

Abstract
Stemlike cells have been isolated by their ability to efflux Hoechst 33342 dye and are called the side population (SP). We evaluated the effect of axitinib on targeting cancer stemlike cells and enhancing the efficacy of chemotherapeutical agents. We found that axitinib enhanced the cytotoxicity of topotecan and mitoxantrone in SP cells sorted from human lung cancer A549 cells and increased cell apoptosis induced by chemotherapeutical agents. Moreover, axitinib particularly inhibited the function of adenosine triphosphate (ATP)-binding cassette subfamily G member 2 (ABCG2) and reversed ABCG2-mediated multidrug resistance (MDR) in vitro. However, no significant reversal effect was observed in ABCB1-, ABCC1- or lung resistance–related protein (LRP)-mediated MDR. Furthermore, in both sensitive and MDR cancer cells axitinib neither altered the expression of ABCG2 at the mRNA or protein levels nor blocked the phosphorylation of AKT and extracellular signal-regulated kinase (ERK)1/2. In nude mice bearing ABCG2-overexpressing S1-M1-80 xenografts, axitinib significantly enhanced the antitumor activity of topotecan without causing additional toxicity. Taken together, these data suggest that axitinib particularly targets cancer stemlike cells and reverses ABCG2-mediated drug resistance by inhibiting the transporter activity of ABCG2.
INTRODUCTION
Axitinib is an oral, potent, small-molecule adenosine triphosphate (ATP)-competitive multitargeted tyrosine kinase inhibitor (TKI). It inhibits cellular signaling by blocking vascular endothelial growth factor receptor 1 (VEGFR-1), VEGFR-2 and VEGFR-3; platelet derived growth factor receptor (PDGFR); and c-KIT (CD117) (1–3). These receptor TKs are transmembrane proteins at the cell surface that play critical roles in the transduction of extracellular signals to the cytoplasm. It has been reported that these receptors are important in signaling pathways and the development of a number of tumors (4,5). Inhibition of these TKs blocks signal transduction pathways that affect many of the processes involved in tumor cell proliferation, progression, metastasis and angiogenesis. In preclinical and clinical studies, axitinib has been shown to inhibit angiogenesis, vascular permeability and blood flow. In phase II studies, axitinib showed single-agent activity in a variety of tumor types, including non–small cell lung cancer (6), advanced renal cell carcinoma (7) and thyroid cancer (8).
ATP-binding cassette (ABC) drug transporter proteins can use the energy derived from ATP hydrolysis to extrude numerous structurally and mechanistically unrelated anticancer drugs, which play a key role in the development of multidrug resistance (MDR). Overexpression of ABC transporters is a significant hindrance to successful cancer chemotherapy (9). There are 49 ABC transporter family genes in the human genome, which are divided into seven subfamilies on the basis of amino acid sequence similarities and phylogeny (10). Among them, the ABC transporter–subfamily B member 1 (ABCB1/MDR1/P-glycoprotein, P-gp), subfamily C member 1 (ABCC1/MRP1) and subfamily G member 2 (ABCG2/BCRP) are considered to be the most important transporters to confer MDR to tumor cells.
ABCG2/BCRP, also called MXR (mitoxantrone resistance associated protein) and ABCP (placenta-specific ABC transporter), was identified independently from drug-selected human breast cancer cells (MCF-7/AdrVp) (11), human colon carcinoma cells (S1-M1-80) (12) and human placenta (13). The human ABCG2 gene is located on chromosome 4, band 4q21-4q22 and encodes a 72.6-kD membrane protein composed of 655 amino acids (14). ABCG2 can transport a wide range of anticancer agents such as doxorubicin (Dox), toptecan, SN-38, mitoxantrone and methotrexate as well as fluorescent dyes such as Hoechst 33342 (15). Wild-type ABCG2, with an arginine at position 482 (R482), facilitated efficient transport of mitoxantrone, but not rhodamine 123 or Dox. MCF7/AdVp3000 and S1-M1-80 cells expressing R482T and R482G variants of BCRP/ABCG2, respectively, transported rhodamine 123 and Dox while also maintaining their ability to transport mitoxantrone (16,17). Therefore, certain kinds of single-nucleotide polymorphisms of ABCG2 can alter its function, and consequently affect the disposition of substrate drugs.
Malignant stemlike cells have been identified in various malignant tumors, ranging from leukemia to solid tumors. Like normal stem cells, these cancer stem-like cells (CSCs) are able to self-renew, differentiate and proliferate extensively. The cancer mass originates from rare stemlike cells that can transfer the disease to immunodeficient mice. This finding suggests that these CSCs are responsible for the relapse of cancer following conventional or targeted cancer therapy and that eradication of these CSCs might be necessary to cure the disease permanently. However, it appears likely that CSCs are not effectively ablated by most current therapeutic strategies, leaving the potential for disease progression or relapse. Several recent studies have provided insight into the signaling pathways underlying the CSC phenotype and have also suggested approaches to eliminate CSCs (18,19). The side population (SP) phenotype cells, believed to be CSCs, are present in diverse tumor types and over-express ABCG2, producing inherent drug resistance (20,21). Currently, ABCG2 is considered to be a molecular marker for the SP cells (22).
