

Abstract
NOL7 is a candidate tumor suppressor gene that localizes to 6p23, a chromosomal region frequently associated with loss of heterozygosity in a number of malignancies including cervical cancer (CC). Re-expression of NOL7 in CC cells suppresses in vivo tumor growth by 95% and alters the angiogenic phenotype by modulating the expression of VEGF and TSP1. Here, we describe the determination of two NOL7 transcriptional start sites (TSS), the cloning of its regulatory promoter region, and the identification of transcription factors that regulate its expression. Using 5′ Rapid amplification of complementary DNA ends (RACE), two transcriptional start sites were identified. Deletion analysis determined that the essential elements required for the optimal promoter activity of NOL7 were 560 bp upstream of its translation start site. In silico analysis suggested that the promoter region contained potential binding sites for the SP1, c-Myc and RXRα transcription factors as well as an overall GC content of greater than 60%. Chromatin immunoprecipitation (ChIP) confirmed that SP1, c-Myc and RXRα bound to the NOL7 promoter region. Finally, we demonstrate that NOL7 expression was positively regulated by c-Myc and RXRα. These results demonstrate that the NOL7 promoter region possesses each of the key elements of a TATA-less promoter. In addition, the positive regulation of NOL7 by c-Myc and RXRα provides additional mechanistic insights into the potential role of NOL7 in CC and other malignancies.
Keywords: NOL7, Transcription, TATA-less promoter, c-Myc, RXRα
1. Introduction
Cervical Cancer (CC) is the second most common malignancy in women worldwide after breast cancer (American Cancer Society, 2008). HPV infection is observed in 99% of CC cases. However additional cooperating genetic alterations are required for malignant transformation (Lazo, 1999; Walboomers et al., 1999; Kaufmann et al., 2002; Branca et al., 2006; Narisawa-Saito et al., 2008). NOL7 is a novel tumor suppressor gene which localizes to 6p23, a region frequently lost in CC (Janet S. Rader, 2000; Chatterjee et al., 2001; Mazurenko et al., 2003; Hasina et al., 2006; American Cancer Society, 2008). In addition, 6p23 loss is also observed in other malignancies including hormone refractory breast cancer, leukemias, lymphomas, osteosarcomas, retinoblastoma and nasopharyngeal carcinomas (Fleischman et al., 1983; Hoyle et al., 1988; Jadayel et al., 1995; Nemani et al., 1996; Mutirangura et al., 1997; Liao et al., 1998; Nagai et al., 1999; Chen et al., 2000; Nakase et al., 2000; Shao et al., 2000; Achuthan et al., 2001; Giagounidis et al., 2001; Lung et al., 2001; Batanian et al., 2002; Starostik et al., 2002; Fan and Rizkalla, 2003; Amare Kadam et al., 2004; Lim et al., 2004; Takeshita et al., 2004; Gasowska-Giszczak et al., 2005). NOL7 is a 29 kDa protein that localizes to both the nucleus and nucleolus. Reintroduction of NOL7 into CC tumor cells alters the angiogenic phenotype by modulating the expression of VEGF and TSP1, thereby inhibiting in vivo tumor growth (Hasina et al., 2006). Allelic loss of NOL7 has been identified in 40% of CC cell lines and tumor samples by fluorescent in situ hybridization (FISH) analysis. Similarly, NOL7 mRNA expression was also reduced in 38% of CC cell lines (Hasina et al., 2006).
Nothing is known regarding the transcriptional activation or regulation of NOL7. Therefore, the aim of this work was to identify and characterize the promoter region of the human NOL7 gene. We have determined two transcriptional start sites of NOL7 and defined the promoter region immediately upstream of these sites. In silico examination of the promoter region predicted SP1, c-Myc and RXRα as potential transcription factors. Further, we validated these predictions, showing that SP1, c-Myc and RXRα bind to the NOL7 promoter and that c-Myc and RXRα positively regulate NOL7 expression.
2. Materials and Methods
2.1. Cell Culture
HeLa and HEK293T cell lines (ATCC) were cultured in DMEM (Invitrogen) supplemented with 10% FBS, 100 μg/ml penicillin-streptomycin (Gemini Bioproducts) and maintained at 37°C in a 5% CO2–95% air environment in humidified incubators.
2.2. 5′ RACE Assay
Total RNA was extracted from cell lines using TRIzol® reagent (Invitrogen) and quantified spectrophotometrically. The 5′ RACE kit (Invitrogen, 18374–058) was used according to the manufacturer’s instructions. Briefly, cDNA was synthesized using the RT Primer 5′-GTCTCCCGCACTCGCCGCTC-3′ and a dCTP tail was added to it. This “C-tailed cDNA” was then amplified using a nested PCR primer 5′-CCTTCCTCGTCTTCCTCCAG-3′ and the primer provided in the kit, using cycling parameters: 94 °C for 1 min, (94 °C for 30s, 55 °C for 30s, 72 °C for 1min) 30 cycles, 72 °C for 7 min. The primer design was based on the NOL7 mRNA sequence (NCBI ID: NM_016167). The 5′-RACE PCR products were resolved on 2% agarose gel, the bands were excised, cloned in to pCR®4TOPO sequencing vector (Invitrogen) and sequenced.
2.3. PCR Amplification and Cloning
Human chromosome 6 cosmid clone, LA0634c7 (LANL Human Chromosome 6 Library, HGMP Resource Centre) spanning base pairs 13,545,500 to 13,585,060 (NCBI ID: NT_007592.15) was used as a template for amplifying the NOL7 upstream fragments, using primers described in Supplementary figure 1. PCR products were digested with BglII and HindIII restriction enzymes (New England Biolabs) and resolved on a 2% agarose gel. The bands were excised, purified (Gel Extraction Kit, Qiagen) and cloned into the pGL3 basic vector (Promega) digested with the same enzymes, to obtain the deletion constructs described in Figure 3. pSV-β-Galactosidase control vector was purchased from Promega. For c-Myc cloning, SuperScript® III Kit (Invitrogen) was used to generate cDNA from HeLa RNA as per the manufacturer’s instructions. The c-Myc coding region (NCBI ID: NM_002467.3) was amplified from the HeLa cDNA using primers, 5′-GGCACTTTGCACTGGAACTT-3′ and 5′-CGCACAAGAGTTCCGTAGCT-3′ and cloned into pCDNA3.1 DEST40 vector (Invitrogen, 12274-015), which has a c-terminal V5 tag using the Gateway cloning protocol. PCR was performed using Hi Fi Phusion Master Mix (New England Biolabs) and cycling parameters; 98°C for 30s, (98°C for 10s, 64°C for 30s, 72°C for 3mins) 35 cycles and 72°C for 10 mins. All clones were verified by sequencing and the plasmid DNA was extracted using the Hi-Speed Maxi Prep Kit (Qiagen).
