

Abstract
The uptake and clearance of apoptotic cells by macrophages and other phagocytic cells, a process called efferocytosis, is a major component in the resolution of inflammation. Increased concentrations of extracellular histones are found during acute inflammatory states and appear to contribute to organ system dysfunction and mortality. In these studies, we examined the potential role of histones in modulating efferocytosis. We found that phagocytosis of apoptotic neutrophils or thymocytes by macrophages was significantly diminished in the presence of histones H3 or H4, but not histone H1. Histone H3 demonstrated direct binding to macrophages, an effect that was diminished by preincubation of macrophages with the opsonins growth arrest–specific gene 6 (Gas6) and milk fat globule–epidermal growth factor (EGF) 8 (MFG-E8). Incubation of histone H3 with soluble αvβ5 integrin and Mer, but not with αvβ3, diminished its binding to macrophages. Phagocytosis of apoptotic cells by alveolar macrophages in vivo was diminished in the presence of histone H3. Incubation of histone H3 with activated protein C, a treatment that degrades histones, abrogated its inhibitory effects on efferocytosis under both in vitro and in vivo conditions. The present studies demonstrate that histones have inhibitory effects on efferocytosis, suggesting a new mechanism by which extracellular histones contribute to acute inflammatory processes and tissue injury.
INTRODUCTION
The uptake and clearance of apoptotic cells by macrophages and other phagocytic cells, a process called “efferocytosis,” is a major component in the resolution of inflammatory states (1,2). Ingestion and elimination of cells undergoing apoptosis prevents the release of their intracellular contents, a proinflammatory and potentially harmful event for surrounding tissue. Moreover, engulfment of apoptotic cells decreases local inflammation through enhancing release of antiinflammatory cytokines and suppressing production of proinflammatory mediators by the phagocytic cells (3,4). Impairment in efferocytosis of neutrophils is associated with acute lung injury and cystic fibrosis, and diminished uptake of lymphocytes correlates with severity in systemic lupus erythematosus (5).
Engulfment of apoptotic cells requires the recognition of “eat-me” signals on the surface of the dying cell through interaction with specific receptors expressed on the phagocyte (3–5). One of the best characterized eat-me signals is phosphatidylserine, a phospholipid situated on the inner leaflet of the cell membrane of viable cells that is exposed on the cell surface during the early stages of apoptosis. Several receptors on macrophages, including Tim4, BAI1 and RAGE, bind directly to phosphatidylserine on the apoptotic cell surface (6–8). An additional mechanism by which phagocytic cells associate with apoptotic cells is through interaction with opsonins (such as milk fat globule–epidermal growth factor [EGF] 8 [MFG-E8] and growth arrest–specific gene 6 [Gas6]) that bridge phosphatidylserine with macrophage-specific receptors, including integrins. Blockade of receptors involved in the recognition of apoptotic cells diminishes the ability of the phagocyte to engulf target cells. For example, the late mediator of sepsis (high mobility group box protein 1 [HMGB1]) can bind to αVβ3 integrins on the macrophage, thereby inhibiting efferocytosis (9–11).
Histones are essential components of eukaryotic nucleosomes and play important roles in the regulation of deoxyribonucleic acid (DNA) repair, gene transcription and chromatin condensation. However, whereas histones are usually intranuclear in location, they can also be found in the cytoplasm or even in the extracellular milieu (12–16). Although how nuclear histones become extracellular is unclear, it is suggested that they can be actively secreted by activated inflammatory cells and be passively released by apoptotic or necrotic cells (17). Additionally, neutrophil extracellular traps may be another source of extracellular histones (15). Extracellular histones appear to contribute to organ system dysfunction in acute inflammatory conditions, including sepsis (18). Patients with severe sepsis and septic shock have elevated plasma levels of histone-containing nucleosomes, suggesting that nucleosomes might be involved in the pathogenesis of multiple organ dysfunctions (13,18,19). In addition, blood concentrations of histones were found to increase dramatically after lethal intravenous injection of Escherichia coli in baboons (18). The intravenous injection of histones in mice resulted in a state mimicking septic shock and was associated with pulmonary lesions that were similar to those present in acute lung injury, including neutrophil accumulation, microvascular thrombosis and fibrotic changes (18). The in vivo effects of histones on lung and other organ dysfunction were reversed by treatment of mice with antibodies against H3 histones or by administration of activated protein C (APC), which cleaves histones (18).
The mechanisms by which extracellular histones contribute to acute inflammatory processes and tissue injury remain incompletely characterized. In the present studies, we show that histones have inhibitory effects on efferocytosis, diminishing the uptake of apoptotic cells by macrophages under both in vitro and in vivo conditions.
MATERIALS AND METHODS
Reagents
Custom cocktail antibodies and negative selection columns for bone marrow neutrophil isolation were from Stem Cell Technologies (Vancouver, BC, Canada). Penicillin-streptomycin, Brewer thioglycollate, bovine serum albumin (BSA), calf thymus DNA and PKH26 Red Fluorescent Cell Linker Kit were from Sigma-Aldrich (St. Louis, MO, USA). Fluorescein isothiocyanate (FITC)-conjugated anti-CD11b antibody, APC-conjugated anti-CD90.2 antibody and APC-conjugated Annexin V were from BD Bioscience (San Jose, CA, USA). Cytotoxicity Detection Kit (LDH [lactate dehydrogenase]), as well as calf histone H1 and H3, was from Roche Applied Science (Indianapolis, IN, USA). Recombinant human APC (Xigris®) was from Eli Lilly (Indianapolis, IN, USA). Anti-histone H3 and anti-Gas6 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Recombinant Mer, αvβ3, αvβ5 and anti-Mer antibody were from R&D Systems. Antibodies to αvβ3 and αvβ5 were from Millipore (Billerica, MA, USA).
Mice
Male C57BL/6 mice were purchased from NCI Frederick. Mice were housed and studied at the University of Alabama at Birmingham (Birmingham, AL, USA) by using Institutional Animal Care and Use Committee–approved protocols. Experiments were performed using 8- to 10-wk-old mice.
