

Abstract
Hassawi rice (Oryza sativa L.) is a landrace adapted to the climate of Saudi Arabia, characterized by its strong resistance to soil salinity and drought. Using high quality sequencing reads extracted from raw data of a whole genome sequencing project, we assembled both chloroplast (cp) and mitochondrial (mt) genomes of the wild-type Hassawi rice (Hassawi-1) and its dwarf hybrid (Hassawi-2). We discovered 16 InDels (insertions and deletions) but no SNP (single nucleotide polymorphism) is present between the two Hassawi cp genomes. We identified 48 InDels and 26 SNPs in the two Hassawi mt genomes and a new type of sequence variation, termed reverse complementary variation (RCV) in the rice cp genomes. There are two and four RCVs identified in Hassawi-1 when compared to 93–11 (indica) and Nipponbare (japonica), respectively. Microsatellite sequence analysis showed there are more SSRs in the genic regions of both cp and mt genomes in the Hassawi rice than in the other rice varieties. There are also large repeats in the Hassawi mt genomes, with the longest length of 96,168 bp and 96,165 bp in Hassawi-1 and Hassawi-2, respectively. We believe that frequent DNA rearrangement in the Hassawi mt and cp genomes indicate ongoing dynamic processes to reach genetic stability under strong environmental pressures. Based on sequence variation analysis and the breeding history, we suggest that both Hassawi-1 and Hassawi-2 originated from the Indonesian variety Peta since genetic diversity between the two Hassawi cultivars is very low albeit an unknown historic origin of the wild-type Hassawi rice.
Introduction
Rice (Oryza sativa L.) is a grass species of plants, providing staple food for over half of the world populations. Aside from being one of the top three major cereal crops, rice is also widely used as a model plant for genetic studies [1]. The cultivated rice has two main subspecies, indica and japonica, which are estimated to be separated about 0.05–0.44 mya [2]. The Hassawi rice (Oryza sativa L.) is a landrace adapted to the climate of Eastern Saudi Arabia. It is characterized by strong adaptability to soil salinity and drought [3], [4]. However, it bears some undesired characteristics such as susceptibility for lodging, delayed maturity, and photoperiod sensitivity. In Saudi Arabia, there are two Hassawi cultivars, which provide a staple carbohydrate source [3]. One of the cultivars, Hassawi-1 is the wild-type originated from an indica ancestor and the other cultivar Hassawi-2 is a hybrid between Hassawi-1 and IR1112 (according to the breeding record, its maternal parent is IR262-43-8-11, a cultivar originated from Peta, an indica variety from Indonesia). However, until now, few studies have been carried out on genetics and genomics about this valuable rice variety and its related cultivars, and there has been a limited literature about the genetic background of those Hassawi rice. The organelle genome sequences of Hassawi rice are very helpful for understanding the inheritance of this cultivar and its future breeding research.
The complete chloroplast (cp) and mitochondrion (mt) genomes of both indica and japonica are published [2], [5]–[7], and a comparative analysis showed that the gene order and essential gene content are highly conserved for most cp genomes [8]. In contrast, plant mt genomes are known to be more complex than those of chloroplasts. The mt-encoded genes are highly conserved, but their gene order, genomes structure, and genome size are highly variable among plant species [2], [9], [10]. Genetic markers have played a major role in our understanding of heritable traits, serving as landmarks for genes and their variations. With the increasing application of next-generation sequencing technologies, there is a rapid growth in information on genetic polymorphisms [11] albeit minor hindrance of sequencing errors [12]. As the most abundant genetic markers, the discovery of both single nucleotide polymorphism (SNP) and insertion or deletion (InDel) is of paramount importance for marker-assisted crop breeding and genetic studies. Simple sequence repeats (SSRs), also known as microsatellites, are also abundant across plant organellar genomes. SSR-based markers can be developed to be a useful tool in determining the maternal origin of rice varieties and for phylogenetic studies [13]. SNPs, InDels, and SSRs of organellar genomes are all invaluable as genetic markers for plant genetics.
We recently began to sequence the genomes of two Hassawi cultivars using next generation sequencing platforms (both 454 GS FLX and SOLiD 4.0). Using our recently published procedure for plant organellar genome assembly [14], we finished both cp and mt genomes of Hassawi-1 and Hassawi-2. Our in-depth comparative analysis of the organellar genomes revealed the genome variations of SNP, InDel, SSR, reverse complementary variation (RCV) and repeats among Hassawi-1, Hassawi-2, indica 93–11 and japonica Nipponbare. Based on those sequence variation analysis and the breeding history, we confirmed that both Hassawi-1 and Hassawi-2 were originated from the Indonesian variety, which provide important information for future genetic studies of this unique rice variety.
Results and Discussion
Genome Assembly Results
The original raw data from the whole genome shotgun sequencing projects contain a large amount of reads from cp and mt genomes, which can be assembled into complete genome sequences independently [2], [15]. We have developed an efficient procedure for plant organellar genome assembly, which based on whole genome data from the 454 sequencing platform [14]. Using this procedure, we have successfully assembled the complete cp and mt genome sequences of Boea hygrometrica from the whole genome sequencing data [16]. With the same method, we used this procedure to assemble the cp and mt genomes of both Hassawi-1 and Hassawi-2.
Using 454 sequencing platform, We totally got 7 runs (2.37 Gbp)and 11 runs (3.49 Gbp) for Hassawi-1 and Hassawi-2, respectively (our unpublished data). The raw data quality was good with 95% bases above Q40 and with the peak of the average read quality above 30 for both Hassawi-1 and Hassawi-2. There were totally 419,015 reads (136 Mbp) and 550,696 reads (207 Mbp) filtered as reads belong to cp genome in Hassawi-1 and Hassawi-2, respectively. Using Roche Newbler software, we de novo assembled those reads and constructed the contig graphs for whole cp genome. There were 32 and 131 contigs with total length 134,448 bp and 134,459 bp composed complete cp genome of Hassawi-1 and Hassawi-2, respectively (Figure S1 and S2). The assembly of mt genome was more complex than cp genome. Using all the sequencing raw data, we assembled 278,498 and 235,389 contigs with average length 1,061 bp and 1,417 bp in Hassawi-1 and Hassawi-2, respectively. With the same method of assembling mt genome of Boea hygrometrica [14], [16], we filtered and construed contig graphs belong to mt genome with the reference of mt genomes of other rice ( Table 1 ). There were 117 and 213 contigs with total length 454,820 bp and 454,894 bp composed complete mt genome of Hassawi-1 and Hassawi-2, respectively (Figure S3 and S4). In order to assess the cp and mt assembly quality for both Hassawi-1 and Hassawi-2, we mapped both the 454 raw data and the SOLiD mate pair data (data unpublished) with different insert size to the assembled cp and mt genomes. The result showed that in all assembly, there were no gap between two connecting contigs and all the contig order were supported by mate pair reads ( Figure 1 ).
