

Abstract
Notch signaling is of high importance for growth and survival of various cell types. We now analyzed the protein expression of two key components of the Notch signaling pathway (Notch-1, Jagged-1) in spontaneously immortalized (HaCaT) and in malignant (SCL-1) human keratinocytes, using western analysis. We found that Notch-1 and its corresponding ligand Jagged-1 are expressed in both cell lines, with no marked change following UV-B treatment. Moreover, treatment of both cell lines before or after UV-B irradiation with 1,25-dihydroxyvitamin D3, the biologically active form of vitamin D, and/or epigenetic modulating drugs (TSA; 5-Aza) did not result in a marked modulation of the protein expression of Notch-1 or Jagged-1. Under the experimental conditions of this study, treatment with 1,25(OH)2D3 protected human keratinocytes in part against the antiproliferative effects of UV-B-radiation. In conclusion, our findings do not point at a differential expression of these two key components of Notch signaling in non-malignant as compared to malignant human keratinocytes, indicating that alterations in their expression are not of importance for the photocarcinogenesis of human squamous cell carcinomas. Moreover, our findings do not support the hypothesis that modulation of Notch signaling may be involved in the photoprotective effect of 1,25-dihydroxyvitamin D3, that we and others reported previously. Additionally, we demonstrate that epigenetic modulating drugs (TSA, 5-Aza) do not markedly modulate the expression Notch-1 or Jagged-1 in UV-B-treated human keratinocytes in vitro.
Keywords: 1,25-dihydroxyvitamin D3; Notch; epigenetics; keratinocytes; skin cancer; vitamin D
Introduction
UV-radiation is the most important environmental risk factor for non-melanoma skin cancer, causing photocarcinogenesis of basal cell carcinomas (BCC) and cutaneous squamous cell carcinomas (SCC). These types of skin cancer are the most common malignancies in Caucasian populations with annual incidence rates in the United States of nearly 1 million and 250,000 cases, respectively.1-5 In contrast to BCCs that in general do not metastasize, SCCs are characterized by biologically more aggressive growth and exert metastatic potential with frequencies approaching 12.5%.3-5 In many cases, they develop from characteristic precursor lesions (actinic keratoses), that are nowadays accepted to represent in situ carcinomas. Mutations in genes involved in the sonic hedgehog (SHH) signaling pathway have been shown to be of high importance for the carcinogenesis of both human and mouse BCC.5-9 Until today, no gene mutation that uniquely underlies human SCCs is known,8,9 although UV-induced mutations in the p53 gene are a key event in the photocarcinogenesis of SCCs. We and others have shown previously that the biologically active vitamin D metabolite 1,25-dihydroxyvitamin D [1,25(OH)2D] and its analogs have the capacity to protect cultured human keratinocytes in part against the hazardous effects of UV-radiation.10-17
It was the aim of this study to gain more insights into the role of vitamin D- and Notch-signaling pathways for the photocarcinogenesis of SCC, using cultured non-malignant (HaCaT) and malignant (SCL-1) keratinocytes as an in vitro model to investigate potential characteristic changes during the process of malignant transformation of human keratinocytes. Understanding the role of Notch signaling for the photocarcinogenesis of SCC is of high relevance, given the enormous impact of Notch signaling for cellular processes that are implicated in carcinogenesis, including cellular proliferation/differentiation. Decades ago, the Notch signaling pathway was first described as the key regulator of ectodermal specification and neurogenesis in Drosophila.5,18 More recently, it has been shown that Notch signaling is of critical importance not only for the embryonic development but also for the growth of various cell types, including human keratinocytes.5,18-25 It has now convincingly been demonstrated that the Notch pathway regulates a broad variety of cellular processes, including differentiation, proliferation, angiogenesis, apoptosis and cell fate decisions, that are of high importance for organogenesis and tissue homeostasis and that are involved in skin carcinogenesis.5,18-29 Underlining its importance, Notch signaling is highly conserved during evolution, developmentally regulated, and cell type dependent. In general, Notch signaling regulates developmental processes though binary decision, lateral inhibition and boundary formation.5,30 Notch-mediated cell-cell communication is mediated via the coordinated and differential expression of Notch receptors and their corresponding ligands on the cell surface.5,18,19,24 At present, four evolutionary conserved transmembrane Notch receptors (Notch1–4) are known in mammals, that are activated by five corresponding Notch ligands of the Delta (Delta-like 1, 3 and 4) and Jagged (Jagged1 and 2) families. Ligand-induced Notch receptor activation causes cleavage of the Notch intracellular domain (NICD). The NICD then translocates to the nucleus and forms a ternary complex with the transcriptional coactivator, mastermind-like (MAML) protein, as well as the DNA-binding protein, CSL, which regulates DNA-binding specificity and target gene expression.5,23,24,31,32 At present, only a few Notch target genes are known, including transcriptional repressors such as basic-helix-loop-helix proteins of the hairy and enhancer of split (Hes) and Hes-related transcription factor (Hrt) families.5,25,33
In healthy and diseased human skin, Notch receptors and their ligands are differentially expressed and localized in the different cell layers of the viable epidermis.5,26,34 However, many of their specific functions are until today not completely understood.5 In healthy “normal” skin Notch1 and the corresponding ligands Delta1 and Jagged1 are expressed in all viable cell layers of the epidermis, with the strongest detection of Delta1 and Jagged 1 in keratinocytes of the epidermal basal layer.5 It was reported that signaling mediated by Delta/Notch is increased when cells undergo a normal and regulated differentiation program, as in keratinocyte layers of the normal adult human epidermis.5,34 In contrast, Notch signaling has been reported as reduced in psoriasis vulgaris and other hyperproliferating skin diseases.5,34 Moreover, loss of Notch1 in young mice has been shown to cause hyperproliferation of the basal epidermal layer and to change expression of many markers involved in cellular differentiation and proliferation, including p21 (reduced) and Gli2 (elevated).5,9 In epidermal epithelial cells, activation of Notch1 induces p21 expression in a CSL-dependent manner, resulting in cell cycle withdrawal and terminal differentiation.5,22 Additionally, Notch1 stimulates caspase 3 activity, that promotes terminal differentiation of embryonic keratinocytes.5,23 However, the potential physiological/pathophysiological function and the regulation of the Notch pathway in the photocarcinogenesis of SCCs are largely unknown. While it has been demonstrated that other molecular pathways modulate the activity of the Notch pathway,5,35 little is known about the impact of UV-B or vitamin D compounds on Notch signaling. We and others have previously reported that 1,25-dihydroxyvitamin D and analogs protect human keratinocytes against the hazardous effects of UV-radiation.10-17 Furthermore, increasing evidence now clearly indicates that the vitamin D endocrine system is implicated in pathogenesis and progression of various malignancies, including the photocarcinogenesis of human cutaneous SCC.15 It is well known that 1,25-dihydroxyvitamin D3, the biologically most active natural vitamin D metabolite, and its analogs, act via binding to a corresponding intranuclear receptor (VDR), present in target tissue cells.36 In various normal and malignant cell types the effects of 1,25-dihydroxyvitamin D3 include the regulation of cell growth and differentiation.37-39 It can be speculated whether pharmacologic regulation of vitamin D- or Notch-signaling may represent a new target for cancer prevention and therapy. Modulation of Notch and/or vitamin D-signaling pathways may represent a potential novel strategy to prevent the photocarcinogenesis of and to treat cutaneous SCCs. It was the aim of this study to gain further insights into the role of Notch- and vitamin D signaling pathways for photocarcinogenesis of SCCs by analyzing the protein expression of two key components of Notch signaling (Notch1, Jagged 1), and their modulation by treatment with UV-B and/or 1,25-dihydroxyvitamin D3, and epigenetic modulating drugs (TSA, 5-Aza) in non-malignant (HaCaT) and in malignant (SCL-1) keratinocyte cell lines in vitro.
