

Abstract
The NF-κB family member p65 is central to inflammation and immunity. The purpose of this study was to identify and characterize evolutionary conserved genes modulating p65 transcriptional activity. Using an RNAi screening approach, we identified chaperonin containing TCP1 subunit η (CCTη) as a regulator of Drosophila NF-κB proteins, Dorsal and Dorsal-related immunity factor (Dif). CCTη was also found to regulate NF-κB-driven transcription in mammalian cells, acting in a promoter-specific context, downstream of IκB kinase (IKK). CCTη knockdown repressed IκBα and CXCL2/MIP2 transcription during the early phase of NF-κB activation while impairing the termination of CCL5/RANTES and CXCL10/IP10 transcription. The latter effect was associated with increased DNA binding and reduced p65 acetylation, presumably by altering the activity of histone acetyltransferase CREB-binding protein (CBP). We identified p65 lysines (K) 122 and 123 as target residues mediating the CCTη-driven termination of NF-κB-dependent transcription. We propose that CCTη regulates NF-κB activity in a manner that resolves inflammation.
Introduction
Nuclear factor κB (NF-κB) proteins comprise a family of eukaryotic transcription factors that control the expression of a large number of genes regulating inflammation and immunity as well as developmental processes including cellular growth and apoptosis [1]. Unfettered NF-κB activation has been associated with the pathogenesis of a number of inflammatory diseases [2]. In their active form, NF-κB proteins are nuclear homo- or hetero-dimeric complexes composed of p65/RelA, RelB, cRel, p105/p50, and p100/p52. The prototypical and most ubiquitously expressed NF-κB dimer is composed of p50 and p65 subunits where p65 is the main transcriptional activator [3].
Under homeostatic conditions, NF-κB activity is constitutively repressed by its interaction with cytoplasmic NF-κB inhibitors (IκB) [4]. While inducible degradation of IκB molecules is a central mechanism regulating p65 transcriptional activity, posttranslational modifications are also important for its activity [5], [6]. These include phosphorylation, which modulates DNA binding, interactions with other proteins as well as p65 stability [5]. Phosphorylation often precedes other posttranslational modifications such as acetylation and ubiquitination [7]–[10]. For example, p65 phosphorylation at serine (S) 276 facilitates interaction with CREB-binding protein (CBP)/p300 and diminishes histone deacetylase 1 (HDAC1) binding, leading to p65 acetylation [11].
Lysine (K) acetylation is mostly a nuclear event [6], controlling p65 transcriptional activity [12]–[15] as well as the duration of NF-κB activation via regulation of DNA binding [13], [16] and association with IκBα [13]. Specific p65 residues may be preferentially targeted by different histone acetyltransferases (HAT), which include CBP, p300 and p300/CBP-associated factor (P/CAF) [13], [14], [16]. The acetylation status of p65 is controlled by the opposing activities of HATs and HDACs including HDAC1 [17], HDAC3 [12], [16], SIRT1 [18], and SIRT2 [19]. Beside acetylation, p65 lysine residues can be modified by methylation and ubiquitination resulting in altered transcriptional activity or proteasomal degradation [20]–[24].
NF-κB proteins and the signal transduction pathways leading to their activation are highly evolutionary conserved. As in mammals, Drosophila p65 homologues Dorsal and Dif are constitutively inhibited by the IκB like molecule Cactus [25], [26]. Toll receptor activation leads to Cactus degradation, Dorsal and Dif nuclear translocation and transcription of NF-κB dependent genes such as Drosomycin and Cecropin [27]–[29]. Little is known about the impact of posttranslational modifications on Dorsal and Dif transcriptional activity. Yet, several phospho-acceptors as well as lysine residues targeted by posttranslational modifications in p65 are conserved in Dorsal and Dif. Supporting this view, phosphorylation of Dorsal S312 and S317, which correspond to p65 S276 and S281, can control Dorsal nuclear translocation [30], [31].
We set out to identify evolutionary conserved genes regulating NF-κB-dependent gene transcription using a functional RNAi based gene screen in Drosophila cells. We reveal the chaperonin containing TCP1 subunit eta (CCTη) as a novel gene regulating NF-κB acetylation and activity.
Materials and Methods
Cell Culture
Drosophila S2 cells were kindly provided by Monica Bettencourt Dias (Instituto Gulbenkian de Ciência, Oeiras, Portugal) [32] and were grown at 25°C in Schneider’s Drosophila medium (Invitrogen). HeLa and HEK293 cells were obtained from ATCC. Mouse embryonic fibroblasts (MEF) isolated from Rela −/− mice were kindly provided by Dr. Amer Beg (Moffitt Cancer Center, Tampa, FL) and have been previously described [33]. Rela −/− MEF were retrovirally transduced with human RelA wt, K122/123R, K221R or K310R mutants essentially as described [34]. Cells were grown at 37°C in DMEM (MediaTech) supplemented with 10% FBS, 100 units/ml penicillin G and 100 µg/ml streptomycin B (all Atlanta Biologicals) in a humidified atmosphere containing 5% CO2.
Plasmid Constructs
Drosomycin-luciferase (Drs-luc) and Toll 10b expression vectors were kindly provided by Dr. Mika Ramet (University of Tampere, Tampere, Finland) and were described elsewhere [35]. Dorsal and Dif were amplified from the original pAct-dl and pAct-Dif vectors (kind gift from Dr. Ylva Engstrom, Stockholm University, Stockholm, Sweden [29]) by PCR (Dorsal 5′-GAT CCT CGA GAT GTT TCC GAA CCA GAA CAA TGG AG-3′ and 5′-TCG ATC TAG ACG TGG ATA TGG ACA GGT TC-3′; Dif 5′-GAT CCT CGA GAT GTT TGA GGA GGC TTT C-3′ and 5′-TCG ATC TAG ATT TGA ATG GCT GAA TTC CCA AG-3′) and cloned (EcoRV/XbaI) and (XhoI/XbaI) into pAc5.1/V5-HisA vector (Invitrogen), respectively. NF-κB luciferase reporter construct, i.e. κB-luc, has been described elsewhere [36]. Scrambled, CCTη, CCTα and CCTζ shRNA constructs were cloned by inserting annealed oligonucleotides containing a non-targeting (5′-GAG TGT TTG AGT TTG AGA TCC-3′) or a unique 19–21 bp sequence derived from the mRNA transcript of CCTη (5′-GCC ACA AAC ATT CTC AAC A-3′), CCTα (5′-AAA TAC TAA GGC TCG TAC GTC-3′) or CCTζ (5′-AAG TCT GTG GCG ATT CAG ATA-3′) gene between the unique BglII and HindIII restriction sites of the pSUPER vector (OligoEngine). The N-terminal myc-tagged p65 expression vector has been described [37]. C-terminal HA-tagged CBP was expressed from a pcDNA3 vector. p65 K122/123R, K221R and K310R expression vectors were generated by site-directed mutagenesis and cloned into the pLXIH vector as described [34]. All constructs were verified by automated DNA sequencing using dye termination chemistry.