Isolation and Induction of Apoptosis in Neutrophils
Mouse neutrophils were purified from bone marrow cell suspensions essentially as described previously (20). In brief, bone marrow cells were incubated with 20 μL primary antibody cocktail specific to the cell surface markers F4/80, CD4, CD45R, CD5 and TER119 for 15 min at 4°C. Anti-biotin tetrameric antibody complexes (100 μL) were then added to the cells and incubated for 15 min at 4°C, followed by incubation with 60 μL colloidal magnetic dextran iron particles for 15 min at 4°C. The entire cell suspension was then placed into a column surrounded by a magnet. The T cells, B cells, red blood cells, monocytes and macrophages were captured in the column, allowing the neutrophils to pass through as a result of negative selection. The cells were then pelleted and washed. Neutrophil purity, as determined by Wright-Giemsa–stained cytospin preparations, was >97%. Cell viability, as determined by trypan blue exclusion, was consistently >98%. Apoptosis was induced by heating 6 × 106 cells/mL serum-free RPMI-1640 media at 43°C for 60 min, followed by incubation at 37°C in 5% CO2 for 150 min. Using this method, >75% of the neutrophils were apoptotic (early and late apoptotic) as demonstrated by Annexin V and propidium iodide staining (Supplementary Figures S1A and B).
Induction of Apoptosis in Thymocytes
Mouse thymocytes were labeled using a PKH26 Red Fluorescent Dye Linker kit, according to the manufacturer’s instructions before apoptosis induction. To induce apoptosis, labeled thymocytes at 6 × 106 cells/mL were resuspended in RPMI-1640 containing 5% fetal bovine serum (FBS) and 1 μmol/L dexamethasone and incubated at 37°C in 5% CO2 for 12 h. At this time point, >80% of the thymocytes were apoptotic, as demonstrated by Annexin V and propidium iodide staining (Supplementary Figures S1C and D).
Isolation and Preparation of Mouse Peritoneal Macrophages
Peritoneal macrophages were elicited from 8- to 10-wk-old mice by intraperitoneal injection of 1 mL 3% thioglycollate. Cells were harvested 4 d later by peritoneal lavage. The 0.5 × 106 macrophages were plated on coverslips in 24-well plates in RPMI-1640 media containing 5% FBS (Atlanta Biologics, Atlanta, GA, USA). After 1 h at 37°C, nonadherent cells were removed by washing with medium. Macrophages were cultured in RPMI-1640 media containing 5% FBS at 37°C and maintained under the same conditions with changes of media every 3 d. One hour before the phagocytosis assay, the macrophages were washed three times with fresh serum-free medium.
In Vitro Efferocytosis Assays
Phagocytosis of apoptotic neutrophils (efferocytosis) was determined by adding 1.5 × 106 apoptotic neutrophils suspended in 600 μL RPMI-1640 medium to each well of a 24-well plate containing macrophage monolayers on coverslips followed by incubation at 37°C for 120 min. FBS was included at a final concentration of 5% during the incubation of macrophages with apoptotic cells. Non-ingested cells were removed by washing three times with ice-cold phosphate-buffered saline (PBS). Cells on coverslips were fixed in 100% methanol and then stained by using the Wright-Giemsa method. Phagocytosis was evaluated by a blinded observer by counting at least 300 macrophages per slide from duplicate experiments. The phagocytosis index was calculated as the ratio of macrophages containing at least one ingested apoptotic cell.
Phagocytosis of apoptotic thymocytes was determined by adding 1 × 106 apoptotic thymocytes suspended in 300 μL RPMI-1640 medium to each well of a 24-well plate containing macrophage mono-layers by incubation at 37°C for 120 min. FBS was included at a final concentration of 5% during the incubation of macrophages with apoptotic cells. Noningested cells were removed by washing three times with ice-cold PBS. Cells were collected in PBS containing 1% albumin, FITC-conjugated CD11b (macrophage marker) antibody and APC-conjugated CD90.2 (thymocyte marker) antibody. Flow cytometry was performed. The phagocytic index was calculated as the ratio of FITC+PKH26+APC– cells to all cells gated. Engulfed thymocytes are not accessible to the APC-conjugated CD90.2 antibody. Therefore, FITC+PKH26+APC– cells are macrophages that have engulfed PKH-labeled thymocytes, whereas the APC+PKH+FITC+ cells were macrophages, which thymocytes are adherent to but are not engulfed by.
In Vivo Efferocytosis Assay
Mice were anesthetized with isofluorane, and then 10 × 106 apoptotic thymocytes in 50 μL sterile PBS were injected intratracheally. Two hours later, the mice were sacrificed and bronchoalveolar lavage was performed using 1 mL sterile PBS containing 5 mmol/L EDTA (ethylenediaminetetraacetic acid). Samples were treated as described above, and flow cytometry was performed to determine phagocytic index.
Phagocytosis of E. Coli
Phagocytosis of E. coli was determined by adding FITC-labeled heat-inactivated E. coli suspended in 300 μL RPMI-1640 medium to each well of a 24-well plate containing macrophage monolayers by incubation at 37°C for 20 min. Non-ingested E. coli was removed by washing seven times with ice-cold PBS. Flow cytometry was performed.
Solid-Phase Enzyme-Linked Immunosorbent Assay (ELISA) or Histone Binding
The 96-well plates were precoated with Histone H3 (1 μg/mL in PBS) for 12 h. The wells were then washed three times and blocked with 1% BSA in PBS. Soluble Mer, integrin αvβ5, or integrin αvβ3 were added to the wells at increasing concentrations (0, 0.05, 0.1, 0.5, 1 and 5 μg/mL) and incubated at room temperature for 1 h. The wells were then washed three times. Specific antibodies for Mer, integrin αvβ5 or integrin αvβ3 were added to the wells and incubated for 2 h, followed by washing and addition of appropriate peroxidase-conjugated secondary antibodies. After final washing, TMB solutions were added to the wells and the plates read at A450.
