

Abstract
Massively parallel sequencing technology and the associated rapidly decreasing sequencing costs have enabled systemic analyses of somatic mutations in large cohorts of cancer cases. Here we introduce a comprehensive mutational analysis pipeline that uses standardized sequence-based inputs along with multiple types of clinical data to establish correlations among mutation sites, affected genes and pathways, and to ultimately separate the commonly abundant passenger mutations from the truly significant events. In other words, we aim to determine the Mutational Significance in Cancer (MuSiC) for these large data sets. The integration of analytical operations in the MuSiC framework is widely applicable to a broad set of tumor types and offers the benefits of automation as well as standardization. Herein, we describe the computational structure and statistical underpinnings of the MuSiC pipeline and demonstrate its performance using 316 ovarian cancer samples from the TCGA ovarian cancer project. MuSiC correctly confirms many expected results, and identifies several potentially novel avenues for discovery.
The continued advancement of DNA sequencing technologies (Mardis 2011) now allows for the rapid sequencing of large sets of cancer cases (matched tumor and normal samples) for the purposes of mutation discovery. This technological progression has shifted the emphasis in cancer genomics from the analysis of a single patient sample to that of hundreds of patient samples across a broad range of tumor types. Such an expansion of scope facilitates the ascertainment of recurrent mutations within genes and functional pathways. Additionally, the increasing scope permits correlating mutations and pathways with clinical phenotypes where appropriate clinical data exist. The outcome of such correlation can include the identification of prognostic or diagnostic markers or the identification of actionable targets for developing therapeutic options that may inform clinical trials development.
To this end, we present a packaged suite of comprehensive, user-friendly tools designed to determine mutational significance in cancer (MuSiC). The primary goal of MuSiC is to separate the significant events which are likely drivers for disease from the passenger mutations present in mutational discovery sets using a variety of statistical methods. This package provides unique practical advantages over existing software and requires a few basic input elements: mapped reads in BAM format, predicted or validated single nucleotide variants (SNVs) and indels in mutation annotation format (MAF), a set of regions of interest (typically the boundaries of coding exons), and any relevant numeric and/or categorical clinical data. Usage is straightforward. With a single command, a user can (1) apply statistical methods across the cohort to identify significantly mutated genes and (2) identify significantly altered pathways and gene sets, (3) investigate the proximity of amino acid mutations within the same gene, (4) search for gene-based or site-based relationships and correlations between the mutations themselves, (5) correlate mutations to clinical features, and (6) cross-reference the findings with relevant databases, such as Pfam (Finn et al. 2010), COSMIC (Forbes et al. 2008), and OMIM (McKusick 1998). These functions can be accessed individually or run as an automated serial implementation.
To illustrate performance, we tested the MuSiC suite on an exome capture data set consisting of 316 cases of ovarian carcinoma (OV) that were previously described by the TCGA Research Network (The Cancer Genome Atlas Research Network 2011). The original analysis provided statistically supported lists of significantly mutated genes, therapeutically targetable copy number amplifications in several genes (e.g., MECOM, MAPK1, CCNE1, and KRAS), evidence of overlaps between DNA methylation clusters and gene expression subtypes, and confirmation of involvement of the NOTCH and FOXM1 signaling pathways in serous ovarian cancer pathophysiology. Using MuSiC, we complement the previous results with detailed descriptions of the correlations between mutation spectra and clinical data. We found evidence of mutual exclusion between mutations in two major tumor suppressors, TP53 and RB1, and strong statistical support for the correlation of germline BRCA1 point mutations and age at disease onset (Hall et al. 1990; Miki et al. 1994). MuSiC also helps us confirm the importance of mutations in the P53 DNA-binding domain (Sigal and Rotter 2000).
Results
Overview
The development of MuSiC was motivated by the rapidly expanding numbers of mutation data sets from a wide variety of tumor types. It is imperative during post-discovery analysis to separate the significant, or “driver,” mutations from the passenger mutations to more accurately pinpoint the key genes and pathways critical for disease initiation and progression. MuSiC is designed precisely to streamline this process into an easily accessible high-throughput software exercise.
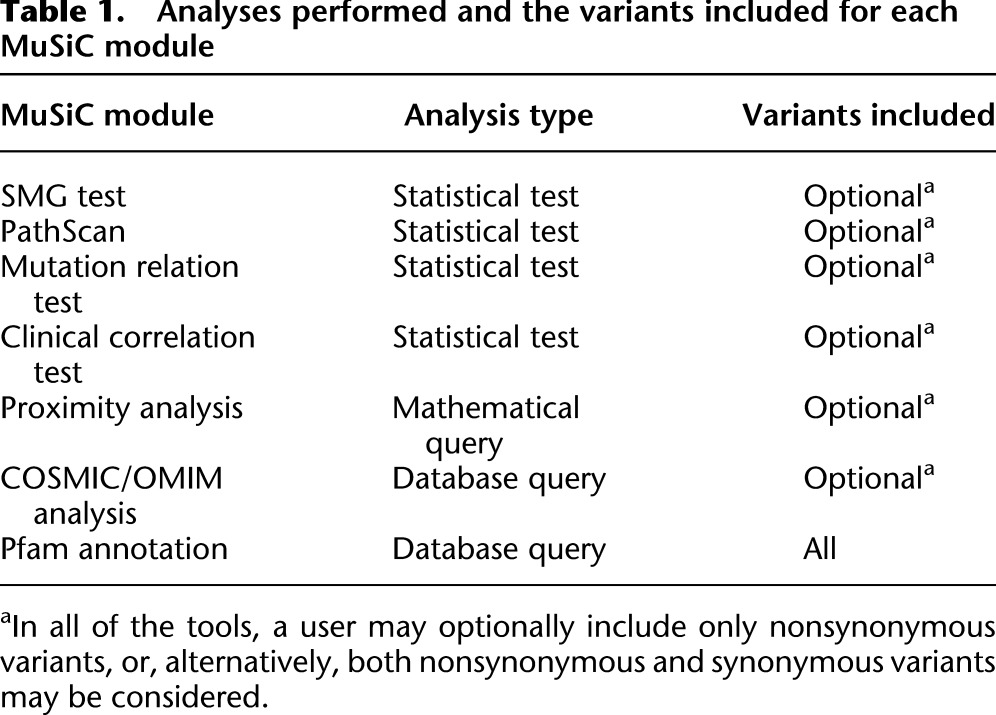
MuSiC currently consists of seven analysis modules and an eighth execution module, “MuSiC Play,” which runs each analysis module sequentially (Fig. 1). MuSiC Play parses the input and output of each of the individual modules and then produces a composite summary of all executed modules. Table 1 lists the type of analysis performed and the types of variants considered by each individual MuSiC module. More detailed descriptions of the specific analysis algorithms performed by each module are given below.