Figure 3.

2.4. In Silico Analysis of NOL7 Promoter Elements
The EMBOSS-Isochore program was used to identify regions within the NOL7 promoter that were particularly GC-rich (Bernardi, 1995; Pesole et al., 1999; Bernardi, 2000). TESS, Genomatrix and Alggen Promo predication softwares were used to identify the transcription factor binding sites within the promoter (Werner, 2000; Farre et al., 2003; Schug, 2003). These programs were inputted with the NOL7 promoter sequence, as identified by the luciferase assays (13,555,060 to 13,555,590 base pairs, NCBI ID: NT_007592.15). Sequences of the NOL7 upstream region among different species were analyzed using MegAlign software (DNASTAR, Inc). The NOL7 upstream region was defined as the genomic sequence separating the SIRT5 and NOL7 genes in the individual species.
2.5. Transfections
Cells were plated in 6 well plates (BD Biosciences) such that they were 70–75% confluent and co-transfected with 1μg of each promoter construct and 0.5 μg pSV-β-Galactosidase vector using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s protocol. All transfections were performed in duplicate at a given time and repeated a total of three times. Cells were lysed 24 hours after transfection and the cell lysates were used for the luciferase and β-galactosidase assays. For c-Myc transfection, cell lines were transfected with equal amounts of either pDEST40-c-Myc or pCDNA3.1 DEST40 vector for 24, 48 and 72hrs. Total RNA and protein were extracted from the cells after each of these time points.
2.6. Luciferase and β-Galactosidase Assays
Luciferase and β-Galactosidase assays were performed using luciferase and β-Galactosidase assay reagents respectively (Promega) as per the manufacturer’s guidelines. Luciferase activity was read using 20/20n Single Tube Luminometer (Turner Biosystems). The β-gal values were used to normalize the luciferase activity and either the pGL3-3.1 or pGL3-2.1 constructs were used to calibrate the normalized luciferase activity in the other constructs.
2.7. Retinoic Acid Treatment
Cells were grown to 70–75% confluence in DMEM supplemented with 10% FBS, after which they were switched to DMEM containing 1% BSA (Sigma), to eliminate artifacts caused by retinoids known be present in serum in small quantities. After growing cells overnight in the 1% BSA medium, they were treated with 1 μM of ATRA (Sigma) or 9-cis RA (Sigma) or DMSO for 4 hrs. After treatment, the cells were used to generate chromatin preparations or to extract total RNA for quantitative RT PCR as described below. The ATRA treatment was repeated three separate times.
2.8. Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed for the transcription factors c-Myc, RXRα and SP1. Cell lines untreated or treated with DMSO or ATRA or 9-cis RA were fixed with 1% formaldehyde, quenched using 1.25M Glycine, resuspended in SDS lysis buffer and sonicated (10 second pulses × 5 at 50% duty cycle, output 5, Branson sonifier) to generate chromatin sizes between 200–1000bp. ChIP assay was carried out using EZ-ChIP assay kit (Upstate Biotechnology), according to the manufacturer’s instructions. The 100 μl of soluble DNA fraction was saved as input (10%). 5 μg of c-Myc (Abcam, ab56), RXRα (Millipore, MAB5478), SP1 (Millipore, 17-601), Mouse IgG (Millipore, 12-371B) and Rabbit IgG (Millipore, 17-601) antibodies were used for the ChIP assay. Each ChIP assay was repeated twice. 2 μls of eluted DNA from ChIP and input reactions was used for PCR with primers 537F 5′-TAGAGCGCATTTCTTCCCAT-3′ and 1004R 5′-GCGCTAGACCGTCTGACCT -3′ using program 98°c for 30s, (98°c for 20s, 62°c for 30s and 72°c for 30s) 19 cycles and 72°c for 5 mins. A second PCR was performed using 1–2 μls of the amplicon as a template, this time using nested primers A3F 5′-TCTTCCCTGCCTTGAAATCAA-3′ and A3R 5′-GGCAGTGGGCGTGTTTCT-3′. The products were resolved on 2% agarose Gel.
2.9. Quantitative RT-PCR
Endogenous NOL7 mRNA expression was determined by quantitative RT PCR (PRISM 7900HT, Applied Biosystems), using Ag-Path ID RT PCR Master mix (Ambion). The TaqMan assays for NOL7 and GAPDH were purchased from Applied Biosystems (Hs00982304_m1 and Hs99999905_m1 respectively). Gene expression was quantified using the relative quantification method, according to the manufacturer’s instructions (Biosystems). Briefly, GAPDH cycle threshold (Ct) value of each sample was used to normalize NOL7 Ct value for that sample, to calculate its “delta Ct” value. Further, the NOL7 delta Ct values in the ATRA treated or c-Myc transfected cells were normalized to their respective untreated delta Ct values, to calculate delta delta Ct (ddCt). The ddCt values were then used to estimate the relative fold change in NOL7 mRNA expression.
2.10. Western Blotting
Cells transfected with pDEST40-c-Myc or pCDNA3.1 DEST40 vectors for various time periods were resuspended in 500–750 μl of lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100), and sonicated (10 second pulses × 5 at 30% duty cycle, output 3, Branson sonifier). Protein concentration was measured using Bio-Rad protein assay reagent and a Synergy HT microplate reader (BioTEK Instruments). 30 μg of protein was separated on 10% SDS-PAGE gels (Pierce) and transferred to Immunoblot-PVDF membrane (Bio-Rad). The blot was blocked and then incubated with V5 (Invitrogen, R960-CUS) or Actin (Abcam, ab8227) antibodies, followed by the appropriate secondary antibodies. Blots were developed with SuperSignal West Dura Extended Duration Substrate (Pierce) and exposed to film (Denville Scientific).