Table 1. General features of cp and mt genomes among the four rice cultivars.
| Size (bp) | GC (%) | Gene number (total/protein/tRNA/rRNA) | Coding (%) | Repeat (%) | SSR (%) | Cp-derived1 (%) | ||
| Chloroplast | Hassawi-1 | 134448 | 39.0 | 136/91/37/8 | 54.9 | 1.2 | 4.3 | – |
| Hassawi-2 | 134459 | 39.0 | 136/91/37/8 | 54.9 | 1.2 | 4.3 | – | |
| 93–11 | 134496 | 39.0 | 136/91/37/8 | 54.9 | 1.2 | 4.4 | – | |
| Nipponbare | 134525 | 39.0 | 136/91/37/8 | 54.9 | 1.2 | 4.4 | – | |
| Mitochondrion | Hassawi-1 | 454820 | 43.9 | 102/69/27/6 | 15.9 | 54.2 | 3.6 | 5.9 |
| Hassawi-2 | 454894 | 43.8 | 101/67/28/6 | 15.6 | 54.2 | 3.6 | 5.9 | |
| 93–11 | 491515 | 43.8 | 93/54/33/6 | 12.4 | 58.5 | 3.5 | 6.9 | |
| Nipponbare | 490520 | 43.9 | 84/56/22/6 | 10.8 | 59.7 | 3.6 | 6.9 | |
Note: 1. Cp-derived stands for sequences that are homologous to those of rice cp genomes.
Figure 1. Circular representation of the cp and mt genome assemblies of both Hassawi-1 and Hassawi-2.

Circle display (from the outside): (1) physical map scale in kilobase pairs (in cp genome, LSC region in blue, SSC region in green, and IRs regions in red); (2) read depths of the 454 sequencing data in plum (step size: 100 bp in cp genome and 200 bp in mt genome; cp assembly: range 200–1325 in Hassawi-1 and range 200–2230 in Hassawi-2; mt assembly: range 0–500); (3) SOLiD mate-pair read validation with the 0.5–1 kb insert library in purple (insert size 600–800 bp and step size 100 bp in the cp assembly and 450 bp in the mt assembly); (4) SOLiD mate-pair read validation with 1–3 kb library in orange (insert size 1400–1600 bp and step size 150 bp in the cp assembly and 700 bp in the mt assembly). The high variance in read depth of the mt genome results from the regions of cp-derived sequences. This figure is generated by the Circos program.
Genome Features of the Two Hassawi Rice Cultivars
The cp genome is in general composed of a single circular molecule with a quadripartite structure, which includes a large single copy region (LSC) and a small single copy region (SSC), separated by two copies of inverted repeats (IRs) [17], [18]. The Hassawi-1 cp genome has 134,448 bp in length and a GC content of 39%. 58.8% of its genes are located in LSC region (80,513 bp, covering 59.9% of the cp genome). All rRNA and 16 tRNA genes reside in IR regions (41,590 bp, covering 30.9% of the cp genome). The SSC region (12,345 bp, 9.2%) includes most NADH oxidoreductases. In plant cp genomes, gene content, order, and organization are highly conserved and their inheritance is always maternal, which is different from nuclear genomes [17], [19]. Gene number (136), coding fraction (54.9%), and repeat content (1.2%) of the two Hassawi rice are identical ( Table 1 ). The sequence alignment of Hassawi-1 to 93–11 and Nipponbare shows excellent colinearity ( Figure 2 ). Other than what is in IR regions, we failed to identify any sequence repeats greater than 1 kb in the Hassawi cp genomes.
Figure 2. Dot matrix alignment of cp genomes between Hassawi-1 and 93-11 (top) and between Hassawi-1 and Nipponbare (bottom).

The blue and red lines show direct and reverse matches, respectively. The red lines indicate IR regions in cp genomes.
The mt genome is much larger and more complex than the cp genome [20]. Moreover, mt genomes of seed plants are unusually variable in size at least in an order of magnitude, and much of these variations occur within a single family [21]. In this study, we observed that the length of Hassawi mt genomes is obviously different from those of 93–11 and Nipponbare ( Table 1 ). The wild-type Hassawi rice has a circular DNA molecule with 454,820 bp in length, which is smaller than both 93–11 (491,515 bp) and Nipponbare (490,520 bp). However, compared to 93–11 and Nipponbare, the mt genome of Hassawi-1 has a larger coding region (15.9%) and smaller repeat region (54.2%). Unlike cp genomes, most mt genomes are non-coding or functionally unknown. The functional genes in plant mt genomes are conserved between Hassawi and other varieties, such as NADH dehydrogenase and cytochrome c oxidase. The mt genomic structure of Hassawi-1 is very different from that of 93–11 and Nipponbare. Alignment plots among them show multiple recombination events ( Figure 3 ). There are some large repeats (>10 kb) in the rice mt genomes, which were also found in other seed plants [22], [23]. The percentage of cp-derived sequences in the Hassawi mt genomes is lower than that of other varieties.
Figure 3. Dot matrix alignment of mt genomes between Hassawi-1 and 93-11 (top) and between Hassawi-1 and Nipponbare (bottom).

The blue and red lines show direct and reverse matches, respectively.
Cp Genome Variations
There are two basic categories of sequence variations with regard to varieties and subspecies when assessing polymorphisms in organellar genomes; one is intraspecific or intravarietal, where variations within a variety or subspecies are identified, and the other is interspecific or intervarietal, where variations between two varieties or subspecies are defined [2], [5]. Comparing cp genomes of Hassawi-1 and Hassawi-2, we detected eleven insertion and five deletion events ( Table 2 ), which resulted in an 11-bp difference overall. Among all 16 InDel events, we detected one deletion and two insertions as intravarietal InDels based on an analysis of the sequencing reads. All these InDels are located in intergenic regions. Three InDels (D-2, I-4 and I-4 with positions of 36,491, 80,517, and 134,446 in Hassawi-1, respectively) are larger than 1 bp and are all located in LSC region and they are candidate genetics marks for distinguishing wild-type Hassawi rice from its hybrid. We did not observe any SNP between Hassawi-1 and Hassawi-2 ( Figure 4 ).
Table 2. InDels of cp genomes between Hassawi rice and its hybrid.
| InDel1 | Position in Hassawi-1 | Position in Hassawi-2 | Sequence | Intravarietal InDel2 | Gene location | Region |
| I-1 | 8069 | 8076 | A | Y | trnS-GCU/psbD | LSC |
| D-1 | 8214 | 8220 | T | N | trnS-GCU/psbD | |
| I-1 | 17031 | 17038 | A | N | psbM/petN | |
| D-1 | 17734 | 17740 | T | Y | petN/trnC-GCA | |
| I-1 | 18586 | 18593 | A | Y | trnC-GCA/rpoB | |
| D-1 | 31973 | 31979 | T | N | atpI/atpH | |
| D-2 | 36491 | 36496 | TT | N | trnR-UCU/psaB | |
| I-1 | 41448 | 41453 | A | N | psaA/ycf3 | |
| I-1 | 46123 | 46129 | T | N | trnT-UGU/trnL-UAA | |
| I-1 | 46432 | 46439 | A | N | trnT-UGU/trnL-UAA | |
| D-1 | 53261 | 53267 | A | N | atpB/rbcL | |
| I-1 | 78351 | 78358 | T | N | rpl16/rps3 | |
| I-4 | 80517 | 80525 | TCTT | N | rpl22/rps19 | |
| I-1 | 82608 | 82620 | T | N | rpl2/rpl23 | IRA |
| I-1 | 132357 | 132370 | A | N | rpl23/rpl2 | IRB |
| I-4 | 134446 | 1 | GAAA | N | rps19/psbA | LSC |
Hassawi-1 is set to be the reference.