Figure 1.

MuSiC flow diagram. MuSiC modules can either be implemented individually with various required input files or may be implemented in serial via one command where four inputs are used to execute the entire package of tools.
Table 1.
Analyses performed and the variants included for each MuSiC module
Significantly mutated gene tests
We use the concept of “significantly mutated genes” (SMG) to describe genes that show a significantly higher mutation rate than the background mutation rate (BMR) when multiple mutational mechanisms (coding indel and single nucleotide substitution, splice site mutation, etc.) are considered. Specialized measurements of the BMR may also be considered; BMRs in MuSiC are optionally calculated across the entire sample set, across particular subgroups of similarly mutated samples, or for each sample individually. For each BMR subgroup considered and for each category of mutational mechanism, the mutation rates are compared to the appropriate BMR, and a single P-value summarizing all considerations is generated for each gene. We refer to this summarization procedure as the significantly mutated gene (SMG) test.
We assessed multiple methods of calculating summarized P-values, including a convolution test (CT), a Fisher's combined P-value test (FCPT), and the likelihood ratio test (LRT), using a partially simulated data set (this data set and the associated test simulations are described in the Supplemental Material). By this approach, we determined that the P-value distribution obtained using the CT method most closely resembled the uniform distribution expected under the null (in this case, the null is such that no gene is truly significantly mutated), while the FCPT and LRT methods produced slightly inflated or deflated P-values, respectively (Supplemental Fig. S1). During the SMG test, a false discovery rate (FDR) also is calculated. We evaluate our SMG test results by establishing a P-value or FDR threshold (threshold typically 0.2 or less for FDR), and then appropriately filtering the test output.
The results of MuSiC's SMG analysis for the ovarian cancer data set were previously reported (The Cancer Genome Atlas Research Network 2011). Briefly, there were 12 genes found to be significantly mutated in the data set. The CT, FCPT, and LRT P-values for these genes as well as the BMR for each mutational mechanism category in the ovarian data set are displayed in Figure 2. BRCA1 and BRCA2 are known ovarian cancer risk genes (King et al. 2003; Pal et al. 2005). In addition to 27 (BRCA1) and 25 (BRCA2) germline nonsense, splice site, and indel mutations, 11 and 10 nonsynonymous somatic mutations were discovered in this data set in BRCA1 and BRCA2, respectively (The Cancer Genome Atlas Research Network 2011).
Figure 2.

Mutation rates and SMGs in the OV data set. (A) The cohort-wide background mutation rates for all seven mutational mechanism categories are plotted for the OV data set. The overall BMR is also plotted, combining all types of mutations. (B) −Log10(P) for the top 12 OV SMGs are plotted for all three SMG tests in order of decreasing convolution test P-value.
Significantly mutated pathway/gene set analysis
To identify known cellular pathways with significant accretions of somatic mutations in ovarian tumors, we integrated the PathScan algorithm (Wendl et al. 2011) as a module of the MuSiC pipeline. PathScan treats pathways as groups of genes defined by databases such as KEGG (Kanehisa and Goto 2000), BioCarta (Nishimura 2001), and Reactome (Joshi-Tope et al. 2005), with the KEGG definitions currently set as the default implementation. PathScan can be configured, however, to assess any grouping of genes, including groupings from nonpathway databases such as Pfam (Finn et al. 2010).
Using PathScan, we analyzed the OV somatic mutation data set in two ways. First, the entire data set was analyzed regardless of the frequency of mutation in specific genes. Secondly, due to the overwhelming abundance of TP53 mutations, we also performed the analysis using identical parameters but excluding TP53 mutations.
The most significant pathways identified in the first analysis were a collection of KEGG cancer pathways including “Thyroid Cancer” (hsa05216), “Bladder Cancer” (hsa05219), “Basal Cell Carcinoma” (hsa05217), “Nonsmall Cell Lung Cancer” (hsa05223), and “Melanoma” (hsa05218). In the midst of those significant cancer pathways sits the “p53 Signaling” pathway (hsa04115) at a P-value of 2.62 × 10−126. Also, MuSiC found the “Apoptosis” pathway (hsa04210), including not only TP53 mutations but also nine phosphoinositide 3-kinase mutations, to be affected. This latter group of mutations includes two PIK3CA mutations, previously implicated in both breast and ovarian cancers (Levine et al. 2005).
Table 2.