3. Results
3.1. Identification of NOL7 transcription start sites (TSS)
As a first step towards defining the promoter region of NOL7, 5′ RACE was performed to identify the gene’s TSS, which is designated as the first nucleotide copied at the 5′ end of the corresponding mRNA (Sandelin et al., 2007). 5′ RACE PCR, followed by nested amplification, was performed using total RNA isolated from HeLa cells. This yielded two product sizes of 232 and 143 bp (Fig. 1A). Analysis of these sequences demonstrated that the products corresponded to two distinct transcripts starting at −32 and +60 with respect to the NOL7 translation start site which will be referred to as +1 throughout the remainder of this report (Fig. 1B). Conventionally, transcriptional initiation is believed to occur from a single focused “TSS” (Bjornsdottir and Myers, 2008; Anish et al., 2009). Therefore, the presence of two distinct transcriptional start sites suggests that NOL7 transcription may be regulated by “non-traditional” mechanisms.
Figure 1.

3.2. Cloning and Identification of NOL7 Promoter Region
In order to compare the genomic architecture of NOL7 across different species and identify possible conserved regions, the sequences located upstream of the NOL7 gene were aligned and analyzed in four species (human, chimpanzee, mouse and dog). The genes SIRT5 and RANBP9 flank NOL7 at its 5′ and 3′ ends respectively in each species, suggesting conservation of genomic architecture (Fig. 2A). However, no specific region conservation was observed in the sequence 5′ upstream of the NOL7 gene (Fig. 2B). Therefore, in order to accurately identify the proximal promoter region, we cloned the 5′ flanking region of the human NOL7 gene. We generated an initial set of three luciferase reporter gene constructs containing deletions of the NOL7 upstream region (Fig. 3A+B). The longest construct of the three constructs was 3.1kb (pGL3-3.1) in length, which extended −3162 bp upstream of the NOL7 translation start site and 421 bp downstream of the SIRT5 stop codon (NCBI ID: NM_012241.3) (Fig. 3B). Further, the 3.1 kb region was divided into 1.2 and 2.1 kb regions (pGL3-1.2 and 2.1 respectively; Fig. 3B). To determine the functional significance of the regulatory domains of the NOL7 promoter region, equal amounts of the three constructs, and the promoter less pGL3 basic vector, were individually transfected in HEK293T cells along with the β-Galactosidase vector. The 1.2 kb region did not demonstrate any luciferase activity when compared to the pGL3 basic control vector. Conversely, the 2.1 kb and the 3.1 kb constructs dramatically increased promoter activity (Fig. 3B). To further characterize the promoter region, we generated four additional deletion constructs by creating 5′ and 3′ truncations of the 2.1 kb region as depicted in Fig. 3C. The proximal constructs pGL3-1P and pGL3-1.5P failed to demonstrate significant luciferase activity. Conversely, both of the distal constructs, pGL3-1.5 D and pGL3-1D, induced relative luciferase activity that was comparable to pGL3-2.1. Finally, the distal 2.1 kb region was further characterized by the generation of four additional constructs approximately 0.5 kb in length, that sequentially spanned the entire distal 2.1 kb region. The pGL3-0.5(4) construct demonstrated luciferase activity that was equivalent to the activity expressed by either the pGL3-2.1 or the pGL3-3.1. Conversely, the pGL3-0.5 (1), pGL3-0.5 (2) and pGL3-0.5(3) failed to demonstrated significant luciferase activity (Fig. 3C). These results demonstrated that the genomic region occupying the −560 to −30 nucleotides upstream of the first NOL7 translation start site contain the essential elements required for the optimal promoter activity of the NOL7 gene.
Figure 2.

3.3. Identification of regulatory elements in the promoter region
In silico analysis of the promoter by the TESS, PROMO and Genomatrix programs (Werner, 2000; Messeguer et al., 2002; Farre et al., 2003) demonstrated that this region lacked the major eukaryotic promoter elements, such as TATA and CCAAT boxes, implying that the NOL7 may have a TATA-less promoter. It is now believed only 10–20% of human protein-coding genes have promoters that contain TATA boxes (Anish et al., 2009). TATA-less promoters are characterized by the presence of multiple TSSs, CpG islands and SP1 transcription factor binding sites (Zhu et al., 2008; Anish et al., 2009). Because NOL7 has two TSSs at positions −32 and +60 respectively (Fig. 1) and lacked the major eukaryotic promoter elements, we searched for the presence of CpG islands immediately upstream of the TSS. The overall GC content of the promoter region was estimated to be greater than 60% by the EMBOSS-isochore program (Bernardi, 1995; Pesole et al., 1999; Bernardi, 2000) (Fig. 4A). Using the EMBOSS-CpG plot program it was determined that the NOL7 genomic region contains a large CpG island about 1120 nucleotides in length, containing 111 CpG dinucleotides (13,554,908 to 13,556,028 bp, NCBI ID: NT_007592) (Rice et al., 2000). Promoter hypermethylation is a common mechanism by which tumor suppressor genes can be silenced in various malignancies including CC (Dong et al., 2001). However, bisulfite sequencing of the NOL7 promoter region from genomic DNA derived from normal cervical epithelium, CC cell lines and CC tumor samples failed to identify persistent sites of methylation (Mankame, et al, in submission). This data suggest that NOL7 expression was not regulated by an epigenetic mechanism. However the presence of a CpG island further supports the hypothesis that NOL7 may be regulated by a “TATA-less promoter”. Finally, a search of transcription factor consensus motifs identified two potential SP1 binding sites in the promoter (Fig. 4A+B). To verify that the SP1 transcription factor physically occupied the SP1 sites identified within the NOL7 promoter, a ChIP assay was performed. Immunoprecipitation of cross-linked chromatin from HEK 293T and HeLa cells with anti-SP1 polyclonal antibody followed by PCR amplification of the region (the sequence between 13,555,323 and 13,555,447 of NCBI ID: NT_007592) demonstrated that endogenous SP1 protein specifically binds to this region of the NOL7 promoter in both HEK 293T and HeLa cells (Fig. 4C). The data presented above confirms that the NOL7 promoter region possesses each of the key features of a classical “TATA-less” promoter.
Figure 4.