Intravarietal InDels are detected based on the raw sequencing reads.
Figure 4. Circos diagram illustrating SNP and InDel distributions in cp genomes of Hassawi-1 and the other three cultivars.

The first circle (from outside) displays genomes (color-coding) and genes (blocks). The second circle displays genomic regions including SSC, LSC, IRA, and IRB. The connecting lines inside the circles show SNPs (blue) and InDels (red) between two genomes.
We also compared the cp genome of Hassawi-1 with those of 93–11 and Nipponbare. Between the Hassawi-1 and 93–11 cp genomes, there are 40 deletions, 37 of which are 1-bp deletions and the longest is a 6-bp one in an IRA region. Most of them are interspecific deletions. The cumulative length attributable to InDels is 49 bp, which is consistent with the overall length difference between Hassaw-1 and 93–11. However, we did not find any insertions or SNPs between Hassawi-1 and 93–11. Comparing to the Nipponbare cp genome, we identified 71 deletions, 40 insertions, and 110 SNPs in Hassawi-1; the longest deletion is 69 bp (D-69, position 8,551) and the longest insertion is 32 bp (I-32, position 17,734). We also identified 13 co-segregated SNPs, which are all located in LSC region ( Table 3 ). Half of these co-segregation SNPs are transversions between GC and CG. The co-segregated SNPs represent the best candidate molecular markers devoid of sequencing errors [12]. The co-segregating SNPs, S-2 (position 27,469 in Hassawi-1) is located in rpoC2, which is a key diagnostic variation between two switchgrass ecotypes [17]. The intersubspecific polymorphism rates between Hassawi-1 and Nipponbare are 0.082% and 0.083% for SNPs and InDels, respectively.
Table 3. InDels and SNPs in cp genomes of Hassawi-1 when compared to 93–11 and Nipponbare.
| Reference | InDel/SNP | Position inreference | Position inHassawi-1 | Sequence | IntravarietalInDel |
| 93–11 | D-2 | 49224 | 49208 | TT | N |
| D-4 | 78379 | 78351 | TTTT | N | |
| D-6 | 80552 | 80517 | TCTTTT | N | |
| Nipponbare | I-7 | 5014 | 5014 | CCTTTAT | N |
| D-69 | 8548 | 8551 | GAATCCTATTTTTGTTCTTATACCCATGCAATAGA GAGCGAGTGGGAAAAGGGAGGTTACTTATTTTTT | N | |
| D-4 | 12671 | 12605 | GAGG | N | |
| S-2 | 12819 | 12750 | TA->CC | N | |
| D-2 | 14009 | 13940 | CA | N | |
| D-6 | 17389 | 17317 | ATAGAA | N | |
| S-2 | 17746 | 17669 | GG->AA | Y | |
| I-32 | 17810 | 17734 | TTAACAAATTCTTAGAGTATTTCTGGTAGAAT | Y | |
| S-2 | 27516 | 27469 | CG->GC | N | |
| S-2 | 39771 | 39724 | GC->CG | N | |
| S-2 | 41920 | 41871 | GC->CG | N | |
| S-3 | 40687 | 40640 | ACT->GAA | N | |
| I-2 | 43607 | 43553 | GT | N | |
| D-5 | 46099 | 46038 | TTATA | N | |
| D-2 | 46184 | 46117 | TT | N | |
| D-6 | 46550 | 46479 | AAGAAA | N | |
| D-2 | 49283 | 49208 | TT | N | |
| I-2 | 50641 | 50566 | GC | N | |
| I-15 | 53857 | 53782 | CGAATTCCTATAGTA | N | |
| S-2 | 57028 | 56968 | TT->AG | N | |
| S-2 | 57040 | 56979 | CT->AA | N | |
| D-11 | 57053 | 56991 | AAAAAAAAGTT | N | |
| I-5 | 57644 | 57572 | TAAAG | N | |
| I-5 | 60861 | 60794 | TTGTA | N | |
| S-2 | 64660 | 64594 | CG->GC | N | |
| S-2 | 64688 | 64622 | CG->GC | N | |
| D-2 | 65614 | 65547 | TT | N | |
| I-2 | 68610 | 68541 | AT | N | |
| S-2 | 70224 | 70156 | GC->CG | N | |
| S-2 | 70307 | 70239 | AG->GA | N | |
| D-6 | 75976 | 75909 | TTAATT | N | |
| S-2 | 77793 | 77723 | CG->AC | N | |
| D-5 | 78423 | 78351 | TTTTT | N | |
| I-3 | 104518 | 104437 | ACA | N | |
| I-6 | 110509 | 110431 | ATAACT | N |
Note: Only InDels larger than 1bp and co-segregating SNPs are shown.
Mt Genome Variations
We compared the mt genome of Hassawi-1 with Hassawi-2 as well as between the representative indica and japonica varieties ( Table 4 ). The cumulative length difference attributable to InDels is 74 bp, which is the total difference in length between Hassawi-1 and its hybrid. There are 39 insertion and only 9 deletion events in addition to 26 base substitutions. The deletion rate for mt genome in Hassawi-2 is nearly 4 times lower than that of the insertion. There is the same rate (0.003%) between transitions and transversions in mt genome of Hassawi-2. We found a large insertion (I-44, 44bp) within the intergenic region between rrn18 and rpl2 in Hassawi-2, which could be used as a useful marker distinguish the two Hassawi cultivars.
