

Abstract
Whole-genome shotgun assembly has been a long-standing issue for highly polymorphic genomes, and the advent of next-generation sequencing technologies has made the issue more challenging than ever. Here we present an automated pipeline, HaploMerger, for reconstructing allelic relationships in a diploid assembly. HaploMerger combines a LASTZ-ChainNet alignment approach with a novel graph-based structure, which helps to untangle allelic relationships between two haplotypes and guides the subsequent creation of reference haploid assemblies. The pipeline provides flexible parameters and schemes to improve the contiguity, continuity, and completeness of the reference assemblies. We show that HaploMerger produces efficient and accurate results in simulations and has advantages over manual curation when applied to real polymorphic assemblies (e.g., 4%–5% heterozygosity). We also used HaploMerger to analyze the diploid assembly of a single Chinese amphioxus (Branchiostoma belcheri) and compared the resulting haploid assemblies with EST sequences, which revealed that the two haplotypes are not only divergent but also highly complementary to each other. Taken together, we have demonstrated that HaploMerger is an effective tool for analyzing and exploiting polymorphic genome assemblies.
Whole-genome shotgun (WGS) assembly for highly polymorphic organisms, such as many outbred insects and marine animals, is difficult and does not usually reach the same level of quality as assemblies for organisms with low levels of polymorphism (Aparicio et al. 2002; Dehal et al. 2002; Holt et al. 2002; Jones et al. 2004; Vinson et al. 2005). This difficulty has become even more challenging with the application of next-generation sequencing (NGS) technologies (Pop 2009). On the one hand, short read lengths and new types of sequencing errors exacerbate the difficulties of polymorphic WGS assemblies; on the other hand, NGS technologies have stimulated an increase in genome projects targeting polymorphic organisms.
Several assembly and pre-assembly strategies can be used to handle the heterozygosity of polymorphic genomes. Inbreeding prior to genome sequencing reduces heterozygosity, although the efficacy is much lower than theory predicts (Barriere et al. 2009). Clone-by-clone (BAC and fosmid) sequencing is a well-known method for resolving haplotypes and becomes more promising when combined with NGS technologies (Kitzman et al. 2011). In addition, assembly algorithms can be adjusted to better accommodate polymorphic data. For highly polymorphic genomes, one effective algorithm is to force different haplotypes to assemble separately by imposing a strict overlap requirement on reads (Vinson et al. 2005). For less polymorphic genomes, a feasible strategy is to force different alleles into one consensus by allowing promiscuous overlaps of reads (Aparicio et al. 2002; Dehal et al. 2002). Recently, Donmez and Brudno (2011) proposed a new algorithm for polymorphic assembly that featured a haplotype-aware Bayesian approach for error correction and a novel graph-based method for mate-pair analysis.
Although assembly and pre-assembly strategies may improve the quality of polymorphic assemblies, they do not resolve allelic relationships between haplotypes. Usually, allelic relationships in a polymorphic assembly are inferred at the post-assembly stage: all-against-all alignments are first created for an assembly, and then allelic relationships are determined by examining the alignments. In theory, if allelic relationships are correctly reconstructed, a reference haploid assembly can be created from the polymorphic assembly. First, allelic relationships are used to separate different allelic sequences into subassemblies, where each subassembly contains at most one allele for every genomic locus, which is termed a haploid assembly. Second, for each locus, a single allelic sequence from one of the haploid assemblies is elected as the representative for the locus, and the combination of all representative sequences comprise the so-called reference haploid assembly. Depending on operational preferences, the derived reference haploid assembly may exceed the original assembly in certain attributes, including contiguity, continuity, and accuracy.
In reality, a polymorphic assembly could be highly fragmented and contain numerous assembly errors. In such an assembly, less polymorphic alleles tend to collapse together, whereas divergent alleles remain separate, resulting in fractured contigs and scaffolds. In addition, mate-pairs are prone to linking different haplotypes by error, which causes switches (or splices) from one haplotype to another. When complicated by sequencing errors, repeats, and sequence duplication, mate-pair errors and the presence of multiple alleles can further confuse the assembly software and lead to excessive assembly errors, including insertions, deletions, inversions, translocations, tandem misassemblies of alleles, and misjoining of different genome portions. Therefore, post-assembly refinements for polymorphic assemblies are inherently messy, and a clean computational solution is difficult. In fact, early studies relied on manual inspection of the BLASTN alignments to curate reference haploid assemblies (Small et al. 2007; Putnam et al. 2008; Denoeud et al. 2010). Moreover, present NGS technologies do not mitigate the problem, but instead make it worse, because a high-sequencing error rate and short read length obscure the differences (SNPs and indels) between alleles and aggravate the extent of mate-pair errors.
Here we present HaploMerger, an easy-to-use automated pipeline for streamlining the post-assembly refinement operations for polymorphic diploid assemblies (Fig. 1). HaploMerger features a LASTZ-ChainNet approach for whole-genome alignments and a novel graph-based structure for describing and untangling the homologous/allelic relationships in a diploid assembly. The LASTZ alignment tool inherits its strategy from BLASTN (and its sensitivity) and gains more specificity by implementing colinearity checking, recursive search, soft masking, dynamic masking, and a genome-specific scoring matrix (Altschul et al. 1997; Schwartz et al. 2003; Harris 2007). LASTZ alignments are chained by the ChainNet algorithm, which has at least two intended advantages for HaploMerger: a balance between sensitivity and specificity that is achieved by using a k-dimensional tree, a red-black tree, a genome-specific scoring matrix, and a gap-scoring matrix; a flexible chain-net alignment structure that accommodates inversions, translocations, duplications, large insertions/deletions, and overlapping alignment gaps (Kent et al. 2003). HaploMerger uses a novel graph-based structure called the diploid genome assembly (DGA) graph to describe and store the inter-relationships between alleles or homologs in a diploid genome assembly (Fig. 2). The DGA graph accommodates all chain-net alignment structures and permits operations that are necessary for the reconstruction of allelic relationships and the creation of reference haploid assemblies.
Figure 1.
