

Abstract
Curation of a high-quality gene set is the critical first step in genome research, enabling subsequent analyses such as ortholog assignment, cis-regulatory element finding, and synteny detection. In this project, we have reannotated the genome of Caenorhabditis briggsae, the best studied sister species of the model organism Caenorhabditis elegans. First, we applied a homology-based gene predictor genBlastG to annotate the C. briggsae genome. We then validated and further improved the C. briggsae gene annotation through RNA-seq analysis of the C. briggsae transcriptome, which resulted in the first validated C. briggsae gene set (23,159 genes), among which 7347 genes (33.9% of all genes with introns) have all of their introns confirmed. Most genes (14,812, or 68.3%) have at least one intron validated, compared with only 3.9% in the most recent WormBase release (WS228). Of all introns in the revised gene set (103,083), 61,503 (60.1%) have been confirmed. Additionally, we have identified numerous trans-splicing leaders (SL1 and SL2 variants) in C. briggsae, leading to the first genome-wide annotation of operons in C. briggsae (1105 operons). The majority of the annotated operons (564, or 51.0%) are perfectly conserved in C. elegans, with an additional 345 operons (or 31.2%) somewhat divergent. Additionally, RNA-seq analysis revealed over 10 thousand small-size assembly errors in the current C. briggsae reference genome that can be readily corrected. The revised C. briggsae genome annotation represents a solid platform for comparative genomics analysis and evolutionary studies of Caenorhabditis species.
The nematode Caenorhabditis briggsae is the most extensively studied sister species of the model organism Caenorhabditis elegans (Hillier et al. 2005). C. elegans has been the platform for a remarkable list of groundbreaking discoveries such as the cloning and functional analysis of the first microRNA (miRNA) genes (Ambros 2004) and the elucidation of key components of the programmed cell death (PCD) pathway (Horvitz 2003). These discoveries have reshaped the landscape of biomedical research, much of which has benefited greatly from comparative analysis between C. elegans and C. briggsae. These two organisms are both hermaphrodites that share remarkable similarity in morphology and development programs (Gupta et al. 2007), though they have diverged from their common ancestor ∼100 million years ago (MYA) (Coghlan and Wolfe 2002; Stein et al. 2003). By using confocal microscopy and fluorescence-labeling-based automated single-cell lineage tracing technology (Bao et al. 2006), these two nematode species were found to possess almost identical cellular developmental programs (Zhao et al. 2008). To facilitate genome-wide investigation of the molecular mechanisms underlying morphology and development of these two species, the C. briggsae genome was sequenced through whole-genome shotgun sequencing (Stein et al. 2003), after the sequencing of the C. elegans genome (The C. elegans Sequencing Consortium 1998). C. elegans was the first metazoan with a genome that was subjected to whole-genome sequencing and the only genome of a multicellular organism that has been completely sequenced and assembled with no remaining gaps. As a demonstration of the value of the C. briggsae genome sequencing project, the annotation of the C. briggsae genome enabled the discovery of 1300 new genes in C. elegans (Stein et al. 2003). Because of this and the possibility of many more potential discoveries such as the identification of cis-regulatory elements and studies on the molecular evolution of gene families, publication of C. briggsae genome and accompanying annotation was immediately established as an excellent platform for comparative genomics by the C. elegans research community (Gupta and Sternberg 2003).
While the C. elegans genome has been extensively annotated through combined approaches of bioinformatics gene finding, whole transcriptome analysis, and molecular studies of individual genes by the C. elegans research community, the C. briggsae genome has been less well annotated, limiting its value as a comparative genomic platform. The C. elegans genome has been extensively annotated using an ab initio gene finding program Genefinder (Spieth and Lawson 2006), then exploiting transcription expression evidence including expression sequence tags (ESTs) (Kohara 1996; Shin et al. 2008), open reading frame sequence tags (OSTs) (Reboul et al. 2003; Lamesch et al. 2004; Wei et al. 2005), serial analysis of gene expression (SAGE) tags (Ruzanov et al. 2007; Nesbitt et al. 2010; Ruzanov and Riddle 2010), RNA-seq results (Hillier et al. 2009; Allen et al. 2011), as well as translational expression evidence (Shim and Paik 2010). C. elegans genome annotation has been further improved through genome-wide RACE analysis (Salehi-Ashtiani et al. 2009) and the determination of 3′ UTRs for the entire C. elegans genome (Mangone et al. 2010). In contrast, C. briggsae gene models have mostly been limited to bioinformatics predictions that have not been experimentally validated. For example, in the most recent WormBase release WS228, only 0.2% of the C. briggsae gene models are fully confirmed, compared with 47.8% of the C. elegans gene models. A more completely defined C. briggsae gene set would greatly facilitate an array of research including accuracy in assigning true orthologs (Stein et al. 2003), effective identification of synteny blocks (Vergara and Chen 2010), understanding of operon evolution (Blumenthal et al. 2002; Stein et al. 2003; Qian and Zhang 2008), let alone the understanding of numerous individual genes in these two species.
In this study, we undertook the task of refining the C. briggsae gene set, by taking advantage of recent progress in bioinformatics and genomics tools to establish its full potential as an accurate platform of comparative genomics. First, we have recently developed an effective homology-based gene finder genBlastG (She et al. 2011). Because the C. elegans gene annotation has been improved substantially after the initial annotation of the C. briggsae genome (Stein et al. 2003), we use the improved C. elegans gene models as queries to improve their homologous C. briggsae gene models. Second, the tremendous increase of high-throughput DNA sequencing coupled with decreased cost facilitated deep sequencing of transcriptomes, i.e., RNA-seq (Hillier et al. 2009; Wang et al. 2009). We used RNA-seq of C. briggsae transcriptome to validate and refine C. briggsae gene models. This resource has enabled us to annotate trans-splicing leaders, 5′ UTRs, 3′ UTRs, as well as operons in C. briggsae. Our revised annotation of the C. briggsae genome provides an excellent platform for more accurate comparative analyses between C. elegans and C. briggsae.
Results
Reannotation using genBlastG revised 6715 C. briggsae gene models
In this study, the primary focus of this project is to produce a high-quality C. briggsae gene set, which can already provide a good starting point for researchers. A comprehensive and correct definition of C. briggsae alternative isoforms needs an extensive sampling of different cell and tissue types at different developmental stages, which needs much more time to accomplish. The primary reason that enabled us to make this decision to focus on producing one isoform for each gene is that there is an urgent need to have a high-quality C. briggsae gene set. In the decade since the first C. briggsae gene set was annotated (Stein et al. 2003), only a small portion of the models has been supported by expression evidence. And each gene only has a single gene model. Thus, it is highly needed to build a gene set with confirmed gene models. Therefore, for each C. elegans gene with alternative isoforms, we used only the longest isoform as query. The reason why we decided to use the longest isoform of each C. elegans query is simply because the longest ones contain more coding information, which helps us annotate more coding information for C. briggsae genes. Using 20,335 C. elegans peptide sequences (from WormBase WS215) as queries, we predicted 16,285 C. briggsae gene models. These genBlastG-defined C. briggsae gene models were then compared with C. briggsae gene models available in WS215 to evaluate their quality. Three major types of revisions to the WormBase C. briggsae gene models were made (Fig. 1A–C). First, 5387 WormBase-derived C. briggsae gene models were replaced by their corresponding genBlastG-defined gene models. These replacements were made because these genBlastG-defined gene models had substantially higher similarity (measured using percentage identity, PID, at the protein sequence level) to their corresponding orthologous C. elegans genes. Improvements facilitated by the genBlastG-predicted gene models involved extending or truncating gene models, or by adding or removing exons (Fig. 1A). Second, based on homology with C. elegans orthologs, 282 C. briggsae gene models were obtained by merging multiple gene predictions that represented part of the corresponding C. elegans ortholog. All merging cases are supported by RNA-seq results as described later. In other words, each C. briggsae gene model group corresponds to a single putative orthologous gene model in C. elegans. Most merging was made between two adjacent gene predictions, while in rare cases merging of multiple adjacent gene models was required (Fig. 1B). Third, we split a number of WormBase C. briggsae gene models into two or more gene models based on the genBlastG-defined gene models. This resulted in 954 revised gene models. In most cases, a single gene prediction was split into two independent gene models, while in some cases, three independent gene models were generated (Fig. 1C). Additionally, a small number of WormBase C. briggsae gene models needed to be modified by a series of merging and splitting steps. Specifically, these gene models were merged with their adjacent gene models and split into two or more gene models, resulting in 92 gene models in this category. Furthermore, we uncovered 1091 novel gene models in previous intronic or intergenic regions (Fig. 1D). For WormBase C. briggsae genes that did not have C. elegans orthologs, we reasoned that they might be species-specific and therefore kept them in the revised gene set. This approach resulted in a “hybrid C. briggsae gene set,” containing 23,276 gene models (Supplemental data 1–3). Of these, 7806 gene models were defined by genBlastG and 15,470 were originally defined in WormBase (WS215). Of the 7806 genBlastG-defined gene models, 6715 were revised gene models, while the rest were novel gene models missed in previous annotations. Some revised C. briggsae gene models were longer than their corresponding orthologs in C. elegans due to mutations that eliminated stop codons (Supplemental Fig. 1).
