

Abstract
A two-microelectrode voltage clamp and optical measurements of membrane potential changes at the transverse tubular system (TTS) were used to characterize delayed rectifier K currents (IKV) in murine muscle fibers stained with the potentiometric dye di-8-ANEPPS. In intact fibers, IKV displays the canonical hallmarks of KV channels: voltage-dependent delayed activation and decay in time. The voltage dependence of the peak conductance (gKV) was only accounted for by double Boltzmann fits, suggesting at least two channel contributions to IKV. Osmotically treated fibers showed significant disconnection of the TTS and displayed smaller IKV, but with similar voltage dependence and time decays to intact fibers. This suggests that inactivation may be responsible for most of the decay in IKV records. A two-channel model that faithfully simulates IKV records in osmotically treated fibers comprises a low threshold and steeply voltage-dependent channel (channel A), which contributes ∼31% of gKV, and a more abundant high threshold channel (channel B), with shallower voltage dependence. Significant expression of the IKV1.4 and IKV3.4 channels was demonstrated by immunoblotting. Rectangular depolarizing pulses elicited step-like di-8-ANEPPS transients in intact fibers rendered electrically passive. In contrast, activation of IKV resulted in time- and voltage-dependent attenuations in optical transients that coincided in time with the peaks of IKV records. Normalized peak attenuations showed the same voltage dependence as peak IKV plots. A radial cable model including channels A and B and K diffusion in the TTS was used to simulate IKV and average TTS voltage changes. Model predictions and experimental data were compared to determine what fraction of gKV in the TTS accounted simultaneously for the electrical and optical data. Best predictions suggest that KV channels are approximately equally distributed in the sarcolemma and TTS membranes; under these conditions, >70% of IKV arises from the TTS.
INTRODUCTION
Voltage-dependent delayed rectifier K channels (KV) are known to play a crucial role in skeletal muscle physiology; they are responsible for the downstroke phase of the action potential (AP) that rapidly reestablishes the resting membrane potential after the opening of Na channels. The overall properties of KV currents have been mostly studied in muscle fibers from the frog (Adrian et al., 1970; Adrian and Marshall, 1976) and the rat (Duval and Léoty, 1980; Pappone, 1980; Beam and Donaldson, 1983a,b) and to a much lesser extent in fibers from the mouse (Brinkmeier et al., 1991; Hocherman and Bezanilla, 1996). The studies in mouse fibers have limitations derived from the fact that they have been performed using several configurations of the patch-clamp technique. For example, when on-cell or excised patch configurations were used (Hocherman and Bezanilla, 1996), no information was obtained about K channels potentially located in the transverse tubular system (TTS) membranes or about the ensemble properties of currents from the entire muscle cell. Alternatively, attempts to evaluate the properties of KV currents (IKV) using the whole-cell patch-clamp configuration (Brinkmeier et al., 1991) suffer from technical limitations possibly related to the large magnitude of the currents. Consequently, a more detailed characterization of IKV in the mouse is timely. The application of the two-microelectrode voltage-clamp technique in short fibers from the foot muscles of the mouse (flexor digitorum brevis [FDB] or interosseous muscles) is currently accepted as the most adequate approach to investigate the electrophysiological properties of muscle fibers without the aforementioned limitations (Friedrich et al., 1999; Ursu et al., 2004; DiFranco et al., 2011a; Fu et al., 2011).
It is generally postulated that IKV in adult mammalian muscle fibers display decaying phases that result from channel inactivation and/or K accumulation in the lumen of the TTS, indirectly implying that a fraction of KV channels may be located in the TTS. Thus, though the presence of IKV contributions arising from both the TTS and surface membranes has been suggested for rat skeletal muscle (Duval and Léoty, 1980; Beam and Donaldson, 1983a), no specific information regarding the KV channel distribution is available in the literature.
The identification of KV channels in skeletal muscle has been undertaken mostly using molecular biology and biochemical approaches. Using Northern blotting analysis, several types of KV channels have been identified in adult mice, including members of the Shaker (e.g., KV1.1, KV1.4, KV1.5, and KV1.7) and Shaw (KV3.1 and KV3.4) subfamilies (Lesage et al., 1992; Kalman et al., 1998; Vullhorst et al., 1998) and members of the slowly activating and inactivating KV subfamily (KV7.2, KV7.3, and KV7.4; Iannotti et al., 2010). Nevertheless, only KV3.4 and KV1.5 have been reported to be expressed (as proteins) in rat and human muscles (Abbott et al., 2001; Bielanska et al., 2009). Interestingly, recent reviews about ionic channel genes expressed in skeletal muscle membranes suggest that only KV1.4, KV3.4, and KV7.4 may be functionally important in this tissue, but no evidence supporting this statement is given (Jurkat-Rott et al., 2006; Kristensen and Juel, 2010). Although the currents carried by KV isoforms expressed in heterologous systems have been studied (Po et al., 1993; Abbott et al., 2001), limitations of this approach weaken the implications for native KV currents in adult muscle fibers. For example, it is well known that KV channels are assembled in vivo from more than one α subunit isoform (Ruppersberg et al., 1990; Po et al., 1993) and that tetramers are regulated by accessory subunits (Abbott et al., 2001; Pongs and Schwarz, 2010). To our knowledge, there are no published attempts comparing properties of IKV recorded from adult muscle fibers and those from heterologous expression systems, but recent studies about the properties of NaV1.4 channels suggest that results in vitro are not readily applicable to in vivo conditions (DiFranco and Vergara, 2011; Fu et al., 2011). Considering the crucial role that IKV play in skeletal muscle fibers, the scarcity of information is surprising, and the present work aims to fill the gap.
We first determine the kinetic and voltage dependence of IKV and decide whether the currents are transported through one or more KV channels. For this, we compare the properties of IKV in intact and osmotically treated (detubulated) muscle fibers. We also provide evidence about the molecular identity of KV channels that are expressed in the muscles from which the fibers used for electrophysiological measurements are isolated. Finally, we address the question of the relative distribution of KV channels between the TTS and surface membranes. To this end, we simultaneously record IKV and fluorescence transients from fibers stained with the potentiometric indicator di-8-ANNEPS and, as recently reported for other conductances (DiFranco and Vergara, 2011; DiFranco et al., 2011a), use radial cable model simulations of the membrane potential changes in the TTS to predict the relative distribution of KV channels between the surface and TTS membranes within a narrow range.
MATERIALS AND METHODS
Biological preparation
Animal handling followed the guidelines laid down by the University of California, Los Angeles, Animal Care Committee. FDB and interossei muscles from C57BL 3–4-mo-old mice were used. Isolated fibers were obtained by enzymatic dissociation as previously described (Woods et al., 2004; DiFranco et al., 2011a). Two groups of fibers were used in electrophysiological experiments: control fibers, which were maintained intact after isolation (n = 44), and osmotically shocked fibers, which were selected after formamide-based osmotic treatment aimed to disconnect the TTS from the surface membrane (n = 40). From this latter group, only those displaying a specific capacitance of <2.5 µF/cm2 were further analyzed (n = 12). The geometrical and electrical parameters of both intact and osmotically treated fibers are shown in Table 1.
Table 1.
Geometrical and electrical properties of muscle fibers
| Property | Intact fibers (n = 44) | Osmotically treated detubulated fibers (n = 11) |
| Radius (µm) | 25.4 ± 3.7 | 24.8 ± 3.0 |
| Length (µm) | 492 ± 80 | 501 ± 57 |
| Capacitance (μF/cm2) | 5.00 ± 0.49 | 2.3 ± 0.31a |
Values correspond to the mean ± SD.
Significance at P < 0.05.
Solutions
The composition of the solutions (in mM) was as follows. Tyrode: 156 NaCl, 10 MOPS, 2 CaCl2, 10 dextrose, 1 MgCl2, and 4 KCl, pH adjusted with NaOH. NMG-Tyrode: 150 NMG, 5 RbCl, 10 MOPS, 2 CaCl2, 10 dextrose, 1 MgCl2, 4 KCl, and 2 × 10−4 tetrodotoxin (TTX), pH adjusted with HCl. TEA-Tyrode: 145 TEA-OH, 10 MOPS, 10 CsOH, 2 Ca(OH)2, 1 Mg(OH)2, 5 dextrose, 2 × 10−2 nifedipine, 0.4 9-anthacene carboxylic acid (9-ACA), and 2 × 10−4 TTX, pH adjusted with HCl. Formamide-Tyrode: Tyrode added with 2 M formamide. K internal solution: 160 KOH, 20 MOPS, 50 EGTA, 5 ATP-Mg, 5 Na2–creatine phosphate, and 5 of reduced glutathione, pH adjusted with aspartic acid. The high EGTA concentration in the internal solution was used to arrest fiber contraction to avoid movement of artifacts in the optical records. The osmolarity of the aforementioned solutions was 300 ± 5 mosmol/kg H2O. Tris-buffered saline (TBS): 10 Tris-Cl and 150 NaCl. Lysis buffer: 3% SDS, 115 sucrose, and 66 Tris-Cl. Fractionation buffer: 150 KCl, 5 MgSO4, 20 MOPS, and 10 EGTA-K. Microsome storage buffer: 20 MOPS-Tris and 300 sucrose. Stock solutions for 9-ACA (500 mM), nifedipine (50 mM), and isradipine (10 mM) were prepared in DMSO. All solutions were adjusted to pH 7.4. All chemicals were purchased from Sigma-Aldrich.
Osmotic shock treatment
The method was based on the use of formamide (del Castillo and Escalona de Motta, 1978) and modified from that described elsewhere for enzymatically dissociated mouse FDB fibers (Lueck et al., 2010; DiFranco and Vergara, 2011). In brief, dissociated fibers were transferred to a 50-ml glass beaker containing 0.5 ml Tyrode, and 10 ml formamide-Tyrode was added. After 10 min, the solution volume was reduced again to ∼0.5 ml, and 40 ml Tyrode was quickly added. Approximately 10 min after the osmotic shock, the fibers were stained with di-8-ANEPPS as described previously (DiFranco and Vergara, 2011). The criteria used to evaluate the extent of detubulation are described in Results (see Osmotic shock treatment affords a degree …).
Electrophysiology
Experiments were performed under voltage-clamp conditions using a two-microelectrode high voltage amplifier (TEV-200A; Dagan Corporation) as previously described (Woods et al., 2004, 2005; DiFranco et al., 2005, 2011a). To improve the frequency response of the voltage clamp, electrodes were drawn to the largest tip size compatible with the viability of the fibers and had resistances in the range of 6 to 10 MΩ when filled with internal solution. Experiments with detubulated fibers required the use of electrodes in the lowest range of resistance to ensure voltage-clamp stability. Command voltage pulses were digitally filtered at 50 kHz. Fibers were impaled under current-clamp conditions and, after an ∼20-min equilibration period between the pipette solution and the myoplasm, were voltage clamped at a holding potential (VH) of −90 mV. Fibers requiring >12 nA to maintain VH were discarded. Two protocols were used to eliminate capacitive and linear ionic currents. (1) Current records elicited by 40–50-mV pulses was linearly scaled and subtracted from the rest of the records. (2) Current records elicited by any given pulse in TEA-Tyrode were subtracted one-to-one from those in which nonlinear currents were recorded. In contrast to the first protocol, the second protocol subtracts not only linear components, but also nonlinear capacitive currents. Linear membrane capacitance was measured in every fiber as previously described (DiFranco et al., 2011a) for both intact and osmotically treated fibers. Potassium currents were typically expressed in microamperes/centimeters squared. In some cases, results were also expressly given in amperes/farads for comparison, but a general conversion to this unit can be readily made using the mean specific capacitance in Table 1.
The voltage dependence of the peak gKV was adjusted with either a single Boltzmann equation or by the following double function:
where A is the peak [gKV]max and ε is the fraction of conductance contributed by one channel. For statistical analysis, individual datasets obtained at various conditions were fitted, and the means for each parameter were comparatively evaluated. All experiments were performed at room temperature (20–22°C).
Recording of di-8-ANEPPS transients
The optical methodology has been described elsewhere (DiFranco and Vergara, 2011; DiFranco et al., 2005, 2011a). In brief, dissociated fibers stained with di-8-ANEPPS (Biotium; Hayward) were placed on coverslip-bottomed small Petri dishes sitting on the stage of an inverted microscope (IX-71; Olympus) equipped with a standard epifluorescence attachment, a cooled charge-coupled device camera (ST-402ME; Santa Barbara Instrument Group), and a photodetector consisting of a photodiode (UV-001; OSI Optoelectronics) connected to a patch-clamp amplifier (Axopatch 2A; Molecular Devices). For control fibers, only those displaying a sharp sarcomere banding and the distinctive pattern of TTS staining by di-8-ANEPPS (DiFranco et al., 2005, 2007) were used for the experiments. For detubulated fibers, only those displaying predominant peripheral di-8-ANEPPS staining were selected. For control fibers, the illumination disc was adjusted to ∼25 µm in diameter and focused (using a microscope [UPLSAPO 100XO; Olympus] and a 1.4 numerical aperture objective) at the center of the x, y, and z axes of the fiber. For detubulated fibers, the illumination spot was widened to a diameter ∼25% larger than the fiber’s width to include the periphery of the fibers. In either case, the illumination spot was centered at the site where the voltage microelectrode was impaled. Optical signals were low-pass filtered (2 kHz) single sweeps. Optical data are presented as normalized fluorescence changes (ΔF/F).
Radial cable model simulations
The properties of K currents and the impact that they have on membrane potential changes in the TTS were simulated using multistate models and the radial cable model, respectively. Details of model equations and integration methods are described in the Appendix.
Total protein lysates and microsomal fractions
To prepare total protein lysates from foot muscles (muscle lysate [ML]), FDB and interossei muscles from adult mice were minced with a razor blade and homogenized on ice using a tissue grinder (Duall; Kontes) in lysis buffer with one tablet/7 ml of a cocktail of protease inhibitors (Roche). The muscle homogenate was rotated at 4°C for 1 h and then centrifuged at 15,000 g for 15 min. The supernatant was collected and stored at −80°C. For muscle microsomal (MM) preparations, hind limb muscles from two mice were dissected and trimmed of connective tissue and fat. Muscles were finely minced using razor blades, mixed with ice-cold homogenization buffer supplemented with protease inhibitor cocktail (4 ml/g of wet tissue), and homogenized for 30 s (Bio-homogenizer M133/1281; Biospec Products Inc.). The homogenate was centrifuged at 1,500 g for 30 min at 4°C. The supernatant was saved, and the pellet was resuspended, homogenized, and centrifuged again at 1,500 g for 30 min. The two supernatants were combined and centrifuged at 15,000 g for 30 min at 4°C. The new supernatant was centrifuged at 125,000 g for 60 min at 4°C. The final pellet was resuspended in a small volume of storage buffer and stored at −80°C. For both preparations, protein concentration was determined using the Quick Start Bradford Protein Assay (Bio-Rad Laboratories).
