth CJ, Hao JL, Aiguade J. Angew. Chem. Int. Ed. 2001;40:3663–3667. doi: 10.1002/1521-3773(20011001)40:19<3663::aid-anie3663>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; (f) Geisler LK, Nguyen S, Forsyth CJ. Org. Lett. 2004;6:4159–4162. doi: 10.1021/ol048581a. [DOI] [PubMed] [Google Scholar]; (g) Nguyen S, Xu JY, Forsyth CJ. Tetrahedron. 2006;62:5338–5346. [Google Scholar]; (h) Forsyth CJ, Xu JY, Nguyen ST, Samdal IA, Briggs LR, Rundberget T, Sandvik M, Miles CO. J. Am. Chem. Soc. 2006;128:15114–15116. doi: 10.1021/ja066971h. [DOI] [PubMed] [Google Scholar]; (i) Li YF, Zhou F, Forsyth CJ. Angew. Chem. Int. Ed. 2007;46:279–282. doi: 10.1002/anie.200601963. [DOI] [PubMed] [Google Scholar]
- 20.(a) Sasaki M, Iwamuro Y, Nemoto J, Oikawa M. Tetrahedron Lett. 2003;44:6199–6201. [Google Scholar]; (b) Ishikawa Y, Nishiyama S. Tetrahedron Lett. 2004;45:351–354. [Google Scholar]; (c) Ishikawa Y, Nishiyama S. Heterocycles. 2004;63:539–565. [Google Scholar]; (d) Ishikawa Y, Nishiyama S. Heterocycles. 2004;63:885–893. [Google Scholar]; (e) Oikawa M, Uehara T, Iwayama T, Sasaki M. Org. Lett. 2006;8:3943–3946. doi: 10.1021/ol0613766. [DOI] [PubMed] [Google Scholar]; (e) Yadav JS, Joyasawal S, Dutta SK, Kunwar AC. Tetrahedron Lett. 2007;48:5335–5340. [Google Scholar]; (f) Yadav JS, Venugopal C. Synlett. 2007:2262–2266. [Google Scholar]; (g) Li X, Li J, Mootoo DR. Org. Lett. 2007;9:4303–4306. doi: 10.1021/ol701866v. [DOI] [PubMed] [Google Scholar]
- 21.(a) Nicolaou KC, Li YW, Uesaka N, Koftis TV, Vyskocil S, Ling TT, Govindasamy M, Qian W, Bernal F, Chen DYK. Angew. Chem. Int. Ed. 2003;42:3643–3648. doi: 10.1002/anie.200351825. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Chen DYK, Li YW, Qian WY, Ling TT, Vyskocil S, Koftis TV, Govindasamy M, Uesaka N. Angew. Chem. Int. Ed. 2003;42:3649–3653. doi: 10.1002/anie.200351826. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Pihko PM, Bernal F, Frederick MO, Qian WY, Uesaka N, Diedrichs N, Hinrichs J, Koftis TV, Loizidou E, Petrovic G, Rodriquez M, Sarlah D, Zou N. J. Am. Chem. Soc. 2006;128:2244–2257. doi: 10.1021/ja0547477. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Chen DYK, Li YW, Uesaka N, Petrovic G, Koftis TV, Bernal F, Frederick MO, Govindasamy M, Ling TT, Pihko PM, Tang WJ, Vyskocil S. J. Am. Chem. Soc. 2006;128:2258–2267. doi: 10.1021/ja054748z. [DOI] [PubMed] [Google Scholar]
- 22.(a) Nicolaou KC, Vyskocil S, Koftis TV, Yamada YMA, Ling TT, Chen DYK, Tang WJ, Petrovic G, Frederick MO, Li YW, Satake M. Angew. Chem. Int. Ed. 2004;43:4312–4318. doi: 10.1002/anie.200460695. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Koftis TV, Vyskocil S, Petrovic G, Ling TT, Yamada YMA, Tang WJ, Frederick MO. Angew. Chem. Int. Ed. 2004;43:4318–4324. doi: 10.1002/anie.200460696. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Koftis TV, Vyskocil S, Petrovic G, Tang WJ, Frederick MO, Chen DYK, Li YW, Ling TT, Yamada YMA. J. Am. Chem. Soc. 2006;128:2859–2872. doi: 10.1021/ja054750q. [DOI] [PubMed] [Google Scholar]