PathScan results for the OV data set
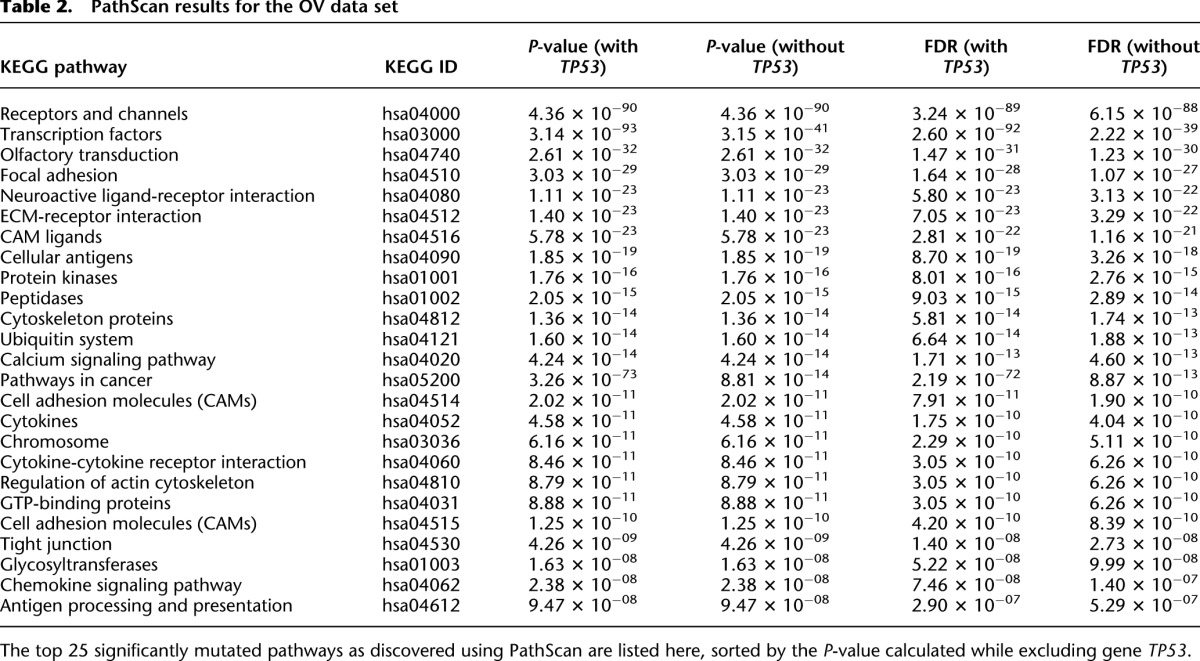
In the second analysis where TP53 mutations were excluded, the collection of KEGG cancer pathways was no longer identified as the most significant pathways in the OV data set. Instead, for instance, this analysis identified the environmental information processing class “Receptors and Channels” pathway from the KEGG Brite database (hsa04000) as the most significant (P = 4.36 × 10−91) (Table 2). Limiting the analysis scope to the KEGG Pathway database, a similar pathway was identified from the environmental information processing class, the “Neuroactive Ligand-Receptor Interaction” pathway (hsa04080). This pathway incurred 266 mutations across the OV data set yielding a P-value of 2.5 × 10−11. The “Calcium Signaling” pathway (hsa04020, P = 4.9 × 10−8) also rose to the level of significance from the TP53-excluded KEGG analysis, which is interesting due to the role of calcium signaling in many cellular processes including cell death (Crompton 2000). The results from the OV data set feature 266 mutations throughout this pathway, highlighted by 35 mutations in voltage-dependent calcium-channel genes (CACNA1A-H and CACNA1S), and 25 mutations in RYR1 and RYR2 genes, whose expression has been correlated with tumor grade in breast cancer (Abdul et al. 2008).
Mutation relation test
The mutation relation test (MRT) attempts to reveal correlations and mutual-exclusion relationships among significantly and highly mutated genes in a pairwise fashion. Positive correlations suggest that mutations and their associated pathways putatively function synergistically to promote carcinogenesis, while negative correlations imply that the alteration of a single component or pathway may be sufficient, wholly or in part, for carcinogenesis.
An example heat map of the MRT analysis for all 316 OV samples is shown in Figure 3. For this data set, we found examples of both concurrent mutations and also mutually excluded mutations among the genes represented in the heat map. Co-mutators FAT3 and EMR3 (P = 0.0333) are both members of the “Receptors and Channels” KEGG pathway (hsa04000), identified as significantly mutated in the pathway analysis described above. And there is also mild evidence of the mutual exclusion of RB1 and TP53 mutations (P = 0.0141). Both of these genes are tumor suppressors, and both were found previously to be significantly mutated genes in this data set (The Cancer Genome Atlas Research Network 2011). This result is potentially meaningful if one considers the possibility that RB1 mutations could be driver events that act independently from TP53. It is well known that the rate of TP53 mutations in ovarian cancer is very high (Ahmed et al. 2010), including the OV data set used in this analysis (The Cancer Genome Atlas Research Network 2011). However, for the few cases that do not have a driver mutation in TP53, we speculate, based on our mutual exclusion results, that mutations in RB1 may represent an independent path to ovarian adenocarcinoma. Of course, due to the small numbers of mutations present in RB1 and EMR3 (six and five mutations, respectively), additional data would be required to confirm any hypotheses generated using these results.
Figure 3.

Mutation relation analysis. A heat map showing mutations in highly mutated genes for all 316 OV samples. Dotted-line boxes highlight concurrent nonsynonymous EMR3 and FAT3 mutations (two concurrent mutations out of five nonsynonymous mutations from EMR3 and 18 from FAT3, P = 0.0333) and mutually exclusive nonsynonymous RB1 and TP53 mutations (297 mutually exclusive mutations out of six nonsynonymous mutations from RB1 and 299 from TP53, P = 0.0141).
Clinical correlation test
The clinical correlation test (CCT) can be used to determine relationships between clinical phenotypes and observed mutations. The input clinical data may be represented in either numeric or categorical (“class”) formats. For example, in the OV data set, we obtained clinical data for 315 of the 316 OV samples; the numeric clinical data consisted of the patients' ages at disease diagnosis and also their survival periods (in days), and the categorical clinical data for the OV data set included information about a sample's race, tumor stage, tumor grade, the outcome of the primary therapy, and, lastly, their vital status. For both data types, the goal of the CCT is to determine whether specific mutations/genes are associated with a particular clinical feature. As these associations can sometimes be biased by covariate clinical features, MuSiC also offers a generalized linear model (GLM) analysis option within the CCT. This tool allows users to define any number of clinical traits as covariates to discovered mutations and, subsequently, to eliminate any possible biases introduced to the phenotype/mutation associations by the covariates' effects.