Transient Transfection and Reporter Assay
S2 cells were cultured in 12-well plates (5×105 cells per well) and a day after exposed to 1 µg of total DNA (150 ng Drs-luc, 5 ng Toll 10b, 50 ng Dorsal, 50 ng Dif; the total amount of DNA was kept constant by using empty vector) and 5 µl of Cellfectin transfection reagent according to manufacturer’s instructions (Invitrogen) in Drosophila SFM medium (Invitrogen) for 12 h at 25°C. Cells were washed and transferred into 96-well plates containing lacZ dsRNA (900ng per 5×104 cells). After incubation for three days, firefly (Luc) and Renilla (Ren) luciferase activity were measured using Dual-luciferase Reporter Assay System (Promega). HEK293 cells were grown in 12-well plates and transfected at 90% confluency. Cells were exposed to 1 µg of total DNA (300 ng of reporter plasmid, 500 ng of shRNA plasmid and 40 ng of CMV enhancer/β-gal control plasmid; the total amount of DNA was kept constant by using empty vector) and 1 µl of Lipofectamine 2000 reagent according to manufacturer’s instructions (Invitrogen) in DMEM for 3 h. After addition of FBS to a final concentration of 10%, cells were incubated (three days, 37°C), stimulated with TNF (10 ng/ml, 6 h), lysed with passive lysis buffer (Promega) and supernatants were assayed for luciferase and β-gal activity as described elsewhere [36].
RNA Interference, Transient Transfection and Reporter Assay
S2 cells were cultured in 12-well plates and transfected as above (150 ng Drs-luc reporter plasmid, 5 ng Toll 10b plasmid; the total amount of DNA was kept constant by using empty vector). Cells were washed, diluted to 1×106 cells/ml and a total of 10 µl of cells were added to the dsRNA-containing 384-well plate (250 ng dsRNA/well), resulting in a final concentration of 1×104 cells per well. Plates were gently centrifuged, cells were incubated (30 min, room temperature) and 30 µl of Schneider’s medium containing 15% FBS was added. Plates were sealed to prevent evaporation and incubated for three days at 25°C. Luciferase (Luc) and Renilla (Ren) activity was measured using Dual-luciferase Reporter Assay System according to manufacturer’s instruction (Promega). 89 wells not containing dsRNA on each experimental plate were used to calculate the average Luc and Ren activities. The positive control in screen and data processing, dsRNA of DIAP1 inhibitor of apoptosis, led to a very significant reduction of Luc and Ren signals in two out of three experimental plates that were considered for further analysis. Experimental data was normalized by setting the average Luc and Ren values of wells not containing dsRNA to 1. dsRNA treatments that modulated Luc expression (values lying outside the mean ±2SD boundaries) with no effect on Ren values (values lying within the mean ± SD boundaries) in two experiments were considered indicative for putative candidate genes.
dsRNA Synthesis and Screen Confirmation
dsRNAs targeting putative genes identified in the screen were generated by in vitro transcription of a PCR-generated DNA template (RNAi probes, FlyBase) containing the T7 promoter sequence on both ends (RiboMAXTM Large Scale RNA Production System-T7, Promega) and purified using Mini Quick Spin RNA Columns (Roche). Transiently transfected S2 cells (150 ng Drs-luc, 50 ng Dorsal, 50 ng Dif, 5 ng Toll 10b per 5×105 cells) were exposed to dsRNA (900 ng per 5×104 cells) in 96-well plate, incubated for three days and Luc and Ren activity were measured.
CCTη Knockdown by Small Interfering RNA (siRNA)
siRNA-mediated knockdown of CCTη was performed in a 12-well plate format in HeLa (6×104 cells per well), HEK293 (3×105 cells per well) and MEF (5×104 cells per well) cells. Cells were exposed to 50 nM siGENOME Non-Targeting siRNA #1 or CCTη siRNA and 1 µl/ml of DharmaFECT1 transfection reagent according to manufacturer’s instructions (Thermo Scientific). CCTη siRNA target sequences were human: 5′-GCC ACA AAC ATT CTC AAC A-3′ and mouse: 5′- GCC ACA AAC ATC CTC AAC A -3′.
Gene Expression Analysis
Real-time quantitative PCR (qPCR) was carried out using SYBR Green chemistry (Invitrogen) on a Chromo4 continuous fluorescence monitoring thermocycler (MJ Research) as described previously [34]. Relative transcript levels were determined by normalization to the housekeeping gene HPRT. Primers used for expression analysis in HeLa cells were: IκBα (5′-TCC TGT TGA AGT GTG GGG CTG ATG-3′ and 5′-CCT CCA AAC ACA CAG TCA TCA T-3′), CXCL2 (5′-CAC TCA AGA ATG GGC AGA AAG-3′ and 5′-TCA GGA ACA GCC ACC AAT AAG-3′), IL8 (5′-TCC TGA TTT CTG CAG CTC TGT-3′ and 5′- TGT GGT CCA CTC TCA ATC ACT C-3′), TNF (5′-AGT GCT GGC AAC CAC TAA GAA-3′ and 5′-ATT CCA GAT GTC AGG GAT CAA-3′), CXCL10 (5′-CCT CTC CCA TCA CTT CCC TAC-3′ and 5′-GCT GAT TTG GTG ACC ATC ATT-3′), CCL5 (5′-TGC CCA CAT CAA GGA GTA TTT-3′ and 5′-CCA TCC TAG CTC ATC TCC AAA-3′) and HPRT (5′-TTC TGT GGC CAT CTG CTT AGT-3′ and 5′-GCC CAA AGG GAA CTG ATA GTC-3′). Primers used for expression analysis in MEF were: Cxcl10 (5′-AAG TCA GCC AAT CAG GAC TCA-3′ and 5′-GTT GGC TCG GGA TGT CTC T-3′). Primers for the mouse Hprt gene have been described elsewhere [34]. Expression levels of unstimulated, scrambled siRNA-exposed cells were set to 1, and fold induction for other experimental groups was calculated. CXCL10 mRNA levels of TNF-unstimulated Hela cells were not consistently detectable; expression level of CXCL10 mRNA in CCTη siRNA-exposed cells 16 h after TNF was set to 100 and relative values for other experimental groups were calculated.
Preparation of Subcellular Extracts, Electrophoretic Mobility Shift Assay (EMSA) and Western Blotting
HeLa cells were transfected with siRNA as described above and incubated for three days before stimulation with TNF (10 ng/ml). Preparation of cytoplasmic/nuclear extracts and EMSA were carried out as described previously [37]. Briefly, cells were lysed in hypotonic buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5% TritonX-100, supplemented with protease inhibitors) and centrifuged (600 g, 5 min, 4°C). Supernatant (cytoplasmic fraction) was transferred into a new tube and the remaining pellet (nuclear fraction) was washed once in hypotonic buffer, lysed (20 mM HEPES pH 7.9, 25% glycerol, 1.5 mM MgCl2, 400 mM NaCl, 0.2 mM EDTA, 1 mM PMSF, 1 mM DTT, protease inhibitors), and cleared by centrifugation. Nuclear extracts were incubated with 100,000 cpm of double-stranded [γ-32P]ATP-radiolabled NF-κB oligonucleotide (5′-AGT TGA GGG ACT TTC CCA GGC-3′; 30 min, room temperature; PerkinElmer) and the resulting DNA-protein complexes were separated on a 6% polyacrylamide gel in Tris/glycine/EDTA buffer pH 8.5. Experimental data were analyzed by setting the net intensity of EMSA bands to 100 and calculating the percentage of intensity for each data point. For supershift analysis, labeled DNA-protein complexes were incubated with 1 µg of p65 (sc-8008x, Santa Cruz) or p50 (sc-114x, Santa Cruz) specific antibodies or non-immune rabbit IgG (I5006, Sigma) (1 h, 4°C) prior to separation. For western blotting, proteins contained in cytoplasmic and nuclear fractions were resolved by electrophoresis on 10% SDS-polyacrylamide gels, transferred to PVDF membranes and detected with antibodies against IκBα (IMG-127A, Imgenex), p65 (sc-372, Santa Cruz), p50 (sc-7178, Santa Cruz), CCTη (sc-13889, Santa Cruz), GAPDH (MAB374, Millipore) and Sp1 (sc-59, Santa Cruz). Primary antibodies were detected using HRP-conjugated secondary antibodies (Santa Cruz).