Figure 1.

Examples of genBlastG-revised C. briggsae gene models. (A) Revision: The WormBase C. briggsae gene model CBG09754 has three exons and two introns. The genBlastG-revised gene model, JNC_CBG06430, has two new exons incorporated. Protein sequence encoded by the revised C. briggsae gene model displays very high PID (99.1%) with the protein sequence encoded by its C. elegans ortholog ben-1/tbb-5 (C54C6.2). ben-1 encodes a redundant beta-tubulin in C. elegans and it is the only benzimidazole-sensitive beta-tubulin in C. elegans (Driscoll et al. 1989). (B) Merge: Based on homology, three WormBase C. briggsae gene models (CBG08435, CBG08437, and CBG08438) were merged to form a single-gene model JNC_CBG02055, which is orthologous with C. elegans gene lrk-1 (T27C10.6) with high identity (92.63%). LRK-1 is the C. elegans ortholog of the familial Parkinsonism gene LRRK2 (previously known as PARK8) that is required for polarized localization of synaptic vesicle (SV) proteins (Sakaguchi-Nakashima et al. 2007). (C) Split: Based on homology, a single WormBase C. briggsae gene model CBG09729 is split into three separate gene models JNC_CBG16717, JNC_CBG16718, and JNC_CBG16719, which are homologous with three fully confirmed C. elegans gene models F32D8.14 (PID = 91.63%), F32D8.4 (PID = 70.55%), and F32D8.12a (PID = 90.11%), respectively. (D) Novel C. briggsae gene model: Based on homology, genBlastG builds a novel C. briggsae gene model JNC_CBG14647, which is orthologous with the fully confirmed C. elegans gene model F54F4.16 (PID = 90.62%).
RNA-seq analysis and gene model validation
Not all C. elegans gene models have been fully confirmed by transcript evidence. C. briggsae gene models curated using C. elegans gene predictions as queries in homology-based gene finding can therefore inherit defects from their C. elegans query gene models. Additionally, C. briggsae gene models can show differences because of bona fide evolutionary divergence. Thus C. briggsae gene models based on homology alone might not reflect real evolutionary difference. For example, two separate genes in one species may have merged into a single gene in another species. Indeed, inspection of the hybrid C. briggsae gene set, which was curated based on homologous gene model definition, revealed a substantial number of C. briggsae gene models that appeared defective. For example, many annotated gene models, especially those directly inherited from the WormBase C. briggsae gene set, do not have annotated stop codons, and a large number of gene models have extremely small annotated introns (i.e., ≤10 bp). Taken together, gene models in the hybrid C. briggsae gene set, including the gene models revised based on homology, need to be validated using transcript information to ensure their quality.
To help build an accurate set of C. briggsae gene models, we undertook an RNA-seq approach (Wang et al. 2009) to validate the hybrid gene models. This method has recently been used to examine the C. elegans gene set (Hillier et al. 2009). In this project, we sequenced two C. briggsae transcriptome libraries with the aim of sampling a wide range of transcripts using the Illumina Genome Analyzer II paired-end DNA sequencing technology, with read length of 42 bp: an L1 stage transcriptome and a mixed-stage (from L1 to young adults) transcriptome. Both transcriptomes were prepared using the C. briggsae AF16 strain, the same strain used in the C. briggsae genome sequencing project (Stein et al. 2003). From this, we obtained 15.5 million paired-end reads for the L1 transcriptome, and 17.5 million paired-end reads for the mixed-stage transcriptome. Together the data accounted for 33.0 million Illumina paired-end reads that could be used for validating and revising C. briggsae gene models. The RNA-seq analysis procedure is illustrated in Figure 2.
Figure 2.

RNA-seq analysis flowchart.
We aligned Illumina reads to the reference C. briggsae genomic DNA sequence using the short read alignment program MAQ (Li et al. 2008). Both ends of 14.0 million read pairs (42.4% of the total reads) were successfully mapped to the reference genome sequences (WormBase, WS215). Aligning using different programs including SSAHA2 (Ning et al. 2001) and BWA (Li and Durbin 2009) designed for short reads yielded similar results (data not shown). These read pairs were mapped either to internal genomic regions within the same exon, or internal genomic regions of adjacent exons. Single ends of some (3.1 million or 9.1%) read pairs were mapped to the C. briggsae reference genomic DNA regions, but their mates were not aligned. All mapped reads thus provided useful information for confirmation or improvement of the C. briggsae gene models. Additionally, the 3.1 million single end reads, with mates not mapped to the C. briggsae genome, were used specifically to define and validate introns. For clarity, we name a putative intron defined by Illumina reads as an “Illumina intron,” while genomic segments in which all bases were covered by Illumina read alignments without gaps are termed “Illumina exons” (see Methods). Applying SuperSplat (Bryant et al. 2010), we have identified 78,252 Illumina introns (Fig. 2), 59,560 of which were supported by two or more independent Illumina reads (Supplemental data 4).
Next, we used the Illumina introns and Illumina exons to validate C. briggsae gene models and to further improve gene models by systematically comparing them with introns and exons in the C. briggsae hybrid gene set defined above. Based on their relationship with predicted protein-coding gene models, an Illumina intron can be categorized as an intragenic intron if it overlaps with a gene and is entirely nested within a predicted gene model, or as a boundary (i.e., intergenic) intron that indicates the presence of a protein-coding gene that has not been previously annotated. In the cases where full gene models are not defined, they are annotated as genelets (Hillier et al. 2009). We have found 2384 such genelets. Some of these may be missing parts of existing gene models, but this observation suggests a fair number of missing protein-coding gene models that have not been defined. As the first step in validating and revising C. briggsae gene models, we focused on internal introns and exons of genes. Protein-coding genes in eukaryotic genomes consist of coding exons (for simplicity, we use the term “exons” in this paper), introns, and untranslated regions (5′ and 3′ UTRs). Because genomic sequences spanning introns and exons are complementary in gene models, gene model definition is essentially equivalent to intron definition. Once introns are defined, exons are readily defined. For gene models that are computationally predicted, such as those in the C. briggsae hybrid gene set, exons and introns may be correct, defective, or entirely missing. Thus, when compared with Illumina introns, introns in predicted gene models can be confirmed, modified, or removed (Fig. 2). Additionally, novel introns may be introduced to the gene models. Furthermore, introns in predicted gene models can be spurious and therefore removed if their existence is in conflict with transcript reads. Finally, alternative introns that overlap with each other can be identified as well. Through comparison of predicted intragenic Illumina introns with predicted introns of gene models in the C. briggsae hybrid gene set (based on WS215 and genBlastG version 135), we validated 59,137 predicted introns (Fig. 3A), created 2111 new introns by revising predicted introns (Fig. 3B), curated 716 novel introns (Fig. 3C), and removed 461 spurious predicted introns.
Figure 3.

RNA-seq revisions of C. briggsae gene models. (A) An intron is confirmed if the predicted intron in the hybrid gene model is supported by an RNA-seq-defined Illumina intron. (B) An intron is replaced by an RNA-seq-defined Illumina intron if the predicted intron overlaps with the Illumina intron but is not identical to the Illumina intron. (C) A new intron is created if an Illumina intron overlaps with a coding region. (D) A predicted intron is removed if it overlaps with an Illumina exon and it is not supported by any Illumina intron and it does not overlap with any Illumina intron. (E) A 3′ intron is created if an Illumina intron overlaps with the 3′ most exon. A new 3′ exon is also created that contains a stop codon. (F) A 5′ intron is created when an Illumina intron overlaps with the 5′ most exon. A new 5′ exon is also created that contains a start codon.
