

Abstract
Genotypic differences greatly influence susceptibility and resistance to disease. Understanding genotype-phenotype relationships requires that phenotypes be viewed as manifestations of network properties, rather than simply as the result of individual genomic variations1. Genome sequencing efforts have identified numerous germline mutations associated with cancer predisposition and large numbers of somatic genomic alterations2. However, it remains challenging to distinguish between background, or “passenger” and causal, or “driver” cancer mutations in these datasets. Human viruses intrinsically depend on their host cell during the course of infection and can elicit pathological phenotypes similar to those arising from mutations3. To test the hypothesis that genomic variations and tumour viruses may cause cancer via related mechanisms, we systematically examined host interactome and transcriptome network perturbations caused by DNA tumour virus proteins. The resulting integrated viral perturbation data reflects rewiring of the host cell networks, and highlights pathways that go awry in cancer, such as Notch signalling and apoptosis. We show that systematic analyses of host targets of viral proteins can identify cancer genes with a success rate on par with their identification through functional genomics and large-scale cataloguing of tumour mutations. Together, these complementary approaches result in increased specificity for cancer gene identification. Combining systems-level studies of pathogen-encoded gene products with genomic approaches will facilitate prioritization of cancer-causing driver genes so as to advance understanding of the genetic basis of human cancer.
Integrative studies of viral proteins have identified host perturbations relevant to viral disease aetiology4,5. We examined whether such a strategy extended systematically across a range of tumour viruses could shed light on cancers even beyond those directly caused by these pathogens. Our hypothesis is inspired by classical examples where DNA tumour virus proteins physically target the products of RB1 or TP53, two well-established germline-inherited and somatically-inactivated tumour-suppressor genes6. We propose that viruses and genomic variations alter local and global properties of cellular networks in similar ways to cause pathological states. Models derived from host perturbations mediated by viral proteins representing the virome7 should serve as surrogates for network perturbations that result from large numbers of genomic variations, or the variome8 (Fig. 1a).
Figure 1. Systematic mapping of binary interactions and co-complex associations between viral and host proteins.

a, The virome-to-variome network model proposes that genomic variations (point mutations, amplifications, deletions or translocations) and expression of tumour virus proteins induce related disease states by similarly influencing properties of cellular networks. b, Experimental pipeline for identifying viral-host interactions. Selected cloned viORFs were subjected to yeast two-hybrid (Y2H) screens, and introduced into cell lines for both tandem-affinity purification followed by mass spectrometry (TAP-MS) and microarray analyses. Numbers of viORFs that were successfully processed at each step are indicated in red. c, Left panel: network of binary viral-host interactions identified by Y2H. Right panel: subsets of human target proteins that have significantly more (red dots) or less (black dots) viral interactors than expected based on their degree in HI-2. d, Network of co-complex associations of E6 viral proteins from six HPV types (hexagons, coloured according to disease class) with host proteins (circles). Host proteins that associate with two or more E6 proteins are coloured according to the disease class(es) of the corresponding HPV types. Circle size is proportional to the number of associations between host and viral proteins in the E6 networks. Distribution plots of 1,000 randomised networks and experimentally observed data (green arrows) for the number of host proteins targeted by two or more viral proteins in the corresponding sub-networks (left histogram), or the ratio of the probability of a host protein being targeted by viral proteins from the same class to the probability it is targeted by viral proteins from different classes (right histogram). Insets: representative random networks from these distributions.
We used an integrated pipeline to systematically investigate perturbations of host interactome and transcriptome networks induced by the gene products of four functionally related yet biologically distinct, families of DNA tumour viruses: Human Papillomavirus (HPV), Epstein-Barr Virus (EBV), Adenovirus (Ad5) and Polyomavirus (PyV) (Fig. 1b and Supplementary Table 1 and Supplementary Fig. 1).
Applying a stringent implementation of the yeast two-hybrid (Y2H) system9, 123 viral open reading frames (viORFs) were screened against a collection of ~13,000 human ORFs10 resulting in a validated viral-host interaction network of 454 binary interactions involving 53 viral proteins and 307 human proteins (Fig. 1c and Supplementary Table 2). Analysis of our binary interaction map identified 31 host target proteins that exhibited more binary interactions with viral proteins than would be expected given their “degree” (number of interactors) in our current binary map of the human interactome network (HI-2)11 (Fig. 1c and Supplementary Table 3 and Supplementary Note 1), suggesting a set of common mechanisms by which different viral proteins rewire the host interactome network.