Determination of Histone H3 Cytotoxicity
To assess the cytotoxic effect of histone H3 on macrophages, cells were exposed to histone H3 at increasing concentrations (0, 1, 5 and 10 μg/mL) in RPMI-1640 media containing 5% FBS for 1 h. LDH released into the media was measured using a Cytotoxicity Detection Kit (Roche) according to the manufacturer’s instructions. Maximal LDH release was obtained by cell lysis in 2% Triton X-100 in PBS.
ELISA
Macrophages were treated with 10 μg/mL BSA or histone H3, or 10 ng/mL LPS or 1 μg/mL PamCSK3 in the presence of 1 μg/mL BSA or histone H3. Interleukin-6 levels in the culture supernatants were determined by ELISA according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA).
Statistical Analysis
Data are presented as means ± SD for each experimental group. One-way ANOVA followed by analysis with a Bonferroni correction for multiple comparisons was performed for comparisons among multiple groups, and the Student t test was used for comparisons between two groups. A p value <0.05 was considered significant.
All supplementary materials are available online at www.molmed.org.
RESULTS
Histones Inhibit Efferocytosis
To examine the potential participation of histones in efferocytosis, we performed efferocytosis assays in the presence of increasing doses of histone H3, one of the core histones. As shown in Figure 1A, the phagocytosis of apoptotic neutrophils by macrophages was significantly diminished in the presence of histone H3. Histone H3 also decreased the ability of macrophages to ingest apoptotic thymocytes (Figure 1B), indicating that the inhibitory effect of histone H3 on efferocytosis is a general phenomenon and not specific for apoptotic neutrophils. A representative image of phagocytosis assays with apoptotic neutrophils and a representative diagram for assays with apoptotic thymocytes are shown in Supplementary Figures S2 and S3.
Figure 1.

Histones inhibit efferocytosis. Peritoneal macrophages were incubated with apoptotic neutrophils (A) or apoptotic thymocytes (B) in medium containing 10 μg/mL BSA (control) or histone H3 at increasing concentrations (1, 5 or 10 μg/mL) for 60 min. Efferocytosis assays were then performed as described in Materials and Methods. The percentage of macrophages that phagocytosed apoptotic cells for the control group is shown above the bar. Fold changes were calculated by dividing the percentage of macrophages that phagocytosed apoptotic cells for the experiment groups by that of the control groups. (C) Peritoneal macrophages were exposed to 10 μg/mL BSA (control), histone H3 (1, 5 or 10 μg/mL) or medium for 60 min and then levels of LDH (optical density [O.D.]) in the culture supernatants were determined. (D) Histones H3 and H4, but not histone H1, inhibit efferocytosis. Macrophages were incubated for 60 min with apoptotic thymocytes in media containing BSA (control) or histones H1, H3 or H4 (10 μg/mL), and then efferocytosis assays were performed. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the control group. The percentage of macrophages that phagocytosed apoptotic cells for the control group is shown above the bar. Fold changes were calculated by dividing the percentage of macrophages that phagocytosed apoptotic cells for the experiment groups by that of the control groups.
Histones have been found to be cytotoxic to endothelial cells (18). Therefore, it was possible that the inhibitory effect of histone H3 on efferocytosis was simply a result of its cytotoxicity to macrophages. To rule out this possibility, macrophages were incubated with increasing doses of histone H3, as used in the efferocytosis assays, and cytotoxicity was determined by measuring LDH levels in the media. We found that histone H3, at the doses used in the efferocytosis assays, caused no cytotoxicity to macrophages (Figure 1C). Of note, the concentrations of histone H3 used in these experiments were comparable to those observed in the serum of patients and animals with sepsis.
To determine if other core histones, such as histone H4, have effects on efferocytosis similar to the effects produced by histone H3, we performed efferocytosis assays in the presence of histone H4. We found that histone H4, like histone H3, diminished the ingestion of apoptotic thymocytes by macrophages (Figure 1D). However, histone H1, a linker histone, demonstrated no effects on efferocytosis (see Figure 1D). These results indicate that the inhibitory effects of histones on efferocytosis are not a general phenomenon, but rather are limited to specific histones, including H3 and H4.
To determine if the inhibitory effects of histones are specific to phagocytosis of apoptotic cells, we determined if histone H3 regulates bacteria phagocytosis and found that histone H3 does not inhibit, but slightly increases, macrophages to ingest E. coli (Supplementary Figure S4). Histone H3 used in the study is purified from calf thymus and likely contains genomic DNA contaminants. To rule out the possibility that the observed effects of histones on phagocytosis of apoptotic cells are caused by DNA, we performed similar experiments in the presence of increasing doses of calf thymus genomic DNA. We found that DNA does not affect the activity of histone H3 (Supplementary Figure S5). We did not find that histone H3 stimulates macrophages or affects the inflammatory response of macrophages to TLR2 or TLR4 activation (Supplementary Figure S6), suggesting that the effects of histone H3 on phagocytosis are not a result of macrophage activation.
Histone H3 Inhibits Efferocytosis by Binding to Macrophages
Macrophages recognize and engulf apoptotic cells through engagement of cell surface receptors, such as integrins αvβ3 and αvβ5, as well as Mer receptor tyrosine kinase (4,6,10,21). To determine if the inhibitory effects of histone H3 on efferocytosis originate from direct interaction with macrophages, we preincubated macrophages with histone H3 for 1 h and then washed the cells three times before performing efferocytosis assays. As shown in Figure 2A, preincubation of macrophages with histone H3 significantly diminished the ability to ingest apoptotic thymocytes. These data suggest that histone H3 exerts its inhibitory effect on efferocytosis through direct interaction with macrophages.
Figure 2.