Immunoprecipitation and Protein Acetylation
HEK293 cells were grown in 12-well plates and transfected with calcium phosphate. Cells were exposed to 1.6 µg of total DNA (50 ng of myc-tagged p65, 200 ng of HA-tagged CBP expressing construct and 1 µg of shRNA plasmid; the total amount of DNA was kept constant by using empty vector), incubated for three days and stimulated with TNF (10 ng/ml, 30 min). Cells were lysed (50 mM HEPES pH 7.9, 250 mM NaCl, 1% NP-40, 1 mM EDTA, protease inhibitors) and centrifuged (10 min, 16000 g, 4°C). Supernatants were incubated with 15 µl c-myc-conjugated agarose beads (2 h, 4°C, Sigma). For CBP detection, cells were transfected with CCTη or scrambled shRNA plasmid, stimulated with TNF (10 ng/ml, 30 min) and cell extracts were incubated with 1 µg CBP antibody (o/n, 4°C, sc-369, Santa Cruz) and 20 µl Protein-A Sepharose beads (1 h, 4°C, GE Healthcare). For CCTη detection, HEK293 cells were exposed or not to CCTη or scrambled siRNA and stimulated with TNF (10 ng/ml) for indicated times. Nuclear extracts were incubated with 1 µg TCP-1η antibody (sc-13889, Santa Cruz) and 20 µl Protein-A Sepharose beads. Precipitated proteins were washed in lysis buffer, resuspended in 2x SDS sample buffer, resolved by SDS-PAGE and subjected to western blotting with antibodies detecting acetylated lysine (9814S, Cell Signaling), p65 (sc-372, Santa Cruz), CBP or CCTη Primary antibodies were detected using Protein-A-HRP conjugates (Millipore) or respective secondary antibodies. Purity of nuclear extracts was confirmed using anti-GAPDH antibody (MAB374, Millipore).
Results
Inhibition of CCTη Expression Modulates NF-κB-Dependent Reporter Activity
To identify evolutionary conserved genes regulating the NF-κB pathway we employed a dsRNA library targeting 265 putative Drosophila kinases and kinase regulatory proteins. Drosomycin-driven luciferase (Drs-luc) expression can be induced by a constitutively active form of Toll (Toll 10b) as well as by Drosophila p65 homologues Dorsal and Dif [35], [38]. Drs-luc expression increased significantly in Drosophila S2 cells co-transfected with Toll 10b, Dorsal or Dif, as compared to cells transfected with Drs-luc alone (Fig. 1A). We used the same assay to perform an RNAi based gene screen in S2 cells transiently transfected with Drs-luc reporter, Toll 10b expression construct and a constitutively active pAct5-Renilla reporter. Firefly and Renilla luciferase values were plotted and dsRNA treatments that significantly modulated firefly luciferase expression (values lying outside the mean ±2SD) with no effect on Renilla luciferase values were further validated (Fig. 1B). We identified chaperonin containing Tcp-1 subunit η (CCTη), an evolutionary conserved protein not previously implicated in the regulation of NF-κB signaling. We confirmed that CCTη regulates NF-κB activity in a reporter assay in S2 cells. CCTη dsRNA inhibited Toll 10b-, Dorsal- and Dif-induced Drs-luc expression, as compared to control LacZ dsRNA (Fig. 1C).
Figure 1. Identification of CCTη as a novel gene regulating NF-κB transcriptional activity.

A: S2 cells were transiently transfected with the Drs-luc reporter plus or minus Dorsal, Dif or Toll 10b and exposed to LacZ dsRNA. Illustrated data represent mean firefly luciferase normalized to Renilla luciferase units ± SEM (n≥11 derived from four independent experiments). B: S2 cells were transiently transfected with the Drs-luc reporter plus Toll 10b and seeded into wells of a 384-well plate containing 265 Drosophila kinases and kinase regulatory subunits. Firefly and Renilla luciferase values from two independent experimental plates were plotted. Control wells not containing dsRNA are illustrated with the light gray diamonds. C: S2 cells were transiently transfected as in A and exposed to CCTη or LacZ dsRNA. Illustrated data represent mean firefly luciferase normalized to Renilla luciferase units ± SEM (n = 9, derived from three independent experiments). D–F: HEK293 cells were transiently transfected with κB-luc reporter together with CCTη, CCTα, CCTζ or scrambled (sc) shRNA, incubated for three days and stimulated with TNF (10 ng/ml, 6 h) or left untreated (control). Results shown are mean normalized firefly luciferase units ± SEM (n≥7, derived from three independent experiments).
To investigate whether CCTη controls mammalian NF-κB signal transduction pathway we assessed its effect on TNF-induced NF-κB reporter activity. HEK293 cells were transiently transfected with a κB-luc reporter and CCTη shRNA or scrambled shRNA as a control. In contrast to S2 cells, CCTη knockdown in mammalian cells enhanced TNF-driven reporter activity, as compared to scrambled shRNA transfected controls (Fig. 1D). The same result was observed when two other CCT subunits, CCTα and CCTζ, were targeted by shRNA (Fig. 1E, 1F), suggesting the involvement of the functional chaperonin CCT complex in the regulation of NF-κB activity in mammalian cells.
CCTη Modulates NF-κB-Dependent Gene Expression
The canonical NF-κB signal transduction pathway can be activated via different receptors, including TNF and IL-1 receptors, that initiate distinct signaling cascades converging at the level of the IκB kinase (IKK) complex [39]. To assess whether CCTη regulates both pathways we analyzed mRNA expression of NF-κB target genes by qPCR in HeLa cells transfected with CCTη or scrambled siRNA and stimulated with either TNF or IL-1β. We found that CCTη affects NF-κB-dependent gene expression in a promoter-specific context. Whereas IκBα and CXCL2 mRNA expression was reduced at 1 h after TNF stimulation in CCTη siRNA transfected cells, expression of TNF, IL-8, CXCL10 and CCL5 mRNA increased at 3 and/or 16h after TNF stimulation, as compared to scrambled siRNA transfected controls (Fig. 2A). Similarly to TNF stimulation, CCTη knockdown led to increased IL-8 and CXCL10 mRNA expression 3 and 16 h after IL-1β addition (Fig. 2B). This suggests the involvement of CCTη in initiation as well as termination of NF-κB-driven transcription, revealing a multi-faced role in regulating NF-κB-dependent genes, possibly acting downstream of IKKs.