- 23.(a) Nicolaou KC, Frederick MO, Loizidou EZ, Petrovic G, Cole KP, Koftis TV, Yamada YMA. Chem. Asian J. 2006;1:245–263. doi: 10.1002/asia.200600059. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Frederick MO, Petrovic G, Cole KP, Loizidou EZ. Angew. Chem. Int. Ed. 2006;45:2609–2615. doi: 10.1002/anie.200600295. [DOI] [PubMed] [Google Scholar]
- 24.For initial communications of this work, see: Evans DA, Kværnø L, Mulder JA, Raymer B, Dunn TB, Beauchemin A, Olhava EJ, Juhl M, Kagechika K. Angew. Chem. Int. Ed. 2007;46:4693–4697. doi: 10.1002/anie.200701515. Evans DA, Dunn TB, Kværnø L, Beauchemin A, Raymer B, Olhava EJ, Mulder JA, Juhl M, Kagechika K, Favor DA. Angew. Chem. Int. Ed. 2007;46:4698–4703. doi: 10.1002/anie.200701520.
- 25.(a) Ley SV, Lygo B, Wonnacott A. Tetrahedron Lett. 1985;26:535–538. [Google Scholar]; (b) Greck C, Grice P, Ley SV, Wonnacott A. Tetrahedron Lett. 1986;27:5277–5280. [Google Scholar]
- 26.(a) Beau JM, Sinay P. Tetrahedron Lett. 1985;26:6185–6188. [Google Scholar]; (b) Beau JM, Sinay P. Tetrahedron Lett. 1985;26:6189–6192. [Google Scholar]; (c) Beau JM, Sinay P. Tetrahedron Lett. 1985;26:6193–6196. [Google Scholar]
- 27.(a) Evans DA, Coleman PJ, Dias LC. Angew. Chem. Int. Ed. 1997;36:2738–2741. [Google Scholar]; (b) Evans DA, Trotter BW, Cote B, Coleman PJ. Angew. Chem. Int. Ed. 1997;36:2741–2744. [Google Scholar]; (c) Evans DA, Trotter BW, Cote B, Coleman PJ, Dias LC, Tyler AN. Angew. Chem. Int. Ed. 1997;36:2744–2747. [Google Scholar]
- 28.Evans DA, Trotter BW, Coleman PJ, Cote B, Dias LC, Rajapakse HA, Tyler AN. Tetrahedron. 1999;55:8671–8726. [Google Scholar]
- 29.(a) Evans DA, Carter PH, Carreira EM, Prunet JA, Charette AB, Lautens M. Angew. Chem. Int. Ed. 1998;37:2354–2359. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2354::AID-ANIE2354>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M. J. Am. Chem. Soc. 1999;121:7540–7552. [Google Scholar]
- 30.(a) Mukaiyama T, Narasaka K, Banno K. Chem. Lett. 1973:1011–1014. [Google Scholar]; (b) Mukaiyama T, Banno K, Narasaka K. J. Am. Chem. Soc. 1974;96:7503–7509. [Google Scholar]; (c) Mukaiyama T. Angew. Chem. Int. Ed. 2004;43:5590–5614. doi: 10.1002/anie.200300641. [DOI] [PubMed] [Google Scholar]
- 31.Abbreviations: Ac = acetyl, Bn = benzyl, Boc = t-butyloxycarbonyl, CBS = Corey-Bakshi-Shibata, COD = 1,5-cyclooctadiene, m-CPBA = m-chlorperbenzoic acid, Cy = cyclohexyl, DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, Dess-Martin periodinane = 1,1,1-tris(acetoxy)1,1-dihydro-1,2-benziodoxol-3-(1H)-one, DIBALH = diisobutylaluminum hydride, DMAP = 4-dimethylaminopyridine, DMF = N,N-dimethylformamide, DMSO = dimethyl sulfoxide, HMPA = hexamethylphosphoramide, HPLC = high pressure liquid chromatography, LDA = lithium diisopropylamide, LiDBB = lithium di-tert-butyl biphenylide, LiHMDS = lithium hexamethyldisilazide, Mes = 2,4,6-trimethylphenyl, pyr = pyridine, MPLC = medium pressure liquid chromatography, PG = protecting group, PMB = 4-methoxybenzyl, PPTS = pyridinium p-toluenesulfonate, RaNi = Raney Nickel, RT = room temperature, L-Selectride = lithium tri-sec-butylborohydride, TBAF = tetrabutylammonium fluoride, TBAI = tetrabutylammonium iodide, TBDPS = tert-butyldiphenylsilyl, TBS = tert-butyldimethylsilyl, Tebbe reagent = (μ-chloro)(μ-methylene)bis(cyclopentadienyl)(dimethylaluminum) titanium, Teoc = 2-(trimethylsilyl)ethoxycarbonyl, TES = triethylsilyl, TIPS = triisopropylsilyl, TMS = trimethylsilyl, Tr = trityl (triphenylmethyl), trisyl = 2,4,6-trisisopropylbenzenesulfonyl, Ts = p-toluenesulfonyl.
- 32.(a) Evans DA, Olhava EJ, Johnson JS, Janey JM. Angew. Chem. Int. Ed. 1998;37:3372–3375. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3372::AID-ANIE3372>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Johnson JS, Olhava EJ. J. Am. Chem. Soc. 2000;122:1635–1649. [Google Scholar]
- 33.(a) Evans DA, Burgey CS, Paras NA, Vojkovsky T, Tregay SW. J. Am. Chem. Soc. 1998;120:5824–5825. [Google Scholar]; (b) Evans DA, Tregay SW, Burgey CS, Paras NA, Vojkovsky T. J. Am. Chem. Soc. 2000;122:7936–7943. [Google Scholar]
- 34.Evans DA, Kozlowski MC, Murry JA, Burgey CS, Campos KR, Connell BT, Staples RJ. J. Am. Chem. Soc. 1999;121:669–685. [Google Scholar]
- 35.(a) Choi TL, Chatterjee AK, Grubbs RH. Angew. Chem. Int. Ed. 2001;40:1277–1279. doi: 10.1002/1521-3773(20010401)40:7<1277::aid-anie1277>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; (b) Connon SJ, Blechert S. Angew. Chem. Int. Ed. 2003;42:1900–1923. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]
- 36.Wright SW, Hageman DL, Wright AS, McClure LD. Tetrahedron Lett. 1997;38:7345–7348. [Google Scholar]
- 37.Corminboeuf O, Renaud P. Org. Lett. 2002;4:1735–1738. doi: 10.1021/ol0257981. [DOI] [PubMed] [Google Scholar]
- 38.Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 39.Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH. J. Am. Chem. Soc. 1999;121:791–799. [Google Scholar]
- 40.Nakatsuka M, Ragan JA, Sammakia T, Smith DB, Uehling DE, Schreiber SL. J. Am. Chem. Soc. 1990;112:5583–5601. [Google Scholar]
- 41.(a) Garcia J, Lopez M, Romeu J. Synlett. 1999:429–431. [Google Scholar]; (b) Garcia J, Lopez M, Romeu J. Tetrahedron: Asymmetry. 1999;10:2617–2626. [Google Scholar]; (c) Yun HD, Danishefsky SJ. J. Org. Chem. 2003;68:4519–4522. doi: 10.1021/jo0341665. [DOI] [PubMed] [Google Scholar]
- 42.(a) Hirao A, Itsuno S, Nakahama S, Yamazaki N. J. Chem. Soc. Chem. Commun. 1981:315–317. [Google Scholar]; (XX) Itsuno S, Ito K, Hirao A, Nakahama S. J. Chem. Soc. Chem. Commun. 1983:469–470. [Google Scholar]; (b) Corey EJ, Bakshi RK, Shibata S. J. Am. Chem. Soc. 1987;109:5551–5553. [Google Scholar]; (c) Corey EJ, Bakshi RK, Shibata S, Chen CP, Singh VK. J. Am. Chem. Soc. 1987;109:7925–7926. [Google Scholar]; (d) Corey EJ, Helal CJ. Angew. Chem. Int. Ed. 1998;37:1987–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 43.After silylation of the C6-alcohol, the four diastereomers formed in the reduction step were separable by HPLC and allowed for the determination of the enantiomeric excess.