As a result of our combined homology and RNA-seq based improvements, in the C. briggsae gene set of 21,683 intron-containing genes (Supplemental data 5–7), 61,503 (60.1%) introns have been validated, 14,812 (68.3%) genes have at least one intron validated, 7347 (33.9% of all intron-containing genes) genes have all introns validated. At the transcript level, 10,235 genes (or 47.0% of all genes) were found to have 95% or more of their coding sequences supported by Illumina read alignments. This is a remarkable advance because after almost 10 yr since the annotation of the C. briggsae genome (Stein et al. 2003) only 853 (3.9%) genes had been partially confirmed according to the most recent release of WormBase (WS228).
RNA-seq analysis of the C. briggsae gene models demonstrated the value of genBlastG-based gene model improvement. Among the 7806 gene models revised using genBlastG, 3547 (45.3%) gene models have >90% of their entire lengths supported (Supplemental Fig. 2). Among the 1091 novel gene models predicted using genBlastG, 100 (9.2%) gene models have ≥90% of their entire lengths supported, and 791 (72.5%) gene models have varying levels of support (Supplemental Fig. 2). Gene models with no RNA-seq support could be either false predictions by genBlast, or those that are not expressed at given conditions. Much deeper sequencing of C. briggsae transcriptomes under various different stress conditions is needed to resolve this.
In the comparative analysis, we also revealed that 2301 Illumina introns overlap with validated introns from 1176 C. briggsae gene models, suggesting alternative splicing exists in at least these 1176 genes. Surprisingly, we observed that 636 Illumina introns, each supported by two or more independent Illumina reads, cannot be incorporated into the C. briggsae genome without causing frame shifts. The cause of these failed attempts will be explored later.
Gene revision improves ortholog assignment and synteny block prediction
The value of homology-based gene model improvement is demonstrated by the detection of previously missed orthologs between C. elegans and C. briggsae. Before homology-based gene model improvement of C. briggsae annotation by using genBlastG and RNA-seq analysis, 14,167 C. elegans genes were found to have clear orthologs in C. briggsae. After the improvement, the number increased to 15,108. Thus, our C. briggsae genome annotation improvement uncovered almost 1000 putative orthologous relationships between these two genomes. Our revised orthologous relationships support the strong chromosomal synteny between these two species previously observed (Hillier et al. 2007).
Groups of syntenic genes can form clusters named synteny blocks (Ng et al. 2009). Improved C. briggsae gene models can help detect correct synteny blocks between C. briggsae and C. elegans. Using the synteny detection program OrthoCluster (Zeng et al. 2008; Ng et al. 2009; Vergara and Chen 2010) and the revised orthologous relationships between the two gene sets, we detected larger synteny blocks, and compared with those obtained using orthologous relationships, which were obtained using predicted gene models. Larger synteny blocks are usually formed by merging small neighboring blocks, as illustrated (Fig. 4A,B). The size of the largest perfect synteny block (which is the synteny block with all its contained genes having a one-to-one relationship, contains no mismatches, and both order and strandedness of the contained genes are conserved between these two genomes) increased from 21 to 25 genes, spanning a 152,869-bp region (V:10107907–10199687). At the whole genome scale, the percentage of the C. elegans genome covered by synteny blocks increased from 43.2% to 46.0% after our C. briggsae genome reannotation.
Figure 4.

Gene model revision and synteny analysis. (A) genBlastG-based C. briggsae gene model revision enables the discovery of larger synteny blocks between C. elegans and C. briggsae. Three small synteny blocks were merged into one single larger synteny block that contains all their adjacent synteny blocks. (B) Four C. briggsae gene models, which were revised or discovered in the genBlastG-based gene model revision, are shown at the synteny block break points. (C) A synteny block between C. elegans and C. briggsae is broken down into two smaller synteny blocks. (D) Two hybrid gene models were merged to form a single gene model based on an RNA-seq Illumina intron. (E) RNA-seq evidence supports the merger of two hybrid C. briggsae gene models into a single gene model. (RS) Number of supporting reads.
We next observed how RNA-seq based gene model improvement impacts the orthology relationships between gene models of C. briggsae and C. elegans. Contrary to our expectation, we found a slight decrease in number of C. elegans–C. briggsae orthologs, down from 15,108 to 15,013. This change in the orthologous relationships has also impacted the predicted number and size of synteny blocks. Genome-wide percent coverage of synteny blocks in C. elegans had a minor decrease from 45.97% to 45.58%. This minor decrease can also be observed in the average size of perfect synteny blocks. Number of genes in an average synteny block decreased slightly from 3.67 to 3.66 genes and the average genomic size decreased from 15,892 bp to 15,842 bp. One example is shown in Figure 4C,D. However, the largest perfect synteny block was not affected. It is the same perfect synteny block that spans 25 genes. The slight decrease in the number of orthologs and the level of synteny is due to some revisions made to the gene models. This observation suggests that these orthologous C. elegans gene models of the corresponding RNA-seq-revised C. briggsae gene models are defective, because these C. elegans gene models were used as queries in the genBlastG-based gene model improvement. These C. elegans gene models should therefore be revisited and revised. Indeed, the RNA-seq-supported C. briggsae gene model JNC_CBG21419 clearly suggests that the two C. elegans gene models Y34B4A.2 and Y34B4A.11 may need to be merged as a single C. elegans gene model because the C. briggsae gene models JNC_CBG21419 and JNC_CBG21420 are merged by a heavily supported Illumina intron (Fig. 4E). In a separate example (Fig. 5A,B), the merger of two C. elegans gene models, which is suggested by RNA-seq-supported C. briggsae gene model JNC_CBG04472, is also supported by RNA-seq evidence obtained in a separate study (B Uyar and N Chen, unpubl.).
Figure 5.

RNA-seq evidence suggests that revisions are needed for C. elegans gene models. (A) RNA-seq evidence supports the merger of two C. briggsae hybrid gene models (JNC_CBG04473 and JNC_CBG04472) into a single gene model (JNC_CBG04474). (B) The C. elegans orthologs of the two C. briggsae gene models above should be merged into one single gene model. (RS) Number of supporting reads.
C. briggsae operons show high conservation
Operons have been found in many prokaryotic genomes, but in eukaryotes, operons have only been found in a minority of eukaryote clades including trypanosomes (Sutton and Boothroyd 1986), flatworms (Davis 1997), cnidarians (Stover and Steele 2001), and primitive chordates (Vandenberghe et al. 2001). In C. elegans, operons were first observed by Blumenthal and colleagues (Spieth et al. 1993). Operons in nematodes have been known to be closely spaced gene clusters and the genes contained within one operon are co-transcribed into a polycistronic mRNA precursor. Further molecular and genomics studies suggest that operons are a fairly common form of chromosomal organization in C. elegans (Zorio et al. 1994; Blumenthal and Spieth 1996). The signature for detecting C. elegans operons is that polycistronic pre-mRNA is processed by trans-splicing downstream genes via the spliced leader SL2 in the intergenic regions of the operon. Also, the separation of the downstream genes within the operons is usually 100 to 300 bp in length. A genome-wide study using microarray technology discovered ∼15% of the C. elegans genes are contained within operons. Additional trans-splicing sites and operons were annotated by the modENCODE effort through RNA-seq analysis of C. elegans transcriptomes (Allen et al. 2011). Together, these methods identified more than 1000 operons in C. elegans.
Genes in operons play roles in critical processes including transcription, splicing, and translation (Blumenthal et al. 2002; Blumenthal and Gleason 2003), and in facilitating accelerated recovery from growth-arrested states (Zaslaver et al. 2011). Because of the proposed critical functions of operons, it has been expected that trans-splicing and operons are conserved. Thus, C. elegans operons should also occur in C. briggsae. In fact, previous bioinformatics analysis suggests that the configuration of 96% of C. elegans operons is conserved in C. briggsae (Stein et al. 2003). This analysis was supported subsequently by a separate study (Qian and Zhang 2008), which reports that 93.2% of the operons are conserved between C. elegans and C. briggsae. A caveat of these studies is that both annotated operons in C. briggsae based on the conservation of protein-coding regions, without support of transcript-based annotation of splicing leaders. In other words, C. briggsae operons used in these studies are purely hypothetical.