ABCG2 is an ideal target for development of chemosensitizing agents for better treatment of drug-resistant cancers. However, very few compounds have been identified as specific inhibitors of ABCG2. Fumitremorgin C (FTC), a mycotoxin from Aspergillus fumigatus, was reported first (23). However, FTC neurotoxicity prevented any clinical use. Although analogues of FTC, such as Ko132 and Ko143, have been developed with low toxicity, the efficacy and safety of these modulators are still under evaluation in in vivo studies (24). Another less well characterized, but promising, strategy is the application of TKIs, small molecule hydrophobic compounds, which are designed to arrest aberrant signaling pathways in malignant cells. It has been recently shown that the TKIs interact with and modulate the function of ABC transporters such as ABCB1, ABCC1 and ABCG2. The BCR-Abl TKIs imatinib (Gleevec) and nilotinib (Tasigna) interact with ABCB1 and ABCG2 transporters and significantly inhibit their transport activity (25,26). Gefitinib, an epidermal growth factor receptor (EGFR) TKI, has been observed to directly inhibit the function of ABCB1 in MDR cancer cells (27) and reverse ABCG2-mediated MDR in vitro (28). In animal models, gefitinib affected the oral absorption of chemo-therapeutic agents by modulating the function of ABCB1 and ABCG2 (29). In our previous study, we also found that lapatinib and sunitinib antagonized ABCB1- and/or ABCG2-mediated MDR (30,31).
Although axitinib was effective as an oral agent in earlier stages of clinical development, its interaction with ABC drug transporters has not been characterized. The objective of this work was to investigate the interaction of axitinib with ABCB1, ABCC1, ABCC4, ABCG2 and lung resistance–related protein (LRP). We show here that axitinib targets CSCs, increases the efficacy of chemotherapeutic agents and reverses ABCG2-mediated drug resistance by inhibiting the drug efflux function of ABCG2 and increasing the intracellular accumulation of cytotoxic agents in ABCG2-overexpressing cells and SP cells.
MATERIALS AND METHODS
Chemicals and Reagents
3-(4,5-Dimethylthiazol-yl)-2,5-diphenyllapatinibrazolium bromide (MTT), vincristine, topotecan, mitoxantrone, Dox, rhodamine 123, verapamil, MK571 and FTC were products of Sigma Chemical Co (St. Louis, MO, USA). Axitinib was purchased from Selleck Chemicals (Houston, TX, USA). Dulbecco’s modified Eagle’s medium (DMEM) and RPMI 1640 were products of Gibco BRL (Gaithersburg, MD, USA). Anti-mitogen-activated protein kinase (anti-MAP) 1/2 (extracellular signal-regulated kinase [ERK]1/2) (KC-5E01), P-ERK (KC-5E04), P-AKT (KC-5A04) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were purchased from Kangchen Co. (Shanghai, China). AKT antibody (#4685) was a product of Cell Signaling Technology Inc. (Danvers, MA, USA). ABCG2 antibody (MAB4146) was obtained from Chemicon International, Inc. (Temecula, CA, USA). Other routine laboratory reagents were of analytical grade and obtained from commercial sources.
Cell Culture
The following cell lines were cultured in DMEM or RPMI 1640 containing 10% fetal bovine serum at 37°C in the presence of 5% CO2: the human colon carcinoma cell line S1 and its mitoxantrone-selected derivative ABCG2-overexpressing S1-M1-80 cell line were kindly provided by SE Bates (National Cancer Institute, National Institutes of Health [NIH], Bethesda, MD, USA) (32); the human oral epidermoid carcinoma cell line KB and its vincristine-selected derivative ABCB1-overexpressing cell line KBv200 were a gift from Xu-Yi Liu (Cancer Hospital of Beijing, Beijing, China) (33); the human leukemia cell line HL60 and its Dox-selected derivative ABCC1-overexpressing cell line HL60/ADR were purchased from the Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China; the human lung squamous carcinoma cell line SW1573 and its Dox-selected derivative LRP-overexpressing SW1573/2R120 were provided by EAC Wiemer (Department of Medical Oncology, Erasmus Medical Center, Rotterdam, the Netherlands) (34); the murine fibroblasts cell line NIH3T3 and the ABCC4-transfected ABCC4 stable expressing NIH3T3/MRP4-2 cells were kindly provided by Z-S Chen (St John’s University, Queens, NY, USA) (35). The human lung cancer A549 cell line was purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). HEK293/pcDNA3.1, ABCG2-482-G2 and ABCG2-482-T7 cells were established by selection with G418 after transfecting HEK293 with either an empty pcDNA3.1 vector or a pcDNA3.1 vector containing full-length ABCG2 coding either glycine (G) or threonine (T) at the amino acid 482 position, respectively (36). These cells were obtained from SE Bates (National Cancer Institute, NIH) and were cultured in medium with 2 mg/mL G418.