- 44.Langille NF, Dakin LA, Panek JS. Org. Lett. 2003;5:575–578. doi: 10.1021/ol027518n. [DOI] [PubMed] [Google Scholar]
- 45.Parikh JR, Doering WvE. J. Am. Chem. Soc. 1967;89:5505–5507. [Google Scholar]
- 46.Evans DA, Peterson GS, Johnson JS, Barnes DM, Campos KR, Woerpel KA. J. Org. Chem. 1998;63:4541–4544. [Google Scholar]
- 47.Nakamura E, Shimada J, Kuwajima I. Organometallics. 1985;4:641–646. [Google Scholar]
- 48.Nakamura E, Aoki S, Sekiya K, Oshino H, Kuwajima I. J. Am. Chem. Soc. 1987;109:8056–8066. [Google Scholar]
- 49.Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. J. Am. Chem. Soc. 1999;121:12208–12209. [Google Scholar]
- 50.Narasaka K, Ukaji Y, Watanabe K. Bull. Chem. Soc. Jpn. 1987;60:1457–1464. [Google Scholar]
- 51.Williams JM, Jobson RB, Yasuda N, Marchesini G, Dolling UH, Grabowski EJJ. Tetrahedron Lett. 1995;36:5461–5464. [Google Scholar]
- 52.(a) Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]; (b) Levin JI, Turos E, Weinreb SM. Synth. Commun. 1982;12:989–993. [Google Scholar]
- 53.(a) Vong BG, Abraham S, Xiang AX, Theodorakis EA. Org. Lett. 2003;5:1617–1620. doi: 10.1021/ol034243i. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Urpi F, Somers TC, Clark JS, Bilodeau MT. J. Am. Chem. Soc. 1990;112:8215–8216. [Google Scholar]
- 54.(a) Bailey WF, Punzalan ER. J. Org. Chem. 1990;55:5404–5406. [Google Scholar]; (b) Negishi E, Swanson DR, Rousset CJ. J. Org. Chem. 1990;55:5406–5409. [Google Scholar]
- 55.(a) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J. Am. Chem. Soc. 1991;113:4092–4096. [Google Scholar]; (b) Dale JA, Mosher HS. J. Am. Chem. Soc. 1973;95:512–519. [Google Scholar]
- 56.Veysoglu T, Mitscher LA, Swayze JK. Synthesis. 1980:807–810. [Google Scholar]
- 57.(a) Freeman PK, Hutchinson LL. J. Org. Chem. 1980;45:1924–1930. [Google Scholar]; (b) Ireland RE, Smith MG. J. Am. Chem. Soc. 1988;110:854–860. [Google Scholar]
- 58.Ding PY, Ghosez L. Tetrahedron. 2002;58:1565–1571. [Google Scholar]
- 59.(a) Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]; (b) Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277–7287. [Google Scholar]
- 60.Trost BM, Arndt HC, Strege PE, Verhoeven TR. Tetrahedron Lett. 1976:3477–3478. [Google Scholar]
- 61.The selective TES-removal was accompanied by minor loss of the C1 TIPS protecting group. Since the crude mixture of 60 was subjected directly to the PPTS-mediated spirocyclization, 76% of 61 was obtained directly in the spirocyclization event while additional 7% of 61 was obtained after reinstallation of the C1 TIPS substituent.