In this study, we have used RNA-seq reads to annotate trans-splicing sites, splicing leaders, and operons in C. briggsae. We annotated trans-splicing sites and trans-splicing leaders (SLs) through examining misalignments between SL-containing transcript sequences and the C. briggsae genome sequences. Illumina reads (42 bp long) that contain full or partial SL sequences, which are ∼22 bp long, cannot be directly aligned to the reference genome by MAQ when few mismatches were allowed. For reads that contain SL2, we realigned them using cross_match (P Green, pers. comm.), a fast implementation of the Smith-Waterman local alignment algorithm (Smith and Waterman 1981). Altogether, we have annotated 11,617 trans-splicing sites in 8555 C. briggsae gene models (Supplemental data 10,11). Among 11,617 SLs, 8856 are SL1s (in 7871 genes) and 2761 of these were SL2s (including all SLs from SL2 to SL12) (Guiliano and Blaxter 2006) (in 2287 genes). A substantial number of C. briggsae genes allow multiple alternative trans-splicing, a phenomenon also observed in C. elegans (Allen et al. 2011). Reads containing full SL sequences are less frequent than those containing partial SL sequences, suggesting that the number of SL sequences found is an underestimate of the real number.
We next identified candidate operons in C. briggsae following the criteria defined previously (Blumenthal et al. 2002). First, the gene clusters must be closely spaced. The distance between the stop codon of the upstream gene and the start codon of the downstream gene must be <2 kb (Supplemental Fig. 3A). Second, the genes in the clusters must be on the same strand. Third, all the downstream genes of the closely spaced gene cluster must be SL2 trans-spliced. Following these criteria, we annotated 1034 putative operons in C. briggsae (Supplemental data 12,13). These annotated C. briggsae operons are generally evenly distributed on the five autosomes while the operon density on the X chromosome is significantly lower (Fig. 6A); low density of operons on the X chromosome has also been observed in C. elegans (Blumenthal et al. 2002). The operons in the C. briggsae genome have a wide range of sizes, ranging from two to nine genes and with a median size of two genes (Fig. 6B), which is similar to C. elegans operons. In total, these annotated C. briggsae operons contain 2408 genes. The 1034 gene clusters represent the first set of evidence-based annotation of operons in C. briggsae. With this C. briggsae operon set, we reexamined the conservation of operons in these two nematodes. We classified the conservation of operons into three categories (Fig. 6C,D). First, operons are called “conserved” if all the genes of a C. briggsae operon have orthologs in a C. elegans operon and vice versa. Second, operons are called “species specific” if none of the operonic genes in one species have an orthologous operonic gene in the other species. Third, operons are called “divergent” if they are neither entirely “conserved” nor “species specific.” Of the 1105 C. briggsae operons detected in this project, 564 (or 51.0%) were perfectly conserved; 345 (or 31.2%) were divergent; and 196 (or 17.7%) were entirely C. briggsae-specific operons. Our analysis suggests that operons in these two Caenorhabditis species are highly conserved but may not be as conserved as previously reported (Stein et al. 2003; Qian and Zhang 2008).
Figure 6.

Operons in C. briggsae. (A) The white bars indicate the genome-wide distribution of operons in C. elegans and C. briggsae. (B) Size distribution (in number of genes) among all operons annotated in C. elegans and detected in C. briggsae. (C) Size distribution (number of genes) for conserved, divergent, and specific C. elegans operons. (D) Size distribution (number of genes) for conserved, divergent, and specific C. briggsae operons.
Limited conservation of alternative splicing in C. elegans and C. briggsae
Although in this project we did not aim to annotate alternative transcripts in C. briggsae, we did find evidence for 1897 genes with alternative isoforms based on the presence of alternative Illumina introns within genes. In contrast, 2715 C. elegans genes are annotated to have alternative isoforms. These alternatively spliced genes are highly conserved. Specifically, 89.7% of the 1897 putative alternatively spliced C. briggsae genes have orthologs in C. elegans, while 91.9% of the 2715 alternatively spliced C. elegans genes have orthologs in C. briggsae. However, alternative splicing shows limited conservation (Fig. 7)—only 39.6% (752) of the alternatively spliced genes in C. briggsae have C. elegans orthologs that are also alternatively spliced, and only 27.4% of the alternatively spliced genes in C. elegans have C. briggsae orthologs with alternative isoform evidence.
Figure 7.
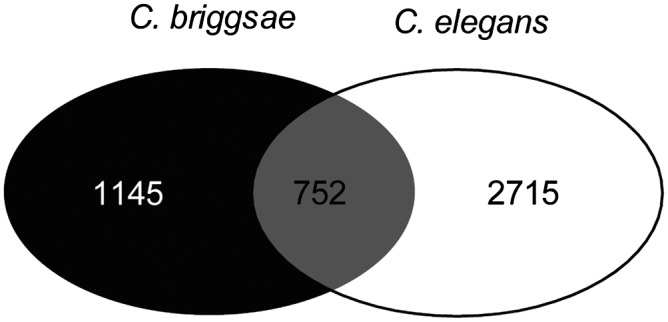
Limited conservation of alternative splicing in C. elegans and C. briggsae.
Two reasons could account for this surprising observation. First, alternative isoforms for many genes, especially those that show very restricted cell-specific expression or are developmentally regulated, may not yet have been discovered. To test this hypothesis, transcriptomes isolated from different tissues or developmental stages, or from worms grown under different conditions including various stress conditions, will need to be sequenced and analyzed. Second, alternative splicing is species specific. In other words, for genes that are alternatively spliced in one species (i.e., C. briggsae), their orthologs in a closely related species (i.e., C. elegans) are not necessarily alternatively spliced. This hypothesis can be tested only after many transcriptomes of both species have been extensively sequenced and analyzed. If evidence supporting this idea is found, such a difference in alternative splicing may help us to understand speciation. Alternative splicing is known to be an important factor in defining species-specific characteristics (Blencowe 2006).
RNA-seq analysis revealed extensive small size errors in the current C. briggsae genome assembly
RNA-seq reads are valuable for validation and fine-tuning gene models (Hillier et al. 2009). Through comparing RNA-seq-defined introns and WormBase introns, we have revised many introns in C. briggsae. However, many Illumina introns cannot be readily used for revising existing introns because their integration would cause frame shifts, as shown above. To explore this, we have examined the alignments of Illumina reads against the C. briggsae genome sequences. We observed both insertions and deletions in the Illumina reads. One possible explanation is that the C. briggsae strain (AF16) used in our transcriptome sequencing project and the C. briggsae strain (another line of AF16) used in the C. briggsae genome sequencing project (Stein et al. 2003) are genetically different due to genetic drift. Such strain differences have been observed between C. elegans strains (Hillier et al. 2008; Flibotte et al. 2010; Sarin et al. 2010). To test this hypothesis, we retrieved Sanger reads used in the C. briggsae genome sequencing projects from NCBI trace archive and aligned these reads using BWA (Li and Durbin 2009) against the reference C. briggsae genome downloaded from WormBase. Genomic differences between the C. briggsae genome sequences and the Sanger reads were identified using SAMTools (Li et al. 2009). To our surprise, we found a large number of misalignments between the Sanger reads and the C. briggsae reference genome sequences. Specifically, we found 7971 insertions (Supplemental data 14), 774 deletions (Supplemental data 15), and 3046 single-nucleotide differences (Supplemental data 16). Although most of the errors are single-base differences, some are much larger. The reason underlying why such differences exist between the WormBase reference sequences and raw Sanger reads generated in the C. briggsae genome sequencing project is not known, but we can argue with confidence that the current C. briggsae reference genome at WormBase contains a large number of errors. This is further supported by the observation that insertions and deletions observed in the alignments of Illumina reads against the WormBase C. briggsae reference genome sequences match perfectly with the insertions and deletions between the Sanger reads and the C. briggsae reference genome sequences. Because hundreds of Illumina introns cannot be incorporated into overlapping genes (as shown above), we expect that up to a few hundred gene models will be further revised after the errors in the C. briggsae reference genome have been fixed.