Table 4. Number and frequency of sequence variations in mt genomes when Hassawi-1 is used as the reference and compared to Hassawi-2, 93–11 and Nipponbare.
| Hassawi-2 | 93–11 | Nipponbare | |
| SNP | 26 (0.006%) | 101 (0.022%) | 233 (0.051%) |
| Transition | 13 (0.003%) | 57 (0.012%) | 99 (0.022%) |
| Transversion | 13 (0.03%) | 44 (0.010%) | 134 (0.029%) |
| InDel | 48 (0.011%) | 63 (0.014%) | 150 (0.033%) |
| Insertion | 39 (0.009%) | 10 (0.002%) | 61 (0.013%) |
| Deletion | 9 (0.002%) | 52 (0.012%) | 89 (0.020%) |
| Total | 74 (0.017%) | 164 (0.036%) | 383 (0.084%) |
Unlike cp genomes, we identified 101 SNPs, 52 deletions, and 10 insertions in the mt genome between Hassawi-1 and 93–11. The presence and absence of deletions and SNPs within mt and cp genomes, respectively, confirm the different evolution characteristics between cp and mt genomes. As in Hassawi-1 and 93–11, deletion events in cp genomes occur at a rate of 0.03%, which is about 2.5 times higher than in mt genomes. The different evolution rate between cp and mt genomes in rice are also reported between 93–11 and a rice cultivar PA64s [24]. The fact that cp genome variation rate is higher than mt genome has also been confirmed based on a comparison between the Hassawi rice and Nipponbare. Between the mt genome of Hassawi-1 and Nipponbare, the intersubspecific polymorphism rate for mt genomes is 0.051% for SNPs and 0.033% for InDels, nearly 1.5 and 2.5 times lower than that of their cp counterparts, respectively. There are a total of 383 intersubspecific polymorphisms (SNPs and InDels) identified between them, and we have not yet found any hotspots among the mt genomes. The transition and transversion rates are almost equal in mt genomes of different rice cultivars. Compared Hasswi-1 with 93–11 and Nipponbare, there are a total of 14 large Indels (>10 bp) ( Table 5 ). There are four common deletions exist in Hassawi-1 (D-19, D-15, D-12, and D-13).
Table 5. Large InDels (>10bp) in mt genomes between Hassawi-1 and 93–11 and between Hassawi-1 and Nipponbare.
| Reference | InDel | Position in reference | Position in Hassawi-1 | Sequence |
| 93–11 | D-19 | 63127 | 195 | CTGATTCAATAATAGAAGC |
| D-15 | 235742 | 106227 | AATGGAAAAAGAGTG | |
| D-12 | 235769 | 106237 | CCCAAAAAAGGC | |
| D-13 | 235791 | 106244 | GAGAAGGAGATAG | |
| D-16 | 243184 | 113610 | CCGTCAGAGGCAGAAG | |
| D-21 | 289897 | 218077 | ATAATTCGACAATTGCTGAGT | |
| I-10 | 360901 | 289056 | ATTGGAGAAT | |
| Nipponbare | D-19 | 62582 | 195 | CTGATTCAATAATAGAAGC |
| I-27 | 206165 | 115095 | GCTTTGCCTGCTTCCTTAGCTACGTCA | |
| D-15 | 234791 | 106227 | AATGGAAAAAGAGTG | |
| D-12 | 234818 | 106237 | CCCAAAAAAGGC | |
| D-13 | 234840 | 106244 | GAGAAGGAGATAG | |
| D-16 | 242238 | 113606 | GAAGCCGTCAGAGGCA | |
| I-12 | 247610 | 404313 | TAATATTCTTAT |
Microsatellites or Simple Sequence Repeats (SSRs)
SSRs, also known as microsatellites, have been used as genetic markers for evolutionary studies on organellar genomes due to their high variability [13], [25]. The complete SSR information on both cp and mt genomes of Hassawi rice and other varieties are summarized in Table 6 . On average, the Hassawi rice cp genome has 4.3% SSR sequences with a density of 43.3 bp/kb. There are similar numbers of SSRs in the cp genome of the indica (9311, Hassawi-1, and Hassawi-2) and japonica (Nipponbare) varieties: 870 and 876 SSRs, respectively. Most SSRs are found in intergenic regions of the cp genome. Compared to 93–11, the Hassawi rice cultivars have their SSRs more in genic and less in intergenic regions of the cp genome; the former one has SSR densities of 1.9/kb and 4.6/kb in genic and intergenic regions, respectively, and the latter two have the same corresponding SSR densities– 2.6/kb and 3.9/kb. As in the mt genome, similar results are observed. The SSR densities of 9311 are 0.5/kb and 4.6/kb in the genic and intergenic regions, respectively, and the corresponding numbers are 0.7/kb and 4.5/kb in the Hassawi-1 mt genome. Compared to cp genomes, mt genomes possess both a lower percentage (3.6%) and a lower density (36 bp/kb) of SSRs in Hassawi rice and its hybrid over the japonica variety. Dinucleotide repeats are dominant in mt genomes whereas mononucleotide repeats are more frequently found in chloroplast genomes. Moreover, the mt genomes possess more tera-, penta-, and hexa-nucleotide repeats than the cp genomes. These findings are consistent with a previous observation [13].
Table 6. Distribution of SSRs in the four rice organellar genomes.
| Chloroplast | Mitochondrion | |||||||
| Repeat motif | 93–11 | Nipponbare | Haasawi-1 | Hassswi-2 | 93–11 | Nipponbare | Hasswi-1 | Hassawi-2 |
| Mono- | ||||||||
| Genic | 154 | 216 | 190 | 190 | 116 | 106 | 149 | 142 |
| Intergenic | 394 | 338 | 358 | 358 | 890 | 897 | 824 | 835 |
| Di- | ||||||||
| Genic | 79 | 135 | 126 | 126 | 118 | 96 | 140 | 134 |
| Intergenic | 190 | 135 | 143 | 143 | 1083 | 1104 | 976 | 981 |
| Tri- | ||||||||
| Genic | 19 | 32 | 29 | 29 | 21 | 19 | 25 | 25 |
| Intergenic | 23 | 10 | 14 | 14 | 248 | 253 | 216 | 216 |
| Tetra- | ||||||||
| Genic | 2 | 3 | 3 | 3 | 2 | 2 | 3 | 3 |
| Intergenic | 8 | 6 | 6 | 6 | 40 | 40 | 33 | 33 |
| Penta- | ||||||||
| Genic | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Intergenic | 1 | 0 | 1 | 1 | 8 | 10 | 11 | 11 |
| Hexa- | ||||||||
| Genic | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 |
| Intergenic | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 870 | 876 | 870 | 870 | 2527 | 2528 | 2378 | 2381 |
| Frequency per kb | 6.5 | 6.5 | 6.5 | 6.5 | 5.1 | 5.2 | 5.2 | 5.2 |
| Density (bp/kb) | 43.6 | 43.9 | 43.3 | 43.3 | 35.4 | 35.6 | 36.0 | 36.1 |
Reverse Complementary Variations (RCVs) in Cp Genome
In addition to SNPs and InDels identified in cp genomes of different rice varieties and cultivars, we also noticed a novel type of sequence variations: reverse complementary variations (RCVs). RCV usually exists as a segment (>1 bp) in one sequence but its reverse complementary form is detected in the other. RCVs in rice have not been reported nor in any other plants. As shown in Table 7 , there are two RCVs between Hassawi-1 and 93–11 and four between Hassawi-1 and Nipponbare. The detail alignments of those RCVs are presented in Figure 5 . Only one of them is intravarietal in Hassawi-1. All of them are in LSC region except R-4 that is in SSC region (position 105,696 in Hassawi-1). Moreover, all RCVs are located in intergenic regions of cp genome except R-4 (position 105696 in Hassawi-1) in ccsA, which do not cause null mutations. The function of two genes (accD and ccsA) involving in RCVs is classified as miscellaneous. The RCV rate between Hassawi-1 and Nipponbare is 0.003%, which is nearly 27 and 28 times lower than those of SNPs and InDels, respectively. The longest RCV in Hassawi-1 is 8 bp (R-8, position 62,457) in intergenic region between psbE and petL. As a unique RCV in Hassawi-1, R-6 (position 55,604, TTTTTC), is a useful genetic marker to distinguish the two major rice subspecies. Since plant chloroplasts have their organelle-specific replication and DNA repair systems, the generation of RCVs may be related to these two systems. Since we did not identify any RCVs between 93–11 and a wild rice Oryza nivara, it is very suggestive that this RCV is either created as very rare event or the mechanism for its generation is developed later in the evolution of rice cp genomes.