Western blotting
Western blot analysis was performed in three total protein MLs, two MM preparations, and two hippocampus lysates (HLs; Delgado and O’Dell, 2005). Gels were loaded with 40, 32, and 35 µg of total protein for ML, MM, and HL samples, respectively. Proteins were separated on 7% SDS-PAGE gels (100 V for 1 h) and transferred to polyvinylidene fluoride membranes (25 V overnight). Membranes were blocked for 1 h in blocking buffer (4% skimmed milk powder and 2% bovine serum albumin in TBS with 0.05% Tween [TBS-T]). Immunoblotting was performed overnight using the following antibodies diluted in TBS-T containing 4% nonfat milk: mouse monoclonal anti-KV1.4 (75–010, clone K13/31, 1:500 dilution), mouse monoclonal anti-KV3.4 (75–112, 1:500 dilution), and rabbit polyclonal anti-KV1.4 (APC-007, 1:200 dilution). The first two antibodies were obtained from the University of California, Davis/National Institutes of Health (NIH) Neuromab Facility, and the third one was obtained from Alomone Laboratories. After three washes for 3 min in TBS-T, membranes were exposed to horseradish peroxidase–conjugated secondary antibody (goat anti–mouse or goat anti–rabbit antibody, as required; MP Biomedicals; 1:2,000 dilution in TBS-T) for 2 h. Enhanced chemiluminescence with Immun-Star horseradish peroxidase substrate (Bio-Rad Laboratories) was used to develop all immunoblots. Images were collected using a charge-coupled device camera attached to a ChemiDoc chemiluminescent detection system and using Quantity One software (Bio-Rad Laboratories).
Data acquisition and statistical analysis
Voltage, current, and fluorescence records were filtered at 10, 5, and 2 kHz, respectively, using multiple pole analogue Bessel filters. Data points were sampled every 30 µs using a data acquisition interface (PCI-6221; National Instruments) and custom software written in LabView (National Instruments). Unless otherwise stated, pooled data are expressed as means ± SEM. Significance was set at P < 0.05. The goodness of the fits comparing single and double Boltzmann functions was evaluated using the Akaike’s Information Criterion (AIC) test (Origin Pro8; OriginLab Corporation).
RESULTS
Delayed rectifier potassium currents (IKV) in intact FDB muscle fibers
Fig. 1 A shows the ionic currents recorded in an FDB fiber under conditions that eliminate the contributions (by specific ion replacements and/or the addition of blockers in the external solution) from Na channels (NaV1.4; by replacing Na by NMG and adding 4 × 10−7 M TTX) and Cl channels (ClC-1, by adding 400 µM 9-ACA) and reduce those from inward rectifier K channels (KIR; by adding 5 mM Rb). The fiber was voltage clamped at a holding potential (VH) of −90 mV (close to the calculated EK of −91 mV), and 50-ms rectangular voltage pulses (from 0 to 200 mV every 10 mV) were applied. The current records were corrected by subtracting scaled linear leak and capacitive currents obtained from a record in response to a 50-mV pulse. Negligible ionic currents were observed in response to hyperpolarizing pulses (not depicted) or to pulses smaller than 70 mV (Fig. 1 A, gray trace). For larger depolarizations, outward currents with typical features of IKV were observed: currents arise after a delay from the pulse onset, and the larger the pulse amplitude, the shorter the delay becomes. A very early nonlinear capacitive current component (gating currents) precedes the delayed ionic component. At every depolarization, IKV grows to an apparent maximum with a rate of rise that increases with the pulse magnitude. For the largest pulse tested (200 mV), a peak IKV of 1,100 µA/cm2 was reached in ∼3.7 ms (Fig. 1 A, orange trace). For physiological depolarizations (to membrane potentials of ∼40 mV), similar to those reached at the peak of an AP, a peak IKV of ∼460 µA/cm2 is attained in ∼5 ms (Fig. 1 A, blue trace). Within the timescale used in Fig. 1 A, only IKV records elicited by relatively large depolarizations (>100 mV) already show evidence of decay during the pulse.
Figure 1.

Delayed rectifier K currents in FDB fibers. (A) IKV recorded in response to depolarizations from VH to membrane potential values ranging from −30 to 110 mV in 20-mV steps (gray to orange traces). The NMG-Tyrode contained no Ca2+ channel blockers. Linear leak and capacitances were removed from each raw current record by subtracting a scaled version of the current record obtained in response to a 50-mV pulse. (B) Voltage dependence of the peak IKV (closed circles) and peak gKV (open circles). For both plots, the circles are connected with straight lines. (C) Voltage dependence of the mean peak IKV obtained from 16 voltage families in 14 different fibers. (D) Voltage dependence of the mean peak gKV (circles), calculated from the data in C. The green trace is a single Boltzmann fit to the data with the following parameters (mean ± SD): A = 4.77 ± 0.9 mS/cm2; V1 = 18.2 ± 6.3 mV; K1 = 20.4 ± 1.5 mV. The black trace is a double Boltzmann fit to the data with the following parameters: A = 4.93 ± 0.9 mS/cm2; ε = 0.36 ± 0.06; V1 = −7 ± 4.7 mV; K1 = 6.7 ± 1.1 mV; V2 = 39 ± 6.6 mV; K2 = 19.9 ± 2.3 mV. The red and blue traces are the independent components of the double Boltzmann function. Error bars represent SEM.
The peak values of the IKV records shown in Fig. 1 A (peak IKV) are plotted as a function of membrane potential in Fig. 1 B (closed circles). It can be observed that peak IKV increases almost linearly with membrane depolarizations for the entire range of voltages explored. Assuming an ohmic behavior for open channels (Hodgkin and Huxley, 1952a), we calculated the peak K conductance (peak gKV) as
The resulting peak gKV values are plotted as a function of the membrane potential in Fig. 1 B (open circles). Notwithstanding the ample voltage range explored, peak gKV did not reach a plateau; a maximum value of ∼5 mS/cm2 was obtained in this fiber for a 200-mV pulse. It is also apparent that the voltage dependence of the data points do not seem to conform to a single sigmoid distribution, but instead there seems to be a noticeable inflection point at ∼25 mV, potentially suggesting the participation of more than one channel’s population. The voltage dependence of the mean peak IKV and gKV, obtained from 16 experiments in 14 fibers, is shown in Fig. 1 (C and D, respectively). The mean membrane potential at which sizable currents were detected is −20 mV, and the mean maximum peak IKV at 110 mV is 990 ± 193 µA/cm2 (mean ± SD), which corresponds to ∼191 ± 37 A/F. The population data in Fig. 1 C display similar features as those from the individual fiber in Fig. 1 B. The peak conductance plot, calculated from mean data in Fig. 1 C, is shown in Fig. 1 D (circles). As observed in Fig. 1 B, a plateau is not reached within the voltage range explored. The maximal peak gKV ((gKV)max) obtained from the mean plot is 5 ± 1 mS/cm2 (mean ± SD), which corresponds to ∼1 ± 0.2 mS/µF. As suggested from data in Fig. 1 B, the voltage dependence of the mean data (open circles) cannot be correctly fitted to a single Boltzmann dependence (Fig. 1 A, green trace). In contrast, a double Boltzmann curve (Fig. 1 D, black trace) adequately predicts the data; the AIC test indicates that this model function is >2 × 108 times better in predicting this mean dataset than the single function. This feature is suggestive that there are at least two populations of K channels contributing to the total IKV records in this preparation. One contribution seemingly arises from a minority population of low threshold channels with a half-activation voltage (V1/2) at approximately −10 mV (Boltzmann fit represented by the red trace in Fig. 1 D), and the other arises from a larger population of higher threshold channels with V1/2 at 40 mV (Boltzmann fit represented by the blue trace in Fig. 1 D). Interestingly, the two single Boltzmann curves in Fig. 1 D are sufficiently distinct to allow us to infer that the low threshold channel (red trace) is fully activated at approximately −5 mV, shows steep voltage dependence, and contributes 40% of the total conductance. In contrast, the high threshold channel (Fig. 1 A, blue trace) displays shallower voltage dependence, is fully activated at a potential >80 mV, and contributes ∼60% of the total current.
IKV records from mouse FDB fibers show a marked decay during long depolarizations
Although the timescale used in Fig. 1 was adequate to illustrate the activation characteristics of IKV, it was insufficient to study the voltage and time dependence of the decay processes. When a longer time window is used, as illustrated in Fig. 2 A, pronounced decays in the currents are observed. Furthermore, it is apparent from IKV records in Fig. 2 A that the decay process is nonmonotonic. Another striking feature of the family of currents in Fig. 2 A is the presence of prominent inward current tails at the end of the pulses, which rapidly decay toward the baseline. Because the fiber was repolarized to −90 mV, a value close to the calculated EK, negligible current contributions through IKV channels would be expected. A possible source for these tail currents is, as suggested previously (Beam and Donaldson, 1983a), that K accumulation had occurred in the TTS and that some of the current contributions arise from this membrane compartment through KV channels (while they close) or through KIR channels. Because 5 mM Rb reduces the latter (Beam and Donaldson, 1983a) while not affecting IKV (unpublished data), at least part of the tail currents may represent the closing of KV channels in the TTS. In addition, Ca2+ current tails, through the prominent CaV1.1 channels in the TTS, could represent an important contribution to the observed currents at the end of the long pulses shown in Fig. 2 A, particularly after large depolarizations. It should be noted that this particular experiment was conducted with 2 mM Ca2+ in the external solution, and no Ca2+ channel blocker was used. In fact, slight distortions suggestive of Ca2+ current contaminations can be observed as minor downward deflections in the IKV traces at large depolarizations (Fig. 2 A, deep blue and orange traces). To remove the possible contributions of putative Ca2+ currents and obtain IKV in complete isolation from other currents, we used known Ca2+ blockers such as isradipine and nifedipine (Lamb and Walsh, 1987; Berjukow et al., 2000; DiFranco et al., 2011b). Fig. 2 B shows IKV records from the same fiber but in the presence of 0.5 µM isradipine. It can be observed that a large fraction of the tail currents was blocked by isradipine. However, in agreement with results obtained with other Ca2+ channel blockers in other preparations (Grissmer et al., 1994; Zhang et al., 1997), Fig. 2 B shows that isradipine also has an important apparent blocking effect on IKV. In our case, ∼40% of the maximal peak current was blocked by the drug. Importantly, the effects of isradipine on IKV records seem to be independent of the entry of Ca2+ through CaV1.1 because IKV reaches a peak at times when activation of the Ca2+ channels is negligible (unpublished data). By comparing the records in Fig. 2 (A and B), it is also noticeable that IKV decays to a larger extent in the presence of isradipine. This is an intriguing observation because the elimination of an inward current component (ICa) would have been expected to have the opposite effect; we suggest that a reasonable explanation for this phenomenon is that the Ca2+ entry through CaV1.1 channels facilitates the activation of a significant outward K current through BK channels in the TTS (Latorre et al., 1983; Tricarico et al., 1997). The voltage dependence of the peak IKV and peak gKV before (black circles) and after exposure to 0.5 µM isradipine (red circles) is shown in Fig. 2 C. It can be seen that, for all voltages tested, isradipine reduces the peak gKV. This suggests this drug blocks the two populations of channels proposed above; in support of this, we found that, as in the case of control peak gKV, double Boltzmann equations were required to accurately fit the data obtained in the presence of isradipine (the AIC test indicates that this function is >9 × 105 better than a single Boltzmann function; Fig. 2 C, red traces). Nevertheless, the results obtained from a population of fibers (Fig. 2 D), which include data from 16 fibers in control conditions and 6 fibers in the presence of 0.5 µM isradipine, suggest that the putative channels may be differentially affected by this drug. Thus, statistical analysis of the mean parameters of the Boltzmann fits to the peak gKV in Fig. 2 D shows that not only (gKV)max, but also the percent contribution of the low threshold channel are significantly reduced by isradipine. These results imply that although both channels are blocked by isradipine, the low threshold channel is more sensitive to the drug that the higher threshold one.
Figure 2.

IKV decays and blocking effect of isradipine. (A) IKV records were obtained in response to 500-ms voltage pulses of 60–200-mV amplitude, with 20-mV increments. (B) IKV from the same fiber as in A, but after exposure to 0.5 µM isradipine. (C) Voltage dependence of peak IKV (closed circles) and peak gKV (open circles) determined from the data in A (black circles, control conditions) and B (red circles, isradipine). The black solid lines are double Boltzmann fits to the control data with the following parameters: A = 4.44 mS/cm2; ε = 0.33; V1 = −3.2 mV; K1 = 6.2 mV; V2 = 44.7 mV; K2 = 17.4 mV. The red solid lines are double Boltzmann fits to the isradipine data with the following parameters: A = 2.68 mS/cm2; ε = 0.31; V1 = −1.8 mV; K1 = 7.0 mV; V2 = 36.7 mV; K2 = 14.8 mV. (D) Mean peak gKV (pooled data) obtained under control conditions (black circles; same data as in Fig. 1 D) and in the presence of isradipine (red circles; n = 6). The red line is a double Boltzmann fit to the isradipine data with the following parameters (mean ± SD): A = 2.90 ± 0.7 mS/cm2 (*); ε = 0.28 ± 0.09 (*); V1 = −1.7 ± 2.8 mV (*); K1 =7.6 ± 1.5 mV; V2 =40 ± 6 mV; K2 = 20.7 ± 3.5 mV. The * indicates statistical significance with respect to the control (P < 0.05). Error bars represent SEM.
Osmotic shock treatment affords a degree of TTS disconnection that allows for better measurements of IKV
The possibility that two populations of K channels are contributing to IKV and the difficulty in eliminating ICa contaminations without affecting IKV complicate the analysis of this current in skeletal muscle fibers. For example, as shown for the Na channel (DiFranco and Vergara, 2011), current contributions from KV channels in the TTS are expected to have a different overall impact on IKV than those at the surface membrane. In addition, though the CaV1.1 conductance is likely to be smaller than that of KV channels, its preferential location at the TTS membranes could be responsible for the distortion seen in IKV records. For these reasons and in the hope that a certain degree of disconnection of the TTS from the sarcolemma affords better isolation of IKV, as shown previously for the Na channel (DiFranco and Vergara, 2011; Fu et al., 2011), we submitted FDB fibers to a formamide osmotic shock.
The extent of TTS disconnection was evaluated by two-photon laser-scanning microscopy (TPLSM) and electrophysiological methods. Fig. 3 A is a TPLSM image that illustrates the typical staining pattern of the surface and TTS membranes in a control FDB muscle stained with di-8-ANEPPS. As described in previous publications from our laboratory (DiFranco et al., 2005, 2007, 2009), the TTS is imaged as paired bands of high fluorescence intensity (two T tubules per sarcomere) running perpendicular to the sarcolemma. These paired bands repeat regularly, with similar intensity, along the longitudinal axis of the fibers (consecutive pairs every 2.9 µm). The inset in Fig. 3 A shows an expanded view of the surface and TTS staining that allows for the identification of the different distances separating the members of each pair of adjacent T tubules with respect to those separating consecutive T tubule pairs. Equivalent images from an FDB fiber stained with di-8-ANEPPS after being osmotically shocked are shown in Fig. 3 B. It is clear that, in contrast to Fig. 3 A, the TTS staining here is reduced to the periphery of the fiber, as indicated by the lack of “internal” staining. The staining pattern displayed in Fig. 3 B is consistent with the notion that osmotic treatment restricted the access of di-8-ANEPPS only to the periphery of the TTS; in other words, the bulk of the TTS is effectively disconnected from the surface membrane. However, TTS “stumps” of variable length can be readily observed up to ∼2–3 µm from the surface membrane. This is better appreciated in tangential sections as shown in the inset of Fig. 3 B, which shows that the pattern of TTS staining is preserved at the periphery of the fiber. The pattern of di-8-ANNEPS staining of the TTS in TPLSM images of control muscle fibers (as in Fig. 3 A) has been consistently established. In contrast, di-8-ANNEPS staining patterns like that illustrated in Fig. 3 B are observed in <15% of osmotically treated fibers; the rest of the treated fibers showed images of TTS staining with intermediate features between those of Fig. 3 (A and B). Membrane capacitance measurements provide support to the TPLSM data. The capacitance of a subpopulation of osmotically treated fibers selected for electrophysiological experiments was as low as 2.3 ± 0.31 µF/cm2 (n = 11); however, the majority of treated fibers that were electrically tested had larger capacitances (unpublished data). It is important to note that, other than differences in capacitance, untreated and osmotically treated fibers had indistinguishable geometrical features (Table 1).