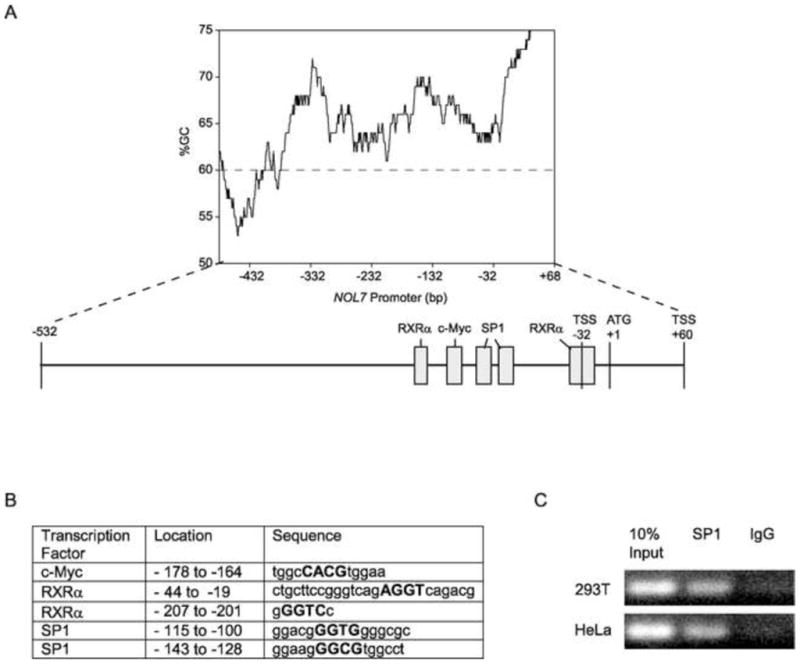
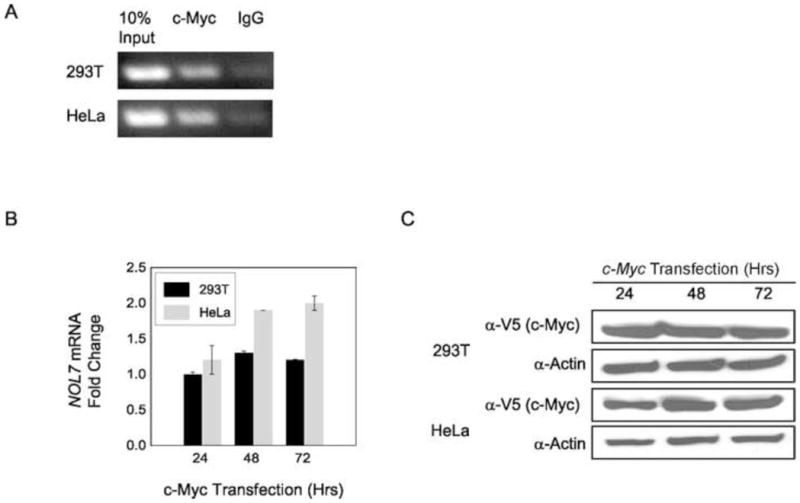
3.4. Regulation of NOL7 expression by c-Myc
In addition, to the SP1 binding site, in silico analysis of the NOL7 promoter regions consistently predicted the presence of conserved c-Myc and RXRα binding sites (Fig. 4A+B). c-Myc is a transcription factor that coordinates diverse intracellular and extracellular programs necessary for growth and expansion of somatic cells and is commonly deregulated or overexpressed in cancer (Soucek et al., 2008). Analysis of the NOL7 promoter region identified the specific conserved c-Myc binding site “CACGTG” (Fig. 4A+B) (Zeller et al., 2006). In addition, NOL7 was identified as a potential c-Myc target gene by a ChIP-ChIP study conducted by another group (Kim et al., 2008). To verify that c-Myc physically occupied the NOL7 promoter region, a ChIP assay was performed. Immunoprecipitation of cross-linked chromatin from HEK 293T and HeLa cells with anti-c-Myc polyclonal antibody followed by PCR amplification of the region (the sequence between 13,555,323 and 13,555,447 of NCBI ID: NT_007592) demonstrated that endogenous c-Myc protein specifically binds to this region of the NOL7 promoter in both HEK 293T and HeLa cells (Fig. 5A). Next, c-Myc was transfected in cells for various time periods to determine whether it acted as a positive or negative regulator of NOL7 expression. Quantitative RT-PCR analysis of c-Myc transfected cells demonstrated an upregulation of endogenous NOL7 mRNA levels in HeLa cells. However this trend was less pronounced in HEK293T cells, although both cell lines exhibited a robust expression of c-Myc protein (Fig. 5B+C).
Figure 5.