Table 7. Reverse complementary variations (RCVs) in cp genomes when Hassawi-1 is compared to 9311 and Nipponbare.
| Reference | RCV1 | Position in reference | Position in Hassawi-1 | Sequence2 | intravarietalRCV | Gene locus | Region |
| 93–11 | R-2 | 15 | 15 | TC->GA | Y | rps19-psbA | LSC |
| R-6 | 55621 | 55604 | GAAAAA->TTTTTC | N | rbcL-accD | LSC | |
| Nipponbare | R-2 | 15 | 15 | TC->GA | Y | rps19-psbA | LSC |
| R-6 | 55665 | 55604 | GAAAAA->TTTTTC | N | rbcL-accD | LSC | |
| R-8 | 62521 | 62457 | CTTGGTCT->AGACCAAG | N | psbE-petL | LSC | |
| R-4 | 105775 | 105696 | AAGC->GCTT | N | ccsA | SSC |
Note: 1. RCV stands for reverse complementary variation.
2. Hassawi specific RCVs are highlighted in bold.
Figure 5. Detail alignments of reverse complementary variations in four cp genomes.

The forward fragment is shown in green and the reverse fragment is shown in purple.
Repeats in Mt Genomes
Plant mitochondria have slow evolutionary rate and rapid rearrangement [26], [27]. Compared with plastid genomes, plant mt genomes are typically rich in large repeats. The extensive use of DNA recombination is an importance process in plant mt genome. Recently, DNA recombination in plant organellar genomes has been confirmed to play an important role in maintaining genome stability [28]. Moreover, there is ample evidence demonstrating that mt genome is important for plant sexual reproduction [29]. In rice, it has been proved that some novel mt genomic rearrangements are unique in cytoplasmic male sterility (CMS), where length variation of mt genome was observed [29]. Such DNA recombination has also been identified between wheat K-type cytoplasmic male sterility line and its maintainer line [26].
Compared to other plants, rice mt genome has a higher content of repetitive sequences; there are 287,556 bp (58.5%) and 293,120 bp repeat sequences (59.7%) in 93–11 and Nipponbare, respectively. The eleven and thirteen large repeats (>1 kb) in indica and japonica are 277,828 bp and 272,688 bp in length, respectively ( Table 8 ). However, the number of large repeats is reduced to six in the mt genomes of Hassawi-1 and its hybrid. Moreover, the lengths of these large repeats had been increased with the longest repeat of 96,168 bp and 96,165 bp in Hassawi-1 and Hassawi-2, respectively. All large repeats in the Hassawi rice match in the forward direction. Compared to 93–11 and Nipponbare, the structure of the Hassawi mt genomes had been re-organized to accumulate more repeats in the large repeat regions ( Figure 6A ). This unusual genomic organization is also detectable by plotting the syntenic regions between Hassawi-1 and either 93–11 or Nipponbare ( Figure 6B ). Longest repeats are clearly identifiable, which are accounted for 78% of the total repeat length in both Hassawi-1 and Hassawi-2. The counts for functional genes in mt genomes are highly conserved among the Hassawi rice and other varieties, but the function of the lost sequences remains unknown. The dynamic genomic rearrangement may represent responses to environment pressures during mt genome evolution of the Hassawi rice and results in an enrichment of large repeats and deletions of some functionally unknown repetitive sequences.
Table 8. Large repeats (>1 kb) in mt genomes of the four rice cultivars.
| Cultivars | Length(bp) | Startposition 1 | Direction1 | Startposition 2 |
| Hassawi-1 | 96165 | 115395 | F | 233158 |
| 6123 | 208572 | F | 363729 | |
| 6030 | 91768 | F | 326335 | |
| 4078 | 90678 | F | 362639 | |
| 2988 | 91768 | F | 208572 | |
| 2988 | 326335 | F | 363729 | |
| Hassawi-2 | 96168 | 115369 | F | 233177 |
| 6124 | 208548 | F | 363754 | |
| 6031 | 91738 | F | 326356 | |
| 4079 | 90648 | F | 362664 | |
| 2989 | 91738 | F | 208548 | |
| 2989 | 326356 | F | 363754 | |
| 93–11 | 46082 | 341366 | F | 422479 |
| 37257 | 0 | F | 95060 | |
| 15084 | 153411 | F | 476431 | |
| 9917 | 37259 | F | 132319 | |
| 8800 | 215462 | F | 274582 | |
| 7214 | 279303 | F | 410259 | |
| 6322 | 145536 | F | 468556 | |
| 4079 | 220183 | F | 410259 | |
| 1551 | 151859 | F | 474879 | |
| 1304 | 149275 | P | 207170 | |
| 1304 | 207170 | P | 472295 | |
| Nipponbare | 45584 | 340412 | F | 421499 |
| 43760 | 0 | F | 94527 | |
| 10305 | 157114 | F | 480215 | |
| 10028 | 147085 | F | 470186 | |
| 5865 | 279707 | F | 410628 | |
| 4078 | 219233 | F | 409280 | |
| 3660 | 219651 | F | 409698 | |
| 3103 | 214512 | F | 273638 | |
| 2730 | 220581 | F | 279707 | |
| 2625 | 144459 | F | 467560 | |
| 2034 | 217616 | F | 276742 | |
| 1347 | 278359 | F | 409280 | |
| 1225 | 45050 | F | 139577 |
Note: 1. F and P stand for forward and palindromic matches, respectively.
Figure 6. Circos diagram illustrating genome rearrangement and repeat distribution of mt genomes between Hassawi-1 and 93–11 (A) and between Hassawi-1 and Nipponbare (B).

The first circle (from outside) displays different genomes (color-coding) and genes (blocks). The second circle displays repeat distribution along genomes. The connecting lines inside the circles join syntenic regions with direct (blue) and reversed matches (red) between two genomes.