Figure 2. CCTη modulates TNF and IL-1β-induced gene expression in a promoter specific way.
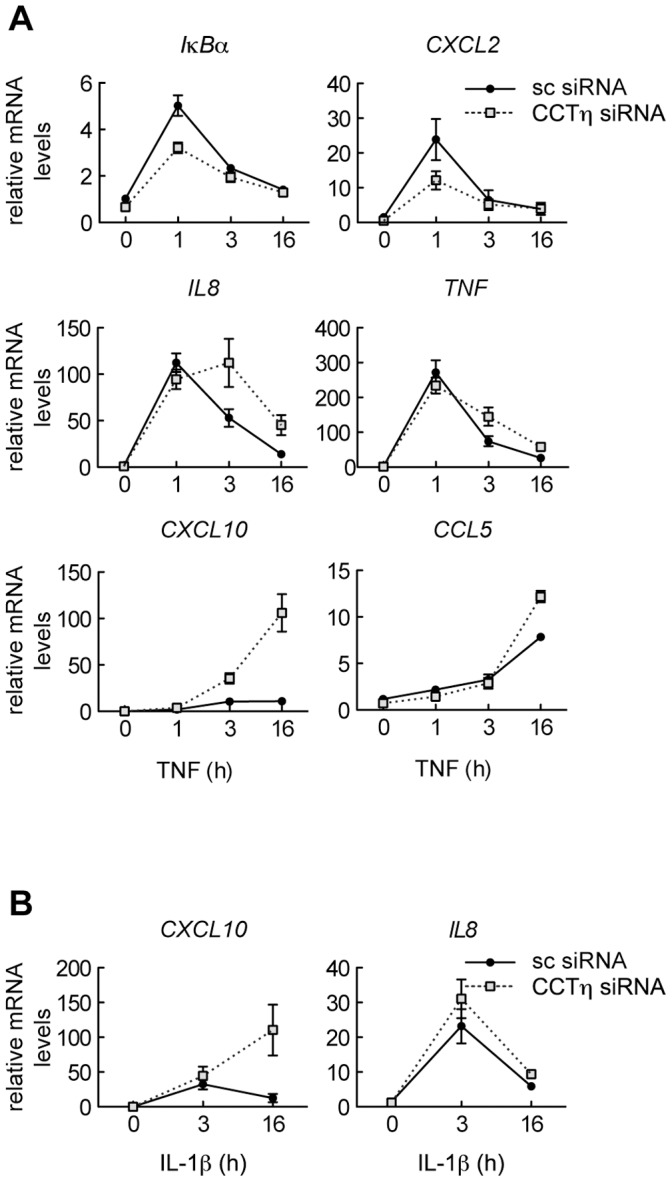
HeLa cells were exposed to CCTη or scrambled siRNA for three days and stimulated with A: TNF (10 ng/ml) or B: IL-1β (10 ng/ml) for indicated times. mRNA levels were analyzed by qPCR. Data represent mean relative mRNA levels ± SEM (n≥3).
CCTη Regulates NF-κB DNA Binding
Proteolytic IκB degradation is required for NF-κB nuclear translocation, binding to the κB enhancer and initiation of transcription [5]. In addition, NF-κB-dependent IκBα re-synthesis is critical for termination of NF-κB signaling pathway [40], [41]. Thus, we investigated whether CCTη modulates IκBα protein levels. CCTη knockdown had no apparent impact on TNF-induced IκBα degradation, as compared to scrambled siRNA transfected cells (Fig. 3A). In accordance, cytoplasmic and nuclear p65 and p50 protein levels in TNF-treated cells were not influenced by CCTη siRNA transfection (Fig. 3A, 3B), suggesting that CCTη exerts its effect independently of IκBα, presumably after p65 nuclear translocation. Indeed, we found that CCTη knockdown was associated with increased NF-κB binding to DNA κB consensus sequence at 3 and 16 h after TNF stimulation, while not affecting binding at 30 min and 1 h, as analyzed by EMSA (Fig. 4A). Resolved DNA protein complexes were identified as p50/p65 heterodimers by specific binding of these complexes to both p50 and p65 specific antibodies (Fig. 4B). Composition of NF-κB heterodimers was not changed at different time points after TNF stimulation or by CCTη knockdown (Fig. 4B), indicating that CCTη does not affect NF-κB dimer formation. This suggests that increased DNA binding at 3 and 16 h after TNF stimulation could account for increased expression of NF-κB target genes at these time points in CCTη siRNA transfected cells. Impaired gene expression at 1 h after TNF, on the other hand, cannot be explained by altered DNA binding. Several post-translational modifications that have been shown to modulate NF-κB transcriptional activity without affecting its DNA binding [6] could account for this effect.
Figure 3. CCTη does not influence NF-κB subcellular distribution.
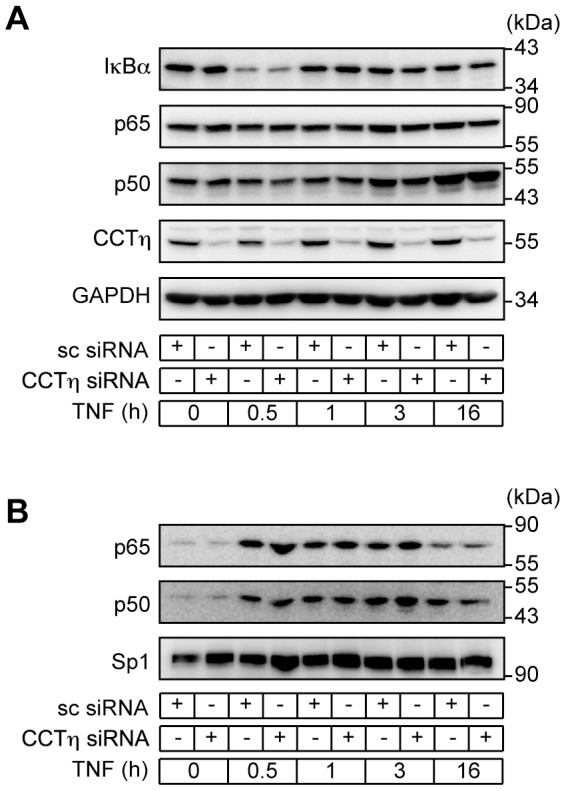
HeLa cells were exposed to CCTη or scrambled (sc) siRNA for three days and stimulated with TNF (10 ng/ml) for indicated times. A: IκBα, p65, p50, CCTη and GAPDH were detected in cytoplasmic extracts and B: p65, p50 and Sp1 were detected in nuclear extracts by western blotting. Immunoblots are representative of three independent experiments.
Figure 4. CCTη knockdown increases NF-κB DNA binding.