- 62.Crouch RD. Tetrahedron. 2004;60:5833–5871. [Google Scholar]
- 63.Taskinen E, Virtanen R. J. Org. Chem. 1977;42:1443–1449. [Google Scholar]
- 64.Rambaud M, Bakasse M, Duguay G, Villieras J. Synthesis. 1988:564–566. [Google Scholar]
- 65.Peterson SL. Ph. D. Thesis. Cambridge: Harvard University; 2006. [Google Scholar]
- 66.(a) Hayward MM, Roth RM, Duffy KJ, Dalko PI, Stevens KL, Guo JS, Kishi Y. Angew. Chem. Int. Ed. 1998;37:192–196. [Google Scholar]; (b) Heathcock CH, McLaughlin M, Medina J, Hubbs JL, Wallace GA, Scott R, Claffey MM, Hayes CJ, Ott GR. J. Am. Chem. Soc. 2003;125:12844–12849. doi: 10.1021/ja030317+. [DOI] [PubMed] [Google Scholar]
- 67.(a) Seebach D, Corey EJ. J. Org. Chem. 1975;40:231–237. [Google Scholar]; (b) Smith AB, Condon SM, McCauley JA, Leazer JL, Leahy JW, Maleczka RE. J. Am. Chem. Soc. 1997;119:947–961. [Google Scholar]
- 68.Bernardi R, Ghiringhelli D. J. Org. Chem. 1987;52:5021–5022. [Google Scholar]
- 69.Shapiro RH, Heath MJ. J. Am. Chem. Soc. 1967;89:5734–5735. [Google Scholar]
- 70.(a) Chamberlin AR, Stemke JE, Bond FT. J. Org. Chem. 1978;43:147–154. [Google Scholar]; (b) Chamberlin AR, Bloom SH. Org. React. 1991;45:1–84. [Google Scholar]
- 71.Mozingo R. Org. Synth. 1955;Collct. Vol. 3:181–183. [Google Scholar]
- 72.Blacklock TJ, Butcher JW, Sohar P, Lamanec TR, Grabowski EJJ. J. Org. Chem. 1989;54:3907–3913. [Google Scholar]
- 73.Casiraghi G, Rassu G. Synthesis. 1995:607–626. [Google Scholar]
- 74.(a) Danishefsky SJ, Deninno MP. J. Org. Chem. 1986;51:2615–2617. [Google Scholar]; (b) Garner P, Park JM. Synth. Commun. 1987;17:189–194. [Google Scholar]
- 75.(a) Kitajima H, Katsuki T. Synlett. 1997:568–570. [Google Scholar]; (b) Evans DA, Johnson DS. Org. Lett. 1999;1:595–598. doi: 10.1021/ol990113r. [DOI] [PubMed] [Google Scholar]; (c) Evans DA, Rovis T, Kozlowski MC, Downey CW, Tedrow JS. J. Am. Chem. Soc. 2000;122:9134–9142. [Google Scholar]; (d) Evans DA, Scheidt KA, Johnston JN, Willis MC. J. Am. Chem. Soc. 2001;123:4480–4491. doi: 10.1021/ja010302g. [DOI] [PubMed] [Google Scholar]
- 76.Ibrahim-Ouali M, Parrain JL, Santelli M. Org. Prep. Proced. Int. 1999;31:467–505. [Google Scholar]
- 77.Horita K, Yoshioka T, Tanaka T, Oikawa Y, Yonemitsu O. Tetrahedron. 1986;42:3021–3028. [Google Scholar]
- 78.(a) Yoshii E, Koizumi T, Kitatsuji E, Kawazoe T, Kaneko T. Heterocycles. 1976;4:1663–1668. [Google Scholar]; (b) Jefford CW, Sledeski AW, Rossier JC, Boukouvalas J. Tetrahedron Lett. 1990;31:5741–5744. [Google Scholar]; (c) Nefkens GHL, Thuring J, Zwanenburg B. Synthesis. 1997:290–292. [Google Scholar]
- 79.Evans DA, MacMillan DWC, Campos KR. J. Am. Chem. Soc. 1997;119:10859–10860. [Google Scholar]
- 80.Evans DA, Wu J. J. Am. Chem. Soc. 2005;127:8006–8007. doi: 10.1021/ja0522130. [DOI] [PubMed] [Google Scholar]