Results
Treatment with 1,25(OH)2D3 protects human keratinocytes in part against the antiproliferative effects of UV-B-radiation
As shown in Figure 1 for HaCaT keratinocytes, treatment with 1,25(OH)2D3 protects under our experimental conditions (crystal-violet staining of cells as described in Materials and Methods) human keratinocytes in part against the antiproliferative effects of UV-B-radiation. Please note that 24 h following treatment with UV-B and 1,25(OH)2D3, 1,25(OH)2D3-mediated effects are more pronounced in cells treated with 100 J UV-B as compared with those treated with 500 J UV-B (Fig. 1). Please also note that epigenetic drugs TSA and 5-Aza as well modulate the proliferation of HaCaT keratinocytes treated with and without UV-B (Fig. 1).

Figure 1. Treatment with 1,25(OH)2D3 protects human keratinocytes in part against the antiproliferative effects of UV-B-radiation. As shown here for HaCaT keratinocytes, treatment with 1,25(OH)2D3 protects human keratinocytes after 24, 48 and 72 h in part against the antiproliferative effects of UV-B-radiation. Proliferation was measured using a dye (CV)-based assay as described in Materials and Methods. Please note that 24 h following treatment with UV-B and 1,25(OH)2D3, 1,25(OH)2D3-mediated effects are more pronounced in cells treated with 100 J UV-B as compared with those treated with 500 J UV-B. Please also note that epigenetic drugs TSA and 5-Aza as well modulate the proliferation of HaCaT keratinocytes treated with and without UV-B.
Protein expression of key components of the Notch signaling pathway in human keratinocyte cell lines
Western analysis revealed that Notch-1 and its corresponding ligand Jagged-1 are expressed in non-malignant (HaCaT keratinocytes) and in malignant (SCL-1 cells) human keratinocytes. Results of representative experiments in HaCaT keratinocytes are shown in Figures 2 and 3.
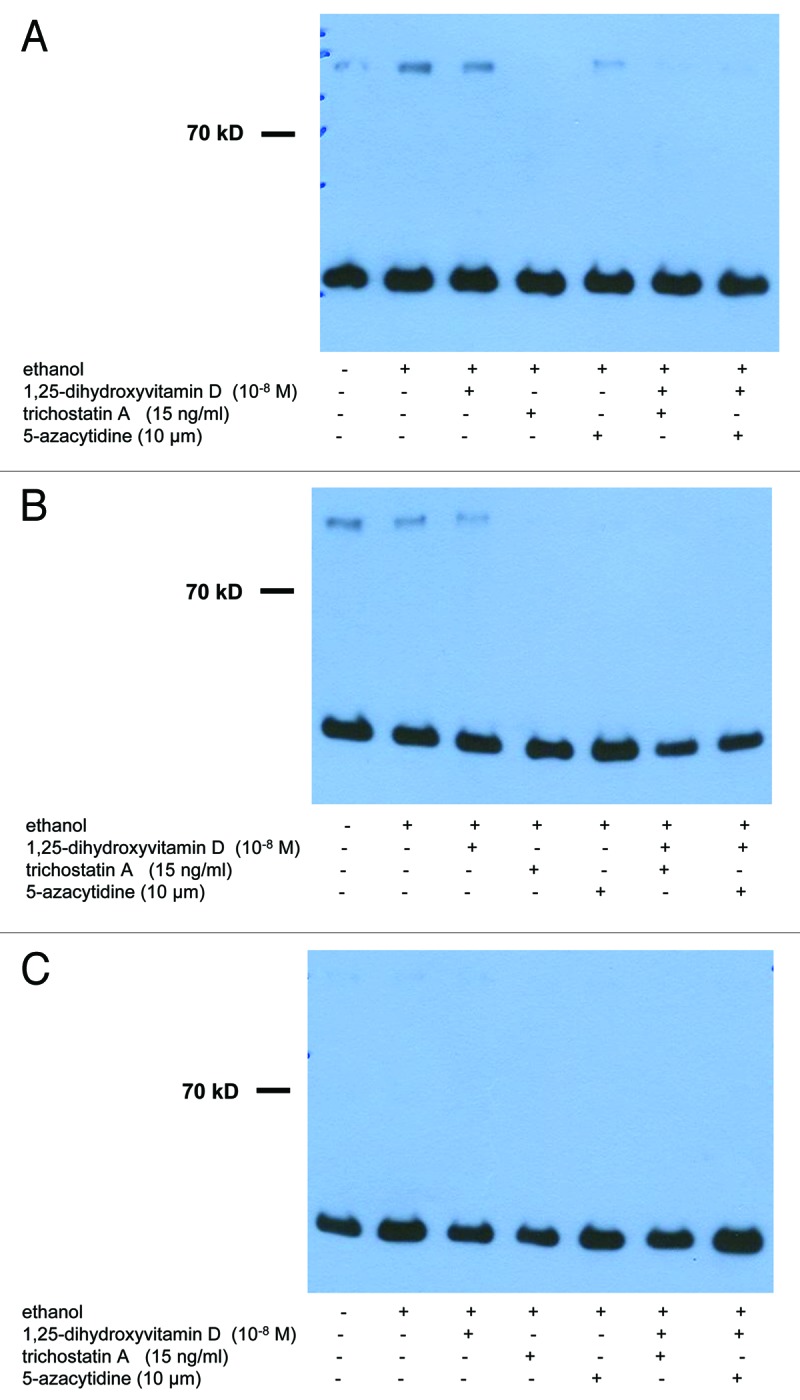
Figure 2. Treatment with UV-B, 1,25(OH)2D3 and epigenetic drugs, alone or in combination, does not exert strong effects on protein expression of Notch-1 in non-malignant and in malignant human keratinocytes. Western analysis revealed that Notch-1 protein is expressed in cultured non-malignant (HaCaT keratinocytes) and malignant (SCL-1 cells) human keratinocytes. Results of representative experiments in HaCaT keratinocytes 48 h after UV-B irradiation are shown here. Please note that treatment with UV-B [(A) 0 J; (B) 100 J; (C) 500 J] and/or 1,25(OH)2D3 (10−8 M) does not exert strong effects on protein expression of Notch-1 after 24, 48 or 72 h. Please also note that treatment with epigenetic modulating drugs (TSA, 5-Aza) does not exert strong effects, alone or in combination with UV-B and/or 1,25(OH)2D3, on protein expression of Notch-1 in HaCaT keratinocytes. Western analysis of GAPDH protein (lower band) served as control.