Histone H3 inhibits efferocytosis by binding to macrophages. (A) Macrophages were preincubated with BSA (10 μg/mL) or increasing doses of histone H3 (1, 5 and 10 μg/mL) for 1 h. The macrophages were then washed with fresh medium to remove unbound proteins, and apoptotic thymocytes were added for 60 min, after which efferocytosis assays were performed. (B) Macrophages were incubated with BSA (control), Chromeo 488–conjugated BSA (BSA-488) or Chromeo 488–conjugated histone H3 (H3-488) (5 μg/mL) for 1 h. The cells were then washed three times with PBS to remove unbound proteins. The quantities of proteins bound to macrophages were determined by a fluorescent plate reader. (C) Peritoneal macrophages were plated on coverslips and incubated with 5 μg/mL Chromeo 488–conjugated histone H3 (Histone H3-488) for 1 h. The macrophages were then washed three times with PBS, and confocal fluorescent microscopy analysis was performed to determine bound histone H3. DAPI was used to stain nuclei. **p < 0.01 compared with the control group.
To determine if histone H3 directly binds to macrophages, Chromeo 488– conjugated histone H3 was incubated with macrophages for 1 h, followed by washing three times to remove unbound histone H3. As shown in Figure 2B, histone H3 demonstrated binding to macrophages, compared with Chromeo 488– conjugated BSA. To further characterize the interaction between histone H3 and macrophages, confocal microscopy analysis was performed after incubation of macrophages with Chromeo 488– conjugated histone H3. As shown in Figure 2C, there were significant amounts of histone H3 bound to the macrophage surface.
Binding of Histone H3 to Macrophages Is Diminished by the Opsonins Gas6 and MFG-E8
The integrins αvβ3 and αvβ5, as well as the Mer tyrosine kinase, are major receptors that mediate efferocytosis through binding to the opsonins MFG-E8 and Gas6 (1,4,21). To determine if there is competitive binding between histone H3 and MFG-E8 or Gas6 to receptors on the macrophage surface, macrophages were preincubated with BSA, MFG-E8 or Gas6. The cells were then washed to remove unbound proteins and incubated with Chromeo 488–conjugated histone H3 or BSA. As shown in Figure 3, preincubation with MFG-E8 and Gas6 diminished the binding of histone H3 to macrophages. These data suggest that histone H3 and opsonins MFG-E8 and Gas6 may interact with the same receptor( s) on the macrophage surface.
Figure 3.
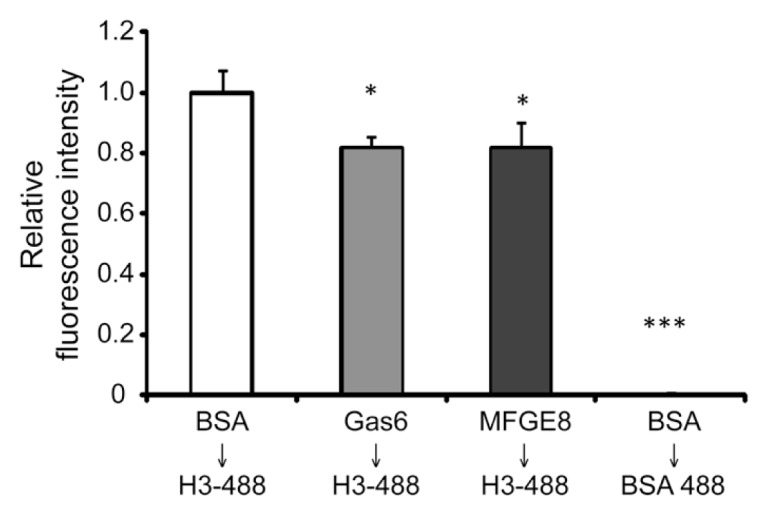
Binding of histone H3 to macrophages is diminished by the opsonins Gas6 and MFG-E8. Macrophages were incubated with BSA (control), Gas6 or MFG-E8 (5 μg/mL) for 1 h and then washed three times, followed by incubation with Chromeo 488–conjugated histone H3 (H3-488) or Chromeo 488–conjugated BSA (BSA-488) (5 μg/mL) for 1 h. The cells were then washed again with PBS to remove unbound proteins. The quantities of proteins bound to macrophages were determined by a fluorescent plate reader. *p < 0.05 and ***p < 0.001 compared with the control group.
Interaction of H3 Histone with Integrin αvβ5 or Mer Receptor Tyrosine Kinase Diminishes Binding to Macrophages
Given our findings that histone H3 competitively interacts with MFG-E8 and Gas6 for binding to the macrophage surface, we used solid-phase ELISAs to determine if there is a direct interaction between histone H3 and the αvβ3 and αvβ5 integrins, as well as with the Mer receptor tyrosine kinase. As shown in Figure 4, histone H3 binds to αvβ3, αvβ5 and Mer.
Figure 4.