- 81.(a) Crabtree R. Acc. Chem. Res. 1979;12:331–338. [Google Scholar]; (b) Crabtree RH, Davis MW. Organometallics. 1983;2:681–682. [Google Scholar]
- 82.(a) Flynn DL, Zelle RE, Grieco PA. J. Org. Chem. 1983;48:2424–2426. [Google Scholar]; (b) Evans DA, Carter PH, Dinsmore CJ, Barrow JC, Katz JL, Kung DW. Tetrahedron Lett. 1997;38:4535–4538. [Google Scholar]
- 83.Evans DA, Dimare M. J. Am. Chem. Soc. 1986;108:2476–2478. doi: 10.1021/ja00269a073. [DOI] [PubMed] [Google Scholar]
- 84.Evans DA, Morrissey MM. J. Am. Chem. Soc. 1984;106:3866–3868. [Google Scholar]
- 85.Evans DA, Bartroli J, Shih TL. J. Am. Chem. Soc. 1981;103:2127–2129. [Google Scholar]
- 86.Omura K, Swern D. Tetrahedron. 1978;34:1651–1660. [Google Scholar]
- 87.(a) Garegg PJ, Samuelsson B. J. Chem. Soc. Perkin Trans. 1980;1:2866–2869. [Google Scholar]; (b) Garegg PJ, Johansson R, Ortega C, Samuelsson B. J. Chem. Soc. Perkin Trans. 1982;1:681–683. [Google Scholar]
- 88.(a) Evans DA, Rieger DL, Jones TK, Kaldor SW. J. Org. Chem. 1990;55:6260–6268. [Google Scholar]; (b) Evans DA, Kaldor SW, Jones TK, Clardy J, Stout TJ. J. Am. Chem. Soc. 1990;112:7001–7031. [Google Scholar]
- 89.(a) Keck GE, Boden EP. Tetrahedron Lett. 1984;25:265–268. [Google Scholar]; (b) Keck GE, Boden EP. Tetrahedron Lett. 1984;25:1879–1882. [Google Scholar]; (c) Takai K, Heathcock CH. J. Org. Chem. 1985;50:3247–3251. [Google Scholar]; (d) Harwood LM, Manage AC, Robin S, Hopes SFG, Watkin DJ, Williams CE. Synlett. 1993:777–780. [Google Scholar]
- 90.(a) Brown HC, Dhar RK, Ganesan K, Singaram B. J. Org. Chem. 1992;57:499–504. [Google Scholar]; (b) Brown HC, Dhar RK, Ganesan K, Singaram B. J. Org. Chem. 1992;57:2716–2721. [Google Scholar]
- 91.Shute RE, Rich DH. Synthesis. 1987:346–349. [Google Scholar]
- 92.Cannizzo LF, Grubbs RH. J. Org. Chem. 1985;50:2386–2387. [Google Scholar]
- 93.At −40 °C, the Tebbe reaction only proceeded to ~60% conversion (55% isolated yield) while the residual starting ketone could be reisolated in high yield. The combined 78% reported yield represents two recyclings of starting material. At higher temperatures, the conversions were higher but the amount of decomposition of the starting ketone was also significant, thus giving an overall lower mass recovery.
- 94.Takai K, Kakiuchi T, Kataoka Y, Utimoto K. J. Org. Chem. 1994;59:2668–2670. [Google Scholar]
- 95.Tebbe FN, Parshall GW, Reddy GS. J. Am. Chem. Soc. 1978;100:3611–3613. [Google Scholar]
- 96.The majority of the sulfone 4 could be recovered (78% based on the 50% coupling yield) as a mixture of 4 and its C21 epimer.
- 97.Lindgren BO, Nilsson T. Acta Chem. Scand. 1973;27:888–890. [Google Scholar]
- 98.(a) Kraus GA, Taschner MJ. J. Org. Chem. 1980;45:1175–1176. [Google Scholar]; (b) Kraus GA, Roth B. J. Org. Chem. 1980;45:4825–4830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.