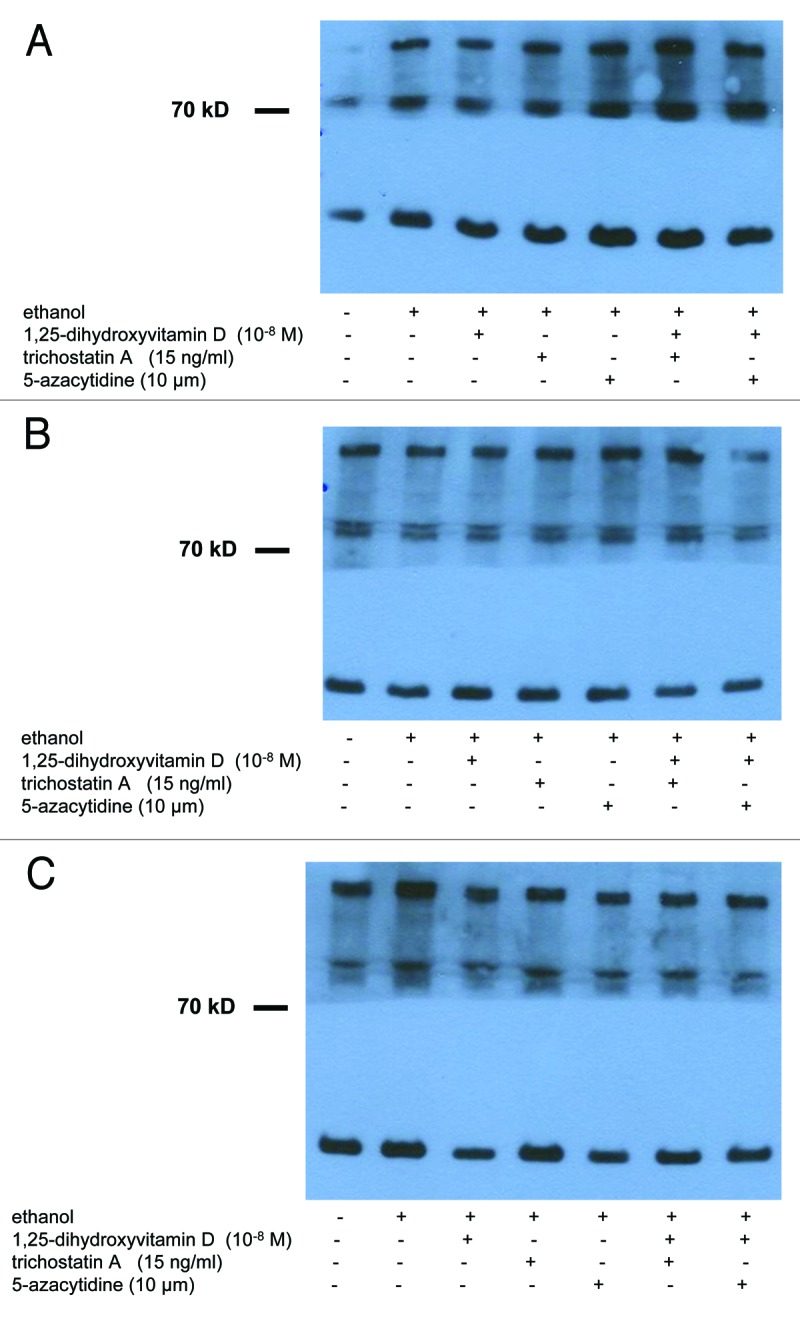
Figure 3. Treatment with UV-B, 1,25(OH)2D3 and epigenetic drugs, alone or in combination, does not exert strong effects on protein expression of Jagged-1 in non-malignant and in malignant human keratinocytes. Western analysis revealed that Jagged 1 protein is expressed in cultured non-malignant (HaCaT keratinocytes) and malignant (SCL-1 cells) human keratinocytes. Results of representative experiments in HaCaT keratinocytes 48 h after UV-B irradiation are shown here. Please note that treatment with UV-B [(A) 0 J; (B) 100 J; (C) 500 J] and/or 1,25(OH)2D3 (10−8 M) does not exert strong effects on protein expression of Jagged 1 after 24, 48 or 72 h. Please also note that treatment with epigenetic modulating drugs (TSA, 5-Aza) does not exert pronounced effects, alone or in combination with UV-B and/or 1,25(OH)2D3, on protein expression of Jagged 1 in HaCaT keratinocytes. Western analysis of GAPDH protein (lower band) served as control.
Treatment with UV-B and/or 1,25(OH)2D3 does not exert strong effects on protein expression of Notch-1 in non-malignant and in malignant human keratinocytes in vitro
Western analysis revealed that treatment with UV-B and/or 1,25(OH)2D3 does not exert strong effects on protein expression of Notch-1 in non-malignant (spontaneously immortalized HaCaT keratinocytes) or in malignant (cutaneous squamous carcinoma cells SCL-1) human keratinocytes. A representative experiment demonstrating protein expression of Notch-1 in HaCaT keratinocytes 24 h following treatment with UV-B [(A) 0 J; (B) 100 J; C: 500 J) alone or together with 1,25(OH)2D3 (10−8 M) is shown in Figure 2. Note that no pronounced visual differences in staining intensity for Notch-1 were seen in protein extracts from controls as compared with those from UV-B- and/or 1,25(OH)2D3-treated cells (Fig. 2). Results in HaCaT keratinocytes were comparable with those in SCL-1 cells (data not shown).