As a proof of principle, we have assessed the well-established relationship between the presence of a BRCA1 germline variant and a patient's age at disease diagnosis (Hall et al. 1990; Miki et al. 1994). In the OV data set, the CCT revealed that patients with germline BRCA1 variants were significantly correlated with earlier disease diagnosis (P = 2.456 × 10−5, Wilcoxon rank sum test), whereas patients with somatic BRCA1 mutations exhibited no such correlation (P = 0.308). A boxplot of the ages at diagnosis for the OV samples (Fig. 4) clearly shows that the mean age of those samples with a germline BRCA1 mutation (51.3 yr) is lower than those samples with either wild-type BRCA1 (60.4) or with a somatic BRCA1 mutation (63.1 yr). Thus, the CCT correctly evaluated the relationship between germline variants in BRCA1 and ovarian cancer susceptibility.
Figure 4.

BRCA1 variant status versus sample age of diagnosis for the OV data set. A boxplot of the age of diagnosis of 315 OV patients grouped by their BRCA1 mutation status. Germline BRCA1 variant status is correlated with a lower age of diagnosis via the CCT (P = 2.456 × 10−5).
Proximity analysis
In certain genes, mutations tend to cluster in close proximity within functional domains. In order to find these dense “clusters” of mutations within a mutation list, we have developed MuSiC's proximity analysis module. This module searches within fixed windows around each mutation, reporting the number of and distances to all neighboring mutations. The size of the fixed windows utilized for searching is user-configurable. In order to determine an appropriate default size for these windows, we have analyzed the distances between all neighboring mutations in version 54 of the COSMIC database (see Methods). Upon finding that over 25% of the nearest-neighbor mutations in COSMIC are within seven (or less) amino acids of each other, we chose to search 7 aa both upstream of and downstream from each OV mutation for dense clusters of variants. TP53, with 302 total nonsilent somatic mutations in the OV data set, dominated the proximity analysis results. The average number of mutations within 7 aa of another TP53 mutation was 4.9, with the densest 14-aa window containing 26 nonsynonymous mutations.
The next-densest group of mutations occurred in DNAH5, where there were four mutations within a space of 3 aa. Several genes have mutations that occur in triplets within a space of 2 aa, including UBR4 and RB1CC1. Both UBR4 and RB1CC1 have relationships with RB1, a gene on the significantly mutated gene list for the OV data set and a gene also found to harbor copy-number alterations in the OV data set (The Cancer Genome Atlas Research Network 2011). UBR4 is a component of the N-end rule pathway that interacts with RB1, and RB1CC1 actually regulates the expression of RB1. RB1 itself has two mutations in close proximity (within 1 aa of each other). These high-density groups of RB1-related mutations, pictured in Figure 5, may support the hypothesis that RB1 could be an additional driver of ovarian cancer.
Figure 5.

Proximity analysis mutation diagrams. These mutation diagrams show recurrent triplet mutations in both UBR4 and RB1CC1, both of which harbor a relationship with tumor suppressor gene RB1.
COSMIC/OMIM query
Using the COSMIC/OMIM module of MuSiC, we attempted to find previously reported mutations matching the query set of somatic OV mutations. This type of analysis can provide a measure of recurrence, as these databases generally contain information about the studies from which their contents were derived, and the COSMIC database deals exclusively with the somatic mutations discovered in cancer studies. A summary of COSMIC/OMIM database comparisons for those significantly mutated genes listed above with at least one database match of any type is presented in Supplemental Figure S2. This summary, however, represents only a subset of all of the information made available via the database queries. We found 15 exact matches in genomic position and nucleotide change between the OV data set and COSMIC, including sites in NF1, RB1, and PIK3CA, all considered significantly mutated genes by the MuSiC pipeline. This type of match is only possible when comparing to the COSMIC database, since OMIM entries contain only amino acid coordinates. We identified another exact match in the FOXG1 gene, which encodes a forkhead transcription factor. Not only was the FOXM1 transcription factor network cited as significantly altered in 87% of samples in the previous TCGA study (The Cancer Genome Atlas Research Network 2011), but, additionally, some forkhead transcription factors were previously identified as therapeutic targets (Moumne et al. 2008; Wang et al. 2010b).
In addition to finding the above COSMIC variants which shared positions and identical nucleotide changes with OV mutations, our comparison of OV mutations to the COSMIC and OMIM databases also identified a large set of database mutations that altered the same amino acid in an identical manner as an OV mutation. Of 233 such matches from COSMIC (76 from OMIM), the overwhelming majority of these, 219 (56), were from the gene TP53. There were 229 (76) other “position” matches, defined as mutations which affect an identically positioned amino acid but which do not cause the same residue change as the previously reported mutation. Most of these matches are from genes that have been previously associated with ovarian cancer (The Cancer Genome Atlas Research Network 2011), such as BRAF, BRCA1, KRAS, and again, TP53.
Pfam annotation
The Pfam annotation module of MuSiC groups genes based on the frequency of mutation in specific protein domains. Grouping mutations by their protein domain can serve to group genes according to putative function, since genes that share a domain are more likely to share related functions. We performed Pfam annotation on the 19,356 somatic variants identified in the 316 OV cases. Supplemental Table S1 reports the number of nonsynonymous mutations, synonymous mutations, and also the number of genes harboring mutations in each Pfam domain with at least five somatic events in the OV data set.
In the analysis of the OV data, many of the frequently mutated domains are also the most prevalent domains in the genome, including the seven-transmembrane G protein-coupled receptor domain, the protein kinase domain, and the zinc finger domain, as illustrated in Figure 6A. This genome-wide abundance is not true, however, for the amply mutated P53 domain. Analysis of this Pfam annotation result correctly confirms the significance of the P53 domain mutations in this cohort. Figure 6A also shows that this domain is recurrently mutated but only in a small number of genes, much different from all of the other domains pictured. And lastly, Figure 6B shows that the P53 domain has an unusually high nonsynonymous:synonymous mutation ratio. All of these details are indicative (correctly) of an important mutation hotspot in this cohort.