Figure 3.

TPLSM evaluation of TTS disruption by osmotic shock treatment. (A) TPLSM fluorescence image of an intact FDB muscle stained with di-8-ANEPPS. The inset is a threefold enlargement of the area defined by the white rectangle. (B) TPLSM fluorescence image of an isolated fiber stained with di-8-ANEPPS after being submitted to a formamide-based osmotic shock. In the area delimited by the rectangle, the plane of the TPLSM section was tangential to the fiber periphery. This area is enlarged threefold in the inset.
Fig. 4 A and its inset show, at two timescales, IKV records obtained from an osmotically shocked fiber in response to depolarizations (in 10-V steps) from −90 to 110 mV. These records share most of the features seen in Fig. 2 A: namely, delayed activation from the pulse onset, increasing rate of rise with membrane depolarizations (Fig. 4 A, inset), and a lowest voltage at which IKV becomes detectable at approximately −20 mV. In addition, Fig. 2 A shows that the decay phases of IKV records are significantly more pronounced than those observed in intact fibers (particularly at large depolarizations). Also, decay of IKV in osmotically treated fibers is monotonic, without the apparent contaminations by ICa-dependent contributions seen in control fibers. This is consistent with the almost complete disappearance of the slow decaying tail currents at the end of the pulses. Consistent with our TPLSM observations and capacitance measurements, we submit that the results in Fig. 4 A can be explained if deeper regions of the TTS were effectively disconnected from the rest of the fiber by osmotic shock treatment. It is interesting to note that although the voltage dependence of the peak IKV for the treated fiber (Fig. 4 C, black circles) is comparable with that obtained in intact fibers (Fig. 1 C), its magnitude is smaller. These features are better illustrated in the peak gKV plot in Fig. 4 C (black circles). Moreover, the voltage dependence of peak gKV still suggests the contribution of two channels to the total currents; this is reinforced in Fig. 4 D by showing that only a double Boltzmann equation (black trace) with a (gKV)max of 2.77 ± 0.02 mS/cm2 (significantly smaller than the 5 mS/cm2 in intact fibers; P < 0.05) can adequately fit the results (Fig. 4 C, black open circles) obtained from several osmotically treated fibers (n = 7). Because the peripheral TTS remnants (together with the sarcolemma) are expected to be under more adequate voltage commands, as compared with the entire TTS in intact fibers, the data in Fig. 4 C support the notion that two populations of channels (with different voltage dependence) contribute to the IKV records.
Figure 4.

IKV records in osmotically treated fibers and isradipine effects. (A) IKV records elicited by 500-ms depolarizations from VH to membrane potentials spanning from −30 to 90 mV every 20 mV. The inset shows the onset of the currents in an expanded timescale. (B) Effects of 1 µM isradipine on IKV. Same fiber and protocol as in A. The onset of the currents is shown in an expanded timescale in the inset. (C) Peak IKV (closed circles) and peak gKV (open circles) determined from the data in A (black circles, control conditions) and B (red circles, isradipine). IKV data points are connected by straight lines. The solid black lines are double Boltzmann fits to the control gKV data with the following parameters: A = 3.2 mS/cm2; ε = 0.21; V1 = −10 mV; K1 = 6 mV; V2 = 31 mV; K2 = 19 mV. The red solid lines are double Boltzmann fits to the isradipine gKV data with the following parameters: A =2.4 mS/cm2; ε =0.2; V1 = −2 mV; K1 = 10 mV; V2 = 31 mV; K2 = 19 mV. (D) Peak gKV plots for a population of fibers maintained in control conditions (black circles; 12 experiments, 11 fibers) and a subpopulation of fibers (n = 4) exposed to 1 µM isradipine (red circles). Both datasets were fitted with double Boltzmann equations. Parameters for control data (black line) were (mean ± SD) A = 2.93 ± 0.5 µA/cm2; ε = 0.29 ± 0.06; V1 = −11.8 ± 3.8 mV; K1 = 5.8 ± 0.7 mV; V2 = 26.5 ± 6.2 mV; K2 = 16.7 ± 3 mV. For this dataset, the fit with a double Boltzmann is >8 × 107 times better than with a single Boltzmann function. Parameters for isradipine data (red line) were A = 2.17 ± 0.7 mS/cm2 (*); ε = 0.22 ± 0.07; V1 = −10.4 ± 2.5 mV; K1 = 7 ± 1 mV (*); V2 = 25.6 ± 4.3 mV; K2 = 16 ± 1.7 mV. The * indicates statistical significance with respect to the control (P < 0.05). The AIC test indicates that the fit to the isradipine data with a double Boltzmann is only 52-fold better than with a single Boltzmann function. Error bars represent SEM.
To determine whether the effects of isradipine depended on the presence of ICa, we next exposed this osmotically treated fiber to 0.5 µM isradipine. Interestingly, we found (Fig. 4 B) that isradipine still blocks IKV in osmotically treated fibers. The effectiveness of the isradipine block on IKV is quantitatively evaluated at every voltage in the peak IKV and peak gKV plots in Fig. 4 C (red circles and traces); it can be observed that addition of isradipine resulted in an overall reduction of the currents and conductances at all the voltages tested. Fig. 4 D shows that on average, the maximal peak gKV is significantly reduced (by 26%) in the presence of 0.5 µM isradipine (black and red circles). By fitting the peak gKV data under control conditions and in the presence of isradipine with double Boltzmann equations (Fig. 4 D, black and red traces, respectively), we found that (as in intact fibers) the blocker tends to affect more prominently the low threshold component of the currents. Nevertheless, probably because of the relatively small number of experiments in which the blocker was tested (n = 4), the difference in ε between control and treated fibers was not significant in this case (P = 0.1); however, the improvement attained by fitting the data to a double Boltzmann instead of a single one is not as marked as under control conditions (only 52-fold vs. >105-fold), thus insinuating a lesser contribution of one channel component to the total current in isradipine.
Kinetic analysis of the current components contributing to IKV in osmotically treated fibers
For the reasons discussed in the previous section, current records from partially detubulated fibers are useful to obtain (with better accuracy than in intact fibers) the voltage dependence of kinetic parameters needed to simulate the presence of components contributing to IKV. Although we tried initially to predict the properties of IKV records using kinetic models contemplating only one channel entity, our attempts to reasonably reproduce their major features (delayed activation, onset kinetics, inactivation, and overall voltage dependence) were unsuccessful. Consequently, we focus on the task of explaining the properties of IKV in the fiber described in Fig. 4, together with data from eight other osmotically treated fibers, assuming that two channels (channels A and B) contribute to IKV. For simplicity, our models (described in the Appendix) assumed that K channel activation followed a sequential scheme including four closed states (C1–C4) and one open state (O); as it is well known, this allows for the delayed activation of the currents akin to the n4 of the Hodgkin and Huxley (HH) formulation (Hodgkin and Huxley, 1952b) and is compatible with the sequential activation of the α subunits in tetrameric K channels. Another simplifying approximation of our model is that the n4-type voltage-dependent activation is sequentially coupled in a voltage-independent fashion with either a single inactivated state (e.g., N type, channel A) or two sequential N-type and C-type inactivated states (channel B). Examples of our resulting simulations are illustrated in Fig. 5 A, and the mean model parameters are included in Table A1 of the Appendix. As an example of the goodness of the fit with the two-channel model, the data in Fig. 5 A (black traces) are superimposed with model simulations (red traces); it can be seen that experimental and simulated currents can be hardly distinguished from each other. The inset in Fig. 5 A illustrates that not only the overall magnitude and decay characteristics of the experimental records, but, most importantly, the activation properties of IKV, are fairly well reproduced by model predictions. It is interesting to note at this point that, to attain this kind of fit to the experimental data, it was necessary to include a low threshold fully inactivating channel (channel A; Fig. 5 B, blue traces) and a high threshold channel that only partially inactivates (channel B; Fig. 5 B, green traces). Inactivation was a required feature for both channels. As expected, the distinct voltage dependence of activation and rate of inactivation between channels A and B determines that the fraction of the total IKV contributed by each channel was voltage and time dependent. For example, the inset in Fig. 5 A illustrates that channel A, by being responsible for most of the current in response to small depolarizations, actually determines the overall delay in activation of IKV. In contrast, the rapid activation kinetics of channel B dominates the IKV records for depolarizing pulses >120 mV. Furthermore, the widely different inactivation features of channel B predict that current contributions during long depolarizations arise exclusively from channel B. The voltage dependence of peak IKV and gKV for the total currents in Fig. 5 A are shown in Fig. 5 C (black circles) and Fig. 5 D (black circles), respectively. The continuous traces are model predictions with the parameters listed in Table 2. Likewise, the corresponding fractions of IKV and gKV contributed by channels A (blue traces) and B (green traces) are shown superimposed in the plots of Fig. 5 (C and D). Fig. 5 C shows that, as expected, currents through channel A at large depolarizations reach an asymptotic dependence (not depicted) that intersects the abscissa at approximately −90 mV (EK). Finally, Fig. 5 D shows that channel A reaches a maximal conductance of ∼1 mS/cm2 at ∼20 mV, whereas the conductance for channel B still does not reach a plateau (∼2 mS/cm2) at potentials as high as 90 mV.
Figure 5.

Predictions of IKV records from an osmotically treated fiber by a two-channel model. (A) Superposition of experimental (black traces) and simulated currents (red traces). Currents were elicited by depolarizations to membrane potentials ranging from −10 to 90 mV in 20-mV increments. The inset displays the onset of currents in an expanded timescale. (B) Individual model current contributions attributed to channel A (blue traces) and channel B (green traces). For every voltage, the currents in A are the sum of the traces in B. The inset displays the currents predicted for channels A and B in an expanded timescale. (C) Voltage dependence of peak IKV from experimental data (circles) and from currents predicted for channels A (blue trace) and B (green trace). The black trace is the sum of the currents from channels A and B. (D) Voltage dependence of peak gKV for the experimental currents (circles) and for the currents thought to be A (blue trace) and B channels (green trace). The black trace was the predicted conductance from the overall model simulations. The model parameters for this particular fiber are listed in Table 2 (see Appendix for nomenclature).
Table A1.
Parameter values for KV channels A and B
| Parameter | Channel A | Channel B |
| (ms−1) | 0.0319 ± 0.009 | 0.049 ± 0.035 |
| (ms−1) | 0.0142 ± 0.004 | 0.346 ± 0.043 |
| (mV) | 2.3 ± 1.8 | 5.7 ± 3.2 |
| kαX (mV) | 13.7 ± 0.9 | 24.2 ± 5 |
| kβX (mV) | 14.1 ± 0.9 | 33.9 ± 5 |
| ΦX (ms−1) | 0.007 ± 0.003 | 0.0105 ± 0.003 |
| ϕX (ms−1) | 1 × 10−5 | 0.0103 ± 0.0016 |
| γB (ms−1) | 0.0021 ± 0.0021 | |
| δB (ms−1) | 1 × 10-6 ± 1.4 × 10−5 | |
| (mS/cm2) | 0.86 ± 0.4 | 1.92 ± 0.4 |
| Percent contribution | 0.31 ± 0.04 | 0.69 ± 0.06 |
| gleak (mS/cm2) | 0.1 | 0.1 |
The values correspond to mean ± SEM calculated from fits to records obtained for 80-, 100-, 120-, 140-, 160-, and 180-mV pulses from nine fibers. Note that the IC state does not exist for channel A. The rate constants α and β for channels A and B had voltage dependences given by Eqs. 2 and 4, respectively. See text for specific deviations from these values.
Table 2.
Parameter values for KV channels A and B in Fig. 5
| Parameter | Channel A | Channel B |
| (ms−1) | 0.0232 | 0.0155 |
| (ms−1) | 0.01 | 0.38 |
| (mV) | 4 | 6.9 |
| kαX (mV) | 14 | 16.7 |
| kβX (mV) | 14.5 | 38 |
| ΦX (ms−1) | 0.004 | 0.0074 |
| ϕX (ms−1) | 1 × 10−5 | 0.0114 |
| γB (ms−1) | 0.0033 | |
| δB (ms−1) | 1 × 10−6 | |
| (mS/cm2) | 1.23 | 2.28 |
| Percent contribution | 0.35 | 0.65 |
Biochemical identification of KV channels in mouse muscle
In an attempt to determine what molecular entities could possibly be responsible for the IKV in adult FDB fibers, we performed immunoblotting experiments aimed to detect inactivating isoforms of known voltage-dependent K channels. To this end, MLs and microsomal preparations were tested with antibodies raised against shaker (KV1.4) and shaw (KV3.4) channels. Two different antibodies recognizing epitopes located at the N terminus (75–010; Neuromab) and C terminus (APC-007; Alomone) of KV1.4 were used; Fig. 6 A shows that both of them recognize one protein band of ∼97 kD in total protein MLs. Furthermore, both antibodies also report one band of comparable molecular weight in MM preparations; the slight difference in migration could be explained by the enhanced presence of SERCA1, a 110-kD membrane protein that is highly enriched in skeletal muscle MM preparations. The aforementioned results are in agreement with similar data reporting that APC-007 recognizes an ∼97-kD band in heart muscle preparations from wild-type animals, which is absent in those from KV1.4-null mice (Guo et al., 2000). Because the presence of KV1.4 has been extensively demonstrated in mouse hippocampus (Sheng et al., 1992, 1993; Maletic-Savatic et al., 1995; Rhodes et al., 1997; Monaghan et al., 2001), as a further control for antibody specificity, we also tested lysates from this tissue with the same antibodies as in muscle preparations. Fig. 6 A shows that both 75–010 and APC-007 recognize two bands in the HL lanes: a prominent ∼95-kD band and a lighter band of ∼85 kD. This is in agreement with previous studies showing the same two bands in mouse hippocampus tissue (Veh et al., 1995; Wickenden et al., 1999; Juiz et al., 2000). Altogether, our results and those of Guo et al. (2000) may suggest contrasting differences in KV1.4 processing between muscle and brain tissues. Western blot analysis also provides supporting evidence for the presence of KV3.4 in FDB muscles. Fig. 6 B shows the results obtained when ML and MM preparations were tested with a monoclonal anti-KV3.4 antibody (raised against amino acids 175–192 of this channel; 75–112, Neuromab). In this case, the antibody detects a single band of ∼95 kD in the ML lane and, similar to what was observed for KV1.4, a slightly lighter band in the MM preparation. Our finding that KV3.4 is expressed in leg and foot muscles is in agreement with a previous study demonstrating the presence of this channel in crude membranes of rat sartorius muscles (Abbott et al., 2001).