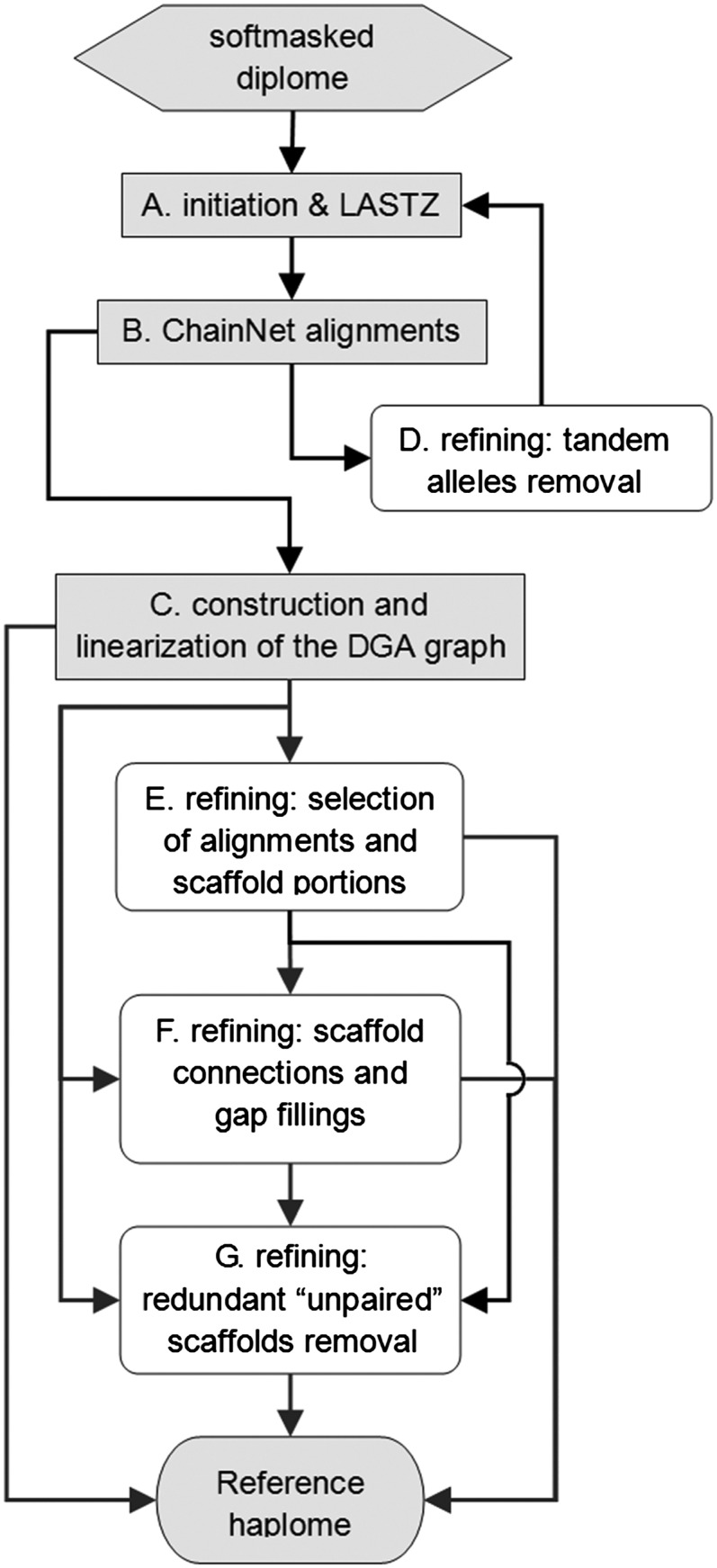
A flowchart of the HaploMerger pipeline. The components required to generate a reference assembly are highlighted in gray. Users are allowed to choose a desired path to finish running the pipeline, to skip some components for a cursory run, or to repeat some components with different parameters.
Figure 2.

A schematic diagram showing the DGA graph-based procedure. This diagram shows how to reconstruct allelic relationships and create the reference haploid assembly for a tiny diploid polymorphic assembly with eight scaffolds. The original diploid assembly (A), is first duplicated into two copies (B); then whole-genome pair-wise alignments are created between two assemblies (C). Based on the alignments, a DGA graph is created (D), from which a reduced linearized DGA graph is subsequently derived (E); guided by the reduced DGA graph, a reference haploid assembly can finally be created (F). This assembly has been included in the HaploMerger package as a simple example for testing. Readers may refer to Supplemental Figure S2 for more details regarding the conversion from the initial complicated DGA graph (D) into the reduced linearized DGA graph (E).
HaploMerger has been tested with simulated genomes, and the software showed excellent performance without inducing new assembly errors. When applied to three real polymorphic diploid assemblies, HaploMerger gave comparable or better results than manual curation. The first two real assemblies are from Ciona savignyi and Branchiostoma floridae, both of which were generated from Sanger reads and accompanied by manually curated reference haploid assemblies (Small et al. 2007; Putnam et al. 2008). The third real assembly is from a single Chinese amphioxus (Branchiostoma belcheri), which was produced in our laboratory using NGS technologies. Moreover, we compared the B. belcheri haploid assemblies (and relevant data produced by HaploMerger) with a large quantity of EST sequences and revealed that the two divergent haplotypes from a single animal are highly complementary to each other. Altogether, we have demonstrated that compared with manual curation, HaploMerger is less error-prone and less time and labor consuming, more flexible and versatile, and importantly, more suitable for large and highly fragmented assemblies.
Results
Simulations
We generated 25 artificial polymorphic genomes containing a pair of 10-Mbp-long chromosomes. Approximately 11× 454 Life Sciences (Roche) reads and 23× Illumina reads were simulated for each genome and assembled with the Celera assembler. As expected, even for such small genomes, the assemblies were heavily fragmented and infested with assembly errors due to the short read length (Supplemental Table S1). HaploMerger was used to create reference haploid assemblies for these data sets, which helped to elevate the scaffold N50 sizes from 0.4–1.2 Mbp to 2.3–8.6 Mbp (Supplemental Table S1). Whole-genome alignments were then generated between the assemblies and their authentic genomes. Through visual inspection of the alignment dot plots, we identified a total of 45 assembly errors (spanning at least 50 kbp, including inversions, insertions, misjoins, and translocations) from all 25 reference assemblies. However, after further examining the alignments between the diploid assemblies and the authentic genomes, we concluded that all errors were inherited from the diploid assemblies; in other words, none of these errors were caused by HaploMerger (Supplemental Table S1).