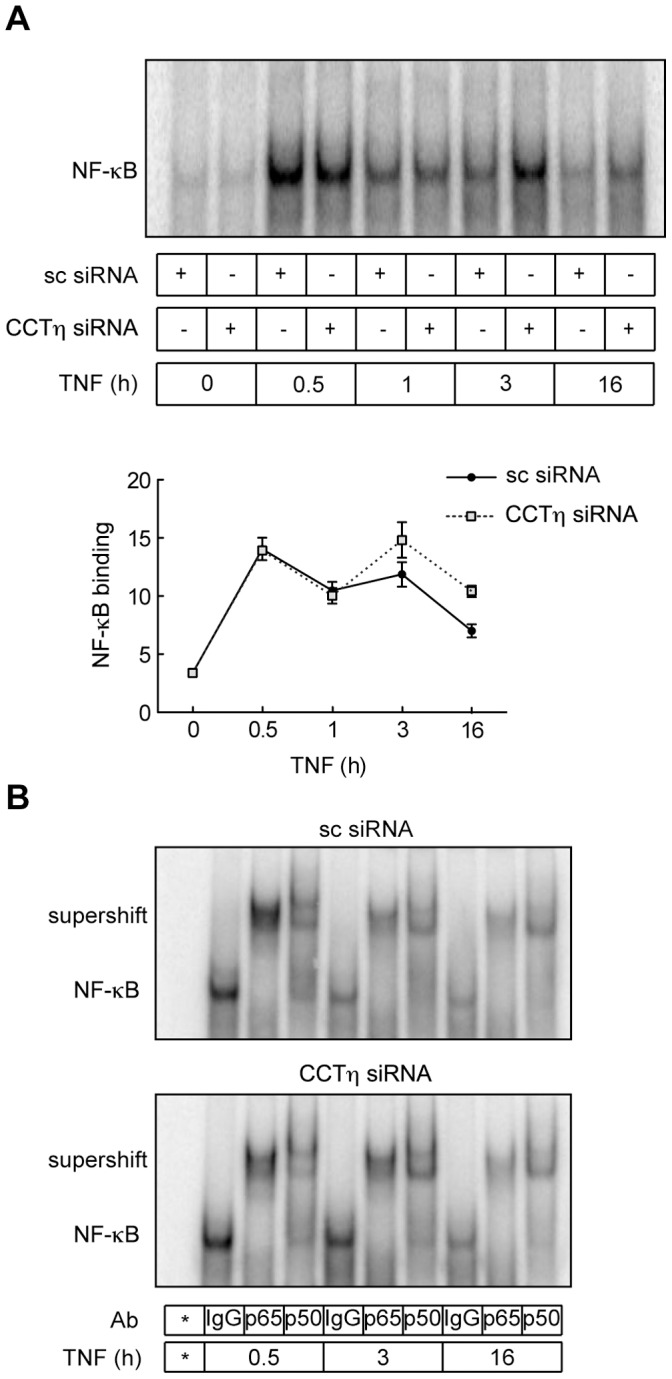
HeLa cells were exposed to CCTη or scrambled (sc) siRNA for three days and stimulated with TNF (10 ng/ml) for indicated times. A: Nuclear extracts were analyzed by EMSA using a κB consensus dsDNA oligonucleotide. Data shown represent mean intensity ± SEM (n = 3). B: Supershift analysis of NF-κB complexes composed of p65/p50 hetero-dimers representative of two independent experiments. Ab, antibody; *, free probe.
CCTη Modulates p65 Transcriptional Activity via a Mechanism that Targets K122/123 Acetylation
CBP/p300 plays a central role in terminating nuclear NF-κB activity by affecting p65 transcriptional activity, IκBα interaction and DNA binding [13], [16]. HDAC3, on the other hand, can antagonize the effect of CBP/p300 by deacetylation of p65 [13], [16]. Given that the chaperonin CCT is required for HDAC3 activity [42], we hypothesized that CCTη regulates NF-κB DNA binding via modulation of HDAC3-dependent p65 deacetylation. If this were the case then knockdown of CCTη should increase p65 binding to DNA κB consensus sequence by increasing p65 acetylation [13]. To test this hypothesis HEK293 cells were transfected with myc-tagged p65, CBP-encoding expression vector and either CCTη- or non-targeting shRNA constructs. p65 acetylation was assessed after immunoprecipitation by immunoblotting with anti-acetyl lysine antibody. Contrary to our expectation, CCTη knockdown decreased CBP-induced p65 acetylation (Fig. 5A), ruling out HDAC3 as a target of the CCTη-mediated effect on NF-κB activity in this experimental system. We next tested whether reduced CBP activity could account for decreased p65 acetylation in CCTη-depleted cells. CBP activity is regulated by several posttranslational modifications and auto-acetylation has been shown to enhance its enzymatic activity [43]. Therefore, we assessed CBP acetylation status in CCTη-depleted cells. CCTη knockdown did not alter CBP protein levels, but reduced CBP acetylation when compared to scrambled shRNA transfected cells (Fig. 5B). This suggests that CCTη regulates NF-κB transcriptional activity through modulation of p65 acetylation by controlling CBP activity.
Figure 5. CCTη regulates p65 transcriptional activity by modulating CBP-dependent p65 acetylation of K122 and K123.

A: HEK293 cells were transfected with CBP, pCDNA3 (control) or myc-p65 and scrambled (sc) or CCTη shRNA, incubated for 3 days and p65 was immunoprecipitated using c-myc-specific agarose beads. Acetylated p65 (Ac-p65) and total p65 were detected using antibodies to acetylated lysine or p65, respectively. B: HEK293 cells were transiently transfected with scrambled (sc) or CCTη shRNA, incubated for three days and endogenous acetylated CBP (Ac-CBP) and total CBP were detected using antibodies to acetylated lysine or CBP after immunoprecipitation, respectively. C: HEK293 cells were transfected or not with scrambled (sc) or CCTη siRNA, stimulated with TNF (10ng/ml) for indicated times and nuclear extracts were prepared. CCTη was immunoprecipitated using TCP-1η antibody. Unrelated EP-1 antibody was used as a goat IgG control (*). D: Rela −/− MEF reconstituted with either WT p65, K310R, K221R or K122/123R mutant were exposed to scrambled (sc) or CCTη siRNA for three days and stimulated with TNF (10 ng/ml) for indicated times. Cxcl10 mRNA levels were analyzed by qPCR as described under “Materials and Methods”. Data represent mean relative mRNA levels ± SEM (n≥3). CCTη and p65 were detected in cell extracts by western blotting (representative of three independent experiment).
While CCT proteins are predominantly localized in the cytoplasm, their nuclear localization and function have recently been reported [44]–[47]. Since p65 acetylation is thought to be a nuclear event [6] and given that CBP/p300 is expressed in the nucleus [48]–[50] we assessed whether chaperonin CCTη was expressed in the nucleus. CCTη was detected in the nucleus of both non-stimulated and TNF stimulated HEK293 cells after immunoprecipitation (Fig. 5C). Specificity was confirmed by lack of nuclear CCTη in CCTη siRNA-transfected cells (Fig. 5C).