The Origin of the Hassawi Rice
According to the breeding record ( Figure 7 ), Hassawi-2 is a hybrid between the wild-type Hassawi-1 and an indica cultivar IR1112 (the International Rice Research Institute, IRRI). There has been a limited literature about the genetic background of both Hassawi rice cultivars. Tracing back to IR1112, we found that IR1112 is a cross between IR262-43-8-11(maternal parent) and IR262-43-8-11/KDM105 (paternal parent), and both of its parental cultivars are descendants of IR262. From a cross of Peta and Peta*2/TN1, IR262 has a maternal inheritance of Peta that is an indica variety from Indonesia. The molecular phylogeny analysis based on whole cp genomes of five rice cultivars including wide rice Oryza. nivara, showed that both Hasswi-1 and Hassawi-2 had a common indica ancestor, which was closely related to O. nivara ( Figure 8 ). Moreover, recent research about resequencing 50 accessions of cultivated and wild rice revealed that indica was very closely related to O.nivara, whereas japonica was closer to Oryza. rufipogon and father from O.nivara [30]. The chloroplasts, together with mitochondria of higher plants, are maternally inherited and have their specific replication and DNA repair systems [24], [31], whereas the nuclear genome is bi-parentally inherited. With uniparental inheritance, organellar genomes are often used for tracing phylogenetic relations [31]. Examining variations in both cp and mt genomes between Hassawi-1 and Hassawi-2 ( Figure 8 ), we conclude that Hassawi-1 and IR1112 are the paternal and maternal parents of Hassawi-2, respectively, and the cp and mt genomes of Hassawi-2 are inherited from Peta in distant origin. Considering the divergence of rice organellar genomes among all the analyzed indica or japonica varieties and between Hassawi-1 and Hasswi-2, we suggest that the wild-type Hassawi rice is a descendant of Peta, which had adapted to the current or similar environment some hundreds or even thousands of years ago and it would be interesting to know the particular history and origin of the Hassawi rice for the sake of both science and civilization studies. Nevertheless, understanding the inheritance of the wild-type Hassawi and its hybrid provides important genetic information for their future breeding as well as genetic and molecular studies.
Figure 7. The origins of Hassawi rice and it’s hybrid.
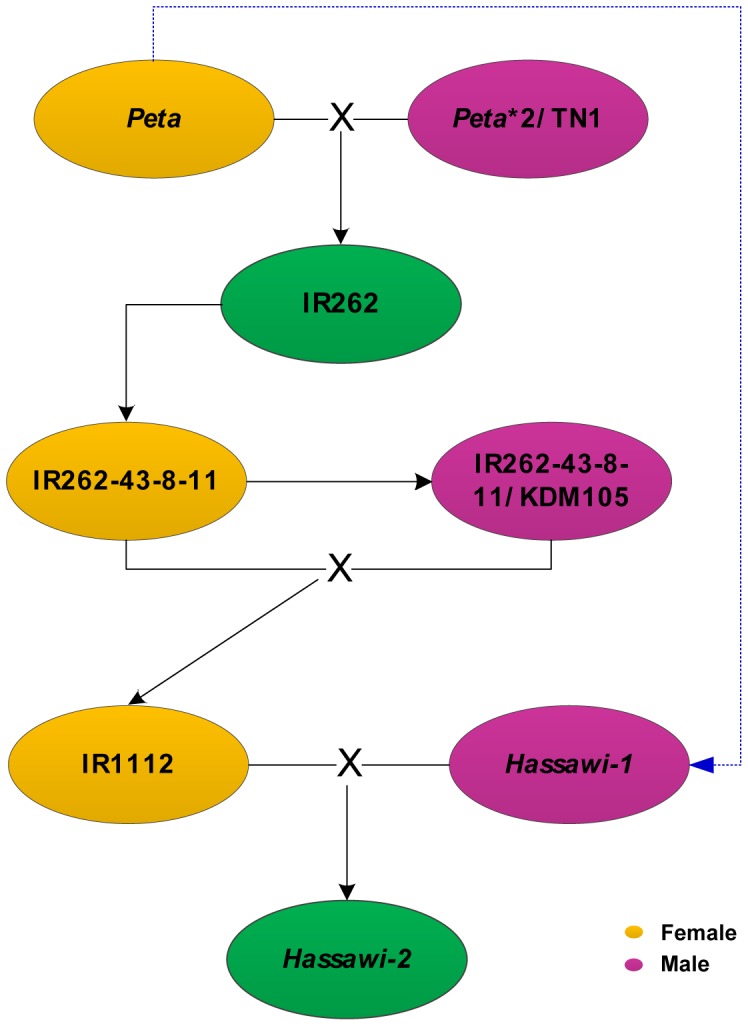
The blue line predicts the origin of wild-type Hassawi rice from the Indonesian variety Peta.
Figure 8. Phylogenetic relationships of 5 rice cultivars as determined from whole cp genomes.

Support values are shown for nodes as maximum likelihood bootstrap. The scale bar denotes substitutions per site. The histogram presents the variations (Insertion, deletion and SNP) in both cp and mt genomes between Hassawi-1 and Hassawi-2.
Conclusion
We report here the complete cp and mt genome assemblies of the wild-type Hassawi and its hybrid, and demonstrate their high degree of conservation in gene content and order among the sequenced rice varieties. Although functional genes among rice mt genomes are also conserved, their gene order, genome structure, and genome size are often variable. Our analyses on sequence variations, including SNPs, InDels, and RCVs in both cp and mt genome assemblies of the Hassawi rice provide detailed genetic information for genetically differentiating the two Hassawi cultivars. A greater number of large and enriched repeats are found in the Hassawi mt genomes as compared to other sequenced rice varieties. This observation is also supported by the distribution of SSRs, which also shows a higher density in the Hassawi rice when compared to those of other rice varieties. Recombination of mt genomes is prevalent in the Hassawi rice and results in complex genome rearrangements. As in the other plant, this phenomenon leads us to believe that such a repeat redistribution in mitochondrial genome may play a role in maintaining genome stability. As a final note, sequence variation data acquired in this study provide strong evidence for the origin of the Hassawi rice and its hybrid: both may be descendents of an Indonesian variety Peta albeit through different routes and at different time in history.
Materials and Methods
Genome Sequencing and Assembly
Both Hassawi rice cultivars were collected from Al-Hassa, Kingdom of Saudi Arabia. We extracted genomic DNA from 50 g young green leaves according to a CTAB-based method [32] and constructed libraries according to the GS FLX Titanium general preparation protocol, started with 5 g purified DNA. The ssDNA libraries were amplified with emulsion-PCR and enriched, and the samples were sequenced on Roche/454 GS FLX platform. In addition, two mate pair libraries for both cultivars were constructed by following SOLiD Library Preparation Guide (SOLiD 4.0). 20 µg or more genomic DNA was used for sequencing in SOLiD 4.0 instrument, which depending on two different insert sizes (500–1000 bp and 1000–3000 bp).