Treatment with UV-B and/or 1,25(OH)2D3 does not exert strong effects on protein expression of Jagged-1 in non-malignant or in malignant human keratinocytes
Western analysis revealed that treatment with UV-B and/or 1,25(OH)2D3 does not exert strong effects on protein expression of Jagged-1 in non-malignant (spontaneously immortalized HaCaT keratinocytes) or in malignant (cutaneous squamous carcinoma cells SCL-1) human keratinocytes. A representative experiment demonstrating protein expression of Jagged-1 in HaCaT keratinocytes 24 h following treatment with UV-B (A: 0 J; B: 100 J; (C) 500 J] alone or together with 1,25(OH)2D3 (10−8 M) is shown in Figure 3. Note that no strong visual differences in staining intensity for Jagged-1 were seen in protein extracts from controls as compared with those from UV-B- and/or 1,25(OH)2D3-treated cells (Fig. 3). Results in HaCaT keratinocytes were comparable with those in SCL-1 cells (data not shown).
Treatment with epigenetic modulating drugs (TSA, 5-Aza), alone or in combination with UV-B and/or 1,25(OH)2D3, does not exert strong effects on protein expression of Notch-1 in non-malignant or in malignant human keratinocytes
Western analysis revealed that treatment with epigenetic modulating drugs (TSA, 5-Aza) does not exert pronounced effects, alone or in combination with UV-B and/or 1,25(OH)2D3, on protein expression of Notch-1 in non-malignant (spontaneously immortalized HaCaT keratinocytes) or in malignant (cutaneous squamous carcinoma cells SCL-1) human keratinocytes (Fig. 2). A representative experiment demonstrating protein expression of Notch-1 in HaCaT keratinocytes 24 h following treatment with UV-B [(A) 0 J; (B) 100 J; (C) 500 J] alone or together with 1,25(OH)2D3 (10−8 M) is shown in Figure 2. Note that no strong visual differences in staining intensity were seen in controls as compared with treated cells (Fig. 2). Results in HaCaT keratinocytes were comparable with those in SCL-1 cells (data not shown).
Treatment with epigenetic modulating drugs (TSA, 5-Aza), alone or in combination with UV-B and/or 1,25(OH)2D3, does not exert strong effects on protein expression of Jagged-1 in non-malignant or in malignant human keratinocytes
Western analysis revealed that treatment with epigenetic modulating drugs (TSA, 5-Aza) does not exert strong effects, alone or in combination with UV-B and/or 1,25(OH)2D3, on protein expression of Jagged-1 in non-malignant (spontaneously immortalized HaCaT keratinocytes) or in malignant (cutaneous squamous carcinoma cells SCL-1) human keratinocytes (Fig. 3). A representative experiment demonstrating protein expression of Jagged-1 in HaCaT keratinocytes 24 h following treatment with UV-B [(A) 0 J; (B) 100 J; (C) 500 J] alone or together with 1,25(OH)2D3 (10−8 M) is shown in Figure 3. Note that no strong visual differences in staining intensity for Jagged-1 were seen in protein extracts from controls as compared with those from treated cells (Fig. 3). Results in HaCaT keratinocytes were comparable with those in SCL-1 cells (data not shown).
Discussion
The hazardous effects of solar or artificial UV (UV) radiation, most importantly of UV-B with a wavelength range between 290 and 320 nm, represent an important etiological factor in the development of non-melanoma skin cancer.2-4 UV-B induces photochemical alterations in the skin that may lead to acute effects including sunburn and immune suppression or to chronic effects such as premature skin aging and non-melanoma skin cancer.2-4 UV-B-mediated biological effects in target cells include the induction of apoptosis12,13 and the production of interleukin-6 (IL-6).11 Apoptosis, as a mode of programmed cell death, is induced following UV-B-irradiation when cellular damage is too severe to be repaired.12,13 To induce apoptosis, UV-B modulates a variety of important cellular signaling pathways that involve various nuclear and cell surface death receptors and the formation of reactive oxygen species (ROS).11 Also involved in this process is the activation of a cascade of cystein proteases called caspases.11 The final effector protease, caspase 3, causes cleavage of several substrates, including PARP, which immediately results in apoptosis.11 This cascade appears to be crucial for executing apoptosis induced by UV-B.11 Furthermore, it has recently been described that C-Jun-NH2-terminal kinase (JNK), a member of the mitogen-activated protein kinases (MAPK), is required for UV-induced apoptosis via the induction of cytochrome c release.11 It has been hypothesized that JNK-dependent apoptosis is mediated through mitochondrial cytochrome c release, which has also been observed as an early event in UV-mediated apoptosis in HaCaT cells.11 On the other hand, UV-B-irradiation strongly induces accumulation of IL-6 mRNA and release of IL-6 protein by human keratinocytes.11 The cytokine IL-6 is an important mediator of the sunburn reaction and of UV-B-dependent immune suppression.12 Furthermore, IL-6 has been implicated in the tumorigenesis of basal cell carcinoma, a frequent neoplasm that can be induced by UV-B radiation.12,13
Our results reported here, demonstrating that 1,25(OH)2D protects cultured HaCaT keratinocytes and SCL-1 cells in part against the antiproliferative effects of UV-B-radiation, are in line with findings published previously by us and other investigators. We and others have previously shown that the biologically active vitamin D metabolite 1,25(OH)2D and its analogs protect human skin cells from UV-induced hazardous effects, including cell death and apoptosis.11-13 However, it is not completely understood, to what extend effects of 1,25(OH)2D on apoptosis, IL-6, PARP, JNK or MAPK are responsible for this protective effect. In previous studies, cytoprotective effects of 1,25(OH)2D on UV-B-irradiated keratinocytes were seen morphologically and using a colorimetric cell survival assay.11 Moreover, using an ELISA that detects DNA-fragmentation, it was shown that pretreatment with 1,25(OH)2D suppressed UV-B-induced apoptotic cell death.11 Pretreatment of keratinocytes with 1,25(OH)2D for 24 h reduced UV-B-stimulated apoptosis by 55–70%. It has been shown that this reduction required relatively high, pharmacological concentrations of 1,25(OH)2D and a preincubation period of several hours.11 Moreover, it has been reported that pretreatment