Since CBP/p300 can acetylate p65 on multiple lysines [13], [14], [16], we assessed whether the effect of CCTη on NF-κB-dependent gene regulation occurs through acetylation on specific p65 lysine residues. To this end, we compared TNF-induced Cxcl10 mRNA expression in p65−/− MEF retrovirally reconstituted with WT p65, K310R, K221R or K122/123R p65 mutants, exposed to scrambled or CCTη siRNA. All MEF cell lines showed comparable Cxcl10 mRNA levels, as assessed 1 h after TNF stimulation (Fig. 5D). Analogous to results obtained in HeLa cells (Fig. 2A), CCTη knockdown in MEF expressing WT p65 increased Cxcl10 expression at 16 h after TNF stimulation, as compared to scrambled siRNA transfected controls (Fig. 5D). Similarly to the WT p65, CCTη knockdown increased p65 K310R mutant activity at the later time point, suggesting that p65 acetylation at this K residue is not required for CCTη effect (Fig. 5D). K221 mutation abolished p65 transcriptional activity 16 h after TNF stimulation (Fig. 5D), possibly by enhancing IκBα binding and leading to p65 nuclear export, as previously reported [13]. Because the effect of CCTη on Cxcl10 expression is only evident at the later phase of NF-κB- dependent transcription (Fig. 5D), we were not able to evaluate a possible effect of CCTη on this K mutant. CCTη knockdown failed to modulate the transcriptional activity of K122/123R p65 mutant (Fig. 5D), suggesting that CCTη facilitates p65 acetylation on lysines 122 and/or 123, reducing p65 DNA binding and gene transcription.
Discussion
CCT is a ubiquitously expressed multimeric protein complex involved in protein folding and assembly of protein complexes in an ATP-dependent manner. Though originally described as a cytosolic protein involved in actin and tubulin folding [51]–[53], CCT is also expressed in the nucleus [44]–[47] (Fig. 5C). CCT can interact with a range of proteins, including Von Hippel-Lindau (VHL)-elongin BC tumor suppressor complex [54], cell-division cycle protein 20 (Cdc20) [55], sphingosine kinase 1 [56], Polo-like kinase 1 [57], HDAC3 [42], huntingtin [58], among others [59]. This suggests the involvement of CCT in various cellular functions beyond cytoskeleton organization, such as cell cycle, transcription, chromatin remodeling and protein degradation.
CCT is composed of eight different subunits (CCTα-θ) that share similar domains and amino acid sequences conserved across species [60]–[62]. Here we identify the chaperonin CCT subunit η (CCTη) as an evolutionary conserved regulator of NF-κB-dependent transcription that exhibits its effect via a mechanism that targets p65 acetylation. While we have focused on the CCTη subunit, it is likely that a functional chaperonin complex is needed to regulate NF-κB transcription. This notion is supported by the observation that knockdown of the CCT subunits α or ζ had comparable effects to that of CCTη in regulating TNF-induced reporter activity (Fig. 1E, 1F), consistent with the notion that depletion of individual CCT subunits reduces the expression and activity of the CCT chaperonin complex [63], [64].
We have established that CCT regulates NF-κB transcriptional activity, presumably at a nuclear level. CCT knockdown modulated both TNF- and IL-1β-driven NF-κB activation (Fig. 2), suggesting that it acts downstream of IKK activation, a common denominator of the signal transduction pathways triggered by cross-linking of the TNF and IL-1 receptors [39]. In addition, CCT knockdown failed to modulate IκBα degradation as well as p65 nuclear translocation (Fig. 3). Consecutively, we found that CCT knockdown increased NF-κB binding to DNA κB consensus sequence (Fig. 4), and we attribute this effect to altered p65 acetylation.
In Drosophila cells, CCT knockdown reduces NF-κB-driven reporter activity, which contrasts with mammalian cells in which CCT depletion promotes NF-κB activity in response to TNF (Fig. 1C–F). This apparent discrepancy could result from opposing functional outcome of NF-κB acetylation in Drosophila vs. mammalian cells. Whereas transcriptional activity of Dorsal and Dif has not been previously shown to be regulated by acetylation, p65 acetylation affects NF-κB-dependent transcription in multiple ways. While K310, K314 and K315 acetylation enhances p65 and hence NF-κB transcriptional activity without affecting DNA binding [13], [14], K218 and K221 acetylation increases NF-κB transcriptional activity by inhibiting its removal from DNA by newly synthesized IκBα [13]. In contrast, K122 and K123 acetylation reduces p65 transcriptional activity by decreasing DNA binding in an IκBα-independent manner [16]. Thus, it is possible that specific lysines are targeted in a timely fashion, and that activatory acetylation might prevail during early stages of the transcriptional response, while inhibitory acetylation will be important for terminating NF-κB activity on genes undergoing prolonged transcriptional activation (e.g. CXCL10). Supporting this notion, CCT knockdown in mammalian cells decreased IκBα and CXCL2 expression, as assessed 1 h after TNF stimulation while increasing that of TNF, IL8, CCL5 and CXCL10, as assessed 3 and/or 16 h thereafter (Fig. 2A), suggesting a different impact of acetylation on early- and late-phase of NF-κB transcriptional activity.
p65 acetylation is controlled via opposing effects of HATs and HDACs [6]. Chaperonin CCT is required for the formation of an enzymatically active HDAC3–SMRT complex [42] that can regulate late phase NF-κB activity by reducing p65 DNA binding, leading to termination of NF-κB dependent transcription [13]. This is consistent with the observation that SMRT and CCT co-localize to the promoter region of NF-κB dependent genes [65]. Based on these findings, one would expect CCT knockdown to enhance p65 activity by increasing p65 acetylation, presumably by decreasing the amount of functionally active HDAC3. This is, however, not the case, as CCT knockdown decreased p65 acetylation (Fig. 5A).
Regulation of p65 activity by acetylation is target residue specific. While K310 deacetylation diminishes p65 transcription without affecting DNA binding, decreased K221 acetylation favors IκBα binding, promoting its removal from DNA and nuclear export [13]. Since p65 deacetylation by CCT knockdown promotes NF-κB-regulated transcription, it is unlikely that CCT knockdown is associated with decreased acetylation of these residues during late phase of NF-κB activation. Consistent with this, CCT knockdown increased transcriptional activity of p65 K310R mutant 16 h after TNF stimulation in a similar manner as observed in MEF expressing WT p65 (Fig. 5D). On the other hand, the p65 K221R mutant showed significantly lower transcriptional activity, as assessed 16 h after TNF stimulation (Fig. 5D), possibly reflecting enhanced nuclear export after de novo IκBα synthesis [13], and was therefore insensitive to CCT knockdown (Fig. 5D). We cannot, however, exclude that CCT knockdown down-regulates K310 acetylation at earlier time points, which could explain transcriptional repression of IκBα and CXCL2 genes as assessed 1 h after TNF stimulation (Fig. 2A).
Acetylation of p65 K122 and K123 inhibits DNA binding, promoting termination of NF-κB-dependent transcriptional response [16]. In line with this, the p65 K122/123R mutant failed to terminate NF-κB-dependent transcription reflected by increased Cxcl10 mRNA, as compared to p65 wt or p65 K310R and K221R mutants (Fig. 5D). Interestingly, the kinetics of mRNA expression of this mutant was similar to that observed in CCT depleted cells expressing WT p65, and was insensitive to CCT knockdown (Fig. 5D). This suggests that CCT regulates late phase NF-κB-dependent transcription by enhancing K122 and K123 acetylation.