To examine both interactome and transcriptome network perturbations directly in human cells, we generated expression constructs fusing each viral ORF to a tandem epitope tag and introduced each construct into IMR-90 normal human diploid fibroblasts. Co-complex associations between viral proteins and the host proteome were identified by tandem affinity purification followed by mass spectrometry (TAP-MS)12. The intersection of two independent TAP-MS experiments yielded 3,787 reproducibly mapped viral-host co-complex associations involving 54 viral proteins and the products of 1,079 host proteins (Supplementary Table 4). Statistically significant overlaps of the Y2H binary and the TAP-MS co-complex datasets with a positive reference set (PRS) of curated viral-host interactions were observed, supporting the quality of the interactome datasets (Supplementary Notes 2 and 3). Host proteins identified as binary interactors or as co-complex members showed a statistically significant overlap (P < 0.001) and a statistically significant tendency to interact with each other in HI-2 (P < 0.001), implying that host targets in the virus-host interactome maps tend to fall in the same “neighbourhood” of the host network1 (Supplementary Fig. 2). Our two complementary interactome datasets highlight specific host biological processes targeted by viral proteins (Supplementary Fig. 3).
To explore the specificity of viral-host relationships, we examined co-complex associations mediated by E6 proteins from six distinct HPV types representing three different disease classes, high-risk or low-risk mucosal, and cutaneous. Multiple host proteins were found to associate with E6 proteins encoded by two or more different HPV types (P < 0.001; Fig. 1d), including the known E6 target UBE3A (E6AP)13. Among these we observed a statistically significant subgroup of host proteins targeted only by E6 proteins from the same disease class (P < 0.001). The transcriptional regulators CREBBP and EP300 were found to associate with E6 proteins from both cutaneous HPV types, but not with those from the mucosal classes. In contrast to E6 proteins, no group of host proteins showed class-specific targeting by HPV E7 proteins (Supplementary Fig. 4). These differential associations reflect how rewiring of virus-host interactome networks may relate to the aetiology of viral diseases.
In addition to targeting protein-protein interactions, viral proteins also functionally perturb their hosts through downstream effects on gene expression. We profiled the transcriptome of the viORF-transduced cell lines to trace pathways through which viral proteins could alter cellular states. Model-based clustering of the ~3,000 most frequently perturbed host genes identified 31 clusters, many of which were enriched (P < 0.01) for specific GO terms and KEGG pathways (Fig. 2a, Supplementary Tables 5 and 6). To uncover transcription factor (TF) binding motifs enriched within the promoters or enhancers of the corresponding genes, a high-confidence map of predicted TF binding sites was generated using cell-specific chromatin accessibility information and consensus TF-binding motifs (Supplementary Fig. 5). We found a densely interconnected set of 92 TFs (Supplementary Fig. 6 and Supplementary Note 4) that either associated with or were differentially expressed in response to viral proteins, and whose target genes were enriched in at least one cluster (Supplementary Fig. 7 and Supplementary Table 7).
Figure 2. Transcriptome perturbations induced by viral protein expression.

a, Heatmap of average cluster expression relative to control. Enriched GO terms and KEGG pathways are listed adjacent to the numbered expression clusters. In cluster C1 eight of the nine transcripts are snoRNAs (denoted with #). Upper dendrogram is shaded by viORF grouping. Grey blocks show which viral proteins associate with the indicated host proteins. b, Schematic shows how the viral protein-TF-target gene network was constructed, with three representative networks shown. Null distribution of average fraction of TF target genes differentially expressed in the corresponding cell lines (histogram), along with observed value (green arrow).
The mean expression change of each cluster revealed three distinct groups of viral proteins (Fig. 2a): Group I included low-risk and cutaneous HPV E6 proteins, Group II contained most of the EBV proteins, and Group III included high-risk HPV E6 and E7 proteins and polyomavirus proteins. Consistent with their ability to associate with RB1, Group III viral proteins increased expression of genes that are involved in cell proliferation and whose promoters are enriched in E2F binding sites (clusters C26 and C31). Steady-state levels of these genes correlate with cellular growth phenotypes (Supplementary Fig. 8 and Supplementary Note 5). Likewise the down-regulation of the p53 signalling pathway likely reflects the ability of Group III proteins to bind to and inactivate p53 (cluster C12).