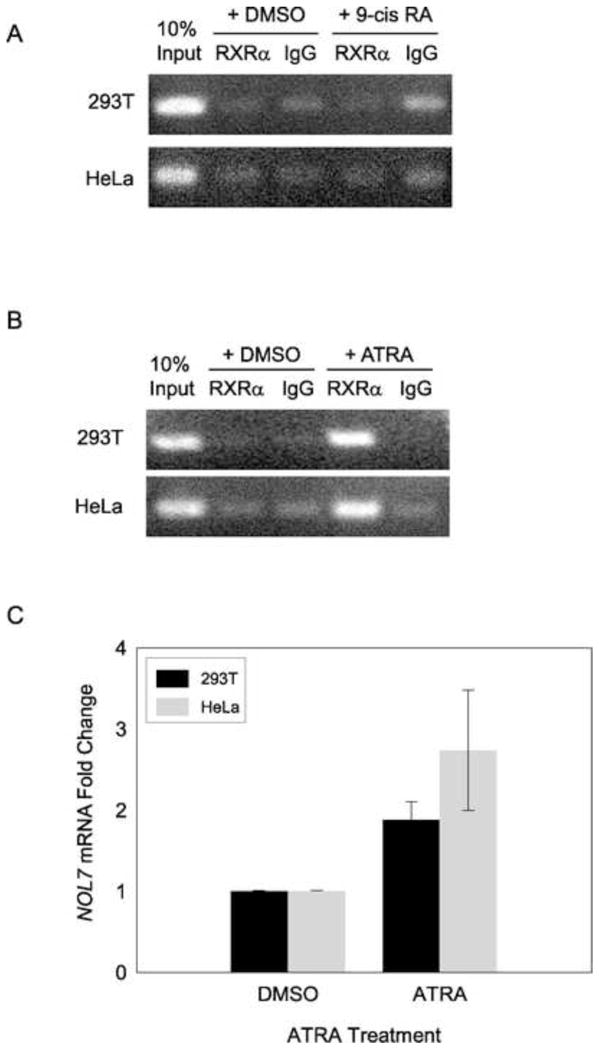
3.5. Regulation of NOL7 expression by Retinoic acid via RXRα
Retinoic X receptor alpha (RXRα) is a nuclear receptor that mediates the biological effects of retinoids by binding to specific sequences in the promoters of target genes and regulating their transcription. 9-cisRA is considered an RXR selective ligand (Chambon, 1996). However, RXRs are known to be obligatory DNA-binding partners for a variety of nuclear receptors, there by broadening the spectrum of their biological activity, to the corresponding nuclear receptor-signaling pathways (Altucci et al., 2007; Desvergne and Gerald, 2007). In addition, recent studies have shown that RXRα is recruited to gene promoters in response to all trans retinoic acid (ATRA) (Saavalainen et al., 2005; Knutson and Clagett-Dame, 2008). As indicated previously, the NOL7 promoter region has two potential RXRα binding sites (Fig. 4A+B). To determine if RXRα bound to the NOL7 promoter upon 9-cis RA treatment, cells were grown in serum free media overnight and then treated with 9-cis RA or DMSO. ChIP analysis of 9-cisRA-treated cells failed to demonstrate a significant association of RXRα with the NOL7 promoter as compared to the DMSO treated cells (Fig. 6A). To determine if NOL7 expression could be alternatively regulated by ATRA, cells were treated with either ATRA or DMSO using the same conditions. ChIP analysis demonstrated binding of RXRα to the NOL7 promoter exclusively in the ATRA treated HEK293T and HeLa cells (Fig. 6B). To determine if ATRA treatment positively or negatively regulated expression of NOL7, its mRNA levels in these cells were evaluated. Quantitative RT-PCR analysis showed a robust upregulation of endogenous NOL7 mRNA (>2 fold) in HEK293T and HeLa cell lines treated with ATRA as compared to DMSO (Fig. 6C). Again this induction was more pronounced in HeLa cells. Thus, these experiments indicated that NOL7 was transcriptionally activated by ATRA treatment, in part by the recruitment of RXRα to its promoter region.
Figure 6.
4. Discussion
HPV infection is believed to be the causative agent in the majority of CC cases. However additional genetic aberrations are required for malignant transformation (Lazo, 1999; Branca et al., 2006; Narisawa-Saito et al., 2008). NOL7 is a candidate tumor suppressor gene, whose expression is commonly lost in CC (Hasina et al., 2006). In order to study the tumor suppressor functions of NOL7, it was necessary to understand the molecular mechanisms behind its transcriptional regulation. This study was designed to identify the promoter region of the NOL7 gene, as well as the potential transcription factors that regulate its expression. Using a combination of bioinformatics and molecular biology techniques we have characterized the NOL7 promoter, provided evidence that it is a TATA-less promoter, and identified c-Myc and RXRα as positive regulators of NOL7 expression.
By generating a series of deletion constructs of the human NOL7 upstream region, its optimal promoter was defined to be the 560 bp region upstream of the initiation codon (Fig. 3). These experiments demonstrated that the highest promoter activity is concentrated within this 560 bp sequence and no enhancers or silencers were found to be present further upstream of this region. Sequence analysis of this region showed that it lacked a “TATA box” which is known to recruit transcriptional machinery needed for efficient expression of a gene (Bjornsdottir and Myers, 2008). Recent bioinformatics studies have suggested that the majority of mammalian gene promoters lack a TATA box, have multiple TSSs and are highly GC rich (Anish et al., 2009). Our 5′ RACE analysis demonstrated that the NOL7 promoter contained two transcriptional start sites (Fig. 1) and had a significantly high GC content (> 60%) (Fig. 4A). Finally, the promoter region contained potential SP1 binding sites and this region was capable of binding SP1 protein as determined by ChIP analysis (Fig. 4). Taken together all these data supported the hypothesis that NOL7 has a TATA-less promoter.
In CC, the integration of HPV DNA near the c-Myc locus has been shown to transcriptionally activate the gene (Peter et al., 2006). RNAi mediated HPV E6 and E7 knockdown resulted in the downregulation of c-Myc and its target genes suggesting that malignant transformation in CC may be mediated in part via the regulation of c-Myc by HPV (Kuner et al., 2007). Our ChIP data demonstrates that c-Myc associated with the NOL7 promoter and that overexpression of c-Myc upregulated endogenous NOL7 mRNA levels, indicating the c-Myc positively regulated NOL7 transcription (Fig. 5). This finding was unexpected considering the oncogenic nature of c-Myc. However it is not unprecedented, as c-Myc is known to transactivate several prominent tumor suppressors such as p53, BRCA1 and PTEN by binding to their respective promoters (Roy et al., 1994; Menssen and Hermeking, 2002; Fernandez et al., 2003). One hypothesis may be that certain cancer related genes respond to deregulated c-Myc levels and are induced in order to activate their specific tumor suppressive pathways. Also of importance is the ability of c-Myc to activate or repress genes depending on the co-activator proteins such as Max or Miz-1 that it associates with (Eilers and Eisenman, 2008). Future studies are required to better understand the relationship between HPV infection, c-Myc and regulation of NOL7 transcription.
ATRA treatment is known to induce growth arrest and cell death in CC cell lines in a dose dependent manner (Arany et al., 2003a; Arany et al., 2003b; Gasowska-Giszczak et al., 2005; Guo et al., 2006). In addition, treatment with ATRA can modulate the angiogenic phenotype in CC and head and neck cancer (Majewski et al., 1994; Lingen et al., 1996). Treatment with ATRA is also known to suppress the transcription of HPV 16, E6 and E7 proteins (Faluhelyi et al., 2004). Additionally, reduced serum levels of ATRA are associated with HPV-mediated progression to invasive carcinoma (Berlin Grace et al., 2006). Endogenous NOL7 mRNA was upregulated greater than two fold in ATRA treated HEK293T and HeLa cells (Fig. 6C). This induction was accompanied by recruitment of RXRα to the NOL7 promoter (Fig. 6B), supporting the hypothesis that RXRα positively regulates NOL7 expression in response to ATRA treatment. Conversely, ChIP experiments demonstrated that RXRα did not associate with the NOL7 promoter upon 9-cis RA treatment (Fig. 6A), suggesting that RXRα may be recruited to the promoter as a heterodimeric complex that does not respond to 9-cis RA (Altucci et al., 2007). Since RXRα is known to heterodimerize with a number of different nuclear receptors, further investigation is required to identify and characterize additional binding partners of RXRα that may exert differential regulation of NOL7 gene expression (Bugge et al., 1992; Desvergne and Gerald, 2007). It will also be interesting to determine if NOL7 plays a role in ATRA mediated growth arrest, cell death and angiogenesis by acting as a downstream effector of ATRA.