CBP/p300 is a major p65 acetyltransferase [13], [14], [16], whose catalytic activity is regulated by a multiplicity of factors [66], including auto-acetylation [43]. CBP/p300 has been shown to acetylate p65 K122 and K123 residues [16]. We found that CCT knockdown reduces acetylation of endogenous CBP (Fig. 5B), implicating reduced CBP activity as a mechanism for diminished p65 acetylation and for the failure to terminate NF-κB signaling.
In conclusion this study identifies the chaperonin CCT as a regulator of NF-κB transcriptional activity via a mechanism that targets p65 acetylation, presumably by altering CBP activity. We propose that CCT might be involved in terminating NF-κB signaling and as such may be important for the resolution of inflammation.
Acknowledgments
The authors thank Gianfranco Racchumi (Weill Cornell Medical College, New York, NY) for performing qPCR, Ylva Engstrom (Stockholm University, Stockholm, Sweden) and Mika Ramet (Institute of Medical Technology, University of Tampere, Tampere, Finland) for providing plasmids used in Drosophila S2 cells studies, Monica Bettencourt Dias (Instituto Gulbenkian de Ciência, Oeiras, Portugal) for Drosophila S2 cells used in this study, Buzz Baum (University College London, London, UK), Nicolas Tapon (Cancer Research UK, London Research Institute, London, UK) and Antonio Jacinto (Instituto de Medicina Molecular, Lisbon, Portugal) for help with plated Drosophila dsRNA used in the gene-screening assay and Amer Beg (Moffitt Cancer Center, Tampa, FL) for providing Rela−/− MEF cells.
Funding Statement
This work was supported by “Fundacão para a Ciência e a Tecnologia”, Portugal grants SFRH/BD/28016/2006 (NP), PTDC/BIA-BCM/101311/2008 and PTDC/SAU-FCF/100762/2008 (MPS), European Community and LSH-2005-1.2.5-1 (MPS), and National Institutes of Health grant HL077308 (JA). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Baeuerle PA, Baltimore D (1996) NF-κB: ten years after. Cell 87: 13–20. [DOI] [PubMed] [Google Scholar]
- 2. Baldwin AS Jr (2001) Series introduction: the transcription factor NF-κB and human disease. J Clin Invest 107: 3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ballard DW, Dixon EP, Peffer NJ, Bogerd H, Doerre S, et al. (1992) The 65-kDa subunit of human NF-κB functions as a potent transcriptional activator and a target for v-Rel-mediated repression. Proc Natl Acad Sci U S A 89: 1875–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thanos D, Maniatis T (1995) NF-κB: a lesson in family values. Cell 80: 529–532. [DOI] [PubMed] [Google Scholar]
- 5. Perkins ND (2006) Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 25: 6717–6730. [DOI] [PubMed] [Google Scholar]
- 6. Huang B, Yang XD, Lamb A, Chen LF (2010) Posttranslational modifications of NF-κB: another layer of regulation for NF-κB signaling pathway. Cell Signal 22: 1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhong H, Voll RE, Ghosh S (1998) Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1: 661–671. [DOI] [PubMed] [Google Scholar]
- 8. Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, et al. (2005) NF-κB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol 25: 7966–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, et al. (2003) Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell 12: 1413–1426. [DOI] [PubMed] [Google Scholar]
- 10. Geng H, Wittwer T, Dittrich-Breiholz O, Kracht M, Schmitz ML (2009) Phosphorylation of NF-κB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep 10: 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhong H, May MJ, Jimi E, Ghosh S (2002) The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol Cell 9: 625–636. [DOI] [PubMed] [Google Scholar]
- 12. Chen L, Fischle W, Verdin E, Greene WC (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293: 1653–1657. [DOI] [PubMed] [Google Scholar]
- 13. Chen LF, Mu Y, Greene WC (2002) Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J 21: 6539–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buerki C, Rothgiesser KM, Valovka T, Owen HR, Rehrauer H, et al. (2008) Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res 36: 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rothgiesser KM, Fey M, Hottiger MO (2010) Acetylation of p65 at lysine 314 is important for late NF-κB-dependent gene expression. BMC Genomics 11: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kiernan R, Bres V, Ng RW, Coudart MP, El Messaoudi S, et al. (2003) Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65. J Biol Chem 278: 2758–2766. [DOI] [PubMed] [Google Scholar]
- 17. Liu Y, Smith PW, Jones DR (2006) Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol Cell Biol 26: 8683–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, et al. (2004) Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23: 2369–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rothgiesser KM, Erener S, Waibel S, Luscher B, Hottiger MO (2010) SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci 123: 4251–4258. [DOI] [PubMed] [Google Scholar]
- 20. Ea CK, Baltimore D (2009) Regulation of NF-κB activity through lysine monomethylation of p65. Proc Natl Acad Sci U S A 106: 18972–18977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang XD, Huang B, Li M, Lamb A, Kelleher NL, et al. (2009) Negative regulation of NF-κB action by Set9-mediated lysine methylation of the RelA subunit. EMBO J 28: 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maine GN, Mao X, Komarck CM, Burstein E (2007) COMMD1 promotes the ubiquitination of NF-κB subunits through a cullin-containing ubiquitin ligase. EMBO J 26: 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saccani S, Marazzi I, Beg AA, Natoli G (2004) Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor κB response. J Exp Med 200: 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanaka T, Grusby MJ, Kaisho T (2007) PDLIM2-mediated termination of transcription factor NF-κB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol 8: 584–591. [DOI] [PubMed] [Google Scholar]
- 25. Kidd S (1992) Characterization of the Drosophila cactus locus and analysis of interactions between cactus and dorsal proteins. Cell 71: 623–635. [DOI] [PubMed] [Google Scholar]
- 26. Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA (1999) Phylogenetic perspectives in innate immunity. Science 284: 1313–1318. [DOI] [PubMed] [Google Scholar]
- 27. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA (1996) The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86: 973–983. [DOI] [PubMed] [Google Scholar]
- 28. Manfruelli P, Reichhart JM, Steward R, Hoffmann JA, Lemaitre B (1999) A mosaic analysis in Drosophila fat body cells of the control of antimicrobial peptide genes by the Rel proteins Dorsal and DIF. EMBO J 18: 3380–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uvell H, Engstrom Y (2003) Functional characterization of a novel promoter element required for an innate immune response in Drosophila. Mol Cell Biol 23: 8272–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Briggs LJ, Stein D, Goltz J, Corrigan VC, Efthymiadis A, et al. (1998) The cAMP-dependent protein kinase site (Ser312) enhances dorsal nuclear import through facilitating nuclear localization sequence/importin interaction. J Biol Chem 273: 22745–22752. [DOI] [PubMed] [Google Scholar]