To investigate additional pathways through which viral proteins perturb TFs to reprogram cellular states we derived a detailed network model containing 58 viral proteins that perturb the activity of 86 TFs, which in turn potentially regulate 30 clusters (Supplementary Fig. 9). This model was predictive of downstream patterns of differential expression (P = 0.003; Fig. 2b) and suggested ways in which viral proteins could regulate many of the biological hallmarks of cancer14 (Supplementary Note 6). For example, we found regulation of several pathways involved in the response to DNA damage (Fig. 2b), including autophagy potentially through NFE2L2 (cluster C3)15, the NFκB-mediated inflammatory response (cluster C23)16, and the type I interferon response through IRF1 (cluster C24; Supplementary Fig. 5)17.
Specific disease outcomes of the three disease classes of HPV might reflect how their respective oncoproteins perturb distinct functional groups of host proteins. Our co-complex map revealed associations between E6 from cutaneous HPVs and MAML1, EP300 and CREBBP. MAML1 forms a transcriptional activation complex that modulates expression of Notch target genes in conjunction with EP300 and CREBBP histone acetyltransferases, the RBPJ transcription factor and the intracellular domain (ICD) of the Notch receptor18. Our transcriptome profiling placed the cutaneous and low-risk HPV E6 proteins in Group I apart from the high-risk HPV E6 proteins in Group III (Fig. 2a), so we investigated these differential perturbations. Both cutaneous HPV5 and HPV8 E6 proteins co-precipitated MAML1 and EP300, while the mucosal HPV E6 proteins did not (Fig. 3a and Supplementary Fig. 10); conversely HPV6b, HPV11, HPV16 and HPV18 E6 proteins associated with UBE3A13, while cutaneous HPV E6 proteins did not.
Figure 3. The Notch pathway is targeted by multiple DNA tumour virus proteins.
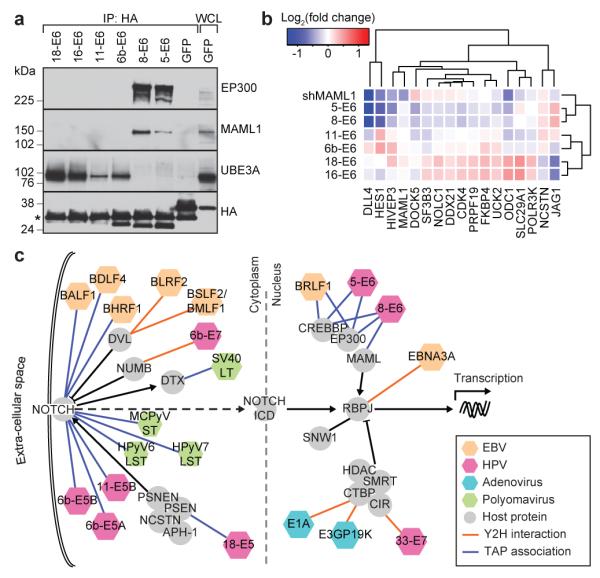
a, Western blots of co-immunoprecipitations of HPV E6 proteins in IMR-90 cells. b, Heatmap of expression of Notch pathway responsive genes in IMR-90 cells upon expression of E6 proteins from different HPV types or upon knockdown of MAML1, relative to control cells. c, Representation of viral protein interactions with components of the Notch signalling pathway (as defined in KEGG).
Notch signalling perturbations can confer either oncogenic or tumour-suppressive effects18. Since both Notch pathway inhibition and HPV8 E6 overexpression promote squamous cell carcinoma19,20, we reasoned that binding of HPV5 and HPV8 E6 to MAML1 might inhibit Notch signalling. To test this, we examined transcript levels of Notch pathway genes and potential Notch target genes with a predicted RBPJ binding site in their promoter across all HPV E6 cell lines as well as in cells depleted for MAML1. Transcript levels of several Notch targets were significantly reduced in IMR-90 cells expressing either HPV5 or HPV8 E6 or depleted for MAML1 (Fig. 3b and Supplementary Fig. 11), These and other results21 indicate that association of HPV5 and HPV8 E6 proteins with MAML1 inhibit Notch signalling. Building on these observations and on the established associations between EBV EBNA proteins and RBPJ22, we observed that viral proteins from all four DNA tumour viruses target proteins of the Notch pathway (P < 0.002; Fig. 3c). Our data highlights the central role of Notch signalling in viral-host perturbations as well as tumourigenesis, and supports observations that implicate MAML1 in cancer pathogenesis23.