We also generated a large artificial polymorphic genome containing a pair of 274-Mbp-long chromosomes. Approximately 11× 454 reads were simulated and assembled. Without Illumina mate-pair reads, the resulting diploid assembly has a better scaffold N50 size (2.2 Mbp) than the small assemblies. This assembly was analyzed with HaploMerger, which helped to reconstruct a reference haploid assembly with greatly improved continuity (Table 1). In this reference assembly, we identified 46 large-scale (>50 kbp) assembly errors. Because the default operation of HaploMerger is to break one scaffold when a long-term colinearity violation is detected, assembly errors may be carried from the diploid assembly to the reference assembly. Therefore, we set HaploMerger to break both scaffolds and then reprocessed the diploid assembly. The new operation removed 22 large-scale assembly errors from the reference assembly and reduced the scaffold N50 size from ∼9.0 Mbp to ∼3.1 Mbp. For the 24 remaining errors, visual inspection confirmed that they were all inherited from the diploid assembly (Supplemental Fig. S3). Taken together, both the small and large genome simulations verified the high efficiency and accuracy of HaploMerger.
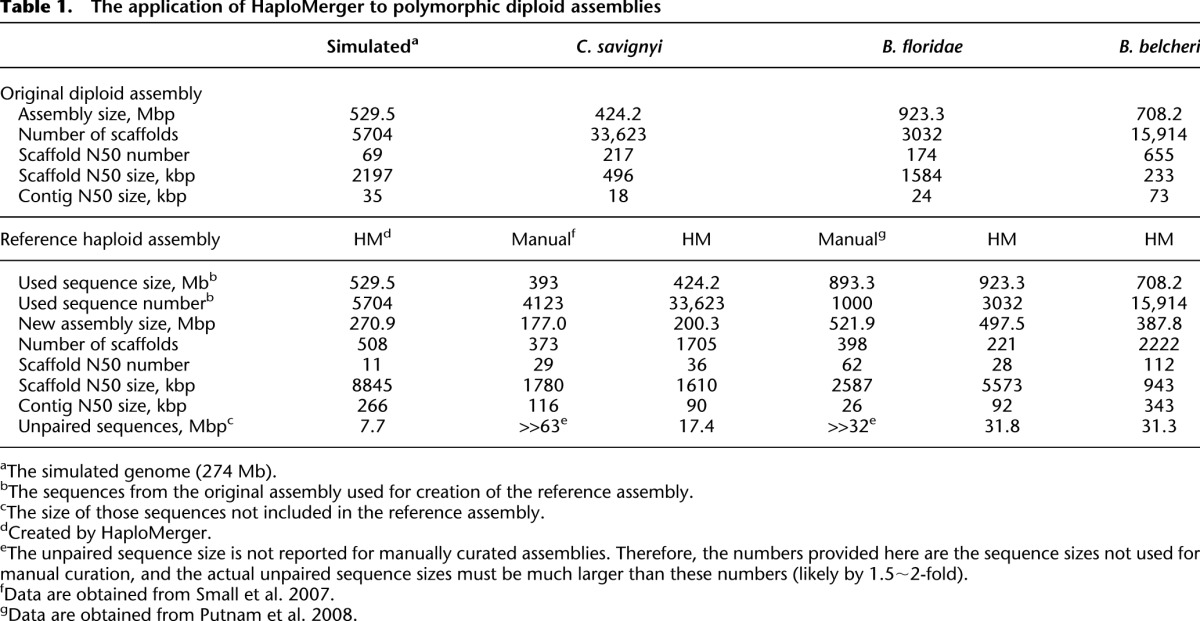
Table 1.
The application of HaploMerger to polymorphic diploid assemblies
Application to three real polymorphic WGS assemblies
We first tested HaploMerger on two real polymorphic WGS assemblies (4%–5% heterozygosity), one for C. savignyi and the other for B. floridae. Both diploid assemblies were released with manually curated reference haploid assemblies (Vinson et al. 2005; Small et al. 2007; Putnam et al. 2008). For these two diploid assemblies, HaploMerger produced reference haploid assemblies with comparable or better continuity than the manually curated references (Table 1). HaploMerger also showed another advantage over manual curation: the ability to process large genomes and large (virtually unlimited) numbers of scaffolds (Table 1). In theory, the inclusion of complete diploid assembly data guarantees better quality for reference assemblies because including more sequences not only helps identify more allele pairs and connects more scaffolds, but also suppresses spurious alignments and incorrect scaffold connections. As a direct result, the HaploMerger output has less unpaired sequences when using complete genome data (Table 1).
HaploMerger includes an algorithm to detect tandem misassembled alleles. The algorithm accommodates poor-quality sequences and ambiguous alignments (Fig. 3D) and in theory could be more sensitive than visual inspection of self-alignment dot plots. To test this, the algorithm was applied to the version 1.5 diploid assembly of the B. floridae genome (kindly provided by Dr. Putnam) (Putnam et al. 2008), in which the algorithm reported 99 additional cases of potential tandem alleles. In addition, HaploMerger reported all 443 cases of tandem alleles that were at least 10-kb long and had been identified in the assembly.
Figure 3.

A schematic diagram showing the algorithm used for detecting tandem alleles. The dot plots (A) and (B) show the self-alignments. An algorithm is used to slice the alignment panel into small cells based on the coordinates for the ends of alignment portions (C). A pair of tandem alleles are detected by the algorithm and shown in detail (D), where length_1, length_2, interval_1, interval_2, coverage_1, and coverage_2 are adjustable parameters used to detect potential cases of tandem assembled alleles.