Discussion
High-quality genome annotation is critical for accurate ortholog assignment, synteny block discovery, and phylogenetic analysis of the molecular evolution of genes and their promoters. It is also important for investigating the molecular evolution of genes including intron gain and loss (Roy and Gilbert 2005), gene family expansion and contraction (Prachumwat and Li 2008), and segmental genome duplication (Jiang et al. 2007). Thus the comparative analysis between the model organism C. elegans and its best studied sister species C. briggsae has been greatly hampered by the limited annotation of the C. briggsae genome. As stated in the most recent WormBase release note (WS228), only 0.2% (or 52) of C. briggsae gene models are fully confirmed and 95.9% lack any confirmation. There are only a very small number of ESTs and experimentally determined mRNAs for the C. briggsae genome in public databases including dbEST database at GenBank. Only 15 C. briggsae gene models are fully supported by these limited numbers of ESTs and mRNAs. In this project, we have attempted to improve and validate the C. briggsae gene models. In the revised C. briggsae gene set, 14,812 (68.3%) genes are at least partially validated, with 7347 (33.9% of all genes with introns) C. briggsae gene models having all of their introns validated. At the transcript (mRNA/cDNA) level, in 47.0% (10,235) of all genes, at least 95% of their entire transcript lengths were validated; 62,727 (60.9%) introns of the C. briggsae gene set are validated. All of these 15 gene models that are supported by ESTs in GenBank are fully supported by RNA-seq data, suggesting a good coverage of the C. briggsae transcriptome by our RNA-seq data. Additionally, we have annotated 5′ UTR sequences for 14,089 C. briggsae genes, and 3′ UTR sequences for 14,089 C. briggsae genes. Furthermore, our RNA-seq analysis also allowed us to successfully identify trans-splicing sites in 8555 C. briggsae gene models; 11,617 SLs were identified, including 8856 SL1s (in 7871 genes) and 2761 SL2s (in 2287 genes). Based on the trans-splicing information, we annotated 1034 operons in the C. briggsae genome. This major advancement in C. briggsae genome annotation, validation, and confirmation will undoubtedly facilitate a far more accurate comparative genomics analysis between these two important nematode models: C. elegans and C. briggsae.
The improvement in C. briggsae genome annotation was made possible partly by the recent, and substantial, progress in the C. elegans genome annotation. This progress is due to a large collection of high-throughput gene annotation projects as well as gene annotation from small-scale projects in the C. elegans research community. We have developed a novel suite of algorithms genBlastA (She et al. 2009) and genBlastG (She et al. 2011), to take advantage of the improved C. elegans genome annotation. This had allowed us to improve the C. briggsae genome annotation through homology-based genome annotation. In particular, genBlastA predicts genomic regions that contain a candidate homologous gene, while genBlastG defines the homologous gene model based on similarity between the query gene and the target gene. This effort resulted in revising 6715 C. briggsae gene models and adding 1091 novel gene models that were entirely missed in previous annotations. Validation and further improvement of the C. briggsae genome annotation was made possible by exploiting RNA-seq data (Hillier et al. 2009; Wang et al. 2009). These data were generated from high-quality C. briggsae transcriptomes and sequenced using Illumina DNA sequencing technology. The power of RNA-seq enabled us to validate gene models, revise defective gene models, as well as detect UTRs and trans-splicing signals, which in turn allowed us to detect operons in C. briggsae.
We demonstrated that the revised C. briggsae genome annotation improved ortholog assignment between C. elegans and C. briggsae and the identification of synteny blocks between these two species. Note that the synteny block results reported here are noticeably different from our previous report based on WormBase WS180 (Vergara and Chen 2010). For example, the largest synteny block reported using the WS180 annotation is 42 genes after gene model improvement (Vergara and Chen 2010), compared with 21 genes (or 25 genes after the improvement) in this study. The difference is due to the incorporation of the nGASP annotations (Coghlan et al. 2008) in recent WormBase releases, suggesting that many nGASP C. briggsae gene models are likely false positives that incorrectly break synteny blocks. Most likely these false annotations will be eliminated through evidence-based gene model improvement.
With transcript evidence-based operons in C. briggsae, we can for the first time compare the conservation and divergence of operons in C. elegans and C. briggsae. In contrast, conservation analyses of operon evolution in previous studies were all carried out between curated C. elegans operons and estimated operons in C. briggsae (Stein et al. 2003; Qian and Zhang 2008). Comparing operons annotated in C. elegans (obtained from WormBase) with C. briggsae operons curated in this project through RNA-seq analysis, we have confirmed that the majority (51.4%) of operons are conserved between these two species. However, we found operon conservation is not as high as previously estimated (i.e., 93%–96% of conservation) because we found at least 153 operons (14.8%) that are entirely C. briggsae specific. The remaining 349 operons (33.8%) are essentially conserved between C. elegans and C. briggsae, but their configurations are not identical and have some level of divergence.
We expect that the C. briggsae genome annotation will be further improved by deeper sequencing of transcriptomes sampled from different cell types at various developmental stages. In particular, alternative isoforms for C. briggsae genes will be defined. In this study, we found a very limited level of conservation of alternatively spliced gene models. For genes that are alternatively spliced in C. elegans, their orthologs in C. briggsae are not necessarily alternatively spliced, and vice versa. This result is interesting and the different use of alternative splicing isoforms may be important for defining species specificity. However, the differences may be also due to lack of depth of transcriptome sequencing for both C. elegans and C. briggsae. Deeper sequencing will be able to help test this hypothesis. Deeper transcriptome sequencing may also find evidence to support or disprove 3072 gene models that currently have no support. Furthermore, deeper sequencing of C. briggsae transcriptomes will also enable us to build novel full-length gene models based on genelets uncovered by RNA-seq analysis.
An unexpected finding of this project is the observation that the current C. briggsae genome assembly harbors over 10 thousand instances of genomic errors. Although the reason for the occurrence of these errors is still unknown, we have identified their coordinates and nature. These errors can be easily corrected in the C. briggsae genome (Supplemental data 14–16), which will allow for the repair of hundreds of gene models that were incorrectly annotated to circumvent the impact of the genomic errors.
Methods
Creation of the hybrid C. briggsae gene set
The hybrid C. briggsae gene set was built by using genBlastG-predicted gene models to replace WormBase C. briggsae gene models if the protein PIDs between genBlastG-predicted gene models and their corresponding C. elegans orthologs were at least 2% higher than the PIDs between WormBase C. briggsae gene models and their corresponding C. elegans orthologs. Because some C. briggsae gene models may overlap just as some of the C. elegans gene models do (Chen and Stein 2006), and because some C. briggsae gene models may still be defective, we allow 5% overlap of coding sequences between two adjacent gene models to ensure that bona fide gene models are not eliminated from the analysis. In this analysis, we used C. briggsae reference genome sequences and gene set from WormBase release WS215 and genBlastG (v135).
Transcriptome library production and deep sequencing
Tissue samples were put through an RNA extraction using TRIzol (Invitrogen, SKU# 10296-028). The cDNA libraries used in this project were created with the Superscript III reverse transcriptase kit (Invitrogen, SKU# 18080-085), and the primer used to initiate reverse transcription was a modified oligo d(T) primer (5′-CCAGACACTATGCTCATACGACGCAGT(16) VN-3′) (Invitrogen). The protocol accompanying the kit was followed, and the samples were treated with Ribonuclease H (Invitrogen, SKU# 18021-014). DNA sequencing was performed on the Illumina cluster station and 1G analyzer (Illumina).
MAQ alignment of Illumina reads to the C. briggsae genome
All parameters are in default except for the following parameters: a = 700 (the maximum insert size allowed for correct pairing of reads); n = 3 (the maximum number of mismatches allowed in the first 28 bp of the Illumina read alignment).
Illumina intron and exon identification
MAQ annotate as “64/192” read pairs whose ends are mapped while their mates are not mapped. Read ends with code 64 are mapped successfully and their mates with code 192 are unmapped to the reference genome. Code 192 reads obtained in MAQ alignments can be used to define splicing sites. We first aligned these code 192 reads to the reference genome by employing the widely used local alignment program cross_match (P Green, pers. comm.). With the obtained genomic regions, we applied SuperSplat (Bryant et al. 2010) to find introns. As SuperSplat does not depend on canonical splice sites, from the putative introns reported by SuperSplat, we select only those introns which have canonical splice sites. Thus, our results may underestimate the number of validated introns because a small fraction (1%–2%) of introns is noncanonical (Sparks and Brendel 2005). Next, the Illumina exons were parsed from the consensus sequence.