We next investigated the extent to which viral proteins globally target host proteins altered in cancer. First we compared our viral targets, identified through binary interaction, co-complex associations and TF binding site analyses, against a gold standard set of 107 high-confidence causal human cancer genes in the COSMIC Classic (CC) gene set24 (Supplementary Table 8). Viral targets were significantly enriched among CC genes (Supplementary Fig. 12; Padj = 0.01). To optimize the stringency of potential cancer enrichment analyses, we restricted the TAP-MS co-complex viral targets to those identified by three or more unique peptides, a choice corresponding to an experimental reproducibility rate greater than 90% (Supplementary Fig. 13). The resulting stringent candidate set of 947 host target genes (the “VirHost” set; Supplementary Table 9), included 16 proteins encoded by CC genes (P = 0.007; Fig. 4a) among which tumour suppressor genes were significantly over-represented (P = 0.03).
Figure 4. Interpretation of somatic cancer mutations using viral-host network models.

a, Schematic describing composition of VirHost (proteins identified by TAP-MS with ≥3 unique peptides, Y2H and TF) and overlap with COSMIC Classic genes. Viral protein (hexagon) perturbations of cancer proteins (circles) classified as oncogenes or tumour suppressors. b, Venn diagram of overlaps of VirHost proteins with COSMIC Classic genes and candidate cancer genes identified through four transposon-based functional genomics screens. c, Venn diagram of overlaps of VirHost proteins with COSMIC Classic genes and with a prioritised set of genes found through somatic mutation analysis. P values: Fisher’s exact test or permutation based. d-f, Venn Diagrams comparing VirHost, GWAS (d), SCNA-AMP (e) and SCNA-DEL (f) data sets for ability to recover COSMIC Classic genes.
As a complementary approach to validate our VirHost gene set, we compiled a list of human orthologues of novel mouse genes implicated in tumourigenesis by in vivo transposon mutagenesis screens25. Our VirHost dataset significantly overlaps with these candidate cancer genes (P < 0.0001) (Fig. 4b and Supplementary Table 10). The 156 candidate genes in the overlap were markedly enriched for CC genes (OR = 13, P = 4 × 10−9) as well as genes implicated in apoptosis, hypoxia response and cell growth pathways (Padj < 0.05 for all). Altogether these observations suggest that our VirHost dataset might point to novel human cancer-associated genes.
Large-scale tumour sequencing efforts have the potential to discover new tumour suppressors and oncogenes. To explore how the VirHost set might be used to interpret these data, we compiled somatic mutations for eight different cancers identified through twelve sequencing projects. Non-synonymous somatic mutations were reported for too many genes (10,543) to permit a useful identification of putative causal cancer genes without further prioritisation. We therefore scored the likely functional effects of these mutations using the PolyPhen2 program26 and generated a cumulative somatic mutation (SM) score for each protein (Supplementary Fig. 14). To compare performance in identifying candidate cancer genes of our VirHost set with that of proteins ranked by SM analysis, we tested a matching number (947) of the top ranked SM candidates for overlap with CC genes (Fig. 4c and Supplementary Table 11). Compared to the 16 cancer genes identified in our VirHost set, SM recovered 23 genes (P = 6 × 10−10). Viral perturbation analysis is comparable to somatic mutation sequencing for identifying cancer genes.
Although both strategies showed significant overlap with the reference COSMIC Classic set, neither by itself suffices to pinpoint causal genes with high specificity. To overcome this difficulty we exploited the orthogonal nature of the VirHost and SM sets (given P = 0.58 for their overlap) by focusing on the 43 proteins at their intersection (the “VirHostSM” subset) (Fig. 4c). Compared to VirHost (OR = 3.7) or SM (OR = 5.8), the VirHostSM set was markedly enriched in CC proteins (5 proteins, OR = 26, P = 3 × 10−6). Pathway analysis of the 43 proteins revealed 12 proteins implicated in the GO pathway linked to “regulation of apoptosis” (OR = 6.0, Padj = 0.017). The intersection also includes plausible contributors to cancer pathogenesis (Supplementary Fig. 15) such as the oxidative stress response transcription factor NFE2L2.