Finally, we applied HaploMerger to a preliminary version of the B. belcheri diploid assembly (4% heterozygosity). This assembly is solely derived from NGS technologies and highly fragmented, with 708-Mbp sequences separated into 15,914 scaffolds, with half of the sequence contained in ∼15,000 scaffolds and a scaffold N50 size of 233 Kb. Therefore, this assembly is more challenging for HaploMerger than the previous two. HaploMerger resolved the allelic relationships of this assembly and created a reference haploid assembly of ∼388 Mbp distributed into 2222 scaffolds, with the greatly improved scaffold and contig N50 sizes (Table 1).
Comparisons between assemblies and EST sequences from Chinese amphioxus
We also assessed the completeness of the genome assemblies of the Chinese amphioxus (B. belcheri) with a set of 52,961 nonredundant protein-coding EST contigs (Supplemental Table S2). At 80% identity and 80% coverage, ∼87% of the EST contigs were mapped to the original diploid assembly. The reference haploid assembly showed similar completeness (86.5%) with a slight deficit of −0.5%. For comparison, based on the analysis of redundant unassembled ESTs, an early study on C. savignyi reported a deficit of −3% between the original diploid assembly and the manually curated reference haploid assembly (Small et al. 2007). We believe that the excellent completeness of the amphioxus reference assembly is largely a direct result of the effective refining algorithms of HaploMerger. Specifically, selecting longer alleles, reclaiming unpaired sequences, and filling N gaps together reduced the deficit from −1.7% to −0.5% (Suppplemental Table S2). It is also worth noting that despite dramatically increasing contig sizes, filling N gaps made little contribution to gene completeness (Supplemental Table S2). By visually examining the sequences, we discovered that the N gaps tended to occur in repeats and intergenic regions and, hence, had a trivial effect on the coding sequences.
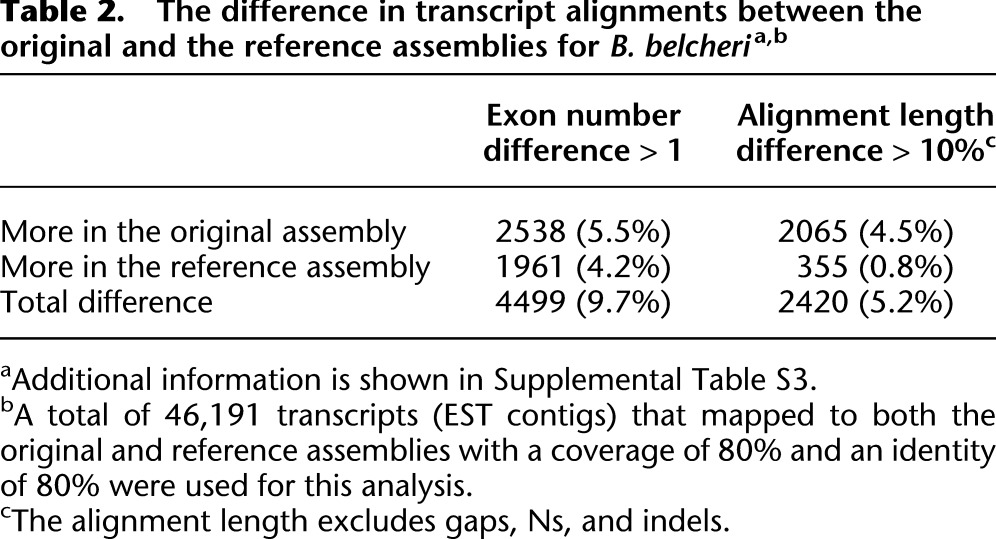
We also compared the relative completeness of each EST contig between the original assembly and the reference assembly (Table 2; Supplemental Table S3). This analysis revealed that the diploid assembly was not always more complete than the reference assembly, suggesting that the improved contiguity and continuity in the reference assembly helps recover some gene structures. Nevertheless, these results suggest that neither the diploid assembly nor the single reference assembly can provide complete gene coverage.
Table 2.
The difference in transcript alignments between the original and the reference assemblies for B. belcheria,b
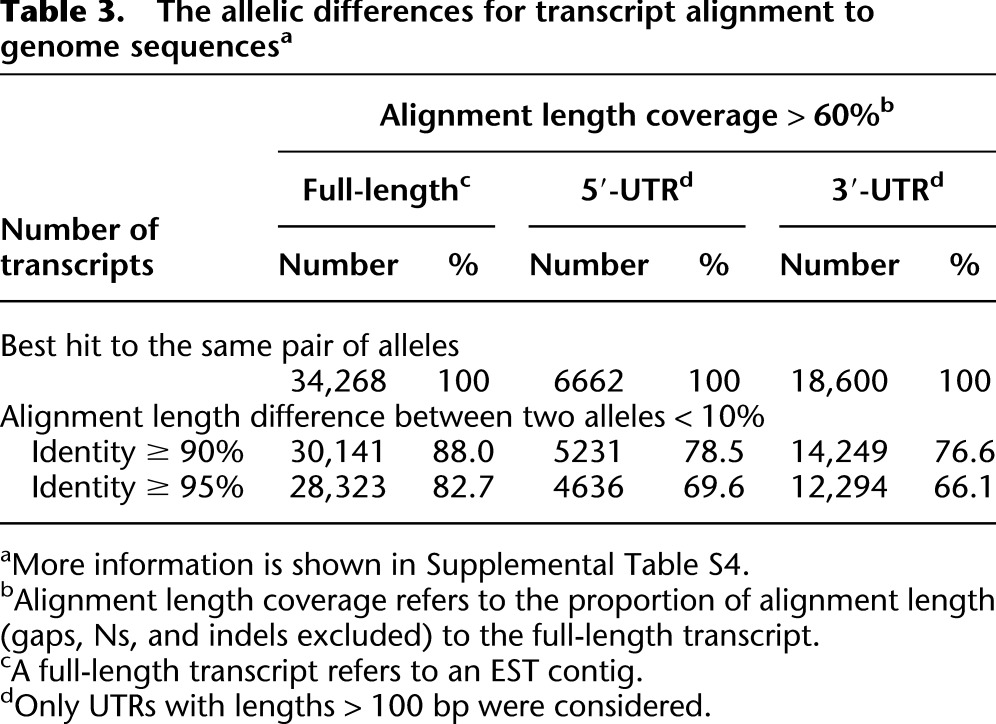
To assess the relative completeness of each EST contig in different alleles, we extracted all of the allele sequences from the N-gap-free alignments (used previously in the simulation) and aligned the EST contigs to them. The results showed that the difference in the average alignment length between two paired alleles corresponding to the same EST contig ranged from 10% to 20% (Table 3; Supplemental Table S4). This length difference corresponds to the difference in completeness between two alleles. This difference may reflect both the difference in sequencing/assembling quality between haplotypes (although the B. belcheri assembly was sequenced to a high depth with good sequence contiguity on the contig level) and the high complementarity between two divergent haplotypes. However, further analysis showed that the difference was even greater in 5′/3′-UTRs than in coding regions. The greater difference in the UTR region is less likely caused by the assembly quality difference between haplotypes.