We extracted cp and mt genome sequence reads from whole genome sequencing data generated from both 454 GS FLX and SOLiD 4.0 platforms and assembled the 454 GS FLX reads based on a protocol we developed recently[14]. For the cp genome assembly, we filtered cp reads from the raw data according to the three known rice cp genome sequences. The clean cp reads were assembled into contigs by using Newbler (v2.6). In the mt genome assembly, we first assembled the raw data with Newbler, and then used Blast tool to filter for the mt contigs that were aligned to the known rice mt genomes. The mt contigs are not usually clean enough as they often contain cp genome sequences. We also used information on unknown mt contigs and read coverage for the removal of cp sequence contaminations. At the end, we validated the organellar genome assemblies with SOLiD sequencing data.
Genome Annotation
We used DOGMA for cp genome annotation (Dual Organellar GenoMe annotator)[33] and manually corrected start and stop codons. We annotated mt genome based on aligning sequences to the known rice mt genomes using NCBI BlastX and BlastN tools. We carried out all BlastN and BlastX searches using the blastall executable (version 2.2.25) with default settings (e-value 1e-10). Protein-coding genes, rRNAs, and tRNAs were identified by using the plastid/bacterial genetic code. We also used tRNAscan-SE[34] to corroborate tRNA boundaries identified by BlastN.
Genome Comparison Analysis
SSRs were identified and localized by using the Simple Sequence Repeat Identification Tool (SSRIT) [35] that identifies perfect nucleotide repeats of mono-, di-, tri- tetra-, tetra-, penta-, and hexa-nucleotides, and those equal or greater than three repeat units were collected except monomers. Intersubspecific polymorphisms were first identified based on the MUMmer package (v3.06) [36]. The results were then acquired by using a custom-designed Perl script and confirmed through careful visual inspection. Intravarietal polymorphisms were identified by using Newbler (v2.6) and Bioscope (v1.3) software, for 454 data and SOLiD data, respectively. We carried out repeat sequence analysis using the REPuter web-based interface (http://bibiserv.techfak.uni-bielefeld.de/reputer) [37], including forward, palindromic, reverse, and complemented repeats with a minimal length of 50 bp. Cp-derived sequences are identified with BlastN search of mt genomes against annotated cp genomes (Identity ≥80%, E-value ≤1e-5, and Length ≥50 bp). The cp-derived sequences were then aligned to all known plant mt genomes by using BlastN (Identity ≥80%, E-value ≤1e-5, and Coverage ≥50%). The syntenic regions of cp and mt genomes between different cultivars were detected by using Nucmer of the MUMmer package (v3.06) [36] with 50-bp exact minimal match. The annotated cp and mt genome features including gene coordinate, genome structures in cp genomes, repeats in mt genomes and different genome variations were used to draw genome maps using Circos software [38].
Phylogenomic Analysis
The whole cp genomes of five rice cultivars were aligned using the program MAFFT version 6 [39] and adjusted manually where necessary. The unambiguously aligned DNA sequences were used for phylogenetic tree construction. Maximum likelihood method analysis was performed with PhyML v3.05 [40] under GTR (General time Reversible) model of nucleotide substitution to construct phylogenetic tree. 1,000 bootstrap replications were used to estimate the confidence of brand points. We obtained the best tree after heuristic search with the help of Modelgenerator [41].
Accession Numbers
The complete sequence of the four genomes was deposited to GenBank with accession numbers: JN861109, JN8611010, JN8611011, and JN8611012 for Hassawi-1 chloroplast, Hassawi-2 chloroplast, Hassawi-1 mitochondrial, and Hassawi-2 mitochondrial genomes, respectively. Other sequences used for comparative analysis are: NC_008155, NC_001320, NC_005973, NC_007886, and NC_011033 from 93–11 chloroplast, Nipponbare chloroplast, O. nivara chloroplast, 93–11 mitochondrial, and Nipponbare mitochondrial genomes, respectively.
Supporting Information
The cp genome assembly of Hassawi-1 from 454 sequencing reads. The large single copy, the small single copy, and the inverted repeats are shown in blue, green, and red, respectively. The boxes stand for contigs and the lines indicate the link (overlapping) between two contigs. The numbers in the boxes show contig name, length, and read depth. The numbers on lines are reads spanning two contigs. This figure was generated with Graphviz (http://www.graphviz.org/).
(PDF)
The cp genome assembly of Hassawi-2 from 454 sequencing reads. The large single copy, the small single copy, and the inverted repeats are shown in blue, green, and red, respectively. The boxes stand for contigs and the lines indicate the link (overlapping) between two contigs. The numbers in the boxes show contig name, length, and read depth. The numbers on lines are reads spanning two contigs.
(PDF)
The mt genome assembly of Hassawi-1 from 454 sequencing reads. The boxes stand for contigs and the lines indicate the link (overlapping) between two contigs. The numbers in the boxes show contig name, length, and read depth. The numbers on lines are reads spanning two contigs. The boxes in red show contigs with matched the mt genomes of other rice cultivars.
(PDF)
The mt genome assembly of Hassawi-2 from 454 sequencing reads. The boxes stand for contigs and the lines indicate the link (overlapping) between two contigs. The numbers in the boxes show contig name, length, and read depth. The numbers on lines are reads spanning two contigs. The boxes in red show contigs with matched the mt genomes of other rice cultivars.