Figure 6.

Western blot analysis of KV1.4 and KV3.4 expression. (A and B) Immunoblots tested with the specific antibodies anti-KV1.4 (A) and anti-KV3.4 (B). See Materials and methods and Results for details. The results with the 75–010 antibody were obtained from two different blots, as indicated by the vertical line. The results with the APC-007 antibody were obtained from a single blot, but a lane between MM and ML was removed, as indicated by the vertical line.
Simultaneous detection of di-8-ANEPPS transients and IKV in intact FDB fibers
The results from osmotically treated fibers suggested that a substantial fraction of IKV arises from the TTS; nevertheless, quantitating this fraction is difficult because (as shown in Fig. 3 and related text) detubulation by osmotic shocks is incomplete, leading to uncertainties on the extent of TTS contributions that still remain in current records. Alternatively, as we have extensively demonstrated previously (Heiny et al., 1983; Ashcroft et al., 1985; DiFranco and Vergara, 2011; DiFranco et al., 2011a), the relative distribution of a given type of ion channel between the surface and TTS membrane compartments can be quantitatively determined by studying the effects that the current carried by these channels has on the TTS membrane potential, as assessed by potentiometric dyes. Fig. 7 shows, in an experiment done in the presence of 5 µM nifedipine (which has an effect similar to that of 0.5 µM isradipine) to avoid ICa contamination, that the features of di-8-ANEPPS transients are actually altered by the activation of IKV. Fig. 7 A demonstrates that small depolarizations (to less than –50 mV) do not significantly activate IKV (Fig. 7 C, black trace) and result in a step-like fluorescence signal (Fig. 7 A, black trace). This optical response is indistinguishable from that obtained from the same fiber after blocking IKV with TEA (Fig. 7 B, black trace). Likewise, optical transients recorded in response to hyperpolarizing pulses under control or TEA solutions could not be distinguished from each other (unpublished data). In contrast, depolarizations to 70 mV, for instance, which activate a large IKV (Fig. 7 C, orange trace), result in di-8-ANEPPS transients that are depressed early after the pulse onset and later rise in amplitude toward the end of the pulse (Fig. 7 A, orange trace), thus creating a notch (or valley) soon after the onset of the transient. These features are better noticed with an expanded timescale as in the inset of Fig. 7 A (same trace colors). It should be noted that the time course of the increase in fluorescence during the pulse closely resembles that of the decay phase of the IKV trace, clearly suggesting that the activation of the latter may be responsible for the kinetic features of the di-8-ANEPPS fluorescence transient. That this is the case is demonstrated by abolishing IKV with TEA. Under these passive conditions, the largest depolarization elicits a step-like fluorescence response (Fig. 7 B, orange trace). It should be noted that the transients recorded under control and passive conditions (Fig. 7, A and B, orange traces) differ not only in their kinetic features, but also in the final value attained at the end of the pulse; the control transient reaches a significantly smaller value as compared with that in the presence of TEA. These differences can be explained by the relatively large IKV still present at the end of the pulse. Comparative inspection of Fig. 7 (A and B) clearly shows that depolarizations to intermediate values between −60 and 110 mV are associated with fluorescence transients displaying intermediate characteristics between those seen in the black and orange traces. By comparing traces in Fig. 7 (A and B), it can be noticed that the activation of IKV also affects the properties of di-8-ANEPPS records after the end of the pulses. For example, the orange and blue traces in Fig. 7 A remain substantially elevated above baseline after the end of the voltage pulse. It is also clear that these postpulse elevations depend on the magnitude of IKV because it becomes smaller for smaller pulses (Fig. 7 A, red trace) and is effectively eliminated by blocking IKV with TEA (Fig. 7 B). An obvious interpretation of these latter results is that the activation of IKV results not only in a sustained flow of K ions across the membranes of the TTS but also leads to a substantial accumulation of K ions in the lumen of the T tubules. The concomitant changes in EK result in sustained depolarizations of the TTS membranes that are accurately reported in potentiometric dye records for a long time after the command pulses are turned off.
Figure 7.

Effects of IKV activation on di-8-ANEPPS transients. (A) di-8-ANEPPS transients recorded in response to 500-ms depolarizations from VH to membrane potentials spanning −90–70 mV in 40-mV steps. The inset shows the early phases of fluorescence transients in an expanded timescale. (B) Fluorescence transients elicited in the same fiber as in A, and using the same pulse protocol, after exchanging NMG-Tyrode with TEA-Tyrode. (C and D) The currents recorded simultaneously with the optical records in A and B are shown in C and D, respectively. For both fluorescence and current records, green, black, red, blue, and orange traces correspond to responses to depolarization to −90, −50, −10, 30, and 70 mV, respectively.
Model simulations of voltage changes in the TTS associated with the activation of KV channels
We have previously shown that when radial cable model predictions of TTS voltage changes are contrasted with potentiometric dye transients acquired simultaneously with the currents in voltage-clamped muscle fibers, there are enough constrains in the model to allow for quantitative evaluations of conductance distributions between the surface and TTS membrane compartments (Ashcroft et al., 1985; DiFranco and Vergara, 2011; DiFranco et al., 2011a). The specific model used here to simulate IKV and di-8-ANEPPS is described in detail in the Appendix. In brief, it is similar to that used previously for the cases of ClC-1 and NaV1.4 channels but includes instead the multistate models of two K channels (refined with data from osmotically treated fibers) and equations to account for K accumulation. In addition, the relative contribution of radial shells of TTS membrane to the experimental fluorescence transients takes into account a correction that incorporates the optical properties of the 100×, 1.4 numeral aperture objective used, as previously described (Kim and Vergara, 1998). The parameter values used in the model are given in Tables A1 and A2 of the Appendix. Fig. 8 (A and D) shows optical transients and IKV recorded in the absence of Ca blockers, respectively, in response to a family of 25-ms duration depolarization pulses. It can be seen in Fig. 8 A that the optical transients display the typical current-dependent attenuations during the pulse, as described in Fig. 7 A. It can also be observed that at the end of the pulse, the fluorescence signals decay slowly to baseline, in correlation with the relatively slow decaying tail current observed in Fig. 8 D. For the same depolarizations, the simulated mean TTS membrane potential (corrected for optical detection; see Appendix) is shown in Fig. 8 B; the simultaneous model predictions of IKV are shown in Fig. 8 E. It can be seen that the simulated traces quantitatively reproduce most of the kinetic features of both sets of experimental data during each of the depolarizations tested. Nonetheless, the model does not reproduce the slow decay of the optical transients and the tail currents seen experimentally after the end of the pulses; instead, model data show smaller tail currents and postpulse depolarizations. These discrepancies can be explained if we assume that optical records and tail currents result from residual IKIR (not sufficiently blocked by 5 mM Rb) and/or from unblocked Ca2+ currents that are not included in the model. Also, the model does not contemplate the existence of charge movement currents present in the experimental records. It is important to note that, although the model predicts substantive K accumulation in the lumen of the TTS occurring concomitantly with IKV during the pulses, these are not manifested as tail currents and/or postpulse depolarizations. Another noticeable difference between model predictions and data is that for large depolarizations, theoretical IKV traces decay slightly faster than the experimental ones.
Table A2.
Specific parameter values for passive radial cable
| Parameter | Symbol | Value |
| Radius (µm) | a | 20–30 |
| Specific capacitance TTS wall (μF/cm2) | CW | 0.9 |
| TTS lumen conductivity (NMG external) (mS/cm) | GL | 9.5 |
| TTS access resistance (Ωcm2) | Rs | 30–40 |
| Fraction of fiber volume occupied by the TTS | ρ | 0.004 |
| Volume to surface ratio of the TTS (cm) | ς | 1 × 10−6 |
| Tortuosity factor on the TTS | σ | 0.32 |
| K diffusion coefficient (cm2/s) | DK | 1 × 10−5 |
Figure 8.

Radial cable predictions of IKV and TTS membrane potential changes in intact fibers using multistate models of K channels. (A) di-8-ANEPPS transients recorded in the presence of NMG-Tyrode. The fiber was depolarized in 20-mV steps from −90 (VH) to 110 mV. (B) Model predictions of the mean TTS voltage (see Appendix) for the same conditions as in A, and assuming the same value of gKV for the sarcolemma and TTS membranes (0.97 mS/cm2). The model parameters for channels A and B are those in Table A1 of the Appendix. (C) Model predictions of the mean TTS voltage, assuming that gKV (3.88 mS/cm2) is located exclusively at the surface membrane. (D) IKV recorded simultaneously with the optical data in A. (E and F) The predicted currents corresponding to the optical simulations in B and C are shown in E and F, respectively. The same color code is used for electrical and optical data.
We may now ask the hypothetical question of what the predicted TTS voltage changes and currents would look like had all gKV been located at the sarcolemma. To this end, as shown in Fig. 8 F, we account reasonably for the total IKV by including a large gKV (4 mS/cm2) at the sarcolemma. However, as shown in Fig. 8 C, the mean TTS membrane potential changes display step-like responses for all depolarizations that are in sharp contrast with the observed optical responses. Notice that the predicted TTS transients do not rise instantaneously as a result of the relatively slow charging of the large capacitance of the TTS, but their amplitude depends linearly on the amplitude of the command pulse. It should be also observed that the responses shown in Fig. 8 C are quite similar to those obtained in the presence of TEA (e.g., Fig. 7 B). Finally, as expected, no tail currents are predicted by the model when currents do not flow across TTS membranes.
Quantitative evaluation of current-dependent attenuations on di-8-ANEPPS transients
The effects of IKV on TTS membrane potential changes, as assessed by di-8-ANEPPS transients, are further explored in Fig. 9, which shows superimposed optical records obtained in response to two pulse amplitudes (100 and 180 mV; Fig. 9, A and B, respectively) before (red traces) and after (black traces) blocking the corresponding IKV, as shown in Fig. 7 (C and D; same color coding), respectively. The IKV associated with the 100-mV depolarization generates an observable reduction in the magnitude of the optical trace (Fig. 9 A, red trace), which is larger early during the pulse than at the end of the pulse. For the larger pulse, the attenuating effects of the larger current (Fig. 9 D, red trace) on the optical record (Fig. 9 B, red trace) are more prominent early during the pulse, but they remained throughout the duration of the pulse. It should be noted that the magnitude of the depression displays a reversed pattern with respect to the decay of the corresponding IKV record. To quantitatively evaluate the relationship between IKV activation and the transient depression that is observed in di-8-ANEPPS transients (creating a valley or notch) relative to the step-like shapes seen in the absence of current with external TEA, we must implement a normalization procedure. Furthermore, this normalization is necessary to later compare the optical data with model predictions that compute the mean membrane potential changes in the TTS. To this end, we define attenuation as the percent difference between transients recorded before (Fig. 9, A and B, red traces) and after blocking IKV (Fig. 9, A and B, black traces), normalized by the amplitude of the largest transient acquired (e.g., the black trace in Fig. 9 B for this particular experiment). As in this experiment, the normalization is usually done with respect to signals elicited (after making the fibers electrically passive) by 200-mV pulses, a typical maximum stimulus. Attenuation traces calculated this way are shown in green in Fig. 9 (A and B; and in the inset of Fig. 9 B). Several features are salient: (a) their amplitude is significantly larger for the larger depolarization (∼9% for Fig. 9 A and ∼26% for Fig. 9 B), in correlation with the magnitude of the IKV records shown in Fig. 9 (C and D); (b) the time course of the attenuation traces mirrors that of the corresponding IKV records; namely, it has rising and decay phases comparable with those of the current traces; and (c) the attenuation trace reaches a peak with a visible delay with respect to the onset of the pulse, and its peak value (peak attenuation) closely coincides in time (within a millisecond) with the notch (minimum) observed in di-8-ANEPPS transients and with the peak of IKV. The close correlation between the peak of the attenuation trace and the notch in the optical transients itself is better visualized at the expanded timescale of the inset to Fig. 9 B. Altogether, these features suggest a kinetic correlation between the attenuation of optical records and IKV that could possibly be exploited to obtain a mechanistic understanding of their interdependence (see the following paragraph).
Figure 9.

Calculation of IKV-dependent attenuation of fluorescence transients. The attenuations in di-8-ANEPPS transients are exemplified with two pulse amplitudes (100 mV in A and 200 mV in B). (A) The red and black traces are the di-8-ANEPPS transients recorded in the presence of NMG-Tyrode and TEA-Tyrode, respectively. The green trace represents the attenuation (percentage), calculated by subtracting the red from the black traces and dividing the result by the amplitude of the black trace in B. (B) Same as in A, but for a depolarization to 110 mV. The inset shows the initial part of the records in an expended timescale. (C and D) The currents recorded simultaneously with the optical data in A and B are shown (with the same color code) in C and D, respectively. To reduce the increment of noise inherent to the subtraction operation, the attenuation traces (green traces) were digitally filtered.
Another manifestation of the current dependence of optical traces is illustrated in Fig. 10. The red circles in Fig. 10 A correspond to ΔF/F values of the notches in di-8-ANEPPS transients plotted as a function of membrane potential, whereas the black circles show the steady-state value of the transients in the presence of TEA. The depression in magnitude of the transients when the currents are active is seen as markedly sublinear voltage dependence; this dramatically contrasts with the quasilinear behavior in their magnitude when the currents are blocked. For comparison, the voltage dependence of the peak IKV is shown in Fig. 10 B. It can be seen that the departure from the quasilinearity of the optical transients in the absence of currents (Fig. 10 A, compare black with red circles) occurs precisely in the same voltage range at which IKV becomes activated (Fig. 10 B, compare black with red circles). This correlation is reinforced by the fact that the voltage dependence of the peak attenuation (Fig. 10 C) closely resembles that of the current in Fig. 10 B. Fig. 10 D illustrates comparatively (as in Fig. 10 A) the voltage dependence of notch values in di-8-ANEPPS transients before and after IKV block obtained from a population of 10 fibers (without using Ca channel blockers); these conditions allow us to obtain an assessment of the role of IKV with physiological values of K conductance. It must be noted that the activation of ICa, mainly in response to large depolarizations, does not have significant effects on these measurements because the notches in di-8-ANEPPS transients occur too early (within <8 ms) to be affected by currents that typically peak at >50 ms for the largest pulses used here (DiFranco et al., 2011b). Because of the larger peak IKV (Fig. 10 B), reaching mean values up to 960 µA/cm2 for depolarizations to 110 mV, the deviation of the notches (from the more linear behavior when the currents are blocked) is significantly more prominent. In this case, the mean ΔF/F at 110 mV is −0.16, significantly smaller than the −0.24 recorded in TEA. The close correspondence between the activation voltages for IKV and the departure from linearity of di-8-ANEPPS transients are clearly indicated by comparing the plots in Fig. 10 (D and E). However, this notion becomes further stressed by noticing the remarkable similarity between Fig. 10 F, which plots the voltage dependence of the peak attenuation, and the peak IKV plot in Fig. 10 E. The importance of the representation in the form of percent peak attenuation is that, as previously demonstrated (DiFranco and Vergara, 2011; DiFranco et al., 2011a) and discussed later here, it allows for the comparison between model predictions of TTS voltage changes (in millivolts) with experimental data (in Δfarads/farads).
Figure 10.