with 1,25(OH)2D also inhibits mitochondrial cytochrome c release (90%), a hallmark event of UV-B-induced apoptosis.11 Additionally, it has been shown that 1,25(OH)2D affects two important mediators of the UV-response: it reduces c-Jun-NH2-terminal kinase (JNK) activation and interleukin-6 (IL-6) production.11 Analyzing the cleavage of poly (ADP-ribose) polymerase (PARP) further corroborated these observations. As mentioned before, PARP-cleavage is clearly induced by UV-B-irradiation. In recent studies, it has been shown that pretreatment of keratinocytes with 1,25(OH)2D (1 μM for 24h) efficiently, but not completely, inhibited this UV-B-induced PARP-cleavage.11 Apart from these effects, MT-induction may be relevant for the anti-UV-B effects of 1,25(OH)2D. MT acts as a radical scavenger in oxygen-mediated UV-B-injury.11 MTs are a class of small cysteine-rich proteins that bind and exchange heavy metal ions but also have clear scavenging properties for ROS.11 Indeed, part of the UVB-induced damage to cells occurs through the formation of ROS and antioxidative agents such as MT have been reported to be photoprotective.11 MT mRNA expression was clearly induced by 1,25(OH)2D. Following these investigations, the anti-apoptotic effect of 1,25(OH)2D in keratinocytes was confirmed, using cisplatin and doxorubicin as apoptotic triggers.13 In that study, it was demonstrated that 1,25(OH)2D activated two independent survival pathways in keratinocytes: the MEK/extracellular signal regulated kinase (ERK) and the phosphatidylinositol 3-kinase (PI-3K)/Akt pathway.13 Activation of ERK and Akt by 1,25(OH)2D was transient, required a minimal dose of 10−9 M and could be blocked by actinomycin D and cycloheximide. Moreover, inhibition of Akt or ERK activity with respectively a PI-3K inhibitor (LY294002) or MEK inhibitors (PD98059, UO126), partially or totally suppressed the anti-apoptotic capacity of 1,25(OH)2D. Finally, 1,25(OH)2D changed the expression of different apoptosis regulators belonging to the Bcl-2 family. 1,25(OH)2D treatment increased levels of the anti-apoptotic protein Bcl-2 and decreased levels of the pro-apoptotic proteins Bax and Bad in a time- and dose-dependent way.13 The authors concluded that 1,25(OH)2D protects keratinocytes against apoptosis by activating the MEK/ERK and the PI-3K/Akt survival pathways and by increasing the Bcl-2 to Bax and Bad ratio. Taken together, these findings suggested the existence of a photoprotective effect of active vitamin D and created new perspectives for the pharmacological use of active vitamin D compounds in the prevention of UV-B-induced skin damage and carcinogenesis.11-14 It is well known that photocarcinogenesis of skin cancer is caused largely by mutations at sites of incorrectly repaired DNA photoproducts, of which the most common are the cyclobutane pyrimidine dimers (CPDs).40,41 Recently, it has been demonstrated that 1,25(OH)2D protects primary human keratinocytes against the induction of CPDs by UVB.14 This protection required pharmacologic doses of 1,25(OH)2D and an incubation period of at least 8 h before irradiation. It has been speculated that the anti-proliferative capacity of 1,25(OH)2D underlies its protective effect against UVB-induced DNA damage.14 De Haes et al. demonstrated that 19-nor-14-epi-23-yne-1,25(OH)2D (TX 522) and 19-nor-14,20-bisepi-23-yne-1,25(OH)2D (TX 527), two low-calcemic analogs of 1,25(OH)2D, were even 100 times more potent than the parent molecule in inhibiting UV-B-caused DNA damage.14 It was speculated that these molecules therefore may represent promising candidates for the chemoprevention of UV-B-induced skin cancer.14 Other investigators showed that treatment with three different vitamin D compounds [1,25(OH)2D; the rapid acting, low calcemic analog, 1α,25(OH)(2)lumisterol(3) (JN); and the low calcemic but transcriptionally active hybrid analog 1α-hydroxymethyl-16-ene-24,24-difluoro-25-hydroxy-26,27-bis-homovitamin D3 QW-1624F2–2 (QW)] diminished in all skin cell types the numbers of UV-induced pre-mutagenic CPDs from 0.5 h after cessation of UV radiation, which may explain the enhanced cell survival of skin cells.42 In that study, the rapid response antagonist analog 1β,25(OH)2D3 (HL) abolished the photoprotective effects of 1,25(OH)2D while a genomic antagonist, (23S)-25-dehydro-1α-hydroxyvitamin D3-26,23-lactone (TEI-9647), had no effect.42 UV radiation increased p53 expression in human skin cells, while concurrent treatment with 1,25(OH)2D further enhanced this effect several fold, at 3 and 6 h after UV radiation.42 Combined with previously reported lower nitrite levels with 1,25(OH)2D, it has been speculated that this increased p53 expression may favor DNA repair over apoptosis.42 Additionally, it has convincingly been shown that topical application of 1,25(OH)2D or QW suppressed solar simulated UV (SSUVR)-induced pyrimidine dimers in the epidermis of irradiated hairless Skh:HR1 mice, measured 24h after irradiation.42 Furthermore, UV-induced immunosuppression in the mice was markedly reduced by topical application of either 1,25(OH)2D or QW.42 Taken these data together, they convincingly show a protective effect of vitamin D compounds against UV-B-induced photodamage in vitro and in vivo. It is tempting to speculate that the UV-B-induced cutaneous production of vitamin D may represent an evolutionary highly conserved feed-back mechanism that protects the skin from the hazardous effects of solar UV-radiation.
Our results reported here, demonstrating that 1,25(OH)2D protects cultured HaCaT keratinocytes and SCL-1 cells in part against the antiproliferative effects of UV-B-radiation, are in line with the findings published previously that are outlined above.
Next, we analyzed whether co-treatment with epigenetic drugs 5-Aza or TSA modulates biologic effects of UV-B and/or 1,25(OH)2D in cultured human HaCaT keratinocytes or in SCL-1 cells. Interestingly, both compounds alone exerted antiproliferative effects in both cell lines. Antiproliferative effects of combined treatment with 500 J UV-B and 5-Aza or TSA were stronger as compared with treatment with 500 J UVB, 5-Aza or TSA alone. In most cases, antiproliferative effects of combined treatment with UV-B, 1,25(OH)2D and 5-Aza or TSA were slightly smaller as compared with treatment with UV-B and 5-Aza or TSA alone.