Figure 6.

Pfam domains affected by OV mutations. (A) A histogram of the most highly mutated domains in the OV data set next to the number of genes affected in each domain. (B) A stacked bar-graph where the value 100% represents the total number of mutations in a particular Pfam domain. Lighter and darker sections of the bars represent which proportions of the total mutations are nonsynonymous and synonymous, respectively.
The Pfam annotation module output may also be modified slightly and fed into the SMG test algorithm in order to produce a mathematical result describing “significantly mutated domains” (rather than significantly mutated genes) and their associated P-values. For a detailed explanation of this option, please see the Supplemental Material, including Supplemental Table S2. The results presented therein reaffirm the significance of the mutations in the P53 domain, as well as in the other frequently mutated domains.
Discussion
There are several software tools available purporting to determine mutational significance. CHASM (Carter et al. 2010), for instance, uses a machine-learning technique to distinguish driver mutations from passengers based on a driver/passenger mutation training set. Mutation Assessor (Reva et al. 2011) provides a prediction of the functional impact of a mutation based on the specificity of multiple sequence alignments and conservation scores. And tools such as CanPredict (Kaminker et al. 2007) are database-driven, deriving multiple metrics for each variant, starting with stored information and models, and then making use of the metrics through a decision-tree analysis. Other tools, such as ANNOVAR (Wang et al. 2010a), provide detailed annotations of genetic alterations, much like MuSiC's Pfam annotation module. Although not currently publicly available, the “Firehose” pipeline does share some features with MuSiC, including an SMG analysis tool and a pathway analysis tool, PARADIGM (Vaske et al. 2010), but Firehose as a whole is heavily focused on orderly sequencing and quality control rather than post-discovery variant analysis.
MuSiC, on the other hand, is the first available set of combined tools that enables a complete, multidimensional statistical evaluation of next-generation-derived cancer data sets. No other publicly available tool suite currently incorporates clinical data along with coverage data and database references into the determination of the most significant mutations among a large mutation list full of passengers. MuSiC merges several methodological aspects of the above-mentioned tools with many novel additional algorithms, providing the capability of large-cohort, data-driven statistical analysis to the entire research community.
Use of MuSiC is straightforward. The simplicity of the input files and tool updating make this package extremely accessible. MuSiC also accommodates both large and small research organizations; although the entire suite of tools is capable of running sequentially on a single processor, the most CPU-intensive modules offer easily parsable options for parallelizing jobs across multiple machines or across a job cluster.
Future support for the MuSiC package will be devoted to the handling of additional file formats, such as the variant call format (VCF), and also to the development of a graphical user interface (GUI). We intend to design new tools aimed at incorporating a wider variety of biological and variant data types, such as copy alteration data, and 3D protein structures from RCSB's Protein Data Bank (Berman et al. 2000), to be used for improved proximity analysis calculations. We also intend to implement recurrence tests across other data entities, such as significantly mutated transcripts and significantly mutated gene families. We also plan to enhance the assessment of the functional impact of protein mutations in MuSiC through the integration of published solutions, such as Mutation Assessor (Reva et al. 2011) and PolyPhen-2 (Adzhubei et al. 2010), as well as through the design of new modules which take advantage of databases that categorize such effects, such as SIFT (Kumar et al. 2009). And lastly, a fuller integration of the results from various MuSiC modules, many of which are currently considered in isolation, will provide an even more comprehensive picture of cancer genomic mechanisms.
Methods
Sample data set used in this study
Alignment mapping files for the ovarian cancer cohort, as well as all mutational data in mutation annotation format (MAF) are available at The Cancer Genome Atlas (TCGA) website, http://cancergenome.nih.gov/, via dbGaP. The dbGaP study accession number is phs000178.v1.p1.
Significantly mutated gene tests
We describe our calculation of background mutation rates and three methods for calculating summarized P-values below.
Background mutation rate (BMR)
Calculations of BMR, although simply described as the number of mutated bases per total bases, are often controversial. Our method is dependent upon the data available, under the premise that the number of bases having available alignment data should provide the denominator for the BMR calculation, since this number also provides the upper limit to the number of bases available for mutation discovery. Therefore, we first count the number of bases with sufficient aligned read-depth based upon user-defined coverage limits set independently for the tumor and normal BAM files for each sample in the cohort. For the purposes of our algorithm, counts are determined for six specific reference sequence-based denominations, with separate counts for each of A and T bases, CpG-connected C and G bases, and lastly C and G bases not connected as CpG. We also categorize the discovered mutations along the lines of mutational mechanism, with separate categories for AT transitions, AT transversions, CpG transitions, CpG transversions, CG (non-CpG) transitions and transversions, and a seventh “indel” category, for which we use the entirety of the covered space in the sample-set when comparing indel-affected bases versus available bases. In order to calculate the BMR of each mutational mechanism category, the number of mutations found in that category is divided by the total number of bases available in which such a call could have been made.
MuSiC allows for the calculation of BMR and for the subsequent comparisons of mutation rate to BMR, to be performed separately for each subgroup in a user-specified number of subgroups. If the user specifies that only one subgroup is to be considered (the default option), then the entire cohort is used in the calculations of mutation rate and BMR for the seven mutational mechanism categories. In this case, seven P-values are generated for each gene and are summarized using the three methods described below. However, if more than one subgroup is to be considered for a given cohort, the mutation rates and BMRs for the seven mutational mechanism categories are calculated separately for each subgroup (seven mutation rate and BMR calculations per subgroup), with P-values generated for each category within each subgroup. In this case, the number of P-values generated for each gene will equal seven times the number of subgroups to be considered. All P-values for each gene are still summarized using the three methods discussed below.
If the number of subgroups to be considered is greater than one but not equal to the number of samples in the cohort, the BMR for each sample is first calculated individually, and then these per-sample BMR values are clustered using k-means clustering into the number of subgroups specified by the user. These clustered subgroups are then used for all subsequent mutation rate and BMR calculations. Alternatively, if the number of subgroups specified by the user equals the number of samples in the cohort, then each sample is considered independently for each mutation rate and BMR measurement.