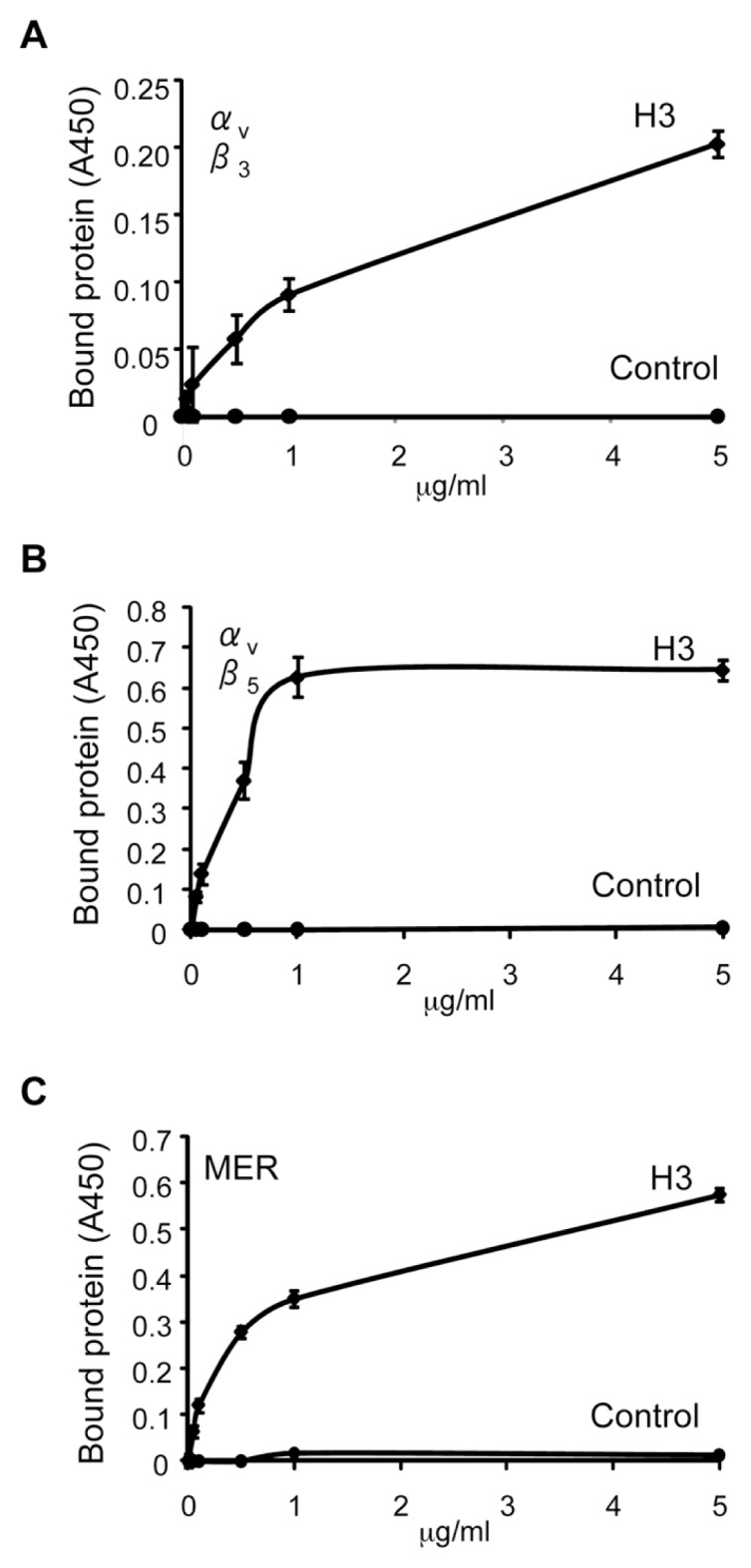
Histone H3 binds to the αvβ5 integrin and to the Mer receptor but not to the αvβ3 integrin. The 96-well plates were precoated with histone H3 or BSA (1 μg/mL) in PBS. The plates were then washed three times with PBS and blocked with PBS containing 1% BSA for 1 h. After removing the blocking solution, the plates were incubated with increasing doses (0, 0.1, 0.5, 1 and 5 μg/mL) of recombinant mouse Mer (A), αvβ5 (B) or αvβ3 (C) dissolved in PBS for 1 h, followed by washing with PBS containing 0.05% Tween 20. Protein bound to the wells was quantified by solid-phase ELISA as described in Materials and Methods. A450, absorbance at 450 nm.
To further characterize the interactions between histone H3 and the αvβ3 and αvβ5 integrins, as well as with Mer, we incubated histone H3 with soluble αvβ3, αvβ5 or the Mer extracellular domain and then examined binding to macrophages. As shown in Figure 5, incubation of histone H3 with soluble αvβ5 and Mer, but not with αvβ3, diminished the binding of histone H3 to macrophages. Taken together, these data suggest that the mechanism by which histone H3 diminishes efferocytosis is through binding to αvβ5 and Mer, thereby interrupting their interactions with phosphatidylserine-associated Gas6 and MFG-E8.
Figure 5.
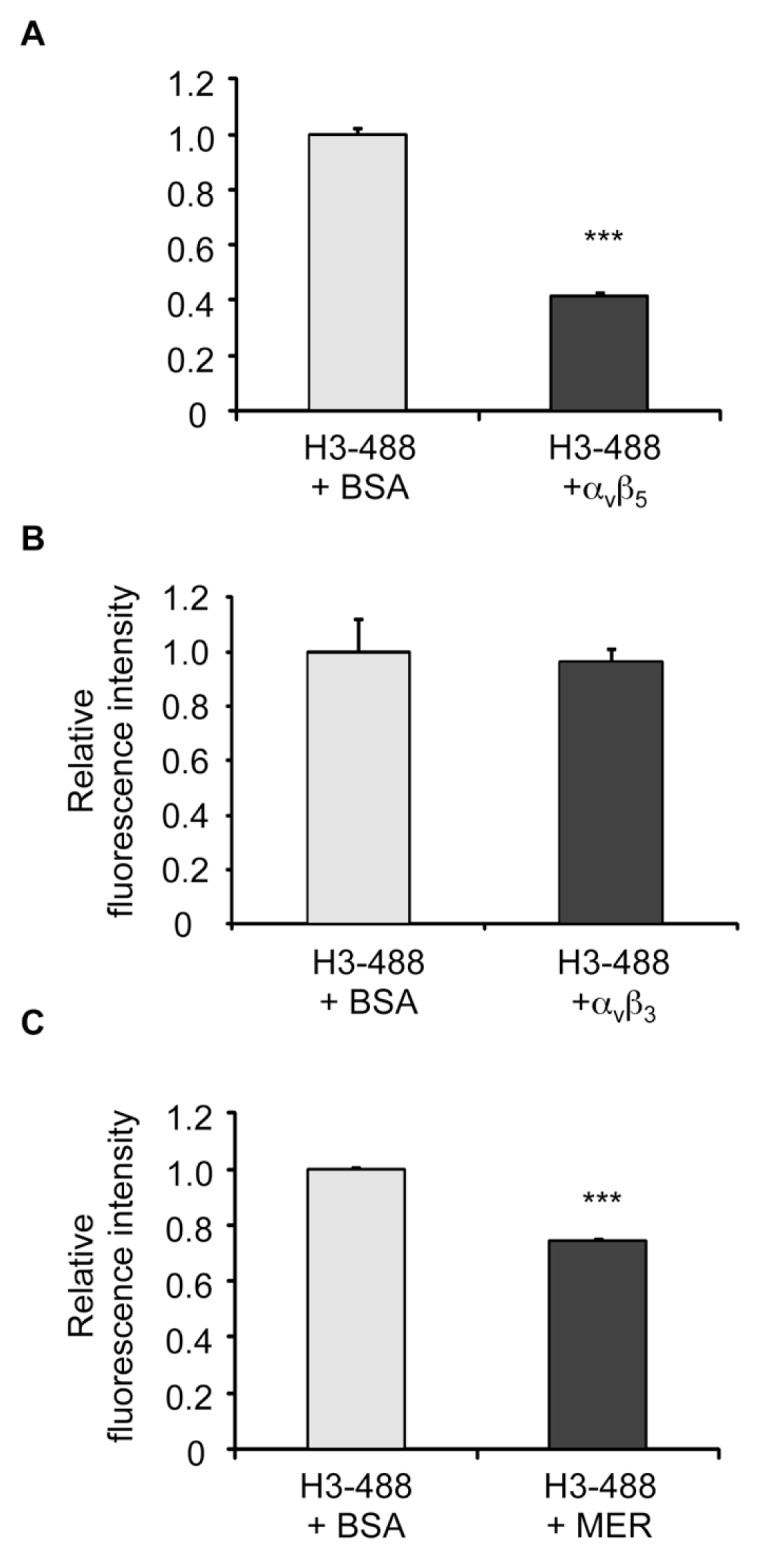