Voltage dependence of the peak attenuation. (A) Red circles: voltage dependence of the minimum (notch) ΔF/F value observed in di-8-ANEPPS transients from a fiber exposed to NMG-Tyrode containing 0.5 µM isradipine. Black circles: steady-state amplitude of di-8-ANEPPS transients recorded from the same fiber after replacing NMG-Tyrode with TEA-Tyrode. (B) Voltage dependence of peak IKV (red circles) and leak current (black circles) measured from the same fiber and conditions as in A. (C) Voltage dependence of the percent peak attenuation calculated from data in A. (D) Red circles: voltage dependence of the mean notch ΔF/F calculated from a family of di-8-ANEPPS transients in 10 fibers exposed to NMG-Tyrode (with no added Ca blocker). Black circles: steady-state amplitude of di-8-ANEPPS transients recorded from the same population of fibers after replacing NMG-Tyrode with TEA-Tyrode. (E) Voltage dependence of the mean peak IKV (circles) determined from the same fiber and conditions as in D. (F) Voltage dependence of the mean percent peak attenuation calculated from data in D. Error bars represent SEM.
Quantitative estimation of KV channel distribution between the surface and TTS membranes
The question that has yet to be addressed is what are the respective contributions of the sarcolemma and the TTS membrane KV currents to the total IKV records that are compatible with the optical data? Although the question is complex, we already ruled out the possibility that all KV channels are in the sarcolemma because this option is generally incompatible with all the optical data presented throughout the paper and is specifically addressed in Fig. 10 (C and F). The other extreme case, that all the KV channels are in the TTS, seems unrealistic because it is unlikely that the sarcolemma is entirely deprived of a rapid repolarization mechanism; also, model simulations suggest that K accumulation in the TTS lumen would be excessive if this was the case (unpublished data). It might seem that the excellent predictions by model simulations of the overall properties of optical and current records in the case of the fiber in Fig. 10 have already provided a definite answer about the issue of gKV distribution. However, this is not necessarily the case because we should explore other possibilities to encompass possible fiber-to-fiber variability in the data. Fortunately, our simultaneous measurements of the voltage dependence of peak attenuation and peak I-V curves from multiple fibers (Fig. 10, E and F) allow us to complete the task. Fig. 11 shows the results from model simulations using various (surface/TTS) gKV ratios to generate a range of attenuation profiles while constraining the total peak IKV dependence to be constant. The resulting simulated I-V plots (Fig. 11, D–F, stars) are superimposed with the mean experimental data (Fig. 11, D–F, circles) for comparison. Fig. 11 A shows the superposition of the experimental peak attenuation (black circles) with that predicted by model simulation assuming a 50:50 (gKV-S/gKV-TTS) ratio (red circles). It can be seen that this assumption provides a very adequate prediction of the maximum attenuation (∼30%; Fig. 11 A) and of the maximal peak IKV (∼960 µA/cm2; Fig. 11 D) obtained from the population of fibers. Notably, because the TTS represents the largest of the two membrane compartments in the muscle fiber, an equal distribution of gKV between the TTS and surface membranes results in a predicted maximal IKV arising from the TTS of ∼690 µA/cm2 (at 110 mV). This represents ∼72% of the total current at this membrane potential, whereas the surface membrane contributes the other ∼28% (∼270 µA/cm2). Fig. 11 (B and E) shows the results of simulations in which we limited the density of KV channels in the TTS membranes to be ∼43% of that at the surface membrane (70:30 ratio). It can be seen that this ratio predicts that the maximal TTS voltage attenuation (Fig. 11 B, green circles) is ∼23%, significantly smaller than the one observed experimentally. Thus, although the peak I-V plot of the total current (Fig. 11 E, stars) was similar to the one shown in Fig. 11 D, the TTS component (Fig. 11 E, closed triangles) with a maximum of ∼541 µA/cm2 seems to be too small to explain the observed peak attenuation in the mean population of fibers. In spite of this, this current component would still contribute a substantial proportion (>50%) to the total current recorded at the same membrane potential. These types of comparisons illustrate the great sensitivity afforded by the optical measurements to discriminate between channel distributions that would give the same total current but have very different balances between the surface and TTS current contributions. Without the information provided by the potentiometric dye records about the associated changes in TTS membrane potential, it would be impossible to decide which one is correct. Likewise, a 30:70 gKV-S/gKV-TTS ratio (Fig. 11, C and F) predicts a peak overshoot of ∼34% (Fig. 11 C, blue circles), which is larger than those recorded. In this case, the current component arising from the TTS (Fig. 11 F , closed triangles) has a maximum of ∼808 µA/cm2, which not only seems to be too large to explain the optical data but would also account for >84% of the total current at that potential. This is also incompatible with detubulation experiments. The suggestion from this analysis comparing experimental data and model simulations at a fixed total current is that the overall distribution of KV channels between the surface and TTS of mammalian skeletal muscle fibers is probably constrained within the range between 40:60 and 60:40 (gKV-S/gKV-TTS).
Figure 11.

Model predictions of overshoot and currents for various gKV distributions between the surface and TTS membranes. (A–C) Voltage-dependent peak attenuations calculated from model predictions of the TTS membrane potential using the following gKV-S/gKV-TTS ratios: 50:50 (A, red circles), 70:30 (B, green circles), and 30:70 (C, blue circles). The values of gKV were 1.26 mS/cm2 for both surface and TTS membranes in A; 2.03 mS/cm2 at the surface membrane and 0.87 mS/cm2 at the TTS membranes in B; and 0.71 mS/cm2 at the surface membrane and 1.66 mS/cm2 at the TTS membranes in C. In every panel, the black circles are the mean experimental attenuation values shown in Fig. 10 F. (D–F) The mean peak I-V plot shown in Fig. 10 E (black circles) is superimposed with model peak current predictions for 50:50 (A, red circles), 70:30 (B, green circles), and 30:70 (C, blue circles) gKV-S/gKV-TTS ratios, respectively. In every panel, stars represent the total current, closed triangles represent the TTS current component, and open triangles represent the surface membrane current component. The fiber radius used in model simulations was the mean for all the experiments (24 µm), and Rs was 40 Ωcm2. The rest of the model’s parameters are listed in Tables A1 and A2 of the Appendix. Error bars represent SEM.
DISCUSSION
Though it is well accepted that IKV plays a crucial role in muscle excitability by being the principal component responsible for the repolarization phase of the AP, there is a great shortage of information in the literature about the detailed properties of gKV in mammalian skeletal muscle fibers. The paucity is even more striking for mouse muscle fibers, which is unfortunate because this is the animal choice for models of human diseases, including mutations that affect KV channels. Furthermore, up until now, there was uncertainty about the presence of gKV in the TTS, which is a very relevant issue regarding the possibility that K accumulation in the TTS lumen during bouts of sustained activity would possibly lead to depolarization of the muscle fibers, eventually compromising their excitability. This paper, by providing a quantitative description of the properties of gKV, including the involvement of TTS, not only fills in gaps of understanding about the physiological properties of this conductive pathway in murine skeletal muscle fibers but also opens new avenues for the investigation of its alterations in diseased conditions. Experience accumulated during the last decade suggests that short muscle fibers isolated from the toes of adult mice are practical and reliable preparations for electrophysiological studies, using a two-microelectrode voltage-clamp technique (Woods et al., 2004, 2005; Ursu et al., 2005; Lueck et al., 2010; DiFranco et al., 2011a,b; Fu et al., 2011). We have recently demonstrated that this classical electrophysiological approach can be combined with optical measurements of TTS membrane potential changes and model simulations to determine, with minimal invasion, the quantitative allocation of particular conductances in the TTS and surface membrane (DiFranco and Vergara, 2011; DiFranco et al., 2011a). In this work, we use this approach to investigate overall properties of IKV in muscle.
IKV in intact FDB muscle fibers
The voltage- and time-dependent IKV recorded from intact and detubulated fibers display all the canonical features that traditionally have been ascribed to KV channels: a distinct activation potential, outward rectification, voltage-dependent delayed onset, voltage-dependent rate of rise, and incomplete inactivation. The amplitude of IKV recorded from intact fibers was large; for instance, for depolarizations similar to those reached at the peak of the AP (∼45 mV), peak IKV can reach values of ∼500 µA/cm2, which is smaller, but within range, of the maximal Na current for this preparation (unpublished data). No quantitative comparative measurements of IKV have been provided for mouse muscle fibers because most previous work was performed using either on-cell or excised-patch configuration of patch clamp (Brinkmeier et al., 1991; Hocherman and Bezanilla, 1996) or whole-cell patch clamp; the latter method yielded K currents estimated to be ∼10% of the magnitude of those reported in this paper (Brinkmeier et al., 1991). In contrast, our mean peak IKV values of up to 990 µA/cm2 at 110 mV are in reasonable agreement with results reported for rat muscle fibers using other voltage-clamp approaches (Duval and Léoty, 1980; Pappone, 1980; Beam and Donaldson, 1983a). Also, the main kinetic features of our IKV records are similar to those recorded from fast muscle fibers of mouse and rat, showing a single peak (Duval and Léoty, 1980) and incomplete inactivation during 500-ms pulses (Duval and Léoty, 1980; Beam and Donaldson, 1983a; Brinkmeier et al., 1991; Hocherman and Bezanilla, 1996). A relatively large fraction of IKV does not inactivate even for 1-s pulses (unpublished data), but we have observed that the inactivation process is cumulative. Furthermore, we determined that a resting period of 7–10 s was sufficient to avoid this phenomenon; consequently, the preparation was allowed to rest for at least this long between pulses during the acquisition of a family of records. More importantly, the decay kinetics of IKV records is a complex process involving at least two time constants; although this may be suggestive of more than one channel contribution, we preferred to use a more direct approach to investigate this issue. Interestingly, a two–time constant decay was also found in ensemble currents of membrane patches from mouse fibers (Hocherman and Bezanilla, 1996).
A complication found in our experiments was the contamination by ICa in IKV records from intact fibers and its eventual elimination by the use of typical Ca2+ channel blockers. In reality, ICa’s are small relative to the magnitude of IKV, but they do manifest as small downward deflections during the decay phases of IKV records elicited by depolarizations larger than 140 mV and longer than 100 ms (Fig. 2 A). Prominent ICa tails also dominate the current records after the end of the pulses. It could have been argued that removal of Ca2+ ions from the extracellular solution would have eliminated ICa contaminations. However, we avoided this approach mainly for two reasons: the cells became leaky, probably because of deterioration of the membrane seal around the microelectrode tips, and the voltage dependence of K channels was expected to be substantially shifted as a result of changes in surface charge screening (Hille et al., 1975; DiFranco et al., 2011b). We have found that Mg2+ is a poor replacement of Ca2+ toward these ends. A reasonable replacement for Ca2+ is Ba2+, but it could not be used because it blocked IKV and is permeable through Ca2+ channels. When we tried large concentrations of external Cd2+ (>1 mM), we found that the leak currents increased to levels incompatible with the experiments (unpublished data); also, this manipulation resulted in a significant rightward shift of the voltage dependence of IKV. For these reasons, we took the approach of testing dihydropyridine derivatives (e.g., nifedipine and isradipine) and other organic blockers of CaV1.1. Unfortunately, we found that, as extensively described in Results, these drugs also block IKV.
By analogy with other inactivating currents, we studied the voltage dependence of IKV by plotting the peak currents in response to every pulse as a function of the membrane potential reached. We found that peak IKV plots follow the typical quasilinear dependence for membrane potentials more positive than −10 mV (Fig. 1 D). Assuming a linear driving force (V-EK), we calculated peak gKV and studied its voltage dependence. A novel observation is that the peak gKV versus membrane voltage graphs cannot be accurately predicted by a single Boltzmann equation; there is a marked inflection at ∼25 mV. Instead, the data could be well fitted by double Boltzmann equations with distinct parameters (Fig. 1, B and D; and Fig. 2 D). The suggestion from these fits is that IKV currents contain the contributions of two KV channels, one (channel A) with low threshold for activation and steep voltage dependence and the second with a high activation threshold and less steep voltage dependence (channel B). The double Boltzmann fit of peak gKV plots from intact fibers under control conditions (Fig. 1 D) suggests a channel A/channel B amplitude ratio of ∼0.4 at the time the current peaks. As mentioned previously, peak gKV calculated from peak currents are not significantly distorted by ICa contaminations because this current activates very slowly (DiFranco et al., 2011b); for example, at 4 ms, when IKV peaks for a pulse to 110 mV (Fig. 1 A), we have measured that ICa is not larger than 8 µA/cm2 (not depicted), which is negligible compared with the >900-µA/cm2 peak IKV. It must be noted that, although the data are well fitted with double Boltzmann equations, we cannot discard the possibility that more than two channels actually contribute to IKV. Nevertheless, our findings in fibers isolated from the FDB muscle, a typical fast type in the mouse (González et al., 2000), differ from reports that in fast rat fibers, K currents are contributed by one channel type (Duval and Léoty, 1980; Beam and Donaldson, 1983a); instead, they are compatible with the case in soleus (slow) fibers that suggest two components (Duval and Léoty, 1980).
IKV from osmotically treated fibers
Our laboratory and another group have recently shown that the osmotic shock treatment, by disconnecting a significant fraction of the TTS from the surface membrane, can be used to generate a simpler muscle model preparation to identify the properties of the Na conductance under voltage-clamp conditions (DiFranco and Vergara, 2011; Fu et al., 2011). The results presented here strongly reinforce this concept, but for KV channels. It must be kept in mind, however, that in our hands, the formamide osmotic shock procedure yields fibers from within the same batch treatment with variable degrees of detubulation; furthermore, even within the same fiber, there are zones showing more TTS stumps than others (unpublished data). We also found that, judging by the capacitance and di-8-ANEPPS staining, detubulation was never complete. For these reasons, we routinely ascertained that every electrophysiological experiment was performed in fibers affording the best degree of TTS disconnection. For this, we carefully selected fibers showing di-8-ANEPPS staining only at the periphery throughout most of their length (Fig. 3) and with capacitances <2.6 µF/cm2. In the best cases, peripheral rings of TTS remnants spanning 2–3 µm in thickness were still observed, a result that explains why even this selected population of fibers had a mean capacitance of 2.25 µF/cm2, which is significantly larger than the theoretical ideal of 0.9 µF/cm2 ascribed to the sarcolemma (see Appendix).
A clear effect of the osmotic shock treatment is a significant reduction of ∼40% (P < 0.05) in the peak IKV and gKV values compared with those in intact fibers, with no apparent impairment of other electrical features. This result unquestionably indicates that a fraction of KV channels is located in regions of the TTS disconnected by the procedure. What it does not necessarily imply is that only 40% of the current arises from the TTS compartment; this will be discussed in the section Quantitative evaluation of the relationship between attenuation and currents. Another difference between IKV recorded from control and treated fibers is the significant reduction in the tail currents in the latter, which is expected because CaV1.1 channels are mostly located in the TTS. Aside from these differences, we found that IKV from osmotically shocked fibers (Figs. 4 and 5) display features similar to those from intact fibers (Figs. 1 and 2), which readily indicates that at least a significant portion of the decay in the IKV records is a bona fide inactivation process; namely, IKV in fibers with a significantly reduced diffusional volume (TTS lumen), and thus with a reduced possibility for K accumulation, still display substantial decays. These results are concurrent with those obtained using the on-cell configuration of patch clamp (Brinkmeier et al., 1991) in which currents recorded from surface patches of membrane still show inactivation with similar kinetics as those shown here. In addition, the properties of IKV in osmotically treated fibers seem to be more compatible with those described for fibers from the soleus than the iliacus (fast) muscles in the rat (Duval and Léoty, 1980).