Table 3.
The allelic differences for transcript alignment to genome sequencesa
Discussion
Reference haploid assemblies can never replace the original polymorphic diploid assembly. This is not only because the reference assemblies may be less complete or contain excessive switches between haplotypes, but also because two divergent haplotype sequences from a single organism could be highly complementary to each other. Our EST analyses showed that for at least a subset of transcripts, the difference between two alleles could be much higher than the nominal average polymorphism rate. The situation is even worse in 5′/3′-UTRs than in coding regions and likely the worst in nontranscribed regions. In fact, this complementarity is unexpected, but not surprising. An early study documented that polymorphic outbred worms suffer from inbreeding depression under laboratory conditions, and 10%–30% heterozygosity persists after 20 generations of inbreeding (Barriere et al. 2009). Even in organisms with low heterozygosity, such as humans, the complementarity between alleles from one individual has been recognized in many genetic diseases.
Nevertheless, at the post-assembly stage, it is essential to determine allelic relationships to better facilitate the use of a polymorphic assembly. After all, a reference haploid assembly has several advantages: It is easy and convenient to present and manipulate, it helps detect assembly errors, and it provides better sequence contiguity and continuity. These advantages benefit subsequent gene predictions, structural variation detection, and other annotation efforts. HaploMerger is dedicated to this post-assembly procedure. It features a LASTZ-ChainNet alignment approach and a novel DGA graph-based structure. The LASTZ-ChainNet approach provides high alignment specificity and running efficiency while preserving alignment sensitivity comparable to BLASTN. The DGA graph offers a flexible way to optimize the simplification and linearization of the homologous/allelic relationships inherited in a polymorphic diploid assembly. The DGA graph allows scaffolds to be stitched together in places where breaks occurred due to haplotype/allele separation and can also indicate potential assembly errors that can be addressed if desired. Guided by a traversed DGA graph, several types of reference haploid assemblies, such as assemblies of long contiguity/continuity, assemblies free of long-term violations of colinearity, assemblies with high completeness, or assemblies with minimum switch errors between haplotypes, can be readily derived. We have demonstrated that HaploMerger works fine for the genomes of small and medium sizes (i.e., 10–500 Mbp). However, it can handle very large genomes with millions of scaffolds due to its automated nature, high efficiency, and accuracy. Virtually, HaploMerger has no limitations on genome sizes and scaffold numbers.
Currently, HaploMerger works on the scaffold level at the post-assembly stage and does not rely on the typical information used by assemblers or other assembly analyzers, such as mate-pair graphs and scaffold-contig layouts. Thus, HaploMerger is no replacement for assemblers, scaffold builders, and other assembly analytic tools, but it can be used to aid the assembly of polymorphic genomes. First, HaploMerger can quickly generate a reference haploid assembly for quality assessment. Second, if a hierarchical scaffolding scheme is used, HaploMerger can be used to detect and separate paired alleles before entering the next scaffolding step. Third, HaploMerger can be used to identify several types of potential assembly errors, such as tandem alleles, inversions, translocations, and long-term colinearity violations. Fourth, HaploMerger provides information on overlapping contigs/scaffolds, which can be used to aid genome finishing efforts.
Methods
Perfectly mirrored, reciprocally best, and pairwise whole-genome alignments
As shown in Figures 1 and 2, the first step of the HaploMerger pipeline is to produce pairwise self-alignments for diploid assembly. The local alignment tool LASTZ is used to compute all-against-all alignments (Harris 2007). Because LASTZ requires soft-masked sequences to achieve specificity, which allows for alignment extension but not alignment seeding in the masked regions, the diploid assembly should be soft-masked by one of these programs: WinMasker (Morgulis et al. 2006), TEdenovo/REPET (Flutre et al. 2011), or RepeatModeler/RepeatMasker (Tarailo-Graovac and Chen 2009). We highly recommend preparing a genome-specific scoring matrix for LASTZ. Given a 4%–5% heterozygosity in an assembly, the identity of orthologous alignments is generally within 90%–100%. LASTZ can be used to sample these alignments to compute a specific scoring matrix. The updated matrix should have better sensitivity and specificity in finding true orthologous alignments.
Once soft masking and matrix optimization are completed, we duplicate the assembly into two copies, one labeled “target” and the other “query” (although they are identical assemblies) (Fig. 2A,B). LASTZ is run to produce all-against-all alignments between the target and query. Then, ChainNet is used to chain and net the LASTZ alignments (Kent et al. 2003). The resulting ChainNet alignments are further refined by two more “chain-net” rounds to obtain the reciprocally best pairwise alignments.