In conclusion, we have determined two transcriptional start sites of NOL7, cloned its regulatory promoter region, and identified the transcription factors that regulate its expression. Our data demonstrate that NOL7 contains a TATA-less promoter and is positively regulated by the c-Myc and RXRα transcription factors. This novel regulation of NOL7 by transcription factors that are intimately involved in carcinogenesis provides additional mechanistic insights into the potential role of NOL7 in CC and perhaps other malignancies.
Supplementary Material
Acknowledgments
We would like to thank Dr. Deborah Lang for her advice and assistance with the luciferase assays.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achuthan R, Bell SM, Roberts P, Leek JP, Horgan K, Markham AF, MacLennan KA, Speirs V. Genetic events during the transformation of a tamoxifen-sensitive human breast cancer cell line into a drug-resistant clone. Cancer Genet Cytogenet. 2001;130:166–72. doi: 10.1016/s0165-4608(01)00475-7. [DOI] [PubMed] [Google Scholar]
- Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6:793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- Amare Kadam PS, Ghule P, Jose J, Bamne M, Kurkure P, Banavali S, Sarin R, Advani S. Constitutional genomic instability, chromosome aberrations in tumor cells and retinoblastoma. Cancer Genet Cytogenet. 2004;150:33–43. doi: 10.1016/j.cancergencyto.2003.08.015. [DOI] [PubMed] [Google Scholar]
- American Cancer Society, I. Cancer Facts & Figures 2009. American Cancer Society, Inc; Atlanta: 2008. [Google Scholar]
- Anish R, Hossain MB, Jacobson RH, Takada S. Characterization of transcription from TATA-less promoters: identification of a new core promoter element XCPE2 and analysis of factor requirements. PLoS ONE. 2009;4:e5103. doi: 10.1371/journal.pone.0005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany I, Ember IA, Tyring SK. All-trans-retinoic acid activates caspase-1 in a dose-dependent manner in cervical squamous carcinoma cells. Anticancer Res. 2003a;23:471–3. [PubMed] [Google Scholar]
- Arany I, Whitehead WE, Ember IA, Tyring SK. Dose-dependent activation of p21WAF1 transcription by all-trans-acid in cervical squamous carcinoma cells. Anticancer Res. 2003b;23:495–7. [PubMed] [Google Scholar]
- Batanian JR, Cavalli LR, Aldosari NM, Ma E, Sotelo-Avila C, Ramos MB, Rone JD, Thorpe CM, Haddad BR. Evaluation of paediatric osteosarcomas by classic cytogenetic and CGH analyses. Mol Pathol. 2002;55:389–93. doi: 10.1136/mp.55.6.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin Grace VM, Niranjali Devaraj S, Radhakrishnan Pillai M, Devaraj H. HPV-induced carcinogenesis of the uterine cervix is associated with reduced serum ATRA level. Gynecol Oncol. 2006;103:113–9. doi: 10.1016/j.ygyno.2006.01.057. [DOI] [PubMed] [Google Scholar]
- Bernardi G. The human genome: organization and evolutionary history. Annu Rev Genet. 1995;29:445–76. doi: 10.1146/annurev.ge.29.120195.002305. [DOI] [PubMed] [Google Scholar]
- Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000;241:3–17. doi: 10.1016/s0378-1119(99)00485-0. [DOI] [PubMed] [Google Scholar]
- Biosystems A. Relative Quantitation Using Comparative CT Getting Started Guide. [Google Scholar]
- Bjornsdottir G, Myers LC. Minimal components of the RNA polymerase II transcription apparatus determine the consensus TATA box. Nucl Acids Res. 2008;36:2906–2916. doi: 10.1093/nar/gkn130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branca M, Giorgi C, Ciotti M, Santini D, Di Bonito L, Costa S, Benedetto A, Bonifacio D, Di Bonito P, Paba P, Accardi L, Mariani L, Ruutu M, Favalli C, Syrjanen K. Down-regulated nucleoside diphosphate kinase nm23-H1 expression is unrelated to high-risk human papillomavirus but associated with progression of cervical intraepithelial neoplasia and unfavourable prognosis in cervical cancer. J Clin Pathol. 2006;59:1044–51. doi: 10.1136/jcp.2005.033142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugge TH, Pohl J, Lonnoy O, Stunnenberg HG. RXR alpha, a promiscuous partner of retinoic acid and thyroid hormone receptors. EMBO J. 1992;11:1409–18. doi: 10.1002/j.1460-2075.1992.tb05186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–54. [PubMed] [Google Scholar]
- Chatterjee A, Pulido HA, Koul S, Beleno N, Perilla A, Posso H, Manusukhani M, Murty VV. Mapping the sites of putative tumor suppressor genes at 6p25 and 6p21.3 in cervical carcinoma: occurrence of allelic deletions in precancerous lesions. Cancer Res. 2001;61:2119–23. [PubMed] [Google Scholar]
- Chen Z, Issa B, Brothman LJ, Hendricksen M, Button D, Brothman AR. Nonrandom rearrangements of 6p in malignant hematological disorders. Cancer Genet Cytogenet. 2000;121:22–5. doi: 10.1016/s0165-4608(00)00222-3. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Gerald L. Vitamins & Hormones. Academic Press; 2007. RXR: From Partnership to Leadership in Metabolic Regulations; pp. 1–32. [DOI] [PubMed] [Google Scholar]
- Dong SM, Kim HS, Rha SH, Sidransky D. Promoter hypermethylation of multiple genes in carcinoma of the uterine cervix. Clin Cancer Res. 2001;7:1982–6. [PubMed] [Google Scholar]