A two-channel model of IKV in FDB fibers
The voltage dependence of peak gKV from detubulated fibers also provided valuable information. First, as was the case in control fibers, double Boltzmann equations were required to appropriately fit the data. This is an interesting result because it demonstrates that the two putative channels proposed to explain the data in intact fibers are not exclusively located in the TTS membranes, but instead they may be ubiquitously distributed. In fact, we took advantage of the results in these preparations to obtain the typical behavior of KV channels in an ideal single compartment model. Although this is only an approximation, we reasoned (for reasons already discussed) that gKV in osmotically treated fibers arise from the sarcolemma and peripheral regions of the TTS, where the membrane potential is much closer to that controlled by the voltage-clamp system.
Altogether, the two-channel model described in the Appendix, by quantitatively predicting the voltage-dependent kinetic features of data from treated fibers (Fig. 5), allows us to gain confidence in the proposal that, in skeletal muscle fibers, IKV must emerge from at least two channel contributions, one of which does not inactivate completely. To predict this behavior, we used a general sequential model with the traditional four closed and one open state followed by open channel inactivation processes as described elsewhere (DeCoursey, 1990; Demo and Yellen, 1991; Hoshi et al., 1991; Bett et al., 2011). Although it was conceivable that both channels displayed incomplete (N and/or C) inactivation, the adjustment of the model to the data suggested (in all fibers analyzed) that the scheme illustrated in Fig. 5 (channel A: inactivating, low threshold, and steep voltage dependence; and channel B: incompletely inactivating) was the simplest and strongest. As expected, the voltage dependence of peak gkV in channels A and B provides a mechanistic explanation for the detailed features of peak gKV plots (requiring double Boltzmann fits) both in osmotically treated and intact fibers.
Experiments in osmotically treated fibers also demonstrated that, as in intact fibers, IKV is blocked by the organic Ca channel blockers isradipine and nifedipine (unpublished data). The data suggest that both putative channels (channels A and B) are sensitive to isradipine because double Boltzmann fits were required when fitting peak gKV data in intact and osmotically treated fibers. Nevertheless, as shown by the data in Fig. 2 D and suggested in Fig. 4 D, channel A seems to have an increased sensitivity to the drug. Although further studies will be required to characterize the actual blockage of IKV by these agents and their specific effects on every channel’s contribution to IKV records, our results from both intact and osmotically treated fibers bear important warnings regarding the use of dihydropyridines when studying the properties of KV channels in skeletal muscle and for potential side effects concomitant to their routine use to treat patients with cardiac problems.
The value of an empirical model capable of predicting the kinetic properties of IKV is double; as stated previously, it provides a mechanistic view of what could be reasonable contributions of individual channels to IKV records. In addition, from a practical point of view, it allows us to study the potential implications that the presence of gKV has on the membrane potential of the TTS when the K channel model equations are incorporated into the radial cable model as described in the Appendix.
Western blotting
With the idea in mind that at least two functional KV channels may be contributing to IKV records, we thought it necessary to complement the electrophysiological data with biochemical evidence on the actual expression of KV channels in FDB and interosseous muscles. Although our results are not exhaustive (more channels could be expressed), the Western blot results presented in Fig. 6 convincingly show that the muscles used in electrophysiological studies express both KV1.4 and KV3.4. In fact, we also preliminarily tested the expression KV1.5 and found it in crude extracts (unpublished data). We specifically focused on the expression of KV1.4 and KV3.4 because it is generally suggested that they might be abundant in skeletal muscle; nevertheless, our results seem to be the first proof that they are actually expressed in significant quantities in muscles from the mouse. Several lines of evidence support this claim regarding KV1.4: (a) two different antibodies raised against epitopes at the N terminus (75–010) and the C terminus (APC-007) of this channel identified a unique band in both crude extracts and microsomal preparations from muscle; (b) the specificity of one of these antibodies (APC-007) was previously demonstrated in the heart by comparing results from wild-type and KV1.4-null mice (Guo et al., 2000); (c) our results show that APC-007 recognizes the same ∼97-kD band in skeletal muscle as that reported for wild-type heart muscle (Guo et al., 2000); (d) the 75–010 antibody recognizes an epitope at the N terminus of KV1.4 that is exclusive to this protein, significantly reducing the chance of cross-reactivity with α subunits of the KV1 family (Juiz et al., 2000); this was demonstrated by Neuromab); and (e) the ∼97-kD band detected by both antibodies in muscle preparations coincides with one of the two bands reported in mouse hippocampus extracts, a tissue in which the expression of KV1.4 is generally established (Sheng et al., 1992, 1993; Maletic-Savatic et al., 1995; Veh et al., 1995; Rhodes et al., 1997; Wickenden et al., 1999; Juiz et al., 2000; Monaghan et al., 2001). The ∼97 kD reported in this paper for KV1.4 is not only consistent with that estimated from Western blots in other tissues but is also larger than that strictly deduced from its amino acid sequence (74 kD); this discrepancy is likely caused by in vivo glycosylation of the protein (Sheng et al., 1992; Veh et al., 1995; Juiz et al., 2000).
The evidence for the expression of KV3.4 in skeletal muscle is not as persuasive as for KV1.4 but is sustained mostly by the specificity of the monoclonal antibody 75–112, which has been also demonstrated not to cross react with other KV channels (Neuromab). As an additional evidence of the specificity of this antibody, it has been used to detect changes in the expression of KV3.4 in hippocampus of epileptic rats (Pacheco Otalora et al., 2011). Interestingly, in agreement with our results, a band of ∼95 kD has also been identified (by another antibody) in rat sartorius muscle preparations (Abbott et al., 2001). As with KV1.4, the estimated molecular mass of KV3.4 is larger than the 70 kD predicted from its amino acid sequence (Schröter et al., 1991); the difference has been explained previously on the basis of extensive glycosylation (Cartwright et al., 2007).
Although the biochemical evidence is solid, we do not necessarily imply that there is a strict correspondence between the properties of the two channels (A and B) predicted from our electrophysiological data and those of the channels identified by Western blotting. Against this possibility, it has been shown that when KV1.4 and KV3.4 are expressed in heterologous systems, the recorded currents display properties not identical to either channel A or B (Ruppersberg et al., 1990; Po et al., 1993; Abbott et al., 2001). However, known factors may contribute to the discrepancy: (a) the expression of channels in heterologous systems may result in functional properties widely different from those seen in the muscle fibers, as already shown for the NaV1.4 channel (DiFranco and Vergara, 2011; Fu et al., 2011); and (b) it has been shown that KV channels form heterotetramers (Ruppersberg et al., 1990; Po et al., 1993), associate with regulatory subunits (Abbott et al., 2001; Pongs and Schwarz, 2010), and are under tight regulation in vivo (Covarrubias et al., 1994). As a consequence, it is difficult to predict the voltage dependence, kinetics, and pharmacology of native KV channels in fully developed muscle fibers from data obtained in expression systems (Ruppersberg et al., 1990; Po et al., 1993; Hashimoto et al., 2000; Abbott et al., 2001). In fact, that this may be the case is suggested from expression experiments combining more than one KV isoform and/or regulatory subunit (Lee et al., 1996; Grunnet et al., 2003). Overall, these limitations emphasize that the full characterization of IKV while preserving the normal cellular context, e.g., under the structural and regulatory conditions as they happen in normal cells, represents a valuable approach.
di-8-ANEPPS transients’ association with IKV
As shown in Fig. 7, depolarizations able to activate prominent IKV are associated with the appearance of prominent depressions in di-8-ANEPPS transients. The overall reduction in magnitude of optical signals in the presence of IKV with respect to those when the currents are blocked by TEA illustrates that activation of gKV in the TTS membranes leads to a mean decrease of the TTS membrane potential changes with respect to those imposed by the voltage clamp at the surface membrane. Similar effects were reported previously for amphibian muscle fibers as a result of the activation of KIR currents (Heiny et al., 1983; Ashcroft et al., 1985) and for mammalian fibers in association with ClC-1 currents (DiFranco et al., 2011a), but in response to hyperpolarizing pulses. It is important to note that the kinetic features of di-8-ANNEPS transients associated with the activation of IKV reported here are also qualitatively different from those reported for the other two conductances; whereas the maximal depression in those cases were attained almost instantaneously after the onset of each hyperpolarizing pulse, the depression maxima associated with IKV take time to manifest themselves. As illustrated in the inset to Fig. 7 A, after the onset of depolarizing pulses, di-8-ANEPPS transients initially raised to a peak, decayed rapidly to a minimum as a result of the delayed activation of IKV, and subsequently grew progressively, mainly because of the inactivation of IKV. Altogether, the contrasting kinetic features between di-8-ANEPPS transients reported here, with respect to those associated with the other two conductances, are in agreement with differences between the time- and voltage-dependent activation of gKV with respect to constitutively open conductances.
Probably the most important finding of the present work is precisely that, as illustrated in Fig. 8, the overall properties of di-8-ANEPPS transients, including the time- and voltage-dependent depression associated with the presence of IKV (Fig. 8 A), can be accurately predicted by model simulations (Fig. 8 B) provided that a significant fraction of gKV is included in the TTS membranes (the contrasting case when no channels are placed in the TTS is illustrated in Fig. 8 D). As shown in the Appendix, the radial cable model equations, stipulating the existence of an access resistance (Rs) in series with the TTS, were modified from the original equations of Adrian and co-workers (Adrian et al., 1969; Adrian and Peachey, 1973) to include time- and voltage-dependent KV channels (also described in the Appendix), similar to what was done before for nonregenerative ionic conductances (Heiny et al., 1983; Ashcroft et al., 1985; DiFranco et al., 2011a) and recently with the Na conductance (DiFranco and Vergara, 2011). In the Appendix, we explain how the radial cable elements of the TTS include KV channels with properties extensively described in Fig. 5. In addition, the model contemplates a diffusion equation, modified from Barry and Adrian (1973; Friedrich et al., 2001), to calculate K concentration changes in the lumen of the T tubules resulting from fluxes through KV channels at each radial segment of the TTS. Both the radial cable model and diffusion equations were integrated simultaneously using conventional numerical methods (see Appendix).
Quantitative evaluation of the relationship between attenuation and currents
Similar to our analysis of the attenuation of di-8-ANEPPS in the presence of large ClC-1 chloride currents (DiFranco et al., 2011a), we developed in this paper a method to quantitatively assess the properties of the attenuation in di-8-ANEPPS transients recorded in the presence of relatively large IKV (Figs. 9 and 10). The intrinsic normalization involved in the calculation of the percentage attenuation from experimental records allowed for quantitative comparisons with model predictions of mean TTS potential changes because they were normalized following the same definition. Thus, our results demonstrating the voltage dependence of the experimental peak attenuation was not only to correlate this property with the activation of peak IKV, as illustrated in Fig. 10, but, most importantly, to allow for comparison with radial cable model predictions of the attenuation, as illustrated in Fig. 11. Interestingly, by keeping the IKV predicted by the model constant and varying the gKV-S/gKV-TTS ratios, we could examine plausible scenarios that ultimately allowed us to quantitatively establish that the distribution of gKV in the sarcolemma and TTS membranes (gKV-S/gKV-TTS) that is most compatible with the experimental data must be within a range between 40:60 and 60:40. It is remarkable that this range of channel densities (per unit surface membrane of each compartment), including, of course, the likely possibility that gKV is equally distributed in the surface and TTS membranes, is quite similar to that found previously in our laboratory for ClC-1 (DiFranco et al., 2011a) and NaV1.4 (DiFranco and Vergara, 2011).
Another important inference from the results shown in Fig. 11 is that the magnitude of attenuations seen experimentally can only result if the contribution from the TTS represents a large fraction of the total IKV recorded in intact fibers. In fact, for the very plausible case analyzed, >70% of the total IKV seemingly arises from the TTS. It is important to mention here that the results from these analyses depend on the value of the access resistance to the TTS (Rs) used in model simulations. In this study, we used Rs = 40 Ωcm2, which is the same used previously regarding the analysis of ClC-1 distribution (DiFranco et al., 2011a) but is significantly smaller than the 120–150 Ωcm2 suggested for mechanically isolated amphibian fibers (Adrian and Peachey, 1973; Kim and Vergara, 1998). However, this is slightly larger than the preferred values of 10–20 Ωcm2 recently reported in our analysis of the overshoots associated with the activation of the Na conductance (DiFranco and Vergara, 2011). The reason for the discrepancy with the latter work is that the experiments reported here were all done using NMG as the main cation, which affords a reduced conductivity (to 0.95 Ω/cm) in the external solution, likely increasing Rs.
The question that may arise from our results, and that we can answer now, is how can the ∼56% reduction in capacitance afforded by osmotic treatment result in ∼40% reduction in the mean maximum peak IKV? The answer is given by the model. When gKV is equally distributed between the sarcolemma and the TTS, the majority of the IKV arises from peripheral segments of the TTS. In osmotically treated fibers, these TTS peripheral segments that remain connected undergo voltage changes more similar to the voltage-clamp pulses at the surface membrane and thus contribute in larger proportions to IKV than those of more internal segments of the radial network whose voltage changes are significantly attenuated with respect to the command pulses. di-8-ANEPPS transients recorded from optimally detubulated fibers (unpublished data) indicate that voltage changes in peripheral TTS remnants, together with the sarcolemma, show observable deviations from those commanded by the voltage clamp. These are significantly less pronounced than those observed for the entire TTS network in normal fibers but more than what would be expected for the sarcolemma itself. This is in contrast with the membrane capacitance, which, being measured under passive conditions, reflects more precisely the proportion of the TTS that remains connected.
K accumulation in the lumen of the TTS
As explained previously (also see Appendix), our model incorporates explicit equations for K diffusion in the TTS lumen; this is a highly relevant parameter for understanding the kinetics of decay of IKV records and the repolarization of the TTS during and after long-lasting depolarizations, respectively. We plan to take full advantage of this feature of the model in future experimentation and analysis, but for the purposes of this paper, the changes in the luminal K concentration do not play a considerable role. Nevertheless, as shown in Fig. 8 (A and D), inward tail currents and optical transients slowly returning to baseline are clearly observed after the end of the pulses. Both of these observations could have resulted from K accumulation in the lumen of the TTS and/or from the presence of unblocked Ca2+ channels. Interestingly, model calculations predict significant [K] accumulation in the lumen of the TTS, which, for the largest pulse (to 110 mV) in Fig. 8 D, exceeded 27 mM in the inner elements of the TTS; however, the simulations paradoxically failed to reproduce the slow return to baseline seen experimentally in optical transients (Fig. 8 A). An obvious explanation for this model limitation would be that it lacks Ca2+ conductance. However, the question that has yet to be answered is, why doesn’t the model report the significant increase in luminal [K]? The reason for this stems from the rapid closing of gKV after the end of the pulses and the absence of gKIR in the model. Namely, without gKIR and gKV, the increase in luminal [K] is not being “sensed” by a significant K permeability. The aforementioned reasoning consistently explains the absence of tail currents in model simulations (Fig. 8 E). In contrast, the experimental current records (Fig. 8 D) report small but significant tail currents that could arise from unblocked Ca2+ channels and/or incompletely blocked (by 5 mM Rb) KIR channels; as stated previously, the current model does not reproduce either of them. It is important to mention that changes in luminal [K] do not significantly affect the analysis followed to determine the gKV distribution between the sarcolemma and the TTS (as outlined in Fig. 11) because at the peak of IKV records, the luminal [K] changed <5 mM for the largest currents, even in the innermost radial elements of the TTS.