Due to the heuristic nature of LASTZ and ChainNet, the obtained alignments are not perfectly mirrored. There are two possible scenarios: the alignment of target A to query B differs from that of target B to query A or target A hits to query B, but target B does not hit to query A. We implemented the following algorithm to obtain perfectly mirrored alignments. First, the target and query sequences in the alignments are switched, and thus give a new set of alignments. Now each locus is covered by at most two alignments, the original and the new. Second, all alignments are ranked and examined by scores. Third, for each locus, the higher-ranked alignment is retained, and the second-ranked alignment is discarded. Specifically, if a lower-ranked alignment is completely overlapped with a higher-ranked alignment, it is discarded; if a lower-ranked alignment is partially overlapped with a higher-ranked alignment, the overlapped portion of the lower-ranked alignment is truncated.
Definition of the DGA graph
The perfectly mirrored, reciprocally best, and pairwise whole-genome self-alignments between the target and query can be transformed into a novel data structure termed the diploid genome assembly (DGA) graph (Fig. 2). The DGA graph allows scaffolds to be stitched together in places where breaks occurred due to haplotype/allele separation and helps to detect assembly errors that can be addressed if desired. In definition, the DGA graph is a directed graph. Every node in the graph represents an alignment piece; every node has at most two outward edges and two inward edges, and the direction of the edges represents the 5′ → 3′ sequence direction. For each node, one outward edge and one inward edge represent the direction of the target sequence, and they are therefore termed the target-outward edge and the target-inward edge, respectively. Likewise, the other two edges are the query-outward edge and the query-inward edge. If node A connects to node B by a target-outward edge, it means that the two alignments (A and B) contain the same target sequence and are connected by this target sequence with A on the 5′-upstream and B on the 3′-downstream and that there are no breakpoints on the target sequence portion between A and B. Similarly, other forms of connection follow the same definition.
According to the definition, one can see that the DGA graph can handle all ChainNet alignment structures, including regular alignments (i.e., dovetail overlaps and containment overlaps), large insertions/deletions, inversions, translocations, and overlapping alignment gaps. Because the target assembly and the query assembly are identical and the pairwise alignments are perfectly mirrored (Fig. 2B,C), the derived DGA graph is also symmetrical (Fig. 2D,E). For example, alignment No. 1 and alignment No. 11 are symmetrical to each other (Fig. 2C); thus, node No. 1 and node No. 11 are also symmetrical (Fig. 2D). We know that the breaking and joining of sequences is required for both untangling homologous/allelic relationships in the polymorphic assembly and for creating reference haploid assemblies (Fig. 2, C versus F). The symmetrical structure of the DGA graph simplifies the operations of the breaking and joining of sequences, as exemplified in Figure 2F (note that there is a breakpoint on the scaffold d/d′).
Simplification and linearization of the DGA graph
The initial DGA graph stores all homologous relationships (i.e., alignments or nodes) and all possible connections between scaffolds in the diploid assembly, but it contains numerous conflicts due to spurious alignments and assembly errors. Having reconciled these conflicts, we obtain a reduced DGA graph (Fig. 2E). In definition, the reduced DGA graph contains a subset of nodes from the initial DGA graph; each node has at most one outward edge and one inward edge, and no loops are allowed in the graph. In the reduced DGA graph, all retained alignments (or nodes) are considered orthologous and hence promoted as resolved allelic relationships. Finally, a linearized reference haploid assembly can be derived from the reduced DGA graph (Fig. 2E,F).
We obtained a reduced DGA graph by simplifying and linearizing the initial DGA graph. Simplification means removing nodes from the graph that are unlikely to represent authentic allelic relationships, whereas linearization means removing the nodes or edges that create loops or dichotomous paths. In the graph, loops indicate conflicts between potential allelic relationships, whereas dichotomous paths indicate large-scale rearrangements or misjoins of genome regions from different places. In the graph, deleting a node means removing an alignment, whereas deleting an edge between two nodes means breaking up a scaffold sequence between two pieces of alignment.
Simplification and linearization are performed when traversing the DGA graph. Currently, HaploMerger uses a dynamic greedy algorithm for this task. Technically, an alignment indicates a homologous relationship between the target and the query sequences. Biologically, the higher the score of an alignment, the more likely it is to represent an authentic orthologous/allelic relationship. Based on this assumption, the traversing algorithm starts from the highest-scored nodes to the lowest. For each node, it first assesses adjacent nodes and upstream/downstream sequences to determine whether the node is a spurious alignment. If so, the node is deleted. Second, a tracing algorithm is used to detect whether the node forms loops with previously processed nodes. If so, the current node is deleted because it is the weakest point in the loop. Third, if dichotomous paths are encountered, the contexts including adjacent nodes and their edges are assessed, and based on heuristics, one or more edges will be selected for deletion. The heuristics are set to preserve the contiguity of the original scaffolds and to minimize switching from one haplotype to the other.
To illustrate the algorithmic details, we include a tiny diploid assembly in the HaploMerger package for testing. Figure 2 shows the full process required to turn this diploid assembly into a reference haploid assembly. Supplemental Figure S1 shows the heuristics used to process each node in a DGA graph, whereas Supplemental Figure S2 shows the steps used to traverse, simplify, and linearize the DGA graph shown in Figure 2D.
Long-term colinearity violations
Long-term colinearity violations between two scaffold sequences indicate large-scale genome rearrangements or misjoins of genome regions from different places. As described in the previous section, these events can be reconciled by breaking dichotomous paths during graph traversing. However, HaploMerger offers another layer to detect and handle these colinearity violation events prior to graph traversing (Supplemental Fig. S1). Once a violation event is found, HaploMerger offers four choices: (1) it does nothing but leaves the problem to the subsequent graph traversing; (2) it automatically breaks one of the scaffolds based on heuristics (with ≥50% chance of breaking the problematic scaffold); (3) it automatically breaks both scaffolds (at the expense of losing some scaffold continuity); or (4) it allows the user to determine the solution (users may visually inspect the event, consult a mate-pair graph, or do extra analyses/experiments before passing the decision to HaploMerger).