The BMRs calculated across the entire OV data set for each mutational mechanism category, including the overall BMR across all categories, are plotted in Figure 2A.
Fisher's combined P-value test (FCPT)
FCPT combines all P-values for a particular gene into a statistic, χc, according to Fisher's method (Fisher 1925),
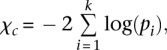 |
where pi is the P-value obtained via binomial distribution for the i-th subgroup mutational mechanism type, and k the number of subgroup mutational mechanism categories for a gene. The final P-value for the entire gene is calculated as the probability of observing a value no less than χc, based on a χ2 distribution with 2k degrees of freedom.
Likelihood ratio test (LRT)
LRT constructs a likelihood ratio-based statistic (χl) for a gene,
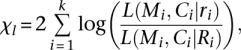 |
where Mi, Ci, Ri, and ri are mutation number, coverage, BMR, and maximum likelihood estimate (MLE) of the mutation rate, respectively, for the i-th subgroup mutational mechanism category of a gene, k is the number of mutation types, and L() is the likelihood of observed mutation number for the i-th subgroup mutational mechanism category, defined as the point probability of observing Mi mutations given a coverage of Ci and a mutation rate of Ri or ri. The final P-value for the entire gene is calculated as the probability of observing a value no less than χl, based on an approximate χ2 distribution with k degrees of freedom.
Convolution test (CT)
CT calculates a summarized log statistic of joint binomial point probability,
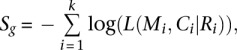 |
where Mi, Ci, Ri, k, and L() are referred to as the same as in the LRT method. Getz et al. (2007) proposed that the P-value for a gene can be calculated by taking one minus a left-tail probability, i.e., the probability of observing a value less than Sg, and the semiexact distribution of Sg can be obtained by a binned histogram-based convolution procedure. This procedure is advantageous for the large amounts of data involved in genome-wide investigation of cohort mutations because it provides exact P-values in a minimum amount of computation time; usually one must choose between precision and length of compute. However, one disadvantage of this convolution procedure is that the two tails of the Sg distribution are mixed, and therefore the directionality is lost. In this case, both improbably large mutation rates and improbably low mutation rates give high values for Sg. To remedy this issue, our CT method modifies the published convolution procedure by excluding genes with mutation rates that are close to or equal to 0. In other words, we exclude genes whose mutation rates are extremely far below the BMR, as they never would be considered “significantly mutated” in practice.
The output file for the SMG test is a compilation of the P-values for each test for each gene under the null hypothesis that the number of mutations seen in the gene is in accordance with those seen in the background. False discovery rates are also reported for each test for each gene.
Significantly mutated pathway/gene set analysis
Sequence data from a single cancer genome do not contain sufficient information to adequately investigate pathway/phenotype associations. This problem requires systematic analysis of larger cohorts. There are several published methods for identifying significantly mutated pathways or gene sets (e.g., Lin et al. 2007; Tarca et al. 2009; Vaske et al. 2010), but we have incorporated a new tool called PathScan into the MuSiC package. PathScan accounts for two important factors other methods neglect: (1) variations in gene lengths and the consequent differences of their mutational likelihoods under the null hypothesis; and (2) distribution of mutations among samples and their proper combination into an overall P-value.
We have configured PathScan in MuSiC to perform pathway analysis using a wide variety of annotated databases, including KEGG (Kanehisa and Goto 2000), BioCarta (Nishimura 2001), and Reactome (Joshi-Tope et al. 2005), as mentioned above. The results described above where TP53 is excluded from the pathway analysis are achieved easily through MuSiC's implementation of a parameter called “genes-to-ignore,” in which the user may provide a comma-delimited list of genes whose mutations should be skipped over when reading the MAF file during the analysis.
Mutation relation test (MRT)
We have developed the MRT to look for any latent relationships among mutated genes. This module is a correlation test for two binary variables to see whether or not any two genes are mutated concurrently (positive correlation) or exclusively (negative correlation). Because the numbers of mutated genes may vary significantly among samples (most have only a few mutations whereas some may have many), and because direct correlation between genes in high-mutation-count samples is not comparable to that in low-mutation-count samples, classic correlation analysis is invalid. To control for this confounder, we calculate P-values in the MRT through a restricted permutation, taking into account the distribution of mutated gene numbers amongt the samples. Permutations are performed, therefore, by first calculating the numbers of mutated genes in individual samples and then randomly permuting the observed mutations, keeping both the number of mutations in each gene and also the distribution of mutated gene number per sample constant. Concurrence and exclusion among mutations are tested separately. This method has been successfully applied to lung cancer data in our previous study (Ding et al. 2008).
Clinical correlation test (CCT)
The CCT module was developed for detecting correlations between mutations and clinical features. Mutation data are again treated as 0-1 variables, where all mutations occurring within the same gene are grouped (the default method), or optionally, where a more strict definition of collapsing is used, grouping only those mutations with identical genomic coordinates and nucleotide changes. Clinical features can either be categorical or numerical variables, where a Fisher's exact test (and optionally, a χ2 test) is used to calculate P-values for categorical clinical data, and a Wilcoxon rank sum test (and optionally, Pearson's correlation) is used to analyze numerical clinical data. The null model for the calculated P-values depends on the type of clinical data being analyzed. For categorical clinical data, the null model is that mutations happen randomly in the various categories represented across the samples, with no preference given to any one category. For numerical clinical data, the null model is that mutations have no effects on a trait (i.e., that mutations will not increase or decrease the value of the numerically reported trait).
A GLM analysis option also exists in which a user may define an analysis model which must include a response variable and a variant, and then any number of covariates may be considered. The variants and covariates may all be clinical features, or, more typically, genes from the mutation list should be listed as variants, with clinical features making up the covariates. The output of the GLM option includes a P-value which indicates association between the response variable and the variant, as well as other standard outputs from a deviance analysis.