- 31. Drier EA, Huang LH, Steward R (1999) Nuclear import of the Drosophila Rel protein Dorsal is regulated by phosphorylation. Genes Dev 13: 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, et al. (2004) Genome-wide survey of protein kinases required for cell cycle progression. Nature 432: 980–987. [DOI] [PubMed] [Google Scholar]
- 33. Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D (1995) Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 376: 167–170. [DOI] [PubMed] [Google Scholar]
- 34. Anrather J, Racchumi G, Iadecola C (2005) cis-acting, element-specific transcriptional activity of differentially phosphorylated nuclear factor-κB. J Biol Chem 280: 244–252. [DOI] [PubMed] [Google Scholar]
- 35. Kleino A, Valanne S, Ulvila J, Kallio J, Myllymaki H, et al. (2005) Inhibitor of apoptosis 2 and TAK1-binding protein are components of the Drosophila Imd pathway. EMBO J 24: 3423–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brostjan C, Anrather J, Csizmadia V, Natarajan G, Winkler H (1997) Glucocorticoids inhibit E-selectin expression by targeting NF-κB and not ATF/c-Jun. Journal of Immunology 158: 3836–3844. [PubMed] [Google Scholar]
- 37. Anrather J, Csizmadia V, Soares MP, Winkler H (1999) Regulation of NF-κB RelA phosphorylation and transcriptional activity by p21(ras) and protein kinase C ζ in primary endothelial cells. Journal of Biological Chemistry 274: 13594–13603. [DOI] [PubMed] [Google Scholar]
- 38. Tanji T, Yun EY, Ip YT (2010) Heterodimers of NF-κB transcription factors DIF and Relish regulate antimicrobial peptide genes in Drosophila. Proc Natl Acad Sci U S A 107: 14715–14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perkins ND (2007) Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol 8: 49–62. [DOI] [PubMed] [Google Scholar]
- 40. Sun SC, Ganchi PA, Ballard DW, Greene WC (1993) NF-κB controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science 259: 1912–1915. [DOI] [PubMed] [Google Scholar]
- 41. Beg AA, Finco TS, Nantermet PV, Baldwin AS Jr (1993) Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IκBα: a mechanism for NF-κB activation. Mol Cell Biol 13: 3301–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guenther MG, Yu J, Kao GD, Yen TJ, Lazar MA (2002) Assembly of the SMRT-histone deacetylase 3 repression complex requires the TCP-1 ring complex. Genes Dev 16: 3130–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, et al. (2004) Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol 11: 308–315. [DOI] [PubMed] [Google Scholar]
- 44. Gerner C, Gotzmann J, Frohwein U, Schamberger C, Ellinger A, et al. (2002) Proteome analysis of nuclear matrix proteins during apoptotic chromatin condensation. Cell Death Differ 9: 671–681. [DOI] [PubMed] [Google Scholar]
- 45. Brown CR, Doxsey SJ, Hong-Brown LQ, Martin RL, Welch WJ (1996) Molecular chaperones and the centrosome. A role for TCP-1 in microtubule nucleation. J Biol Chem 271: 824–832. [DOI] [PubMed] [Google Scholar]
- 46. Roobol A, Carden MJ (1999) Subunits of the eukaryotic cytosolic chaperonin CCT do not always behave as components of a uniform hetero-oligomeric particle. Eur J Cell Biol 78: 21–32. [DOI] [PubMed] [Google Scholar]
- 47. Soues S, Kann ML, Fouquet JP, Melki R (2003) The cytosolic chaperonin CCT associates to cytoplasmic microtubular structures during mammalian spermiogenesis and to heterochromatin in germline and somatic cells. Exp Cell Res 288: 363–373. [DOI] [PubMed] [Google Scholar]
- 48. Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, et al. (1993) Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365: 855–859. [DOI] [PubMed] [Google Scholar]
- 49. Harlow E, Whyte P, Franza BR Jr, Schley C (1986) Association of adenovirus early-region 1A proteins with cellular polypeptides. Mol Cell Biol 6: 1579–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87: 953–959. [DOI] [PubMed] [Google Scholar]
- 51. Gao Y, Thomas JO, Chow RL, Lee GH, Cowan NJ (1992) A cytoplasmic chaperonin that catalyzes beta-actin folding. Cell 69: 1043–1050. [DOI] [PubMed] [Google Scholar]
- 52. Yaffe MB, Farr GW, Miklos D, Horwich AL, Sternlicht ML, et al. (1992) TCP1 complex is a molecular chaperone in tubulin biogenesis. Nature 358: 245–248. [DOI] [PubMed] [Google Scholar]
- 53. Gao Y, Vainberg IE, Chow RL, Cowan NJ (1993) Two cofactors and cytoplasmic chaperonin are required for the folding of alpha- and beta-tubulin. Mol Cell Biol 13: 2478–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feldman DE, Thulasiraman V, Ferreyra RG, Frydman J (1999) Formation of the VHL-elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol Cell 4: 1051–1061. [DOI] [PubMed] [Google Scholar]
- 55. Camasses A, Bogdanova A, Shevchenko A, Zachariae W (2003) The CCT chaperonin promotes activation of the anaphase-promoting complex through the generation of functional Cdc20. Mol Cell 12: 87–100. [DOI] [PubMed] [Google Scholar]
- 56. Zebol JR, Hewitt NM, Moretti PA, Lynn HE, Lake JA, et al. (2009) The CCT/TRiC chaperonin is required for maturation of sphingosine kinase 1. Int J Biochem Cell Biol 41: 822–827. [DOI] [PubMed] [Google Scholar]
- 57. Liu X, Lin CY, Lei M, Yan S, Zhou T, et al. (2005) CCT chaperonin complex is required for the biogenesis of functional Plk1. Mol Cell Biol 25: 4993–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tam S, Geller R, Spiess C, Frydman J (2006) The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol 8: 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dekker C, Stirling PC, McCormack EA, Filmore H, Paul A, et al. (2008) The interaction network of the chaperonin CCT. EMBO J 27: 1827–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kubota H, Hynes G, Carne A, Ashworth A, Willison K (1994) Identification of six Tcp-1-related genes encoding divergent subunits of the TCP-1-containing chaperonin. Curr Biol 4: 89–99. [DOI] [PubMed] [Google Scholar]
- 61. Kubota H, Hynes G, Willison K (1995) The eighth Cct gene, Cctθ, encoding the theta subunit of the cytosolic chaperonin containing TCP-1. Gene 154: 231–236. [DOI] [PubMed] [Google Scholar]
- 62. Kim S, Willison KR, Horwich AL (1994) Cystosolic chaperonin subunits have a conserved ATPase domain but diverged polypeptide-binding domains. Trends Biochem Sci 19: 543–548. [DOI] [PubMed] [Google Scholar]
- 63. Kitamura A, Kubota H, Pack CG, Matsumoto G, Hirayama S, et al. (2006) Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat Cell Biol 8: 1163–1170. [DOI] [PubMed] [Google Scholar]
- 64. Grantham J, Brackley KI, Willison KR (2006) Substantial CCT activity is required for cell cycle progression and cytoskeletal organization in mammalian cells. Exp Cell Res 312: 2309–2324. [DOI] [PubMed] [Google Scholar]
- 65. Hoberg JE, Yeung F, Mayo MW (2004) SMRT derepression by the IκB kinase α: a prerequisite to NF-κB transcription and survival. Mol Cell 16: 245–255. [DOI] [PubMed] [Google Scholar]
- 66. Chan HM, La Thangue NB (2001) p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 114: 2363–2373. [DOI] [PubMed] [Google Scholar]