Splicing leader identification
We detected trans-splicing sites by following these steps: (1) Obtain code 192 reads from MAQ-aligned results. (2) Use cross_match to remap those code 192 reads to the flanking region where their mates are mapped. (3) For each gene model, check the 100-bp region upstream of the 5′ end and find reads that are mapped by MAQ/remapped by cross_match to this region. This number is selected because ∼90% of the known trans-splicing sites are found within a 100-bp region upstream of the start codons (Supplemental Fig. 3B). (4) Align the 5′ end of these read sequences to the 3′ end of the known SL sequences to detect and categorize trans-splicing sites (Guiliano and Blaxter 2006).
RNA-seq-based gene model validation and further revision
Confirmation or addition of new coding exons to the predicted gene models depends on the existence of Illumina reads aligned to the genomic region of interest. Genomic segments in which all bases were covered by Illumina read alignments without gaps, which we termed “Illumina exons,” were obtained by first running MAQ's “assemble” function to get the consensus sequence from the reads mapped to the genome. Read pairs with only single ends mapped to the genome suggest the existence of previously unannotated cis-splicing or trans-splicing events, which can be used to locate the existence of introns or trans-splicing acceptor sites (Blumenthal and Gleason 2003), which is described below. A newly developed program SuperSplat (Bryant et al. 2010) was applied to predict introns defined by such unmappable read mates. We applied SuperSplat to define introns using the 3.1 million code 192 reads. In this project, we name a putative intron defined by Illumina reads a “Illumina intron.” Applying SuperSplat and 3.1 million single ends obtained above, we have identified 78,252 Illumina introns, 59,560 of which are supported by two or more independent Illumina reads (Supplemental data 4).
Next, we used the Illumina introns and Illumina exons to validate C. briggsae gene models and to further improve gene models by systematically comparing with introns and exons in the C. briggsae hybrid gene set defined above. Based on their relationship with predicted protein-coding gene models, an Illumina intron can be categorized as an intragenic intron if it overlaps with a gene and is entirely nested within a predicted gene model. Otherwise, it is categorized as a boundary or intergenic introns. Intergenic introns indicate the presence of protein-coding genes that have not been annotated previously. Because their full gene model is not defined, they are annotated as genelets (Hillier et al. 2009). Protein-coding genes in eukaryotic genomes consist of exons, introns, and untranslated regions (5′ and 3′ UTRs).
As the first step in validating and revising C. briggsae gene models, we focused on the internal components of genes. Because genomic sequences spanning introns and exons are complementary in gene models, gene model definition is essentially equivalent to intron definition. Once introns are defined, exons are readily defined. For gene models that are computationally predicted, such as those in the C. briggsae hybrid gene set, exons and introns may be correct, defective, or entirely missing. When compared with Illumina introns, introns in predicted gene models can be confirmed, modified, or removed. Additionally, novel introns may be introduced to the gene models. Furthermore, introns in predicted gene models can be spurious and can be removed if their existence is in conflict with transcript reads. Finally, alternative introns that overlap with each other can be identified as well.
An intragenic Illumina intron can be a perfect match, a partial match to a predicted intron, or a novel intron. If a predicted intron (in the C. briggsae hybrid gene set) is identical to an Illumina intron that is supported by one or more independent Illumina reads, we annotated this predicted intron as a confirmed (i.e., validated) intron (Fig. 3A). If a validated intron overlaps with one or more different Illumina introns, the intron is recorded as an alternative Illumina intron, which suggests that the corresponding gene has multiple isoforms and is therefore alternatively spliced. Our goal in this study is to identify one transcript per gene, thus the exact structures of alternative isoforms are beyond the scope of this paper. However, the alternative Illumina introns will be valuable for further defining full-length isoforms in the future (Supplemental data 4). Among 102,406 predicted introns in the C. briggsae hybrid gene set (based on WS215 and genBlastG version 135), 59,137 (or 57.5%) predicted introns are confirmed by Illumina introns. These confirmed introns fall into 14,703 protein-coding genes, suggesting that these genes are at least partially confirmed. In other words, because we have detected the existence and expression of 63.2% of the predicted genes in the C. briggsae hybrid gene set, these 14,703 genes are likely real although not necessarily fully defined. Furthermore, out of 21,683 genes (containing at least one intron), 7347 (or 33.9%) gene models have all of their introns fully confirmed.
While the perfect matches validate the corresponding predicted introns, others provide experimental evidence to repair the gene models. Among 59,560 Illumina introns that are supported by split alignments of two or more Illumina reads, we found 10,079 that did not fully match with predicted introns, suggesting that we could make further improvements of the C. briggsae genome annotation. Next, we used these 10,079 Illumina introns to revise the hybrid gene models. Specifically, if a predicted intron overlaps with one Illumina intron that is supported by two or more independent Illumina reads but this predicted intron is different from Illumina intron, it is replaced by an Illumina intron (Fig. 3B). If a predicted intron overlaps with multiple overlapping Illumina introns, the Illumina intron with the highest read support (i.e., the number of split read alignments) is used to replace the predicted intron, while others were recorded as alternative introns. When an Illumina intron is used to replace a predicted intron, the flanking exons were altered to create splice-junctions for the new intron of the revised gene model. Here we enforce that the length difference between the predicted intron (which is to be replaced) and the Illumina intron must be a multiple of 3 so that the reading frame is preserved (i.e., unshifted) and that the introduced coding region does not contain stop codons. Furthermore, we enforce that the newly introduced coding regions must be supported by Illumina exons (at least 90% of the length of the new coding regions must have read support). We found 6617 Illumina introns that overlapped with but were not identical to one or more predicted introns in the C. briggsae hybrid gene set. Among these Illumina introns, 2111 (or 31.90%) were successfully used to replace 2244 predicted introns, while 2301 (or 34.77%) were not incorporated because they overlapped with other Illumina introns, which had higher coverage. These 2301 Illumina introns suggest the existence of alternative splicing in at least 1176 C. briggsae gene models. This type of intron revision affects 2077 (or 8.9%) C. briggsae gene models in the hybrid gene set. The rest of the 2205 Illumina introns were not successfully integrated into the gene models because their integration would cause frame shifts, or there is a lack of support for the flanking exonic regions. The reason for these failures is explored below.
Additionally, among 59,560 Illumina introns that were supported by two or more independent Illumina reads in C. briggsae genome, 878 (or 14.72%) overlapped with annotated hybrid coding exons (but not predicted introns). We attempted to integrate these Illumina introns into gene models as novel introns if their incorporation did not cause a shift in the reading frame (Fig. 3C). In other words, the lengths of such Illumina introns had to be in multiples of 3. If such a novel Illumina intron overlapped with other Illumina introns, only the one supported by the largest number of independent Illumina reads was incorporated. We incorporated 716 out of 878 novel introns into 638 genes. These novel introns range from 39 to 927 bp (average, 51.7 bp) in size. The rest of these novel Illumina introns (162 out of 878) were not incorporated in any gene models because they would have either caused a frame shift (for reasons described later) or have a lower support.
Some predicted introns were not supported by any Illumina intron and the corresponding genomic region was covered by Illumina exons. We reasoned that these introns were likely false annotations and thus removed them from gene models. In the meantime, the corresponding genomic region was converted as part of a coding exon (Fig. 3D). For a predicted intron to be removed, we enforced that the length of the intron was a multiple of 3 so that the incorporation of the new coding region does not shift the reading frame. Altogether, we removed 461 such predicted introns in 392 gene models. These introns range from 18 bp to 1059 bp (average, 106 bp) in size.
Comparisons between hybrid C. briggsae gene models and the Illumina introns suggested that hundreds of gene models needed revision at their ends because they overlapped with Illumina introns at the 3′ or 5′ boundaries. A hybrid gene model was extended at the 3′ end if the following conditions were met (Fig. 3E). First, the gene model lacked an annotated stop codon, which is true for many WormBase C. briggsae gene models that are incorporated into the hybrid gene set. Such hybrid gene models overlapped with Illumina exons at the 3′ end and either contained a stop codon or connected the gene model to a neighboring Illumina intron or a predicted intron. In this project we attempted to identify a stop codon for all gene models. Second, when an Illumina intron overlapped with the 3′ terminal exon of a predicted gene model, which suggested that the predicted gene model should be extended at the 3′ end, the Illumina intron was incorporated into the hybrid gene model. Thus the overlapping terminal exons were reduced, and the hybrid start/stop codons were modified. Third, when an Illumina intron was found in the neighborhood of the hybrid gene model, and an Illumina exon was found to bridge the Illumina intron to the hybrid gene model, the Illumina intron was also incorporated into the hybrid gene model. Accordingly, the terminal exon was extended and the hybrid start/stop codons were modified. Finally, an Illumina exon that could connect a hybrid gene model to a predicted intron of an adjacent predicted gene model suggests that the two adjacent gene models should be merged into a single gene model. Thus, the Illumina exon was incorporated as an exon (or part of an exon) of the new gene model. Illumina introns or predicted introns of neighboring genes were added to the gene model as the extension proceeds. Any Illumina intron that is incorporated must be supported by two or more split read alignments. The extension stops after the first in-frame stop codon was found. The extension was accepted only if the introduced coding exons were supported by Illumina reads up until the new stop codon.