Tandem misassemblies of alleles
As discussed previously, the two alleles in a polymorphic assembly are prone to being misassembled in tandem. In theory, tandem misassemblies can be revealed by searching for mirrored self-alignments; if the involved segment has no corresponding sequence in another allele, it is very likely to be a misassembly. Based on this assumption, we developed an effective algorithm to detect and handle these errors (Fig. 3). This algorithm exploits every advantage of HaploMerger. The use of complete genome data and the reciprocally best alignments guarantee that no other sequences in the assembly can be more similar to the tandem alleles than the tandem alleles are to each other (Fig. 3A). The alignment-refining scheme suppresses fragmented alignments, spurious alignments, and missing alignments that may compromise tandem detection. In operation, this algorithm extracts all of the reciprocally best nontrivial self-alignments for a scaffold and uses their terminal coordinates to slice the alignment panel into small cells. Next, it examines mirrored cells along the diagonal to detect potential tandem alleles (Fig. 3B,C). Finally, the algorithm uses flexible settings for alignment length, coverage, and interval to accommodate poor-quality sequences and ambiguous alignments, which further enhances the sensitivity (Fig. 3D). Once a potential error is detected, HaploMerger offers three choices: (1) it does nothing; (2) it automatically removes the short allele with more Ns; or( 3) it allows the user to determine the solution (users may visually examine the potential error, consult mate-pair graphs, or do extra analyses/experiments before passing the decision to HaploMerger).
Selecting representative alleles, filling N gaps, and removing unpaired sequences
Advanced refinements for reference haploid assemblies are implemented in stage E, F, and G (Fig. 1). In stage E, HaploMerger allows users to decide the representative allele sequence for each allele pair. In stage F, HaploMerger examines every N gap in the representative alleles and tries to replace the N gap-containing sequence with the alternative allele sequence. The default criteria for a successful N gap filling is stringent (but can be adjusted): (1) The alignment length (excluding indels) for an allele pair should be >5000 bp; (2) the N gap should be flanked by >40-bp gap-free alignments; and (3) N gaps implicated in translocations, inversions, and large insertions are excluded from filling because those events represent more radical divergences than SNPs and indels. We should note that selecting longer alleles and filling N gaps may enhance the contiguity and completeness of the reference assembly, but they also cause undesired and excessive switches between haplotypes (i.e., one haplotype splices to another).
Normally, some scaffolds or scaffold portions may exist as single alleles in a diploid assembly, which are called as unpaired sequences and are supposed to be included in the final reference haploid assembly. However, a substantial portion of the “unpaired sequences” classified by HaploMerger is not actually “unpaired”; instead, these sequences are artifact/chimera sequences, repetitive sequences, or allele sequences escaping the initial alignment procedure. Therefore, in stage G, HaploMerger realigns all unpaired sequences to the reference assembly, and those that can be mapped to the reference assembly under given criteria are reclassified as “false unpaired sequences” and removed from the “unpaired” class.
Simulations for polymorphic genome assemblies
The polymorphic diploid assembly of the Chinese amphioxus Branchiostoma belcheri was processed by HaploMerger. A total of 274 Mbp N gap-free alignments for trusted allele pairs (>1000-bp alignment length and >90% alignment identity) were extracted from the HaploMerger outputs. From this alignment pool, we randomly selected 10-Mbp alignments and concatenated the target and query sequences, respectively. By doing so, a small simulated diploid genome was created with a pair of 10-Mbp chromosomes, one from the target and the other from the query. A total of 25 small genomes of 10 Mbp were created. Random sampling without replacement was implemented to ensure no repeated use of any alignment from the pool. In addition, all alignments were concatenated to create a large simulated genome with a pair of 274-Mbp chromosomes.
For each chromosome from the 10-Mbp genomes, we simulated 650,000 454 shotgun reads (350 ± 70 bp), 140,000 3-kb paired-end 454 reads (3000 ± 600 bp), 40,000 8-kb paired-end 454 reads (8000 ± 1600 bp), 40,000 20-kb paired-end 454 reads (20,000 ± 4000 bp), 1,000,000 300-bp mate-pair Illumina reads (115 bp per end), and 1,000,000 500-bp mate-pair Illumina reads (115 bp per end). It should be noted that to induce more assembly errors, the length of each end of the 454 paired-end reads was set to only 104 bp. Reads were randomly sampled from the chromosomes. Sequencing errors were simulated at 1.3%–1.7% for each read. For 454 reads, ∼50% of the error rate represented indels due to homopolymers. Before use, the Illumina reads were subjected to error-correction using Quake (Kelley et al. 2010). In summary, we simulated ∼11× 454 and 23× Illumina reads for each small genome. As for the large genome, only 11× 454 reads were simulated (with the same proportions for the library size as for the small genomes). The Celera assembler version 6.1 was used to assemble the simulated data with the following specific parameters: utgErrorRate = 0.015, overlapper = mer, and unitigger = bog (Miller et al. 2008). Finally, HaploMerger was used to analyze each resulting, soft-masked assembly with the default parameters and a scoring matrix specific to the assembly.
Application to three real polymorphic assemblies
The genome assemblies of the sea squirt Ciona savignyi were previously described (Vinson et al. 2005; Small et al. 2007). The polymorphic diploid assembly (with 4%–5% heterozygosity) was downloaded from http://mendel.stanford.edu/SidowLab/ciona.html. The genome assemblies of the Florida amphioxus Branchiostoma floridae were also previously described (Putnam et al. 2008). The polymorphic diploid assembly (with 4% heterozygosity) was available on http://genome.jgi-psf.org/Brafl1/Brafl1.home.html. The current draft genome of the Chinese amphioxus Branchiostoma belcheri was sequenced from an individual male in our laboratory. Then, a polymorphic diploid assembly (with 4% heterozygosity) was generated from ∼100× raw shotgun and paired-end reads that included both 454 FLX titanium reads (∼30×) and Illumina 115-bp mate-pair reads (∼70×). Both the Newbler and the Celera assembler were used in this task (Myers et al. 2000; Miller et al. 2008; Wheeler et al. 2008). In the current assembly, Illumina reads were only used for gap filling. When these assemblies were ready, HaploMerger was used to analyze the soft-masked versions of each, using the default parameters and a scoring matrix specific to each assembly.