Proximity analysis
In MuSiC, we have defined proximity analysis as an investigation of the density of mutations across a cohort in the amino acid space of a given gene's annotated transcript. The goal of this analysis is to extract evidence of mutations clustered within specific domains, an event which is significant in light of the null model where point mutations occur at random locations throughout the genome. Highly mutated domains across a group of samples might be indicative of an underlying mechanistic association which contributes to the shared condition of the cohort, such as the onset of disease. The specific action taken in the tool is to query the distance (in number of amino acids) between every pair of mutations on a given transcript of a mutated gene within the sample set, and then to determine which mutations fall within “close proximity.” “Close proximity” may be determined by the user as an input to the module, with the units of this parameter being the number of sequential amino acids between two events.
The algorithm operates as follows: First, the amino acid position of each mutation within its respective transcript is determined. Then, for each mutation in the input MAF file, two values are calculated: (1) the number of other mutations on the same transcript within the proximity limit set by the user; and (2) the distance to the closest other mutation in this nearby set. (Note that if more than one mutation occurs within the same amino acid, the distance to the closest mutation in this case would be 0 aa.) Then, for each mutation, we report a few general details from the MAF file in the output, such as the genomic coordinates, reference and variant bases, and the sample in which the mutation was found. We further report the calculated values, such as the amino acid position of the mutation within the transcript, and also (1) and (2) listed directly above.
In the Proximity Analysis Results section, we chose to use 7 aa as the maximum distance from any OV mutation within which to look for clusters of mutations. This default distance was chosen after querying the entire COSMIC mutation database (version 54, comprising 171,473 mutations), and looking for the proximity of mutations found in dense clusters. For each gene present in the database, we calculated the distance between each mutation in that gene and its closest neighboring mutation. As many mutations sharing the exact same genomic coordinates are reported more than once in COSMIC, we only used one instance of each mutational position when calculating nearest-neighbor distances. Through these per-gene measurements, we found that over 25% of all neighboring mutations in COSMIC are within 7 aa of each other (25% thus represents the first quartile of nearest-neighbor distances). We thus set 7 aa as the default range within which to search for clusters of mutations in the proximity analysis module.
COSMIC/OMIM query
In the COSMIC/OMIM tool, a list of mutations is queried for recurrence against both the Catalogue of Somatic Mutations in Cancer (COSMIC) database and the Online Mendelian Inheritance in Man database (OMIM). The usefulness of this tool is twofold; first, recurrent events between these two databases and a list of variations from a cohort provide a quick extension set, especially as the databases are expanded over time, and secondly, we have downloaded entries from the COSMIC database related to mutations that report some type of amino acid transformation and provide this file with the MuSiC package, making this type of query very user-friendly.
The tool functions as follows: For every mutation found in the input MAF file, information related to this mutation is gathered from both the COSMIC and OMIM databases. Relevant information is ascertained by relating the genomic coordinate (COSMIC) or the amino acid change (COSMIC and OMIM) associated with the variant to all database entries. (Note that the MAF used as input must contain an additional field with column header “amino_acid_change,” which is the field searched for to perform the amino acid comparison.) A database entry must lie within the user-specified number of bases (default = 5) or amino acids (default = 2) of the MAF variant to be considered as a “nearby” match. We view the thresholds for “nearby” matches as adequate for taking into account the different definitions of reference sequences and gene transcripts that may be used by different contributors to the databases. Alternatively, “exact” matches are direct overlaps in both the location and base/amino acid change of a variant in the MAF and a mutation found in the database. If only the location (the genomic coordinate or amino acid position) of a variant match a database entry but not the nucleotide or amino acid change, these matches are deemed “position” matches. We currently ignore silent mutations in OMIM queries, as these types of mutations do not affect protein coding. However, the positions of silent mutations are still considered in comparison with the COSMIC database.
All discoveries based on these database queries are appended to the input MAF file as extra columns (one column per database). The information written to the output file by the tool will either describe “exact” or “position” matches in amino acid or base change or will (optionally) give all results from the gene associated with the variant for a “nearby” match, one of which must qualify the site as actually having a “nearby” match. If a variant has no match in the particular database, a “novel” declaration is made for the variant, and (again, optionally) all of the results found from the associated gene will be printed next to the declaration to give the user an idea of what matches are possible for that gene. And lastly, if the gene for a variant is not found in the database, a message noting this circumstance is printed along with a “novel” declaration for the variant.
For each queried database, the tool further prints an output summary which tallies the types of matches found throughout the entire data set using that database. The user can learn, for instance, how many sites matched exactly in both nucleotide and position, or perhaps how many variants matched database entries in only the amino acid category (labeled AA in the summary) while not matching in exact position or nucleotide change (labeled NT).
Pfam annotation
Our Pfam annotation tool supposes the use of Sanger's Pfam database (cited above) as another avenue for defining recurrence within the mutation list of a large group of cancer samples. The Pfam database is a catalog of functional regions (or domains) present within protein sequences. Referencing this list with the locations of somatic mutations across a cohort provides knowledge about which domains are most frequently affected by mutations in a certain disease and, therefore, may provide some insight as to the significance (or insignificance) of the mutations in question.
In order to use the Pfam database, we have translated the catalog's amino acid coordinate system into a genomic position coordinate system. The amino acid sequences chosen to be translated were ranked at every coding and splice site position in the genome, based first on the translational and functional effect predicted by the transcript at a given position, and then secondly on the NCBI status of the particular transcript. The sequence of the “best” transcript chosen at each site was used to recover Pfam domains from Sanger's database. We have incorporated this information into a table used by the tool via a fast-lookup program called Tabix, available in the SAMtools package (Li et al. 2009). The Pfam annotation module uses the genetic coordinates of a variant to append a Pfam annotation domain column to the end of any MAF file.