The gene model extension at the 5′ end was implemented following a similar idea as the extensions at the 3′ end described above (Fig. 3F). The main difference here was that both start and stop codons needed to be examined simultaneously. The start codons were looked at in order to find the 5′ end of the new gene model while the stop codons were looked at to ensure that a premature stop codon was not incorporated into the revised gene model. If an Illumina intron overlapped with the hybrid gene model at the 5′ end, or the Illumina exons at the 5′ end connected the gene model to an upstream Illumina intron or a predicted intron of an upstream neighboring gene model, then the gene model could be extended at the 5′ end. The hybrid gene model was extended and the start codon found upstream was recorded as the new start codon. Extension proceeded until (1) a stop codon was found or (2) Illumina exons or existing coding exons of neighboring gene models did not support the extension. Thus, the most upstream start codon in the genomic region supported by Illumina reads before the first encounter with the stop codon was annotated as the new start codon. As a result of the application of extension procedures to 23,276 gene models in the C. briggsae hybrid set, 762 gene models were extended at either the 3′ or the 5′ end, among which 399 gene models were extended at the 3′ end, 386 gene models were extended at the 5′ end, and 23 gene models were extended at both the 3′ and the 5′ ends.
In addition to coding sequences, we have also annotated UTR sequences based on Illumina reads aligned to the genome. At the 3′ end of the gene model, the Illumina exon that covers the stop codon contains the 3′ UTR, which starts immediately after the stop codon and ends with the end of the Illumina exon. 3′ UTR regions were found for 14,089 genes (Supplemental data 8,9). The Illumina exons immediately upstream of the 5′ end of the gene models contain the 5′ UTRs. For trans-spliced genes, which are described in details below, the 5′ UTR region starts immediately after the trans-splicing acceptor site and ends before the start codons. For the genes that are not trans-spliced, the 5′ UTR is the region between the start codon and the start of the Illumina exon that covers the start codon. We have found 5′ UTR sequences for 14,089 genes (Supplemental data 8,9).
As a result of our combined homology and RNA-seq-based improvements, in the C. briggsae gene set of 21,683 intron-containing genes (Supplemental data 5,7), 61,503 (60.9%) introns have been validated, 14,812 (68.3%) genes have at least one intron validated, and 7347 (33.9%) genes have all introns validated. At the transcript level, 10,235 genes (or 47.0% of all genes) were found to have ≥95% of their cDNA sequences supported by Illumina read alignments. This is a remarkable advance because, after almost 10 yr since the annotation of the C. briggsae genome (Stein et al. 2003), only 853 (or 3.9%) genes are partially confirmed (according to the most recent release from WormBase WS228).
Identification of genomic errors in C. briggsae genome assembly
The raw Sanger sequencing reads and the corresponding quality files were downloaded from NCBI trace archives (ftp://ftp.ncbi.nih.gov/pub/TraceDB/caenorhabditis_briggsae). These reads, which were originally generated by the Genome Sequencing Center at Washington University in St. Louis, were assembled by the C. briggsae genome analysis consortium (Stein et al. 2003). The assembly (cb25 supercontigs) was downloaded from the following WormBase FTP site: ftp://ftp.wormbase.org/pub/wormbase/genomes/c_briggsae/assembly/cb25.agp8/genome_assembly/cb25.agp8.
For detecting potential genomic errors in the current C. briggsae reference genome sequences, we aligned the raw Sangers reads and the cb25 supercontigs separately against the C. briggsae reference genome sequences using the Burrows-Wheeler Alignment tool (BWA) (Li and Durbin 2009). We used the algorithm ‘bwasw’ that was designed for long reads. The resulting SAM alignments were then converted into the BAM format using SAMtools (Li et al. 2009), merged and displayed in a generic genome browser (Stein et al. 2002) for visualization. The variations were generated by BWA and extracted using the ‘pileup’ command from SAMtools (Li et al. 2009). Indels were filtered using the following criteria: read depth ≥5 and >50% of reads supporting the variation. Single nucleotide differences were filtered using the following criteria: read depth ≥5, SNP quality ≥40, reads support >40%, consensus quality (CQUAL) ≥22. A complete list of filtered insertions, deletions, and single nucleotide differences is described in a GFF3 file (Supplemental data 14–16).
Data access
All data that were generated in the course of this research are made publicly available. Paired-end sequencing data of the C. briggsae transcriptomes have been submitted to the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA050228. Processed data are attached as supplementary data and they are being submitted to WormBase for display at WormBase or further analysis by WormBase curators.
Acknowledgments
We thank Nancy Hawkins, Robert Johnsen, and Maja Tarailo-Graovac for critical review of the manuscript. We thank Rong She and Ke Wang for helping us with genBlastG. This study is supported by Discovery Grants from the Natural Science and Engineering Research Council (NSERC) of Canada (to D.L.B. and N.C.). D.L.B. is a Canada Research Chair. N.C. is a Michael Smith Foundation for Health Research (MSFHR) Scholar and a Canadian Institutes of Health Research (CIHR) New Investigator.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.134601.111.
References
- Allen MA, Hillier LW, Waterston RH, Blumenthal T 2011. A global analysis of C. elegans trans-splicing. Genome Res 21: 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V 2004. The functions of animal microRNAs. Nature 431: 350–355 [DOI] [PubMed] [Google Scholar]
- Bao Z, Murray JI, Boyle T, Ooi SL, Sandel MJ, Waterston RH 2006. Automated cell lineage tracing in Caenorhabditis elegans. Proc Natl Acad Sci 103: 2707–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencowe BJ 2006. Alternative splicing: New insights from global analyses. Cell 126: 37–47 [DOI] [PubMed] [Google Scholar]
- Blumenthal T, Gleason KS 2003. Caenorhabditis elegans operons: Form and function. Natl Rev 4: 112–120 [DOI] [PubMed] [Google Scholar]
- Blumenthal T, Spieth J 1996. Gene structure and organization in Caenorhabditis elegans. Curr Opin Genet Dev 6: 692–698 [DOI] [PubMed] [Google Scholar]
- Blumenthal T, Evans D, Link CD, Guffanti A, Lawson D, Thierry-Mieg J, Thierry-Mieg D, Chiu WL, Duke K, Kiraly M, et al. 2002. A global analysis of Caenorhabditis elegans operons. Nature 417: 851–854 [DOI] [PubMed] [Google Scholar]
- Bryant DW Jr, Shen R, Priest HD, Wong WK, Mockler TC 2010. Supersplat—spliced RNA-seq alignment. Bioinformatics 26: 1500–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium 1998. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282: 2012–2018 [DOI] [PubMed] [Google Scholar]
- Chen N, Stein LD 2006. Conservation and functional significance of gene topology in the genome of Caenorhabditis elegans. Genome Res 16: 606–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan A, Wolfe KH 2002. Fourfold faster rate of genome rearrangement in nematodes than in Drosophila. Genome Res 12: 857–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan A, Fiedler TJ, McKay SJ, Flicek P, Harris TW, Blasiar D, Stein LD 2008. nGASP—the nematode genome annotation assessment project. BMC Bioinformatics 9: 549 doi: 10.1186/1471-2105-9-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RE 1997. Surprising diversity and distribution of spliced leader RNAs in flatworms. Mol Biochem Parasitol 87: 29–48 [DOI] [PubMed] [Google Scholar]