Binding of histone H3 to macrophages is diminished by the integrin αvβ5 and Mer, but not by the αvβ3 integrin. Chromeo 488–conjugated histone H3 (H3-488) (5 μg/mL) was incubated with BSA or Mer (A), integrin αvβ3 (B) or integrin αvβ5 (C) (5 μg/mL) for 30 min in RPMI-1640. The mixture of the proteins was then added to macrophages and incubated for 1 h, after which the cells were washed three times with PBS and the quantities of bound protein were determined by a fluorescent plate reader. ***p < 0.001 compared with the 488-H3 + BSA group.
Histone H3 Inhibits Efferocytosis In Vivo
As our experiments demonstrated that the H3 histone inhibited efferocytosis under in vitro conditions, we next determined if this histone has similar effects in vivo. To examine this issue, we injected apoptotic neutrophils or apoptotic thymocytes with BSA or histone H3 (5 μg/mL in 50 μL saline) and then collected bronchoalveolar lavages 1 h later. As shown in Figure 6, phagocytosis of apoptotic neutrophils or apoptotic thymocytes by alveolar macrophages was diminished in the presence of histone H3.
Figure 6.
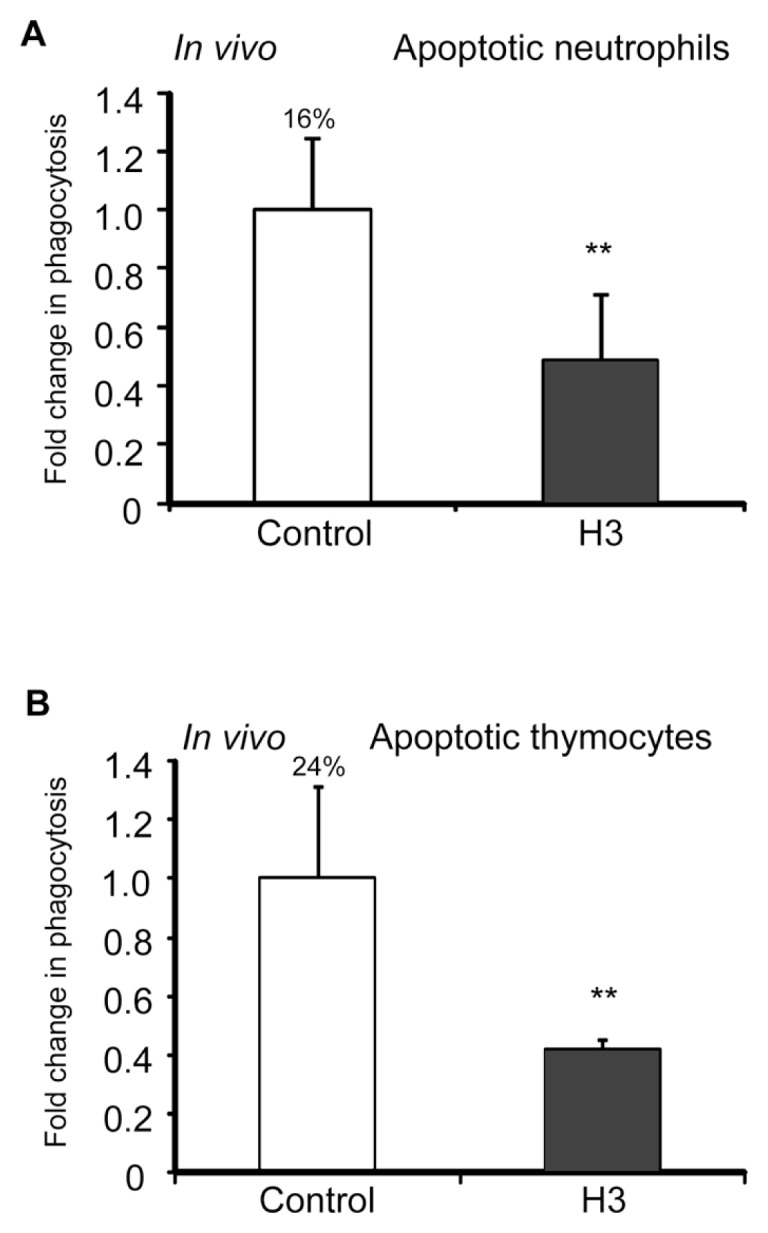
Histone H3 inhibits efferocytosis in vivo. Mice (three in each group) were exposed to the intratracheal administration of 10 × 106 apoptotic neutrophils (A) or apoptotic thymocytes (B) resuspended in 50 μL PBS containing 5 μg BSA (control) or histone H3, and bronchoalveolar lavages were collected 2 h later. Cytospin slides were prepared from the bronchoalveolar lavage fluid, and phagocytosis was determined by microscopy (A) or by flow cytometry (B). **p < 0.05 compared with the BSA group. The percentage of macrophages that phagocytosed apoptotic cells for the control group is shown above the bar.