This tool has been expanded to query a few other protein annotation databases in addition to Pfam. We provide information from the SUPERFAMILY database (Wilson et al. 2009), the SMART database (Schultz et al. 2000), and the Patternscan database in our annotation query. (Patternscan is a new version of the Prosite database [Sigrist et al. 2010].) Patternscan provides information similar to that of the Pfam database regarding protein domains and families at functional sites, but SUPERFAMILY and SMART focus on evolved protein structural families and signaling protein domains, respectively. All of this information will be present in the output file, comma delimited, with the name of the database separated from the annotation result by an underscore.
Software availability
The MuSiC software tools, source code, and reference data are accessible through our website, http://gmt.genome.wustl.edu, and are supported on Ubuntu Linux 10.04 (Lucid Lynx). Installation on all Debian-based systems will initiate automatic updates from our software server. The MuSiC suite is also available on CPAN (http://www.cpan.org) and GitHub (https://github.com) under the namespace “Genome” in both locations, and integration of MuSiC into Galaxy (http://usegalaxy.org) is also available. A summary of benchmarking statistics for each MuSiC module is available in the Supplemental Material.
Acknowledgments
We thank the following people and groups at The Genome Institute: Scott Smith and Joshua F. McMichael from the Analysis Pipeline group for help packaging the software and designing the website; Christopher A. Miller, John W. Wallis, David E. Larson, Christopher C. Harris, and Michael D. McLellan from the Medical Genomics group for constructive discussions; and Devin P. Locke for critical review of the manuscript. This work was funded by grants to R.K.W. from the National Human Genome Research Institute (NHGRI U54 HG003079).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.134635.111.
References
- Abdul M, Ramlal S, Hoosein N 2008. Ryanodine receptor expression correlates with tumor grade in breast cancer. Pathol Oncol Res 14: 157–160 [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed AA, Etemadmoghadam D, Temple J, Lynch AG, Riad M, Sharma R, Stewart C, Fereday S, Caldas C, Defazio A, et al. 2010. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol 221: 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE 2000. The Protein Data Bank. Nucleic Acids Res 28: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H, Samayoa J, Hruban RH, Karchin R 2010. Prioritization of driver mutations in pancreatic cancer using cancer-specific high-throughput annotation of somatic mutations (CHASM). Cancer Biol Ther 10: 582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M 2000. Mitochondrial intermembrane junctional complexes and their role in cell death. J Physiol 529: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. 2008. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. 2010. The Pfam protein families database. Nucleic Acids Res 38: D211–D222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. 1925 Statistical methods for research workers. Oliver and Boyd, Edinburgh, UK. [Google Scholar]
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR 2008. The catalogue of somatic mutations in cancer (COSMIC). Curr Protoc Hum Genet 57: 10.11.1–10.11.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz G, Hofling H, Mesirov JP, Golub TR, Meyerson M, Tibshirani R, Lander ES 2007. Comment on “The consensus coding sequences of human breast and colorectal cancers.” Science 317: 1500 doi: 10.1126/science.1138764 [DOI] [PubMed] [Google Scholar]
- Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC 1990. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 250: 1684–1689 [DOI] [PubMed] [Google Scholar]
- Joshi-Tope G, Gillespie M, Vastrik I, D'Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, et al. 2005. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res 33: D428–D432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminker JS, Zhang Y, Watanabe C, Zhang Z 2007. CanPredict: A computational tool for predicting cancer-associated missense mutations. Nucleic Acids Res 35: W595–W598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28: 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Marks JH, Mandell JB 2003. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302: 643–646 [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081 [DOI] [PubMed] [Google Scholar]
- Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, Boyd J 2005. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res 11: 2875–2878 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Gan CM, Zhang X, Jones S, Sjoblom T, Wood LD, Parsons DW, Papadopoulos N, Kinzler KW, Vogelstein B, et al. 2007. A multidimensional analysis of genes mutated in breast and colorectal cancers. Genome Res 17: 1304–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER 2011. A decade's perspective on DNA sequencing technology. Nature 470: 198–203 [DOI] [PubMed] [Google Scholar]
- McKusick VA. 1998 Mendelian inheritance in man. A catalogue of human genes and genetic disorders. Johns Hopkins University Press, Baltimore, MD. [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. 1994. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266: 66–71 [DOI] [PubMed] [Google Scholar]
- Moumne L, Batista F, Benayoun BA, Nallathambi J, Fellous M, Sundaresan P, Veitia RA 2008. The mutations and potential targets of the forkhead transcription factor FOXL2. Mol Cell Endocrinol 282: 2–11 [DOI] [PubMed] [Google Scholar]
- Nishimura D 2001. BioCarta. Biotech Software and Internet Report 2: 117–120 [Google Scholar]
- Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K, et al. 2005. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104: 2807–2816 [DOI] [PubMed] [Google Scholar]
- Reva B, Antipin Y, Sander C 2011. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res 39: e118 doi: 10.1093/nar/gkr407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P 2000. SMART: A web-based tool for the study of genetically mobile domains. Nucleic Acids Res 28: 231–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal A, Rotter V 2000. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res 60: 6788–6793 [PubMed] [Google Scholar]
- Sigrist CJ, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, Hulo N 2010. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res 38: D161–D166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, Kim CJ, Kusanovic JP, Romero R 2009. A novel signaling pathway impact analysis. Bioinformatics 25: 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM 2010. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 26: i237–i245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H 2010a. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164 doi: 10.1093/nar/gkq [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ahmad A, Li Y, Banerjee S, Kong D, Sarkar FH 2010b. Forkhead box M1 transcription factor: A novel target for cancer therapy. Cancer Treat Rev 36: 151–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendl MC, Wallis JW, Lin L, Kandoth C, Mardis ER, Wilson RK, Ding L 2011. PathScan: A tool for discerning mutational significance in groups of putative cancer genes. Bioinformatics 27: 1595–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D, Pethica R, Zhou Y, Talbot C, Vogel C, Madera M, Chothia C, Gough J 2009. SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res 37: D380–D386 [DOI] [PMC free article] [PubMed] [Google Scholar]