Although several model parameters were adjusted in this paper to explain the various features of the experimental data, the one that needed the most critical attention to predict simultaneously the properties of IKV and the optical data was the relative partitioning of gKV between surface and TTS membranes. An important merit of the current work is precisely that we succeeded in providing a quantitative assessment of the narrow range in the relative distributions of KV channels between these two membrane compartments that can explain the experimental data. This is a fundamental contribution for the future assessment of the electrical properties of skeletal muscle fibers in normal and diseased animals.
APPENDIX
Model equations for IKV as they are observed in osmotically treated fibers
We assume that the currents recorded from osmotically treated fibers arise from surface and peripheral TTS membranes that are under voltage-clamp control. Thus, IKV records acquired from these preparations constitute a reasonable idealization of currents carried by skeletal muscle KV channels as expressed in situ. As suggested in the body of this paper, we must consider the contributions from at least two channel types to account for the properties of IKV records. We call them channels A and B, and we assume that both of them display the general characteristics of Shaker (and/or Sham) K channels; namely, they are multiple-state channels that undergo N- and C-type inactivation processes. The analysis of data obtained in osmotically treated fiber suggests that channel A undergoes complete inactivation, whereas channel B behaves as a more typical N- and C-inactivating channel. Thus, for simplicity, they were modeled as follows.
Channel A.
 |
(SCHEME A1) |
where C0, C1, C2, C3, and O are closed and open states of a typical HH K channel (Hodgkin and Huxley, 1952b) and IN is a terminal inactivated state. The system of differential equations that permit the calculation of the contribution of channel A to the total IKV(V,t) records are
| (A1) |
The rate constants α and β had typical voltage dependences given by the generic formulas
| (A2) |
where ΦA and ϕA are the open channel (voltage independent) forward and reverse inactivation kinetic rate constants, respectively.
Channel B.
Sequential N- and C-type inactivation.
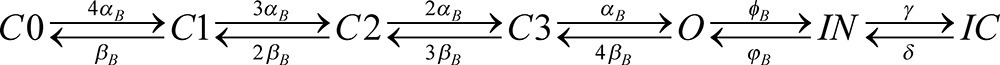 |
(SCHEME A2) |
The system of differential equations that permit the calculation of the contribution of channel B to IKV(V,t) records are
| (A3) |
where C0, C1, C2, C3, O, and IN are the same as for channel A and IC is a C-inactivated state, connected with IN through the voltage-independent rate constants γ and δ. In this case, the rate constants α and β voltage dependences are given by
| (A4) |
Integration of differential equations
Following the HH approach for K channel activation, the initial conditions (t = 0) were established assuming that the open probability at the resting potential (VH) obeys the typical equilibrium equation and that the inactivated states are not populated. Thus,
| (A5) |
IN(0) = 0 for channels A and B; IC(0) = 0 for channel B. In Eq. A5, αX and βX are obtained from Eqs. A2 and A4 for channels A and B, respectively, evaluated at V = VH. From these equations, the probability of all the closed states at rest can be calculated as
The total IKV records were assumed to be generated by the independent contributions of channels A and B; thus,
| (A6) |
Henceforth, the system of simultaneous differential equations was numerically integrated using a fourth order Runge-Kutta algorithm either in a FORTRAN program that calculates the radial cable model predictions or in a Berkeley Madonna (Macey and Oster, version 8.3.18, 2011) program. The parameters , , ΦX, and ϕX (for channels A and B) and γB and δB (for channel B) were determined by simultaneously fitting IKV records obtained at several voltages from osmotically treated fibers with the aid of a Levenberg-Marquardt minimization algorithm least-square fitting routine included in Berkeley Madonna. Mean values are listed in Table A1.
Radial cable model equations for the TTS of mammalian skeletal muscle fibers, including potassium conductance and ion diffusion
The radial cable model equations are essentially the same as those described in detail previously (DiFranco and Vergara, 2011; DiFranco et al., 2011a), which in turn followed the nomenclature published elsewhere (Adrian et al., 1969; Ashcroft et al., 1985; Kim and Vergara, 1998; DiFranco et al., 2007) and the assumption (Adrian and Peachey, 1973) that the lumen of the TTS is separated from the extracellular fluid by an access resistance (Rs, in Ω centimeters squared). The partial differential equation that governs the radial- (r) and time (t)-dependent changes in T tubule membrane potential (u(r,t)) in response to voltage changes at the external boundary is (Adrian et al., 1969)
| (A7) |
where a is the radius of the muscle fiber, R = r/a, T = t/( a2), and ν = a In these equations, the parameters and are the capacitance (in microfarads/centimeters cubed) and conductance (in Siemens/centimeters cubed) of the tubular membrane per unit volume of muscle fiber, respectively ( and ). Also, is the effective radial conductivity (, in Siemens/centimeters). Eq. A1 must be numerically integrated to predict the voltage of TTS cable elements when nonlinear conductances, in parallel with capacitive elements (Adrian and Peachey, 1973; Heiny et al., 1983; Ashcroft et al., 1985; DiFranco and Vergara, 2011; DiFranco et al., 2011a), are assumed to be present. To this end, we replace the term v2u in Eq. A1 with the more general term
where IW(u,r,t) is a generalized current normalized per volume of fiber. Keeping the same definitions for R and T, Eq. A1 is transformed into
| (A8) |
For the simulations in this paper, we assumed that IW(u,R,T) is contributed by a residual (almost negligible) leak current (DiFranco et al., 2011a) but primarily by the flow of K ions across the TTS walls through voltage-dependent K channels generally characterized by gKV. Thus,
| (A9) |
where Ileak(u) and IK(u,R,T) are the leak and KV current per centimeters squared of TTS membrane, respectively. In this paper, we make the assumption that Ileak(u) = gleak(u − VH), where gleak is constant (see Table 1). Also, IKV in Eq. A9 corresponds to the sum of the current contributions of channels A and B as recorded from osmotically treated fibers and summarily calculated from Eq. A6. In addition, unless otherwise noted, we make the further assumption that the proportion of channel A and B contributions is the same in the TTS, as it was obtained in detubulated fibers, an assumption which may or may not be fully justified.
Numerical integration of model equations
TTS voltage.
The TTS cable is assumed to be made of n = 60 radial shells, sealed at the center of the muscle fiber. As described previously (Kim and Vergara, 1998; DiFranco et al., 2007, 2011a,b), at a given time j, the finite differences approximation of the partial differential equation of the T tubular voltage (Eq. A8), while using an implicit Crank-Nicolson algorithm (Crank, 1975; Gerald, 1978), yields the following equation for an arbitrary annulus i:
| (A10) |
where is the current per unit fiber volume (calculated from Eq. A9) flowing through the T tubular element at shell i and at the time interval j during the numerical integration process. Eq. A10 is a recursive formula allowing the calculation of at a time interval δT while knowing . The system of tridiagonal coefficient matrices was solved using an LU (lower and upper) decomposition algorithm (Gerald, 1978). The integration of Eq. A6, updating the value of IKV for every cable element and for the surface membrane, was performed with a fourth order Runge-Kutta algorithm at every time step. The values of the general cable parameters used for the simulations shown in the paper are summarized in Table A2. Specific parameters are given in the figure legends.
Potassium diffusion in the TTS lumen
Changes in luminal potassium concentration in the TTS ([K]o) occurring in response to the current flow across the T tubule walls were calculated from simultaneous integration of the diffusion equation (Barry and Adrian, 1973; Friedrich et al., 2001)
| (A11) |
where DK is the diffusion coefficient of potassium ions in the lumen of the TTS (in centimeters squared/seconds), F is the Faraday constant, IK is the total potassium current (both components added) per centimeter cubed of fiber, and ρ is the fractional volume of the TTS. Using the dimensionless variables R = r/a and τ = σDKt/a2, this equation becomes
| (A12) |
Potassium concentration in the lumen of the TTS.
The finite difference approximation and Crank-Nicolson algorithm used for the integration of Eq. A12 to calculate the luminal K concentration at every shell i and at time interval j follow an analogous procedure to that described for the TTS voltage calculations. For simplicity, let’s denote [K]o= c. Thus, at a given time j, the finite differences approximation of the partial differential equation for [K]o in the TTS (Eq. A12) is
where is the finite difference representation of the luminal K concentration at radial position i at time j and δR is the normalized distance between shells. Also,
| (A13) |
where is the current per unit fiber volume (calculated through the T tubular element at shell i and at the time interval j during the numerical integration process). Eqn. A13 allows us to establish recurrence using the Crank-Nicolson algorithm. If we denote
| (A14) |
At the outside boundary (i = n), we apply Fick’s law to the T tubules’ opening:
From Crank (1975), for the inside (closed) boundary (i = 0),
Eq. A14 is a recursive formula allowing, together with the respective equations of the boundary conditions, for the calculation of at a time interval δT while knowing . The system of tridiagonal coefficient matrices were solved using an LU decomposition algorithm as described for the TTS voltage.
The values of the general cable and K diffusion parameters used for the simulations shown in the paper are summarized in Table A2. Specific conductance parameters are given in the figure legends.
Total current and optical signals
The total ionic currents calculated from the integration of the radial cable equations at each radial cable element of the TTS was the sum of two contributions: (1) a surface membrane component, calculated from the direct numerical integration of Eq. A6 in response to VCOM; and (2) a TTS component, which encompasses the effective sum of currents originated in this membrane compartment; this was calculated from the application of Kirchhoff laws at the external opening of the TTS (Ashcroft et al., 1985; Kim and Vergara, 1998) as
where is the voltage across the outermost segment of the TTS at time interval j.
Under the assumption that di-8-ANEPPS optical signals occur homogenously at superficial and deep regions of the TTS and that changes in its optical properties with the transmembrane voltage occur identically at every submicroscopic region of the TTS, the optical signal obtained within our illumination disk is expected to represent an ensemble mean of the voltage contributed by every cable element in the TTS weighted by the radius of each annulus. This is calculated, for each successive time step, using a numerical trapezoidal integration algorithm based on Simpson’s rule (Gerald, 1978) from the formula
However, as described in detail previously (Kim and Vergara, 1998), the actual contributions of different radial elements to the overall fluorescence transients should be corrected by the optical properties of the objective used both to illuminate the preparation and to collect fluorescence light.
Fig. A1 shows the normalized fluorescence detected from a thin film of rhodamine G (∼10 µm thick) deposited at the bottom of the experimental chamber as the z axis distance was varied in 1-µm steps to slightly above and below the focal plane (z = 0). The continuous line in Fig. A1 was fitted to a Lorentzian function of the form
Figure A1.

Fluorescence intensity depth profile of the objective (USAPO 100XO; Olympus) used in the current experiments. Data points represent normalized mean fluorescence (three trials). The data symbols span the error range.
where F0 = 0.025, A = 119 µm, and the full width at half-maximum = 76 µm. This function was used to correct the weighted mean calculations of the TTS voltage from the radial cable model for depth changes in the fluorescence detection. Of the several illumination/detection algorithms described by Kim and Vergara (1998), we used here the global illumination because it is the one that accurately represents the experimental conditions in the current work.
Acknowledgments
We thank Dr. Thomas O’Dell (Department of Physiology, University of California, Los Angeles, Los Angeles, CA) for providing us with the crude lysate of mouse hippocampus and for the use of his chemiluminescence detection instrument. We also thank R. Serrano for technical support and Carl Yu for help editing the figures.
The University of California, Davis/NIH Neuromab Facility is supported by NIH grant U24NS050606 and is maintained by the Department of Neurobiology, Physiology, and Behavior of the College of Biological Sciences, University of California, Davis. This work was supported by NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases grants AR047664 and AR54816 (to J. Vergara) and the multi-PI grant AR041802.
Edward N. Pugh Jr. served as editor.
Footnotes
Abbreviations used in this paper:
- 9-ACA
- 9-anthacene carboxylic acid
- AIC
- Akaike’s Information Criterion
- AP
- action potential
- FDB
- flexor digitorum brevis
- HL
- hippocampus lysate
- ML
- muscle lysate
- MM
- muscle microsomal
- TBS
- Tris-buffered saline
- TPLSM
- two-photon laser-scanning microscopy
- TTS
- transverse tubular system
- TTX
- tetrodotoxin
References
- Abbott G.W., Butler M.H., Bendahhou S., Dalakas M.C., Ptacek L.J., Goldstein S.A. 2001. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell. 104:217–231 10.1016/S0092-8674(01)00207-0 [DOI] [PubMed] [Google Scholar]
- Adrian R.H., Marshall M.W. 1976. Action potentials reconstructed in normal and myotonic muscle fibres. J. Physiol. 258:125–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrian R.H., Peachey L.D. 1973. Reconstruction of the action potential of frog sartorius muscle. J. Physiol. 235:103–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrian R.H., Chandler W.K., Hodgkin A.L. 1969. The kinetics of mechanical activation in frog muscle. J. Physiol. 204:207–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrian R.H., Chandler W.K., Hodgkin A.L. 1970. Voltage clamp experiments in striated muscle fibres. J. Physiol. 208:607–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft F.M., Heiny J.A., Vergara J. 1985. Inward rectification in the transverse tubular system of frog skeletal muscle studied with potentiometric dyes. J. Physiol. 359:269–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry P.H., Adrian R.H. 1973. Slow conductance changes due to potassium depletion in the transverse tubules of frog muscle fibers during hyperpolarizing pulses. J. Membr. Biol. 14:243–292 10.1007/BF01868081 [DOI] [PubMed] [Google Scholar]
- Beam K.G., Donaldson P.L. 1983a. A quantitative study of potassium channel kinetics in rat skeletal muscle from 1 to 37 degrees C. J. Gen. Physiol. 81:485–512 10.1085/jgp.81.4.485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beam K.G., Donaldson P.L. 1983b. Slow components of potassium tail currents in rat skeletal muscle. J. Gen. Physiol. 81:513–530 10.1085/jgp.81.4.513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berjukow S., Marksteiner R., Gapp F., Sinnegger M.J., Hering S. 2000. Molecular mechanism of calcium channel block by isradipine. Role of a drug-induced inactivated channel conformation. J. Biol. Chem. 275:22114–22120 10.1074/jbc.M908836199 [DOI] [PubMed] [Google Scholar]
- Bett G.C., Dinga-Madou I., Zhou Q., Bondarenko V.E., Rasmusson R.L. 2011. A model of the interaction between N-type and C-type inactivation in Kv1.4 channels. Biophys. J. 100:11–21 10.1016/j.bpj.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielanska J., Hernández-Losa J., Pérez-Verdaguer M., Moline T., Somoza R., Ramón Y Cajal S., Condom E., Ferreres J.C., Felipe A. 2009. Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr. Cancer Drug Targets. 9:904–914 10.2174/156800909790192400 [DOI] [PubMed] [Google Scholar]
- Brinkmeier H., Zachar E., Rüdel R. 1991. Voltage-dependent K+ channels in the sarcolemma of mouse skeletal muscle. Pflugers Arch. 419:486–491 10.1007/BF00370793 [DOI] [PubMed] [Google Scholar]
- Cartwright T.A., Corey M.J., Schwalbe R.A. 2007. Complex oligosaccharides are N-linked to Kv3 voltage-gated K+ channels in rat brain. Biochim. Biophys. Acta. 1770:666–671 10.1016/j.bbagen.2006.11.013 [DOI] [PubMed] [Google Scholar]
- Covarrubias M., Wei A., Salkoff L., Vyas T.B. 1994. Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron. 13:1403–1412 10.1016/0896-6273(94)90425-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crank J.1975. The Mathematics of Diffusion. Oxford University Press, Oxford. 414 pp.