We compared the ability of VirHost to identify CC genes to two other large scale genomic approaches: SCNA (somatic copy number alteration)27 analysis of cancers and GWAS (genome-wide association studies) of cancer susceptibility28. The SCNA deletions (SCNA-DEL) and amplifications (SCNA-AMP) and GWAS sets all significantly overlapped with CC genes, but with lower specificity than the VirHost overlap with CC (OR = 1.9 for SCNA-DEL, 2.1 for SCNA-AMP, and 3.1 for GWAS, versus 3.7 for VirHost) (Fig. 4d-f). The intersections of VirHost with GWAS or SCNA-DEL genes also showed enrichment for cancer genes (Supplementary Table 12). The intersection of VirHost and SCNA-DEL was enriched for genes implicated in apoptosis (GO term “programmed cell death”, 15 genes, Padj = 0.022, OR = 4.3). Conversely, there was no synergy in the intersection of SCNA-AMP and VirHost, perhaps reflecting the preference of viral proteins in targeting tumour suppressors rather than oncogenes (Fig. 4f).
Our systems-level explorations of viral perturbations facilitate the distinction between driver and passenger mutations in cancer genome sequences. Our data indicate that trans-acting viral products and cis-acting genome variations involved in cancer converge upon common pathways.
METHODS SUMMARY
viORF entry clones were generated by PCR-based Gateway recombinational cloning4. After sequence verification viORFs were transferred by in vitro Gateway LR recombinational cloning into expression vectors for Y2H screening9 and for transduction of IMR-90 cells. Y2H screens were carried out against the human ORFeome v5.1 collection of ~13,000 full-length human ORFs10. Total RNA was isolated from IMR-90 cells expressing viORFs and gene expression was assayed on Human Gene 1.0 ST arrays. Microarray data was analysed using R/Bioconductor. Viral proteins and associated host proteins were purified by sequential FLAG and hemagglutinin (HA) immunoprecipitation and analyzed by LC-MS/MS mass spectrometry. Viral-host co-complexes from two independent purifications were analysed. Pathway enrichment was analysed using FuncAssociate29. Assessment of statistical significance for overlap between gene sets was carried out using Fisher’s Exact Test or resampling-based approaches.
A complete description of the materials and methods is provided in the Supplementary Methods.
Supplementary Material
Acknowledgements
The authors wish to acknowledge members of the Center for Cancer Systems Biology (CCSB) and J. Astor, M. Meyerson, W. Kaelin, G. Superti-Furga and S. Sunyaev for helpful discussions. We thank J.W. Harper, W. Hahn, P. Howley, Y. Jacob, M. Imperiale, I. Koralnik, H. Pfister, and D. Wang for reagents. This work was primarily supported by Center of Excellence in Genomic Science (CEGS) grant P50HG004233 from the National Human Genome Research Institute (NHGRI) of the National Institutes of Health (NIH) awarded to M.V. (PI), A.-L.B., J.A.D., E.K., J.M., K.M., J.Q., and F.P.R. Additional funding included Institute Sponsored Research funds from the Dana-Farber Cancer Institute Strategic Initiative to M.V.; NIH grants R01HG001715 to M.V., D.E.H. and F.P.R.; R01CA093804, R01CA063113 and P01CA050661 to J.A.D.; R01CA081135, R01CA066980, and U01CA141583 to K.M.; R01CA131354, R01CA047006, and R01CA085180 to E.K.; T32HL007208 and K08HL098361 to R.C.D.; K08CA122833 to R.B.; F32GM095284 and K25HG006031 to M.P.; Canada Excellence Research Chairs (CERC) Program, Canadian Institute for Advanced Research Fellowship and Ontario Research Fund to F.P.R.; James S. McDonnell Foundation grant 220020084 to A.-L.B. M.V. is a “Chercheur Qualifié Honoraire” from the Fonds de la Recherche Scientifique (FRS-FNRS, Wallonia-Brussels Federation, Belgium).
Footnotes
Additional co-authors are listed alphabetically.