Next, we analyzed whether protein expression of two key components of the Notch signaling pathway, namely Notch-1 and Jagged-1, is modulated by treatment with UV-B and/or 1,25(OH)2D in HaCaT and/or SCL-1 cells. It has been reported previously that Notch-1 deletion in epidermal keratinocytes causes skin carcinogenesis, while in contrast Notch-1 acts in most other tissues as a proto-oncogene.5,43 Recently, the mechanisms underlying the carcinogenesis-promoting properties of Notch-1-deficient skin have been analyzed in mice characterized by a chimeric or global deletion pattern in their epidermis.5,44 Findings of this investigation obtained by deleting Notch-1 either before or after DMBA treatment in the K14CreERT system indicate that loss of Notch-1 is not of importance for the initiating event of multistage skin carcinogenesis.5,45 However, it was demonstrated that Notch-1 loss acts as a skin cancer-promoting event. In this investigation, delaying Notch-1 deletion in K14CreERT mice until after the tumor-promotion stage of carcinogenesis demonstrated that late deletion of Notch-1 contributed to malignant progression of benign papillomas, a phenotype that is found upon loss of p53 but not loss of p21WAF1/Cip1,5,46 a specific Notch-1 target in the skin.5,22 In summary, the authors of this study concluded that the main effect of Notch-1 loss in skin carcinogenesis is to support the initiated cells with a proliferative signal to promote tumor growth and proceed to invasive skin cancer. It has been speculated that this proliferative signal is located downstream of Notch-1 loss and could be delivered from within the initiated cells, supporting Notch-1's role as a classical tumor suppressor in epidermal keratinocytes.5,26 As an alternative pathway, it has been hypothesized that this signal could be sent by the skin microenvironment reacting to Notch-1 loss in the epidermis.5,46-50 The experimental system used by Demehri et al.5,44 was designed to distinguish between these two possibilities. In their investigation, the chimeric pattern of Notch-1 deletion by Msx2-Cre resulted in neighboring territories of Notch-1-expressing and Notch-1-deficient keratinocytes coexisting in the same microenvironment.5,44 Investigating a large number of tumors isolated from DMBA/TPA-treated Msx2-N1CKO mice clearly showed that tumors comprised mostly (> 99%) of Notch-1-expressing cells were as likely to form as tumors comprised predominantly of Notch-1-deleted cells in the same environment.5,44 The authors concluded that epidermal Notch-1 loss generates a non-cell autonomous signal, promoting tumorigenesis from any initiated cell exposed to the microenvironment conditioned by Notch-1-deficient keratinocytes.5,44 These results underline the relevance of the microenvironment as an active contributor to tumor development5,51 by demonstrating that it can be the primary source of proliferative signals to initiated cells.
In summary, these investigations impressively demonstrate the importance of the Notch signaling pathway for the growth characteristics of non-malignant and malignant human keratinocytes. Interestingly, in vitro treatment of HaCaT and SCL-1 cells with UV-B, 1,25(OH)2D, TSA or 5-Aza, did not result in a modulation of the protein expression of Notch-1 and Jagged 1, two key components of the Notch signaling pathway. In conclusion, our findings do not point at a differential regulation of the protein expression of Notch-1 and Jagged 1 in non-malignant or in malignant keratinocytes by treatment with UV-B, 1,25(OH)2D, TSA or 5-Aza. It has been speculated that pharmacologic modulation of Notch signaling could be a promising new target for the treatment of skin cancer.52 It was shown that the topical treatment with the immunomodulatory agent imiquimod selectively upregulates gene expression of Notch-1 and corresponding ligands (Jagged 1, Delta 1) in BCC.52 Imiquimod represents a strong immune response modifier that mediates its effects via activation of toll-like receptor 7 (TLR 7), thereby resulting in an activation of NF-κB,52 elevated synthesis of proinflammatory cytokines and a potent increase of anti-tumor Th1 immunity.52 Interestingly, Jagged 1 is implicated to function as a downstream target of NF-κB activation providing a link between these two signaling pathways.52 Our findings do not support the hypothesis that altered expression of Notch-1 or Jagged 1 may be involved in the photocarcinogenesis of SCCs or may be of importance for the growth characteristics of these tumors. Moreover, our findings do not support the hypothesis that pharmacologic modulation of Notch signaling, e.g., via vitamin D compounds, epigenetic drugs or other agents, could be a promising new target for the treatment of skin cancer. However, one has to keep in mind that our results are very limited and preliminary, and do not allow a definite conclusion on the role of Notch-1 or Jagged 1 for skin photocarcinogenesis. The importance of other factors that influence the role of Notch signaling for skin photocarcinogenesis, including the relevance of the microenvironment as an active contributor to tumor development by representing the primary source of proliferative signals to initiated cells, will have to be analyzed in future studies. These studies will help to clarify the role of Notch signaling for skin carcinogenesis.
Materials and Methods
Cell culture
Non-malignant (spontaneously immortalized HaCaT keratinocytes) and malignant (SCL-1 cells, derived from a cutaneous squamous carcinoma) human keratinocytes were maintained in RPMI 1640 medium (PAA Laboratories) supplemented with 10% fetal calf serum (Biochrom AG) and 1% L-glutamine, as reported previously.40
Cells were treated with UV B (UVB), 1,25(OH)2D3 (Sigma-Aldrich), trichostatin A (TSA, Sigma), and 5-azacytidine (5-Aza, Sigma) in various combinations, in various concentrations, and at different time points. 1,25(OH)2D3 and TSA were dissolved in ethanol as a stock solution (4 mM and 1 mg/ml, respectively) and stored in the dark in -20°C, 5-Aza was dissolved in PBS (10 mM). Media containing varying concentrations of 1,25(OH)2D3, TSA or 5-Aza and their combinations were added to cultured cells 24 h after seeding cells in tissue culture dishes (Greiner Bio-One GmbH), immediately after UV B-irradiation. In experiments involving 1,25(OH)2D3, media containing 1,25(OH)2D3 were added to cultured cells 24 h before or immediately after UV B-irradiation. Cells cultivated with medium alone or cells treated with vehicle (ethanol) alone served as controls.
UV-B irradiation
Immediately before irradiation with a UV-B-lamp (Waldmann), cells were washed with PBS. Cells were then irradiated (0 J, 100 J or 500 J) through a thin film of PBS. UV-B doses were determined using a dosimeter (Waldmann). Twenty-four hours before or immediately after irradiation, cells were provided with fresh medium containing various supplements as outlined above.
Proliferation analysis
The cells were plated into 24-well plates. Proliferation of untreated and treated cells was quantified by crystal violet (CV) dye staining, as reported previously.40 In short, the cells were washed once with PBS and fixed with ethanol (70%) for 30 min at room temperature. The cells were then incubated with a CV solution (1% in 20% ethanol) for 30 min at room temperature and rinsed with water thoroughly. After drying, the dye was extracted with 70% ethanol and its absorbance determined at 550 nm using a microplate reader (Titertek Multiskan Plus MK II, Labsystems).
Protein gel blotting
For protein isolation, cells were lysed in 50 µl of a lysis buffer heated to 100°C and containing 50 mM Tris–HCl (pH 6.8), 100 mM DTT, 2% SDS and 20% glycerol. Samples containing 5 µg of total cellular protein were subjected to 10% SDS–PAGE and transferred to a PVDF membrane (Immobilon-P, Millipore). Signals were detected upon overnight incubation of the membranes with primary antibodies [directed against Notch 1 (EPITOMICS, Burlingame, #1935-1), jagged 1 (clone H-114, Santa Cruz Biotechnology, Inc., sc-8303) or GAPDH (clone FL-335, Santa Cruz Biotechnology, Inc., sc-25778)] followed by a final incubation with a peroxidase-conjugated secondary antibody and Renaissance Enhanced Luminol Reagents (NEN), performed as specified by the supplier.