(PDF)
Funding Statement
The authors thank the Joint Center of Excellence for Genomics, King Abdulaziz City for Science and Technology (KACST) and Chinese Academy of Sciences. KASCT had a role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bajaj S, Mohanty A (2005) Recent advances in rice biotechnology–towards genetically superior transgenic rice. Plant Biotechnology Journal 3: 275–307. [DOI] [PubMed] [Google Scholar]
- 2. Yu J, Tian XJ, Zheng J, Hu SN (2006) The rice mitochondrial genomes and their variations. Plant Physiology 140: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al-Mssallem MQ, Hampton SM, Frost GS, Brown JE (2011) A study of Hassawi rice (Oryza sativa L.) in terms of its carbohydrate hydrolysis (in vitro) and glycaemic and insulinaemic indices (in vivo). Eur J Clin Nutr 65: 627–634. [DOI] [PubMed] [Google Scholar]
- 4. Al-Mssallem IS, Al-Mssallem MQ (1997) Study of glutelin (storage protein of rice) in Al-Hassawi rice grains. Arab Gulf Journal of Scientific Research 15: 633–646. [Google Scholar]
- 5. Tang J, Xia Ha, Cao M, Zhang X, Zeng W, et al. (2004) A Comparison of Rice Chloroplast Genomes. Plant Physiology 135: 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hiratsuka J, Shimada H, Whittier R, Ishibashi T, Sakamoto M, et al. (1989) The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Molecular & General Genetics 217: 185–194. [DOI] [PubMed] [Google Scholar]
- 7. Kadowaki K, Notsu Y, Masood S, Nishikawa T, Kubo N, et al. (2002) The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Molecular Genetics and Genomics 268: 434–445. [DOI] [PubMed] [Google Scholar]
- 8. Kato A, Fujita S, Komeda Y (2000) Identification and characterization of the gene encoding the mitochondrial elongation factor G in rice. DNA Sequence 11: 395–404. [DOI] [PubMed] [Google Scholar]
- 9. Gray MW, Burger G, Lang BF (1999) Mitochondrial Evolution. Science 283: 1476–1481. [DOI] [PubMed] [Google Scholar]
- 10. Mackenzie S, McIntosh L (1999) Higher Plant Mitochondria. The Plant Cell Online 11: 571–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Imelfort M, Duran C, Batley J, Edwards D (2009) Discovering genetic polymorphisms in next-generation sequencing data. Plant Biotechnology Journal 7: 312–317. [DOI] [PubMed] [Google Scholar]
- 12. Barker GL, Edwards KJ (2009) A genome-wide analysis of single nucleotide polymorphism diversity in the world’s major cereal crops. Plant Biotechnology Journal 7: 318–325. [DOI] [PubMed] [Google Scholar]
- 13. Rajendrakumar P, Biswal AK, Balachandran SM, Srinivasarao K, Sundaram RM (2007) Simple sequence repeats in organellar genomes of rice: frequency and distribution in genic and intergenic regions. Bioinformatics 23: 1–4. [DOI] [PubMed] [Google Scholar]
- 14. Zhang T, Zhang X, Hu S, Yu J (2011) An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods 7: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X, Shi X, Hao B (2002) The transfer RNA genes in Oryza sativa L. ssp. indica. Sci China C Life Sci 45: 504–511. [DOI] [PubMed] [Google Scholar]
- 16. Zhang T, Fang Y, Wang X, Deng X, Zhang X, et al. (2012) The complete chloroplast and mitochondrial genome sequences of Boea hygrometrica: insights into the evolution of plant organellar genomes. PLoS ONE 7: e30531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Young HA, Lanzatella CL, Sarath G, Tobias CM (2011) Chloroplast Genome Variation in Upland and Lowland Switchgrass. PLoS ONE 6: e23980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang M, Zhang X, Liu G, Yin Y, Chen K, et al. (2010) The Complete Chloroplast Genome Sequence of Date Palm (Phoenix dactylifera L.). Plos One 5: e12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greiner S, Rauwolf U, Meurer J, Herrmann RG (2011) The role of plastids in plant speciation. Molecular Ecology 20: 671–691. [DOI] [PubMed] [Google Scholar]
- 20. Xue JY, Liu Y, Li LB, Wang B, Qiu YL (2010) The complete mitochondrial genome sequence of the hornwort Phaeoceros laevis: retention of many ancient pseudogenes and conservative evolution of mitochondrial genomes in hornworts. Current Genetics 56: 53–61. [DOI] [PubMed] [Google Scholar]
- 21. Alverson AJ, Wei XX, Rice DW, Stern DB, Barry K, et al. (2010) Insights into the Evolution of Mitochondrial Genome Size from Complete Sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Molecular Biology and Evolution 27: 1436–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kubo T, Nishizawa S, Sugawara A, Itchoda N, Estiati A, et al. (2000) The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNA(Cys)(GCA). Nucleic acids research 28: 2571–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clifton SW, Minx P, Fauron CMR, Gibson M, Allen JO, et al. (2004) Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiology 136: 3486–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian XJ, Zheng J, Hu SN, Yu J (2006) The rice mitochondrial genomes and their variations. Plant Physiology 140: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Powell W, Morgante M, McDevitt R, Vendramin GG, Rafalski JA (1995) Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc Natl Acad Sci U S A 92: 7759–7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu H, Cui P, Zhan K, Lin Q, Zhuo G, et al. (2011) Comparative analysis of mitochondrial genomes between a wheat K-type cytoplasmic male sterility (CMS) line and its maintainer line. Bmc Genomics 12: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palmer JD, Herbon LA (1988) Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. Journal of Molecular Evolution 28: 87–97. [DOI] [PubMed] [Google Scholar]
- 28. Maréchal A, Brisson N (2010) Recombination and the maintenance of plant organelle genome stability. New Phytologist 186: 299–317. [DOI] [PubMed] [Google Scholar]
- 29. Fujii S, Kazama T, Yamada M, Toriyama K (2010) Discovery of global genomic re-organization based on comparison of two newly sequenced rice mitochondrial genomes with cytoplasmic male sterility-related genes. Bmc Genomics 11: -. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu X, Liu X, Ge S, Jensen JD, Hu F, et al. (2011) Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotech advance online publication. [DOI] [PubMed] [Google Scholar]
- 31. Kadowaki K, Nishikawa T, Vaughan DA (2005) Phylogenetic analysis of Oryza species, based on simple sequence repeats and their flanking nucleotide sequences from the mitochondrial and chloroplast genomes. Theoretical and Applied Genetics 110: 696–705. [DOI] [PubMed] [Google Scholar]
- 32. Gawel N, Jarret R (1991) A modified CTAB DNA extraction procedure for Musa and Ipomoea. Plant Molecular Biology Reporter 9: 262–266. [Google Scholar]
- 33. Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20: 3252–3255. [DOI] [PubMed] [Google Scholar]
- 34. Lowe TM, Eddy SR (1997) tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, et al. (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11: 1441–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, et al. (2004) Versatile and open software for comparing large genomes. Genome Biol 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, et al. (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic acids research 29: 4633–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, et al. (2009) Circos: An information aesthetic for comparative genomics. Genome Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, et al. (2010) New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology 59: 307–321. [DOI] [PubMed] [Google Scholar]
- 41. Keane T, Creevey C, Pentony M, Naughton T, Mclnerney J (2006) Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. Bmc Evolutionary Biology 6: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The cp genome assembly of Hassawi-1 from 454 sequencing reads. The large single copy, the small single copy, and the inverted repeats are shown in blue, green, and red, respectively. The boxes stand for contigs and the lines indicate the link (overlapping) between two contigs. The numbers in the boxes show contig name, length, and read depth. The numbers on lines are reads spanning two contigs. This figure was generated with Graphviz (http://www.graphviz.org/).
(PDF)
The cp genome assembly of Hassawi-2 from 454 sequencing reads. The large single copy, the small single copy, and the inverted repeats are shown in blue, green, and red, respectively. The boxes stand for contigs and the lines indicate the link (overlapping) between two contigs. The numbers in the boxes show contig name, length, and read depth. The numbers on lines are reads spanning two contigs.
(PDF)
The mt genome assembly of Hassawi-1 from 454 sequencing reads. The boxes stand for contigs and the lines indicate the link (overlapping) between two contigs. The numbers in the boxes show contig name, length, and read depth. The numbers on lines are reads spanning two contigs. The boxes in red show contigs with matched the mt genomes of other rice cultivars.
(PDF)
The mt genome assembly of Hassawi-2 from 454 sequencing reads. The boxes stand for contigs and the lines indicate the link (overlapping) between two contigs. The numbers in the boxes show contig name, length, and read depth. The numbers on lines are reads spanning two contigs. The boxes in red show contigs with matched the mt genomes of other rice cultivars.
(PDF)