APC Abrogates the Inhibitory Effects of Histone H3 on Efferocytosis In Vitro and In Vivo
It was recently shown that APC cleaves histones, a mechanism that contributes to its protective effects in acute inflammatory conditions, such as sepsis, that are associated with elevated levels of extracellular histones (18). To determine if the ability of APC to cleave histones affects the inhibitory effects of histones on efferocytosis, histone H3 was preincubated without or with APC before efferocytosis assays were performed. As shown in Figure 7A, exposure of histone H3 to APC abolished its inhibitory effects on efferocytosis. Of note, we confirmed that histone H3 was cleaved by APC at the concentrations used in the efferocytosis assays (Figure 7B).
Figure 7.

APC abrogates the inhibitory effects of histone H3 on efferocytosis in vitro and in vivo. (A) Macrophages were incubated for 1 h with apoptotic thymocytes in the presence of 5 μg/mL BSA (control), histone H3, BSA that was preincubated with 200 nmol/L APC or histone H3 that was preincubated with 200 nmol/L APC, after which efferocytosis assays were performed. (B) Histone H3 and histone H3 preincubated with 200 nmol/L APC were resolved by SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis). The gel was stained with Coomassie Blue R-250 to visualize histone H3 and cleaved histone H3. (C) Apoptotic thymocytes were resuspended in 50 μL PBS containing 5 μg BSA (control), histone H3, BSA that was preincubated with 200 nmol/L APC or histone H3 that was preincubated with 200 nmol/L APC and then administered intratracheally for in vivo efferocytosis assays. *p < 0.05 and ***p < 0.001 in comparison with the BSA group. The percentage of macrophages that phagocytosed apoptotic cells for the control group is shown above the bar.
To determine if APC affects the ability of histone H3 to inhibit efferocytosis under in vivo conditions, mice were injected intratracheally with apoptotic thymocytes and BSA or histone H3 that had been preincubated with or without APC. As shown in Figure 7C, preexposure of histone H3 to APC abrogated its inhibitory effects on efferocytosis by alveolar macrophages. Taken together, these data suggest that a potential mechanism for the beneficial effects of APC in acute inflammatory conditions, such as sepsis and acute lung injury, may be partially through cleaving histones and thereby preventing the inhibitory effects of histones on efferocytosis.
DISCUSSION
In these studies, we found that histones, through directly binding to the macrophage surface, diminish the ability of macrophages to ingest apoptotic cells in both in vitro and in vivo settings. Since levels of extracellular histones rise dramatically in acute inflammatory conditions, such as sepsis (18), the present experiments describe a novel mechanism, mediated by extracellular histones, that is likely to contribute to the perpetuation of inflammation. In particular, interactions between extracellular histones and macrophages result in diminished clearance of apoptotic neutrophils and other cell populations that then progress to necrosis with associated release of proinflammatory intracellular contents.
The present experiments suggest that a mechanism by which histones diminish efferocytosis is through inhibiting the potentiating effects of the opsonins Gas6 and MFG-E8 on the uptake of apoptotic cells by macrophages. The in vivo significance of the actions of opsonins in enhancing efferocytosis is highlighted by the ability of therapy with MFG-E8 to diminish the severity of acute lung injury, presumably through enhancing efferocytosis and clearance of apoptotic cells (22). Both Gas6 and MFG-E8 function to bridge phosphatidylserine on the apoptotic cell surface with macrophage ligands, including integrins, such as αvβ3 and αvβ5, and Mer receptor tyrosine kinase (1,4,5,10,23). In these studies, we found that association of histone H3 with either soluble αvβ5 or the Mer extracellular domain, but not with soluble αvβ3, diminished histone binding to macrophages. These results suggest that the ability of histones to interrupt opsoninfacilitated binding between phosphatidylserine- and macrophage-associated αvβ5 and Mer contributes to the inhibitory effects of histones on efferocytosis. Of note, although histones have cytotoxic properties (18), at the concentrations that produced decreases in efferocytosis in the present experiments, histones did not demonstrate any cytotoxic effects on macrophages. Such findings indicate that the inhibitory effects of histones on efferocytosis are due to direct effects on the uptake of apoptotic cells by macrophages and are not secondary to cytotoxicity.
While histones H3 and H4 decreased efferocytosis, no such effect was found with histone H1, indicating that the inhibitory actions of histones on the phagocytosis of apoptotic cells are not generalizable, but rather are only present for specific histones. Previous studies investigating histone involvement in other pathways associated with inflammation have also found greater activity with histones H3 and H4 and relative lack of activity for histone H1. For example, incubation of platelets with histones H3 and H4 stimulated aggregation, whereas histone H1 had no such effect (12). Similarly, while histone H1 demonstrated little cytotoxicity to endothelial cells, histones H3 and H4 were cytotoxic and also contributed to mortality in mouse sepsis models (18).
In the present experiments, concentrations of histone H3 as low as 1 μg/mL inhibited phagocytosis of apoptotic neutrophils. In baboons, circulating levels of histone H3 as high as 15 μg/mL were present 8 h after challenge with E. coli (18). Similarly, elevated plasma concentrations of histone H3 are present in humans with severe sepsis (18). The presence of levels of histone H3 during sepsis that are higher than those required to diminish efferocytosis indicate that the inhibitory actions of histones on efferocytosis described in the present studies are likely to be relevant during pathophysiologic states, including acute lung injury, associated with enhanced inflammation and alteration in apoptotic pathways.