- Eilers M, Eisenman RN. Myc’s broad reach. Genes & Development. 2008;22:2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faluhelyi Z, Rodler I, Csejtey A, Tyring SK, Ember IA, Arany I. All-trans retinoic acid (ATRA) suppresses transcription of human papillomavirus type 16 (HPV16) in a dose-dependent manner. Anticancer Res. 2004;24:807–9. [PubMed] [Google Scholar]
- Fan YS, Rizkalla K. Comprehensive cytogenetic analysis including multicolor spectral karyotyping and interphase fluorescence in situ hybridization in lymphoma diagnosis. a summary of 154 cases. Cancer Genet Cytogenet. 2003;143:73–9. doi: 10.1016/s0165-4608(02)00843-9. [DOI] [PubMed] [Google Scholar]
- Farre D, Roset R, Huerta M, Adsuara JE, Rosello L, Alba MM, Messeguer X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucl Acids Res. 2003;31:3651–3653. doi: 10.1093/nar/gkg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–29. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischman EW, Prigogina EL, Iljinskaja GW, Konstantinova LN, Puchkova GP, Volkova MA, Frenkel MA, Balakirev SA. Chromosomal rearrangements with a common breakpoint at 6p23 in five cases of myeloid leukemia. Hum Genet. 1983;64:254–6. doi: 10.1007/BF00279404. [DOI] [PubMed] [Google Scholar]
- Gasowska-Giszczak U, Darmochwal-Kolarz D, Kwasniewska A, Dziubinska-Parol I, Rolinski J, Oleszczuk J. Apoptosis of HeLa cell lines incubated with retinol. Eur J Obstet Gynecol Reprod Biol. 2005;119:119–22. doi: 10.1016/j.ejogrb.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Giagounidis AAN, Hildebrandt B, Heinsch M, Germing U, Aivado M, Aul C. Acute basophilic leukemia. European Journal Of Haematology. 2001;67:72–76. doi: 10.1034/j.1600-0609.2001.t01-1-00487.x. [DOI] [PubMed] [Google Scholar]
- Guo JM, Xiao BX, Kang GZ, Liu DH, Chen H, Zhang S, Zhang XN. Suppression of telomerase activity and arrest at G1 phase in human cervical cancer HeLa cells by all-trans retinoic acid. Int J Gynecol Cancer. 2006;16:341–6. doi: 10.1111/j.1525-1438.2006.00353.x. [DOI] [PubMed] [Google Scholar]
- Hasina R, Pontier AL, Fekete MJ, Martin LE, Qi XM, Brigaudeau C, Pramanik R, Cline EI, Coignet LJ, Lingen MW. NOL7 is a nucleolar candidate tumor suppressor gene in cervical cancer that modulates the angiogenic phenotype. Oncogene. 2006;25:588–98. doi: 10.1038/sj.onc.1209070. [DOI] [PubMed] [Google Scholar]
- Hoyle CF, Sherrington P, Hayhoe FG. Translocation (3;6)(q21; p21) in acute myeloid leukemia with abnormal thrombopoiesis and basophilia. Cancer Genet Cytogenet. 1988;30:261–7. doi: 10.1016/0165-4608(88)90193-8. [DOI] [PubMed] [Google Scholar]
- Jadayel D, Calabrese G, Min T, van Rhee F, Swansbury GJ, Dyer MJ, Maitland J, Palka G, Catovsky D. Molecular cytogenetics of chronic myeloid leukemia with atypical t(6;9) (p23; q34) translocation. Leukemia. 1995;9:981–7. [PubMed] [Google Scholar]
- Janet S, Rader YL, Huettner Phyllis C, Xu Zhiqiang, Gerhard Daniela S. Cervical cancer suppressor gene is within 1 cM on 6p23. Genes, Chromosomes and Cancer. 2000;27:373–379. [PubMed] [Google Scholar]
- Kaufmann AM, Backsch C, Schneider A, Durst M. HPV induced cervical carcinogenesis: molecular basis and vaccine development. Zentralbl Gynakol. 2002;124:511–24. doi: 10.1055/s-2002-39579. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee JH, Iyer VR. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS ONE. 2008;3:e1798. doi: 10.1371/journal.pone.0001798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson DC, Clagett-Dame M. atRA Regulation of NEDD9, a gene involved in neurite outgrowth and cell adhesion. Archives of Biochemistry and Biophysics. 2008;477:163–174. doi: 10.1016/j.abb.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Kuner R, Vogt M, Sultmann H, Buness A, Dymalla S, Bulkescher J, Fellmann M, Butz K, Poustka A, Hoppe-Seyler F. Identification of cellular targets for the human papillomavirus E6 and E7 oncogenes by RNA interference and transcriptome analyses. J Mol Med. 2007;85:1253–62. doi: 10.1007/s00109-007-0230-1. [DOI] [PubMed] [Google Scholar]
- Lazo PA. The molecular genetics of cervical carcinoma. Br J Cancer. 1999;80:2008–18. doi: 10.1038/sj.bjc.6690635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao SK, Perng YP, Shen YC, Chung PJ, Chang YS, Wang CH. Chromosomal abnormalities of a new nasopharyngeal carcinoma cell line (NPC-BM1) derived from a bone marrow metastatic lesion. Cancer Genet Cytogenet. 1998;103:52–8. doi: 10.1016/s0165-4608(97)00416-0. [DOI] [PubMed] [Google Scholar]
- Lim G, Karaskova J, Vukovic B, Bayani J, Beheshti B, Bernardini M, Squire JA, Zielenska M. Combined spectral karyotyping, multicolor banding, and microarray comparative genomic hybridization analysis provides a detailed characterization of complex structural chromosomal rearrangements associated with gene amplification in the osteosarcoma cell line MG-63. Cancer Genet Cytogenet. 2004;153:158–64. doi: 10.1016/j.cancergencyto.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Lingen M, Polverini P, Bouck N. Retinoic acid induces cells cultured from oral squamous cell carcinomas to become anti-angiogenic. Am J Pathol. 1996;149:247–258. [PMC free article] [PubMed] [Google Scholar]
- Lung ML, Choi CV, Kong H, Yuen PW, Kwong D, Sham J, Wei WI. Microsatellite allelotyping of chinese nasopharyngeal carcinomas. Anticancer Res. 2001;21:3081–4. [PubMed] [Google Scholar]