- Driscoll M, Dean E, Reilly E, Bergholz E, Chalfie M 1989. Genetic and molecular analysis of a Caenorhabditis elegans β-tubulin that conveys benzimidazole sensitivity. J Cell Biol 109: 2993–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flibotte S, Edgley ML, Chaudhry I, Taylor J, Neil SE, Rogula A, Zapf R, Hirst M, Butterfield Y, Jones SJ, et al. 2010. Whole-genome profiling of mutagenesis in Caenorhabditis elegans. Genetics 185: 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiliano DB, Blaxter ML 2006. Operon conservation and the evolution of trans-splicing in the phylum Nematoda. PLoS Genet 2: e198 doi: 10.1371/journal.pgen.0020198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta BP, Sternberg PW 2003. The draft genome sequence of the nematode Caenorhabditis briggsae, a companion to C. elegans. Genome Biol 4: 238 doi: 10.1186/gb-2003-4-12-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta BP, Johnsen R, Chen N 2007. Genomics and biology of the nematode Caenorhabditis briggsae. WormBook: The online review of C. elegans biology 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Coulson A, Murray JI, Bao Z, Sulston JE, Waterston RH 2005. Genomics in C. elegans: So many genes, such a little worm. Genome Res 15: 1651–1660 [DOI] [PubMed] [Google Scholar]
- Hillier LW, Miller RD, Baird SE, Chinwalla A, Fulton LA, Koboldt DC, Waterston RH 2007. Comparison of C. elegans and C. briggsae genome sequences reveals extensive conservation of chromosome organization and synteny. PLoS Biol 5: e167 doi: 10.1371/journal.pbio.0050167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, Fox P, Glasscock JI, Hickenbotham M, Huang W, et al. 2008. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods 5: 183–188 [DOI] [PubMed] [Google Scholar]
- Hillier LW, Reinke V, Green P, Hirst M, Marra MA, Waterston RH 2009. Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res 19: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz HR 2003. Worms, life, and death (Nobel lecture). ChemBioChem 4: 697–711 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE 2007. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet 39: 1361–1368 [DOI] [PubMed] [Google Scholar]
- Kohara Y 1996. [Large scale analysis of C. elegans cDNA]. (Article in Japanese) Tanpakushitsu Kakusan Koso 41: 715–720 [PubMed] [Google Scholar]
- Lamesch P, Milstein S, Hao T, Rosenberg J, Li N, Sequerra R, Bosak S, Doucette-Stamm L, Vandenhaute J, Hill DE, et al. 2004. C. elegans ORFeome version 3.1: Increasing the coverage of ORFeome resources with improved gene predictions. Genome Res 14: 2064–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ruan J, Durbin R 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangone M, Manoharan AP, Thierry-Mieg D, Thierry-Mieg J, Han T, Mackowiak SD, Mis E, Zegar C, Gutwein MR, Khivansara V, et al. 2010. The landscape of C. elegans 3′UTRs. Science 329: 432–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt MJ, Moerman DG, Chen N 2010. Identifying novel genes in C. elegans using SAGE tags. BMC Mol Biol 11: 96 doi: 10.1186/1471-2199-11-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng MP, Vergara IA, Frech C, Chen Q, Zeng X, Pei J, Chen N 2009. OrthoClusterDB: An online platform for synteny blocks. BMC Bioinformatics 10: 192 doi: 10.1186/1471-2105-10-192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Z, Cox AJ, Mullikin JC 2001. SSAHA: A fast search method for large DNA databases. Genome Res 11: 1725–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prachumwat A, Li WH 2008. Gene number expansion and contraction in vertebrate genomes with respect to invertebrate genomes. Genome Res 18: 221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Zhang J 2008. Evolutionary dynamics of nematode operons: Easy come, slow go. Genome Res 18: 412–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboul J, Vaglio P, Rual JF, Lamesch P, Martinez M, Armstrong CM, Li S, Jacotot L, Bertin N, Janky R, et al. 2003. C. elegans ORFeome version 1.1: Experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat Genet 34: 35–41 [DOI] [PubMed] [Google Scholar]
- Roy SW, Gilbert W 2005. Rates of intron loss and gain: Implications for early eukaryotic evolution. Proc Natl Acad Sci 102: 5773–5778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzanov P, Riddle DL 2010. Deep SAGE analysis of the Caenorhabditis elegans transcriptome. Nucleic Acids Res 38: 3252–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzanov P, Jones SJ, Riddle DL 2007. Discovery of novel alternatively spliced C. elegans transcripts by computational analysis of SAGE data. BMC Genomics 8: 447 doi: 10.1186/1471-2164-8-447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N 2007. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol 17: 592–598 [DOI] [PubMed] [Google Scholar]
- Salehi-Ashtiani K, Lin C, Hao T, Shen Y, Szeto D, Yang X, Ghamsari L, Lee H, Fan C, Murray RR, et al. 2009. Large-scale RACE approach for proactive experimental definition of C. elegans ORFeome. Genome Res 19: 2334–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S, Bertrand V, Bigelow H, Boyanov A, Doitsidou M, Poole RJ, Narula S, Hobert O 2010. Analysis of multiple ethyl methanesulfonate-mutagenized Caenorhabditis elegans strains by whole-genome sequencing. Genetics 185: 417–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- She R, Chu JS, Wang K, Pei J, Chen N 2009. GenBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res 19: 143–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- She R, Chu JS, Uyar B, Wang J, Wang K, Chen N 2011. genBlastG: Using BLAST searches to build homologous gene models. Bioinformatics 27: 2141–2143 [DOI] [PubMed] [Google Scholar]
- Shim YH, Paik YK 2010. Caenorhabditis elegans proteomics comes of age. Proteomics 10: 846–857 [DOI] [PubMed] [Google Scholar]
- Shin H, Hirst M, Bainbridge MN, Magrini V, Mardis E, Moerman DG, Marra MA, Baillie DL, Jones SJ 2008. Transcriptome analysis for Caenorhabditis elegans based on novel expressed sequence tags. BMC Biol 6: 30 doi: 10.1186/1741-7007-6-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF, Waterman MS 1981. Identification of common molecular subsequences. J Mol Biol 147: 195–197 [DOI] [PubMed] [Google Scholar]
- Sparks ME, Brendel V 2005. Incorporation of splice site probability models for non-canonical introns improves gene structure prediction in plants. Bioinformatics (Suppl 3) 21: iii20–iii30 [DOI] [PubMed] [Google Scholar]
- Spieth J, Lawson D 2006. Overview of gene structure. WormBook 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieth J, Brooke G, Kuersten S, Lea K, Blumenthal T 1993. Operons in C. elegans: Polycistronic mRNA precursors are processed by trans-splicing of SL2 to downstream coding regions. Cell 73: 521–532 [DOI] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al. 2002. The generic genome browser: A building block for a model organism system database. Genome Res 12: 1599–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, et al. 2003. The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. PLoS Biol 1: e45 doi: 10.1371/journal.pbio.0000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover NA, Steele RE 2001. Trans-spliced leader addition to mRNAs in a cnidarian. Proc Natl Acad Sci 98: 5693–5698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RE, Boothroyd JC 1986. Evidence for trans splicing in trypanosomes. Cell 47: 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe AE, Meedel TH, Hastings KE 2001. mRNA 5′-leader trans-splicing in the chordates. Genes & Dev 15: 294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara IA, Chen N 2010. Large synteny blocks revealed between Caenorhabditis elegans and Caenorhabditis briggsae genomes using OrthoCluster. BMC Genomics 11: 516 doi: 10.1186/1471-2164-11-516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M 2009. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Lamesch P, Arumugam M, Rosenberg J, Hu P, Vidal M, Brent MR 2005. Closing in on the C. elegans ORFeome by cloning TWINSCAN predictions. Genome Res 15: 577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslaver A, Baugh LR, Sternberg PW 2011. Metazoan operons accelerate recovery from growth-arrested states. Cell 145: 981–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Pei J, Vergara IA, Nesbitt MJ, Wang K, Chen N 2008. OrthoCluster: A new tool for mining synteny blocks and applications in comparative genomics. In Proceedings of the Eleventh International Conferences on Extending Database Technology (EDBT'08), Nantes, France [Google Scholar]
- Zhao Z, Boyle TJ, Bao Z, Murray JI, Mericle B, Waterston RH 2008. Comparative analysis of embryonic cell lineage between Caenorhabditis briggsae and Caenorhabditis elegans. Dev Biol 314: 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio DA, Cheng NN, Blumenthal T, Spieth J 1994. Operons as a common form of chromosomal organization in C. elegans. Nature 372: 270–272 [DOI] [PubMed] [Google Scholar]