Author Contributions O.R.-R., G.A., M.A.C., M.G., A.D., Ma.T., F.A., D.B., A.A.C., J.C., M.C., M.D., M.C.F., S.B.F., R.F., B.K.G., A.M.H., R.J., A.K., L.L., R.R., J.M.S., S.W., J.R.-C. and E.J. performed experiments or contributed new reagents. R.C.D., M.P., G.A., T.R., M.A., S.P., A.-R.C., C.F., N.G., T.H., J.C.M., T.R.P., S.R., Y.S., S.S., Mu.T. and J.T.W. performed computational analysis. O.R.-R., R.C.D., M.P., G.A., M.A.C., T.R., M.E.C., D.E.H., K.M., J.A.M., F.P.R., J.A.D. and M.V. wrote the manuscript. A.-L.B., R.B., E.K., M.E.C., D.E.H., K.M., J.A.M., J.Q., F.P.R., J.A.D. and M.V. designed or advised research.
Author Information Microarray data were deposited in the Gene Expression Omnibus database (accession number GSE38467). The authors declare no competing financial interests.
References
- 1.Vidal M, Cusick ME, Barabási AL. Interactome networks and human disease. Cell. 2011;144:986–998. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011;331:1553–1558. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- 3.Gulbahce N, et al. Viral perturbations of host networks reflect disease etiology. PLoS Comput. Biol. 2012 doi: 10.1371/journal.pcbi.1002531. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calderwood MA, et al. Epstein-Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. USA. 2007;104:7606–7611. doi: 10.1073/pnas.0702332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapira SD, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howley PM, Livingston DM. Small DNA tumor viruses: large contributors to biomedical sciences. Virology. 2009;384:256–259. doi: 10.1016/j.virol.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foxman EF, Iwasaki A. Genome-virome interactions: examining the role of common viral infections in complex disease. Nat. Rev. Microbiol. 2011;9:254–264. doi: 10.1038/nrmicro2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Editorial, What is the human variome project? Nat. Genet. 2007;39:423. doi: 10.1038/ng0407-423. [DOI] [PubMed] [Google Scholar]
- 9.Dreze M, et al. High-quality binary interactome mapping. Methods Enzymol. 2010;470:281–315. doi: 10.1016/S0076-6879(10)70012-4. [DOI] [PubMed] [Google Scholar]
- 10.Lamesch P, et al. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89:307–315. doi: 10.1016/j.ygeno.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu H, et al. Next-generation sequencing to generate interactome datasets. Nat. Methods. 2011;8:478–480. doi: 10.1038/nmeth.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou F, et al. Online nanoflow RP-RP-MS reveals dynamics of multicomponent Ku complex in response to DNA damage. J. Proteome Res. 2010;9:6242–6255. doi: 10.1021/pr1004696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brimer N, Lyons C, Vande Pol SB. Association of E6AP (UBE3A) with human papillomavirus type 11 E6 protein. Virology. 2007;358:303–310. doi: 10.1016/j.virol.2006.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc. Natl. Acad. Sci. USA. 2011;108:1427–1432. doi: 10.1073/pnas.1014156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NFκB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka N, et al. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature. 1996;382:816–818. doi: 10.1038/382816a0. [DOI] [PubMed] [Google Scholar]
- 18.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat. Rev. Cancer. 2011;11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 19.Proweller A, et al. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006;66:7438–7444. doi: 10.1158/0008-5472.CAN-06-0793. [DOI] [PubMed] [Google Scholar]
- 20.Marcuzzi GP, et al. Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J. Gen. Virol. 2009;90:2855–2864. doi: 10.1099/vir.0.012872-0. [DOI] [PubMed] [Google Scholar]
- 21.Brimer N, Lyons C, Wallberg AE, Vande Pol SB. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene in press. 2012 doi: 10.1038/onc.2011.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calderwood MA, et al. Epstein-Barr virus nuclear protein 3C binds to the N-terminal (NTD) and beta trefoil domains (BTD) of RBP/CSL; only the NTD interaction is essential for lymphoblastoid cell growth. Virology. 2011;414:19–25. doi: 10.1016/j.virol.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klinakis A, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473:230–233. doi: 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Copeland NG, Jenkins NA. Harnessing transposons for cancer gene discovery. Nat. Rev. Cancer. 2010;10:696–706. doi: 10.1038/nrc2916. [DOI] [PubMed] [Google Scholar]
- 26.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manolio TA. Genomewide association studies and assessment of the risk of disease. N. Engl. J. Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 29.Berriz GF, King OD, Bryant B, Sander C, Roth FP. Characterizing gene sets with FuncAssociate. Bioinformatics. 2003;19:2502–2504. doi: 10.1093/bioinformatics/btg363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.