- Majewski S, Szmurlo A, Marczak M, Jablonska S, Bollag W. Synergistic effect of retinoids and interferon alpha on tumor-induced angiogenesis: Anti-angiogenic effect on HPV-harboring tumor-cell lines. International Journal of Cancer. 1994;57:81–85. doi: 10.1002/ijc.2910570115. [DOI] [PubMed] [Google Scholar]
- Mazurenko NN, Beliakov IS, Bliev A, Guo Z, Hu X, Vinokurova SV, Bidzhieva BA, Pavlova LS, Ponten J, Kiselev FL. Cervical carcinoma progression-associated genetic alterations on chromosome 6. Mol Biol (Mosk) 2003;37:472–81. [PubMed] [Google Scholar]
- Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci U S A. 2002;99:6274–9. doi: 10.1073/pnas.082005599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messeguer X, Escudero R, Farre D, Nunez O, Martinez J, Alba MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics. 2002;18:333–334. doi: 10.1093/bioinformatics/18.2.333. [DOI] [PubMed] [Google Scholar]
- Mutirangura A, Tanunyutthawongese C, Pornthanakasem W, Kerekhanjanarong V, Sriuranpong V, Yenrudi S, Supiyaphun P, Voravud N. Genomic alterations in nasopharyngeal carcinoma: loss of heterozygosity and Epstein-Barr virus infection. Br J Cancer. 1997;76:770–6. doi: 10.1038/bjc.1997.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H, Kinoshita T, Suzuki H, Hatano S, Murate T, Saito H. Identification and mapping of novel tumor suppressor loci on 6p in diffuse large B-cell non-Hodgkin’s lymphoma. Genes Chromosomes Cancer. 1999;25:277–83. [PubMed] [Google Scholar]
- Nakase K, Wakita Y, Minamikawa K, Yamaguchi T, Shiku H. Acute promyelocytic leukemia with del(6)(p23) Leuk Res. 2000;24:79–81. doi: 10.1016/s0145-2126(99)00139-3. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Yoshimatsu Y, Ohno S, Yugawa T, Egawa N, Fujita M, Hirohashi S, Kiyono T. An in vitro multistep carcinogenesis model for human cervical cancer. Cancer Res. 2008;68:5699–705. doi: 10.1158/0008-5472.CAN-07-6862. [DOI] [PubMed] [Google Scholar]
- Nemani M, Bellanne-Chantelot C, Cohen D, Cann HM. Detection of triplet repeat sequences in yeast artificial chromosomes using oligonucleotide probes: application to the SCA1 region in 6p23. Cytogenet Cell Genet. 1996;72:5–8. doi: 10.1159/000134150. [DOI] [PubMed] [Google Scholar]
- Pesole G, Bernardi G, Saccone C. Isochore specificity of AUG initiator context of human genes. FEBS Lett. 1999;464:60–2. doi: 10.1016/s0014-5793(99)01675-0. [DOI] [PubMed] [Google Scholar]
- Peter M, Rosty C, Couturier J, Radvanyi F, Teshima H, Sastre-Garau X. MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors. Oncogene. 2006;25:5985–93. doi: 10.1038/sj.onc.1209625. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–7. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Roy B, Beamon J, Balint E, Reisman D. Transactivation of the human p53 tumor suppressor gene by c-Myc/Max contributes to elevated mutant p53 expression in some tumors. Mol Cell Biol. 1994;14:7805–15. doi: 10.1128/mcb.14.12.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavalainen K, Pasonen-Seppänen S, Dunlop TW, Tammi R, Tammi MI, Carlberg C. The Human Hyaluronan Synthase 2 Gene Is a Primary Retinoic Acid and Epidermal Growth Factor Responding Gene. Journal of Biological Chemistry. 2005;280:14636–14644. doi: 10.1074/jbc.M500206200. [DOI] [PubMed] [Google Scholar]
- Sandelin A, Carninci P, Lenhard B, Ponjavic J, Hayashizaki Y, Hume DA. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat Rev Genet. 2007;8:424–436. doi: 10.1038/nrg2026. [DOI] [PubMed] [Google Scholar]
- Schug J. Baxevanis, AD (Baxevanis, AD)Baxevanis, ADs), Current Protocols in Bioinformatics. J. Wiley and Sons; 2003. Using TESS to Predict Transcription Factor Binding Sites in DNA Sequence. [DOI] [PubMed] [Google Scholar]
- Shao JY, Wang HY, Huang XM, Feng QS, Huang P, Feng BJ, Huang LX, Yu XJ, Li JT, Hu LF, Ernberg I, Zeng YX. Genome-wide allelotype analysis of sporadic primary nasopharyngeal carcinoma from southern China. Int J Oncol. 2000;17:1267–75. doi: 10.3892/ijo.17.6.1267. [DOI] [PubMed] [Google Scholar]
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starostik P, Patzner J, Greiner A, Schwarz S, Kalla J, Ott G, Muller-Hermelink HK. Gastric marginal zone B-cell lymphomas of MALT type develop along 2 distinct pathogenetic pathways. Blood. 2002;99:3–9. doi: 10.1182/blood.v99.1.3. [DOI] [PubMed] [Google Scholar]
- Takeshita A, Naito K, Shinjo K, Sahara N, Matsui H, Ohnishi K, Beppu H, Ohtsubo K, Horii T, Maekawa M, Inaba T, Ohno R. Deletion 6p23 and add(11)(p15) leading to NUP98 translocation in a case of therapy-related atypical chronic myelocytic leukemia transforming to acute myelocytic leukemia. Cancer Genet Cytogenet. 2004;152:56–60. doi: 10.1016/j.cancergencyto.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–9. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Werner T. Computer-assisted analysis of transcription control regions. Matinspector and other programs. Methods Mol Biol. 2000;132:337–49. doi: 10.1385/1-59259-192-2:337. [DOI] [PubMed] [Google Scholar]
- Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, Fu Y, Weng Z, Kuznetsov VA, Sung WK, Ruan Y, Dang CV, Wei CL. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A. 2006;103:17834–9. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, He F, Hu S, Yu J. On the nature of human housekeeping genes. Trends in Genetics. 2008;24:481–484. doi: 10.1016/j.tig.2008.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.