Acknowledgments
This study was supported in part by a grant from the European Sunlight Association (ESA), Brussels, Belgium.
Glossary
Abbreviations:
- 5-Aza
5-azacytidine
- BCC
basal cell carcinoma
- Dll
Delta-like
- DNMTI
DNA methyltransferase inhibitor
- HDACI
histone deacetylase inhibitor
- Hes
hairy and enhancer of split
- Hrt
Hes-related transcription factor
- Jag
Jagged
- MAML
mastermind-like
- miRNA
micro ribonucleic acid
- NICD
Notch intracellular domain
- SCC
squamous cell carcinoma
- TSA
trichostatin A
- VDR
vitamin D receptor
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/dermatoendocrinology/article/19027
References
- 1.Rubin AI, Chen EH, Ratner D. Basal-cell carcinoma. N Engl J Med. 2005;353:2262–9. doi: 10.1056/NEJMra044151. [DOI] [PubMed] [Google Scholar]
- 2.Bagheri MM, Safai B. Cutaneous malignancies of keratinocytic origin. Clin Dermatol. 2001;19:244–52. doi: 10.1016/S0738-081X(01)00179-1. [DOI] [PubMed] [Google Scholar]
- 3.Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:975–83. doi: 10.1056/NEJM200103293441306. [DOI] [PubMed] [Google Scholar]
- 4.Reichrath J, Nürnberg B. Solar UV-radiation, vitamin D and skin cancer surveillance in organ transplant recipients (OTRs) Adv Exp Med Biol. 2008;624:203–14. doi: 10.1007/978-0-387-77574-6_16. [DOI] [PubMed] [Google Scholar]
- 5.Reichrath J, Reichrath S. Notch-signaling and non-melanoma skin cancer: an ancient friend, revisited. Adv Exp Med Biol. 2012;727 doi: 10.1007/978-1-4614-0899-4_20. In press. [DOI] [PubMed] [Google Scholar]
- 6.Green CL, Khavari PA. Targets for molecular therapy of skin cancer. Semin Cancer Biol. 2004;14:63–9. doi: 10.1016/j.semcancer.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH, Jr., Scott MP. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276:817–21. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- 8.Melnikova VO, Ananthaswamy HN. Cellular and molecular events leading to the development of skin cancer. Mutat Res. 2005;571:91–106. doi: 10.1016/j.mrfmmm.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Tsai KY, Tsao H. The genetics of skin cancer. Am J Med Genet C Semin Med Genet. 2004;131C:82–92. doi: 10.1002/ajmg.c.30037. [DOI] [PubMed] [Google Scholar]
- 10.Tremezaygues L, Sticherling M, Friedrich M, Meineke V, Seifert M, Tilgen W, et al. Cutaneous photosynthesis of vitamin D: an evolutionary highly conserved endocrine system that protects against environmental hazards including UV-radiation and microbial infections. Anticancer Res. 2006;26:2743–8. [PubMed] [Google Scholar]
- 11.De Haes P, Garmyn M, Degreef H, Vantieghem K, Bouillon R, Segaert S. 1,25-Dihydroxyvitamin D3 inhibits ultraviolet B-induced apoptosis, Jun kinase activation, and interleukin-6 production in primary human keratinocytes. J Cell Biochem. 2003;89:663–73. doi: 10.1002/jcb.10540. [DOI] [PubMed] [Google Scholar]
- 12.De Haes P, Garmyn M, Verstuyf A, De Clercq P, Vandewalle M, Vantieghem K, et al. Two 14-epi analogues of 1,25-dihydroxyvitamin D3 protect human keratinocytes against the effects of UVB. Arch Dermatol Res. 2004;295:527–34. doi: 10.1007/s00403-004-0451-x. [DOI] [PubMed] [Google Scholar]
- 13.De Haes P, Garmyn M, Carmeliet G, Degreef H, Vantieghem K, Bouillon R, et al. Molecular pathways involved in the anti-apoptotic effect of 1,25-dihydroxyvitamin D3 in primary human keratinocytes. J Cell Biochem. 2004;93:951–67. doi: 10.1002/jcb.20227. [DOI] [PubMed] [Google Scholar]
- 14.De Haes P, Garmyn M, Verstuyf A, De Clercq P, Vandewalle M, Degreef H, et al. 1,25-Dihydroxyvitamin D3 and analogues protect primary human keratinocytes against UVB-induced DNA damage. J Photochem Photobiol B. 2005;78:141–8. doi: 10.1016/j.jphotobiol.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Dixon KM, Norman AW, Sequeira VB, Mohan R, Rybchyn MS, Reeve VE, et al. 1α,25(OH)2-vitamin D and a nongenomic vitamin D analogue inhibit ultraviolet radiation-induced skin carcinogenesis. Cancer Prev Res (Phila) 2011;4:1485–94. doi: 10.1158/1940-6207.CAPR-11-0165. [DOI] [PubMed] [Google Scholar]
- 16.Mason RS, Sequeira VB, Dixon KM, Gordon-Thomson C, Pobre K, Dilley A, et al. Photoprotection by 1alpha,25-dihydroxyvitamin D and analogs: further studies on mechanisms and implications for UV-damage. J Steroid Biochem Mol Biol. 2010;121:164–8. doi: 10.1016/j.jsbmb.2010.03.082. [DOI] [PubMed] [Google Scholar]
- 17.Damian DL, Kim YJ, Dixon KM, Halliday GM, Javeri A, Mason RS. Topical calcitriol protects from UV-induced genetic damage but suppresses cutaneous immunity in humans. Exp Dermatol. 2010;19:e23–30. doi: 10.1111/j.1600-0625.2009.00955.x. [DOI] [PubMed] [Google Scholar]
- 18.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 19.Kolev V, Mandinova A, Guinea-Viniegra J, Hu B, Lefort K, Lambertini C, et al. EGFR signalling as a negaive regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat Cell Biol. 2008;10:902–11. doi: 10.1038/ncb1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moriyama M, Durham AD, Moriyama H, Hasegawa K, Nishikawa S, Radtke F, et al. Multiple roles of Notch signaling in the regulation of epidermal development. Dev Cell. 2008;14:594–604. doi: 10.1016/j.devcel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 21.Lefort K, Dotto GP. Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Semin Cancer Biol. 2004;14:374–86. doi: 10.1016/j.semcancer.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 22.Rangarajan A, Talora C, Okuyama R, Nicolas M, Mammucari C, Oh H, et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001;20:3427–36. doi: 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okuyama R, Nguyen BC, Talora C, Ogawa E, Tommasi di Vignano A, Lioumi M, et al. High commitment of embryonic keratinocytes to terminal differentiation through a Notch1-caspase 3 regulatory mechanism. Dev Cell. 2004;6:551–62. doi: 10.1016/S1534-5807(04)00098-X. [DOI] [PubMed] [Google Scholar]