- DeCoursey T.E. 1990. State-dependent inactivation of K+ currents in rat type II alveolar epithelial cells. J. Gen. Physiol. 95:617–646 10.1085/jgp.95.4.617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Castillo J., Escalona de Motta G. 1978. A new method for excitation-contraction uncoupling in frog skeletal muscle. J. Cell Biol. 78:782–784 10.1083/jcb.78.3.782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado J.Y., O’Dell T.J. 2005. Long-term potentiation persists in an occult state following mGluR-dependent depotentiation. Neuropharmacology. 48:936–948 10.1016/j.neuropharm.2005.01.008 [DOI] [PubMed] [Google Scholar]
- Demo S.D., Yellen G. 1991. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 7:743–753 10.1016/0896-6273(91)90277-7 [DOI] [PubMed] [Google Scholar]
- DiFranco M., Vergara J.L. 2011. The Na conductance in the sarcolemma and the transverse tubular system membranes of mammalian skeletal muscle fibers. J. Gen. Physiol. 138:393–419 10.1085/jgp.201110682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFranco M., Capote J., Vergara J.L. 2005. Optical imaging and functional characterization of the transverse tubular system of mammalian muscle fibers using the potentiometric indicator di-8-ANEPPS. J. Membr. Biol. 208:141–153 10.1007/s00232-005-0825-9 [DOI] [PubMed] [Google Scholar]
- DiFranco M., Capote J., Quiñonez M., Vergara J.L. 2007. Voltage-dependent dynamic FRET signals from the transverse tubules in mammalian skeletal muscle fibers. J. Gen. Physiol. 130:581–600 10.1085/jgp.200709831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFranco M., Quinonez M., Capote J., Vergara J. 2009. DNA transfection of mammalian skeletal muscles using in vivo electroporation. J. Vis. Exp. 32 (JoVE) [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFranco M., Herrera A., Vergara J.L. 2011a. Chloride currents from the transverse tubular system in adult mammalian skeletal muscle fibers. J. Gen. Physiol. 137:21–41 10.1085/jgp.201010496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFranco M., Tran P., Quiñonez M., Vergara J.L. 2011b. Functional expression of transgenic 1sDHPR channels in adult mammalian skeletal muscle fibres. J. Physiol. 589:1421–1442 10.1113/jphysiol.2010.202804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval A., Léoty C. 1980. Comparison between the delayed outward current in slow and fast twitch skeletal muscle in the rat. J. Physiol. 307:43–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich O., Ehmer T., Fink R.H. 1999. Calcium currents during contraction and shortening in enzymatically isolated murine skeletal muscle fibres. J. Physiol. 517:757–770 10.1111/j.1469-7793.1999.0757s.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich O., Ehmer T., Uttenweiler D., Vogel M., Barry P.H., Fink R.H. 2001. Numerical analysis of Ca2+ depletion in the transverse tubular system of mammalian muscle. Biophys. J. 80:2046–2055 10.1016/S0006-3495(01)76178-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Struyk A., Markin V., Cannon S. 2011. Gating behaviour of sodium currents in adult mouse muscle recorded with an improved two-electrode voltage clamp. J. Physiol. 589:525–546 10.1113/jphysiol.2010.199430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerald C.F.1978. Applied Numerical Analysis. Addison-Wesley Publishing Company. 518 pp.
- González E., Messi M.L., Delbono O. 2000. The specific force of single intact extensor digitorum longus and soleus mouse muscle fibers declines with aging. J. Membr. Biol. 178:175–183 10.1007/s002320010025 [DOI] [PubMed] [Google Scholar]
- Grissmer S., Nguyen A.N., Aiyar J., Hanson D.C., Mather R.J., Gutman G.A., Karmilowicz M.J., Auperin D.D., Chandy K.G. 1994. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 45:1227–1234 [PubMed] [Google Scholar]
- Grunnet M., Rasmussen H.B., Hay-Schmidt A., Rosenstierne M., Klaerke D.A., Olesen S.P., Jespersen T. 2003. KCNE4 is an inhibitory subunit to Kv1.1 and Kv1.3 potassium channels. Biophys. J. 85:1525–1537 10.1016/S0006-3495(03)74585-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Li H., London B., Nerbonne J.M. 2000. Functional consequences of elimination of i(to,f) and i(to,s): early after depolarizations, atrioventricular block, and ventricular arrhythmias in mice lacking Kv1.4 and expressing a dominant-negative Kv4 alpha subunit. Circ. Res. 87:73–79 10.1161/01.RES.87.1.73 [DOI] [PubMed] [Google Scholar]
- Hashimoto Y., Nunoki K., Kudo H., Ishii K., Taira N., Yanagisawa T. 2000. Changes in the inactivation of rat Kv1.4 K(+) channels induced by varying the number of inactivation particles. J. Biol. Chem. 275:9358–9362 10.1074/jbc.275.13.9358 [DOI] [PubMed] [Google Scholar]
- Heiny J.A., Ashcroft F.M., Vergara J. 1983. T-system optical signals associated with inward rectification in skeletal muscle. Nature. 301:164–166 10.1038/301164a0 [DOI] [PubMed] [Google Scholar]
- Hille B., Woodhull A.M., Shapiro B.I. 1975. Negative surface charge near sodium channels of nerve: divalent ions, monovalent ions, and pH. Philos. Trans. R. Soc. Lond. B Biol. Sci. 270:301–318 10.1098/rstb.1975.0011 [DOI] [PubMed] [Google Scholar]
- Hocherman S.D., Bezanilla F. 1996. A patch-clamp study of delayed rectifier currents in skeletal muscle of control and mdx mice. J. Physiol. 493:113–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin A.L., Huxley A.F. 1952a. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J. Physiol. 116:449–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin A.L., Huxley A.F. 1952b. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117:500–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. 1991. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 7:547–556 10.1016/0896-6273(91)90367-9 [DOI] [PubMed] [Google Scholar]
- Iannotti F.A., Panza E., Barrese V., Viggiano D., Soldovieri M.V., Taglialatela M. 2010. Expression, localization, and pharmacological role of Kv7 potassium channels in skeletal muscle proliferation, differentiation, and survival after myotoxic insults. J. Pharmacol. Exp. Ther. 332:811–820 10.1124/jpet.109.162800 [DOI] [PubMed] [Google Scholar]
- Juiz J.M., Luján R., Domínguez del Toro E., Fuentes V., Ballesta J.J., Criado M. 2000. Subcellular compartmentalization of a potassium channel (Kv1.4): preferential distribution in dendrites and dendritic spines of neurons in the dorsal cochlear nucleus. Eur. J. Neurosci. 12:4345–4356 [PubMed] [Google Scholar]
- Jurkat-Rott K., Fauler M., Lehmann-Horn F. 2006. Ion channels and ion transporters of the transverse tubular system of skeletal muscle. J. Muscle Res. Cell Motil. 27:275–290 10.1007/s10974-006-9088-z [DOI] [PubMed] [Google Scholar]
- Kalman K., Nguyen A., Tseng-Crank J., Dukes I.D., Chandy G., Hustad C.M., Copeland N.G., Jenkins N.A., Mohrenweiser H., Brandriff B., et al. 1998. Genomic organization, chromosomal localization, tissue distribution, and biophysical characterization of a novel mammalian Shaker-related voltage-gated potassium channel, Kv1.7. J. Biol. Chem. 273:5851–5857 10.1074/jbc.273.10.5851 [DOI] [PubMed] [Google Scholar]
- Kim A.M., Vergara J.L. 1998. Supercharging accelerates T-tubule membrane potential changes in voltage clamped frog skeletal muscle fibers. Biophys. J. 75:2098–2116 10.1016/S0006-3495(98)77652-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen M., Juel C. 2010. Potassium-transporting proteins in skeletal muscle: cellular location and fibre-type differences. Acta Physiol. (Oxf.). 198:105–123 10.1111/j.1748-1716.2009.02043.x [DOI] [PubMed] [Google Scholar]
- Lamb G.D., Walsh T. 1987. Calcium currents, charge movement and dihydropyridine binding in fast- and slow-twitch muscles of rat and rabbit. J. Physiol. 393:595–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre R., Vergara C., Moczydlowski E. 1983. Properties of a Ca2+-activated K+ channel in a reconstituted system. Cell Calcium. 4:343–357 10.1016/0143-4160(83)90013-1 [DOI] [PubMed] [Google Scholar]
- Lee T.E., Philipson L.H., Nelson D.J. 1996. N-type inactivation in the mammalian Shaker K+ channel Kv1.4. J. Membr. Biol. 151:225–235 10.1007/s002329900073 [DOI] [PubMed] [Google Scholar]
- Lesage F., Attali B., Lazdunski M., Barhanin J. 1992. Developmental expression of voltage-sensitive K+ channels in mouse skeletal muscle and C2C12 cells. FEBS Lett. 310:162–166 10.1016/0014-5793(92)81320-L [DOI] [PubMed] [Google Scholar]
- Lueck J.D., Rossi A.E., Thornton C.A., Campbell K.P., Dirksen R.T. 2010. Sarcolemmal-restricted localization of functional ClC-1 channels in mouse skeletal muscle. J. Gen. Physiol. 136:597–613 10.1085/jgp.201010526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M., Lenn N.J., Trimmer J.S. 1995. Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J. Neurosci. 15:3840–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan M.M., Trimmer J.S., Rhodes K.J. 2001. Experimental localization of Kv1 family voltage-gated K+ channel alpha and beta subunits in rat hippocampal formation. J. Neurosci. 21:5973–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco Otalora L.F., Skinner F., Oliveira M.S., Farrell B., Arshadmansab M.F., Pandari T., Garcia I., Robles L., Rosas G., Mello C.F., et al. 2011. Chronic deficit in the expression of voltage-gated potassium channel Kv3.4 subunit in the hippocampus of pilocarpine-treated epileptic rats. Brain Res. 1368:308–316 10.1016/j.brainres.2010.10.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappone P.A. 1980. Voltage-clamp experiments in normal and denervated mammalian skeletal muscle fibres. J. Physiol. 306:377–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Po S., Roberds S., Snyders D.J., Tamkun M.M., Bennett P.B. 1993. Heteromultimeric assembly of human potassium channels. Molecular basis of a transient outward current? Circ. Res. 72:1326–1336 10.1161/01.RES.72.6.1326 [DOI] [PubMed] [Google Scholar]
- Pongs O., Schwarz J.R. 2010. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 90:755–796 10.1152/physrev.00020.2009 [DOI] [PubMed] [Google Scholar]
- Rhodes K.J., Strassle B.W., Monaghan M.M., Bekele-Arcuri Z., Matos M.F., Trimmer J.S. 1997. Association and colocalization of the Kvbeta1 and Kvbeta2 beta-subunits with Kv1 alpha-subunits in mammalian brain K+ channel complexes. J. Neurosci. 17:8246–8258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppersberg J.P., Schröter K.H., Sakmann B., Stocker M., Sewing S., Pongs O. 1990. Heteromultimeric channels formed by rat brain potassium-channel proteins. Nature. 345:535–537 10.1038/345535a0 [DOI] [PubMed] [Google Scholar]
- Schröter K.H., Ruppersberg J.P., Wunder F., Rettig J., Stocker M., Pongs O. 1991. Cloning and functional expression of a TEA-sensitive A-type potassium channel from rat brain. FEBS Lett. 278:211–216 10.1016/0014-5793(91)80119-N [DOI] [PubMed] [Google Scholar]
- Sheng M., Tsaur M.L., Jan Y.N., Jan L.Y. 1992. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 9:271–284 10.1016/0896-6273(92)90166-B [DOI] [PubMed] [Google Scholar]
- Sheng M., Liao Y.J., Jan Y.N., Jan L.Y. 1993. Presynaptic A-current based on heteromultimeric K+ channels detected in vivo. Nature. 365:72–75 10.1038/365072a0 [DOI] [PubMed] [Google Scholar]
- Tricarico D., Petruzzi R., Camerino D.C. 1997. Changes of the biophysical properties of calcium-activated potassium channels of rat skeletal muscle fibres during aging. Pflugers Arch. 434:822–829 10.1007/s004240050471 [DOI] [PubMed] [Google Scholar]
- Ursu D., Schuhmeier R.P., Freichel M., Flockerzi V., Melzer W. 2004. Altered inactivation of Ca2+ current and Ca2+ release in mouse muscle fibers deficient in the DHP receptor gamma1 subunit. J. Gen. Physiol. 124:605–618 10.1085/jgp.200409168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursu D., Schuhmeier R.P., Melzer W. 2005. Voltage-controlled Ca2+ release and entry flux in isolated adult muscle fibres of the mouse. J. Physiol. 562:347–365 10.1113/jphysiol.2004.073882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veh R.W., Lichtinghagen R., Sewing S., Wunder F., Grumbach I.M., Pongs O. 1995. Immunohistochemical localization of five members of the Kv1 channel subunits: contrasting subcellular locations and neuron-specific co-localizations in rat brain. Eur. J. Neurosci. 7:2189–2205 10.1111/j.1460-9568.1995.tb00641.x [DOI] [PubMed] [Google Scholar]
- Vullhorst D., Klocke R., Bartsch J.W., Jockusch H. 1998. Expression of the potassium channel KV3.4 in mouse skeletal muscle parallels fiber type maturation and depends on excitation pattern. FEBS Lett. 421:259–262 10.1016/S0014-5793(97)01577-9 [DOI] [PubMed] [Google Scholar]
- Wickenden A.D., Jegla T.J., Kaprielian R., Backx P.H. 1999. Regional contributions of Kv1.4, Kv4.2, and Kv4.3 to transient outward K+ current in rat ventricle. Am. J. Physiol. 276:H1599–H1607 [DOI] [PubMed] [Google Scholar]
- Woods C.E., Novo D., DiFranco M., Vergara J.L. 2004. The action potential-evoked sarcoplasmic reticulum calcium release is impaired in mdx mouse muscle fibres. J. Physiol. 557:59–75 10.1113/jphysiol.2004.061291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C.E., Novo D., DiFranco M., Capote J., Vergara J.L. 2005. Propagation in the transverse tubular system and voltage dependence of calcium release in normal and mdx mouse muscle fibres. J. Physiol. 568:867–880 10.1113/jphysiol.2005.089318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Anderson J.W., Fedida D. 1997. Characterization of nifedipine block of the human heart delayed rectifier, hKv1.5. J. Pharmacol. Exp. Ther. 281:1247–1256 [PubMed] [Google Scholar]