APC diminishes organ dysfunction in preclinical models of sepsis and acute lung injury and also appears to improve outcome in humans with severe sepsis and septic shock (18,24–28). Whereas the anticoagulant properties of APC were initially thought to be responsible for its benefit, more recent studies indicate that its effects in reducing organ dysfunction and improving survival are likely to arise from antiinflammatory and cytoprotective actions (29–31). APC can cleave histones, thereby reducing their cytotoxic effects in vitro and diminishing the ability of extracellular histones to induce mortality when infused into mice or when released during sepsis (32). In the present experiments, treatment of histone H3 with APC abrogated its inhibitory effects on efferocytosis by peritoneal macrophages under in vitro conditions and by alveolar macrophages in vivo after intratracheal administration of apoptotic thymocytes. Our studies, showing that APC can reverse histone-induced decreases in efferocytosis, therefore provide a new mechanism by which APC may diminish organ dysfunction associated with acute lung injury and other conditions in which decreased efferocytosis contributes to persistent inflammation and tissue injury.
CONCLUSION
Recent studies have described novel extracellular roles in inflammation for proteins, such as HMGB1, that were previously characterized as being localized to the nucleus and associated with DNA (33). Whereas histones have also been demonstrated to be secreted and to contribute to mortality in sepsis through a role postulated to be associated with enhanced endothelial cytotoxicity (18), the present results extend the mechanisms by which histones may contribute to inflammation and tissue injury by demonstrating that they have inhibitory properties in efferocytosis. Although the effects of histones on efferocytosis occur at concentrations lower than those achieved during sepsis, the relative importance of such histone-associated actions on tissue injury and organ dysfunction during inflammatory conditions, such as acute lung injury and sepsis, remain to be explored. Nevertheless, the growing evidence for a direct pathophysiologic role of extracellular histones in inducing and perpetuating organ injury suggests that therapeutic approaches, including the use of APC, will be useful in improving outcomes for conditions associated with increased release of histones into the interstitium and circulation.
Supplemental Data
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants HL105473, HL097218 and HL076206 and American Heart Association (AHA) awards (10SDG4210009 and 11SDG5330014). A Friggeri, E Abraham, and G Liu created conception and design.
A Friggeri, S Banerjee, N Xie, H Cui, M Zerfaoui, E Abraham, and G Liu performed analysis and interpretation. A Friggeri, H Dupont, E Abraham, and G Liu drafted the manuscript.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673–82. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]
- 2.Blasi F, Sidenius N. Efferocytosis: another function of uPAR. Blood. 2009;114:752–3. doi: 10.1182/blood-2009-05-220657. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong A, Ravichandran KS. Phosphatidylserine receptors: what is the new RAGE? EMBO Rep. 2011;12:287–8. doi: 10.1038/embor.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059–70. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207:1807–17. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park D, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–4. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 7.Miyanashi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–9. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 8.He M, et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 2011;12:358–64. doi: 10.1038/embor.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee S, Friggeri A, Liu G, Abraham E. The C-terminal acidic tail is responsible for the inhibitory effects of HMGB1 on efferocytosis. J Leukoc Biol. 2010;88:973–9. doi: 10.1189/jlb.0510262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friggeri A, Yang Y, Banerjee S, Park YJ, Liu G, Abraham E. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol. 2010;299:C1267–76. doi: 10.1152/ajpcell.00152.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu G, et al. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol. 2008;181:4240–6. doi: 10.4049/jimmunol.181.6.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuchs TA, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–5. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pemberton AD, Brown JK, Inglis NF. Proteomic identification of interactions between histones and plasma proteins: implications for cytoprotection. Proteomics. 2010;10:1484–93. doi: 10.1002/pmic.200900818. [DOI] [PubMed] [Google Scholar]
- 14.Monach PA, et al. A broad screen for targets of immune complexes decorating arthritic joints highlights deposition of nucleosomes in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2009;106:15867–72. doi: 10.1073/pnas.0908032106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513–21. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun. 2009;1:194–201. doi: 10.1159/000206974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187:2626–31. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu J, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Margraf S, et al. Neutrophil-derived circulating free DNA (cf-DNA/NETs): a potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock. 2008;30:352–8. doi: 10.1097/SHK.0b013e31816a6bb1. [DOI] [PubMed] [Google Scholar]
- 20.Friggeri A, et al. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol. 2010;299:C1267–76. doi: 10.1152/ajpcell.00152.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207:1807–17. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui T, et al. Milk fat globule epidermal growth factor 8 attenuates acute lung injury in mice after intestinal ischemia and reperfusion. Am J Respir Crit Care Med. 2010;181:238–46. doi: 10.1164/rccm.200804-625OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, et al. Urokinase-type plasminogen activator inhibits efferocytosis of neutrophils. Am J Respir Crit Care Med. 2010;182:1516–23. doi: 10.1164/rccm.201003-0452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esmon CT. Role of coagulation inhibitors in inflammation. Thromb Haemost. 2001;86:51–6. [PubMed] [Google Scholar]
- 25.Abraham E. Effects of recombinant human activated protein C in human models of endotoxin administration. Proc Am Thorac Soc. 2005;2:243–7. doi: 10.1513/pats.200501-004AC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ely EW, et al. Drotrecogin alfa (activated) administration across clinically important subgroups of patients with severe sepsis. Crit Care Med. 2003;31:12–9. doi: 10.1097/00003246-200301000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Bernard GR, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 28.Husari AW, et al. Activated protein C attenuates acute lung injury and apoptosis in a hyperoxic animal model. Shock. 2010;33:467–72. doi: 10.1097/SHK.0b013e3181c69213. [DOI] [PubMed] [Google Scholar]
- 29.Ruf W. New players in the sepsis-protective activated protein C pathway. J Clin Invest. 2010;120:3084–7. doi: 10.1172/JCI44266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kerschen E. Activated protein C targets CD8+ dendritic cells to reduce the mortality of endotoxemia in mice. J Clin Invest. 2010;120:3167–78. doi: 10.1172/JCI42629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao C, et al. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest. 2010;120:1971–80. doi: 10.1172/JCI40380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.