- 24.Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–73. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- 25.Kadesch T. Notch signaling: the demise of elegant simplicity. Curr Opin Genet Dev. 2004;14:506–12. doi: 10.1016/j.gde.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–21. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 27.Kiec-Wilk B, Grzybowska-Galuszka J, Polus A, Pryjma J, Knapp A, Kristiansen K. The MAPK-dependent regulation of the Jagged/Notch gene expression by VEGF, bFGF or PPAR gamma mediated angiogenesis in HUVEC. J Physiol Pharmacol. 2010;61:217–25. [PubMed] [Google Scholar]
- 28.Zhang Y, Li B, Ji ZZ, Zheng PS. Notch1 regulates the growth of human colon cancers. Cancer. 2010;116:5207–18. doi: 10.1002/cncr.25449. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, et al. Down-regulation of Notch-1 is associated with Akt and FoxM1 in inducing cell growth inhibition and apoptosis in prostate cancer cells. J Cell Biochem. 2011;112:78–88. doi: 10.1002/jcb.22770. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Lowell S, Jones P, Le Roux I, Dunne J, Watt FM. Stimulation of human epidermal differentiation by delta-notch signalling at the boundaries of stem-cell clusters. Curr Biol. 2000;10:491–500. doi: 10.1016/S0960-9822(00)00451-6. [DOI] [PubMed] [Google Scholar]
- 31.Lai EC. Keeping a good pathway down: transcriptional repression of Notch pathway target genes by CSL proteins. EMBO Rep. 2002;3:840–5. doi: 10.1093/embo-reports/kvf170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu L, Maillard I, Nakamura M, Pear WS, Griffin JD. The transcriptional coactivator Maml1 is required for Notch2-mediated marginal zone B-cell development. Blood. 2007;110:3618–23. doi: 10.1182/blood-2007-06-097030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–55. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 34.Thélu J, Rossio P, Favier B. Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma, psoriasis and wound healing. BMC Dermatol. 2002;2:7. doi: 10.1186/1471-5945-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weng AP, Aster JC. Multiple niches for Notch in cancer: context is everything. Curr Opin Genet Dev. 2004;14:48–54. doi: 10.1016/j.gde.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Stumpf WE, Sar M, Reid FA, Tanaka Y, DeLuca HF. Target cells for 1,25-dihydroxyvitamin D3 in intestinal tract, stomach, kidney, skin, pituitary and parathyroid. Science. 1979;206:1188–90. doi: 10.1126/science.505004. [DOI] [PubMed] [Google Scholar]
- 37.Danielsson C, Fehsel K, Polly P, Carlberg C. Differential apoptotic response of human melanoma cells to 1 alpha,25-dihydroxyvitamin D3 and its analogues. Cell Death Differ. 1998;5:946–52. doi: 10.1038/sj.cdd.4400437. [DOI] [PubMed] [Google Scholar]
- 38.Hansen CM, Binderup L, Hamberg KJ, Carlberg C. Vitamin D and cancer: effects of 1,25(OH)2D3 and its analogs on growth control and tumorigenesis. Front Biosci. 2001;6:820–48. doi: 10.2741/Hansen. [DOI] [PubMed] [Google Scholar]
- 39.Seifert M, Rech M, Meineke V, Tilgen W, Reichrath J. Differential biological effects of 1,25-dihydroxyvitamin D3 on melanoma cell lines in vitro. J Steroid Biochem Mol Biol. 2004;89-90:375–9. doi: 10.1016/j.jsbmb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Trémezaygues L, Seifert M, Tilgen W, Reichrath J. 1,25-dihydroxyvitamin D(3) protects human keratinocytes against UV-B-induced damage: In vitro analysis of cell viability/proliferation, DNA-damage and -repair. Dermatoendocrinol. 2009;1:239–45. doi: 10.4161/derm.1.4.9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rass K, Reichrath J. UV damage and DNA repair in malignant melanoma and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:162–78. doi: 10.1007/978-0-387-77574-6_13. [DOI] [PubMed] [Google Scholar]
- 42.Dixon KM, Deo SS, Wong G, Slater M, Norman AW, Bishop JE, et al. Skin cancer prevention: a possible role of 1,25dihydroxyvitamin D3 and its analogs. J Steroid Biochem Mol Biol. 2005;97:137–43. doi: 10.1016/j.jsbmb.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 43.Koch U, Radtke F. Notch and cancer: a double-edged sword. Cell Mol Life Sci. 2007;64:2746–62. doi: 10.1007/s00018-007-7164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Demehri S, Turkoz A, Kopan R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell. 2009;16:55–66. doi: 10.1016/j.ccr.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zoumpourlis V, Solakidi S, Papathoma A, Papaevangeliou D. Alterations in signal transduction pathways implicated in tumour progression during multistage mouse skin carcinogenesis. Carcinogenesis. 2003;24:1159–65. doi: 10.1093/carcin/bgg067. [DOI] [PubMed] [Google Scholar]
- 46.Weinberg WC, Fernandez-Salas E, Morgan DL, Shalizi A, Mirosh E, Stanulis E, et al. Genetic deletion of p21WAF1 enhances papilloma formation but not malignant conversion in experimental mouse skin carcinogenesis. Cancer Res. 1999;59:2050–4. [PubMed] [Google Scholar]
- 47.Lee J, Basak JM, Demehri S, Kopan R. Bi-compartmental communication contributes to the opposite proliferative behavior of Notch1-deficient hair follicle and epidermal keratinocytes. Development. 2007;134:2795–806. doi: 10.1242/dev.02868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 49.Vauclair S, Majo F, Durham AD, Ghyselinck NB, Barrandon Y, Radtke F. Corneal epithelial cell fate is maintained during repair by Notch1 signaling via the regulation of vitamin A metabolism. Dev Cell. 2007;13:242–53. doi: 10.1016/j.devcel.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 50.Watt FM, Estrach S, Ambler CA. Epidermal Notch signalling: differentiation, cancer and adhesion. Curr Opin Cell Biol. 2008;20:171–9. doi: 10.1016/j.ceb.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wuest M, Dummer R, Urosevic M. Induction of the members of Notch pathway in superficial basal cell carcinomas treated with imiquimod. Arch Dermatol Res. 2007;299:493–8. doi: 10.1007/s00403-007-0785-2. [DOI] [PubMed] [Google Scholar]