Comparative analyses of the genomic and EST sequences from Chinese amphioxus
We generated two million 454 EST reads and sixty million Illumina EST reads from multiple individuals of B. belcheri and assembled them into ∼90,000 nonredundant EST contigs using Newbler and Abyss (Wheeler et al. 2008; Simpson et al. 2009). Contigs shorter than 150 bp were discarded. FrameDP (Gouzy et al. 2009) was used to correct frame shifts and to identify 52,961 protein-encoding EST contigs. Sim4db/sim4cc was used to align EST contigs to the B. belcheri diploid assembly, the reference haploid assemblies, and the N gap-free paired allele sequences extracted from HaploMerger outputs. Sim4db/sim4cc is able to produce spliced alignments and has excellent performance for polymorphic sequences (Zhou et al. 2009; Walenz and Florea 2011).
Data access
HaploMerger is available as an open-source package from our website: http://mosas.sysu.edu.cn/genome/download_softwares.php. All EST reads are deposited in the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA051482. The reference haploid assembly (version 2.0) for Branchiostoma belcheri is available on our website. The BLAST sever is on http://mosas.sysu.edu.cn/genome/, and the genome browser server is on http://mosas.sysu.edu.cn/genome/gbrowser_wel.php.
Acknowledgments
This work was supported by Project 2008AA092601 (863), projects from the National Natural Science Foundation (31171193, 30901103, and 30730089), 2011CB946101 of the National Basic Research Program (973), projects from the Commission of Science and Technology of Guangdong Province and Guangzhou City, and projects provided by the Sun Yet-sen University Science Foundation.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.133652.111.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, et al. 2002. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297: 1301–1310 [DOI] [PubMed] [Google Scholar]
- Barriere A, Yang SP, Pekarek E, Thomas CG, Haag ES, Ruvinsky I 2009. Detecting heterozygosity in shotgun genome assemblies: Lessons from obligately outcrossing nematodes. Genome Res 19: 470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, et al. 2002. The draft genome of Ciona intestinalis: Insights into chordate and vertebrate origins. Science 298: 2157–2167 [DOI] [PubMed] [Google Scholar]
- Denoeud F, Henriet S, Mungpakdee S, Aury JM, Da Silva C, Brinkmann H, Mikhaleva J, Olsen LC, Jubin C, Canestro C, et al. 2010. Plasticity of animal genome architecture unmasked by rapid evolution of a pelagic tunicate. Science 330: 1381–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez N, Brudno M 2011. Hapsembler: An assembler for highly polymorphic genomes. In RECOMB (ed. V Bafna and CS Sahinalp), Vol. 6577, pp. 38–52. Springer-Verlag, Berlin, Germany. [Google Scholar]
- Flutre T, Duprat E, Feuillet C, Quesneville H 2011. Considering transposable element diversification in de novo annotation approaches. PLoS ONE 6: e16526 doi: 10.1371/journal.pone.0016526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzy J, Carrere S, Schiex T 2009. FrameDP: Sensitive peptide detection on noisy matured sequences. Bioinformatics 25: 670–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS. 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, The Pennsylvania State University. [Google Scholar]
- Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, Wides R, et al. 2002. The genome sequence of the malaria mosquito Anopheles gambiae. Science 298: 129–149 [DOI] [PubMed] [Google Scholar]
- Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, Newport G, Thorstenson YR, Agabian N, Magee PT, et al. 2004. The diploid genome sequence of Candida albicans. Proc Natl Acad Sci 101: 7329–7334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Schatz MC, Salzberg SL 2010. Quake: Quality-aware detection and correction of sequencing errors. Genome Biol 11: R116 doi: 10.1186/gb-2010-11-11-r116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D 2003. Evolution's cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci 100: 11484–11489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman JO, Mackenzie AP, Adey A, Hiatt JB, Patwardhan RP, Sudmant PH, Ng SB, Alkan C, Qiu R, Eichler EE, et al. 2011. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol 29: 59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G 2008. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24: 2818–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgulis A, Gertz EM, Schaffer AA, Agarwala R 2006. WindowMasker: Window-based masker for sequenced genomes. Bioinformatics 22: 134–141 [DOI] [PubMed] [Google Scholar]
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al. 2000. A whole-genome assembly of Drosophila. Science 287: 2196–2204 [DOI] [PubMed] [Google Scholar]
- Pop M 2009. Genome assembly reborn: Recent computational challenges. Brief Bioinform 10: 354–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam NH, Butts T, Ferrier DE, Furlong RF, Hellsten U, Kawashima T, Robinson-Rechavi M, Shoguchi E, Terry A, Yu JK, et al. 2008. The amphioxus genome and the evolution of the chordate karyotype. Nature 453: 1064–1071 [DOI] [PubMed] [Google Scholar]
- Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W 2003. Human-mouse alignments with BLASTZ. Genome Res 13: 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I 2009. ABySS: A parallel assembler for short read sequence data. Genome Res 19: 1117–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small KS, Brudno M, Hill MM, Sidow A 2007. A haplome alignment and reference sequence of the highly polymorphic Ciona savignyi genome. Genome Biol 8: R41 doi: 10.1186/gb-2007-8-3-r41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M, Chen N 2009. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protoc Bioinformatics 25: 4.10.1–4.10.14 [DOI] [PubMed] [Google Scholar]
- Vinson JP, Jaffe DB, O'Neill K, Karlsson EK, Stange-Thomann N, Anderson S, Mesirov JP, Satoh N, Satou Y, Nusbaum C, et al. 2005. Assembly of polymorphic genomes: Algorithms and application to Ciona savignyi. Genome Res 15: 1127–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walenz B, Florea L 2011. Sim4db and Leaff: Utilities for fast batch spliced alignment and sequence indexing. Bioinformatics 27: 1869–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature 452: 872–876 [DOI] [PubMed] [Google Scholar]
- Zhou L, Pertea M, Delcher AL, Florea L 2009. Sim4cc: A cross-species spliced alignment program. Nucleic Acids Res 37: e80 doi: 10.1093.nar/gkp319 [DOI] [PMC free article] [PubMed] [Google Scholar]