Cell Cytotoxicity Test
The MTT assay was used to assess the sensitivity of cells to drugs as previously described (37). Briefly, cells were distributed evenly into 96-well microtiter plates and then various concentrations of axitinib were added to the wells. After 68 h of incubation, MTT (5 mg/mL, 20 μL/well) was added into the cells for 4 h (37°C). Afterward, the medium was discarded, and 200 μL of dimethylsulfoxide was added to dissolve the formazan product from the metabolism of MTT. Optical density was measured at 540 nm with background subtraction at 670 nm by use of the Model 550 Microplate Reader (BIO-RAD, Hercules, CA, USA). The concentration required to inhibit cell growth by 50% (IC50) was calculated from survival curves by use of the Bliss method (38). For reversal experiments, axitinib was added to the medium with full range concentrations of (a) topotecan, mitoxantrone and cisplatin in S1 and S1-M1-80; (b) Dox and cisplatin in KB and KBv200; (c) Dox and cisplatin in HL60 and HL60/ADR; (d) Dox and cisplatin in SW1573 and SW1573/2R120; and (e) 6-mercaptopurine (6-MP) and cisplatin in NIH3T3 and NIH3T3/MRP4-2 cells. Fold of resistance was calculated by dividing the IC50 for the MDR cells by that for the parental sensitive cells. The degree of reversal of MDR (fold reversal) was calculated by dividing the IC50 for cells with the anticancer drug in the absence of axitinib by that obtained in the presence of axitinib.
Animals
Athymic nude mice (BALB/c-nu/nu) of both sexes, 5 to 6 wks old and weighing 18 to 22 g, were bred at the Center of Experimental Animals, Sun Yat-Sen University (Guangzhou, China), and were used for the S1 and S1-M1-80 cell xenografts. Male nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice, 4–5 wks old, were purchased from Beijing HFK Biotechnology Co. Ltd (Changping District. Beijing. PR China) and were used for the tumorigenicity experiments. All animals received sterilized food and water. All experiments were conducted with the approval of the Sun Yat-Sen University Institutional Animal Care and Use Committee.
Tumor Xenograft Experiments
The S1-M1-80 cell xenograft model was established as previously described (39) with slight modification. Briefly, 1 × 107 S1-M1-80 cells were injected subcutaneously into the posterior flank region of the nude mice. The mice were randomized into four groups after the tumors reached a mean volume of about 100 mm3, and then received various treatments: (a) saline (every 4 d × 9, intraperitoneally [IP]); (b) topotecan (every 4 d × 9, IP, 3 mg/kg); (c) axitinib (every 4 d × 9, orally [PO], 25 mg/kg); (d) topotecan (every 4 d × 9, IP, 3 mg/kg) plus axitinib (every 4 d × 9, PO, 25 mg/kg) (axitinib was given 1 h before topotecan administration). The whole administration was divided into three cycles with a 10-d drug-free recovery period between every two cycles.
For the S1 cell xenograft model, 1 × 107 S1 cells were injected subcutaneously into the posterior flank region of the nude mice. The mice were randomized into four groups after the tumors reached a mean diameter of 0.5 cm, and then received various treatments: (a) saline (every 4 d × 4, IP); (b) topotecan (every 4 d × 4, IP, 3 mg/kg); (c) axitinib (every 4 d × 4, PO, 25 mg/kg); (d) topotecan (every 4 d × 4, IP, 3 mg/kg) plus axitinib (every 4 d × 4, PO, 25 mg/kg) (axitinib was given 1 h before topotecan administration). Tumor volumes (V) were calculated by the following formula (38):
In the formula, A is the longer diameter and B is the diameter perpendicular to A. The mouse weight, tumor volume, feeding behavior and activity were recorded every 4 d. Mice were killed when the mean of tumor weights was over 1 g in the control group, and tumor tissue was excised from the mice and weighed. The ratio of growth inhibition (IR) was estimated according to the following formula:
SP Analysis and Sorting
We labeled the cell suspensions with Hoechst 33342 dye using the methods described by Goodell et al. (40) with modifications. Briefly, A549 cells were re-suspended at 1 × 106/mL in prewarmed DMEM with 2% fetal calf serum and 10 mmol/L HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer. Hoechst 33342 dye was added at a final concentration of 5 μg/mL in the presence or absence of FTC (10 μmol/L), and the cells were incubated at 37°C for 90 min with intermittent shaking. At the end of the incubation, the cells were washed with ice-cold phosphate-buffered saline (PBS), centrifuged down at 4°C, and re-suspended in ice-cold PBS. Propidium iodide at a final concentration of 2 μg/mL was added to the cells to gate viable cells. The cells were filtered through a 40-μm cell strainer to obtain a single-cell suspension before sorting. Analyses and sorting were done with fluorescence-activated cell sorting (FACS). The Hoechst 33342 dye was excited at 357 nm and its fluorescence was dual-wavelength analyzed (blue, 402–446 nm; red, 650–670 nm).
Tumorigenicity Experiments
Sorted SP and non-SP cells from A549 cells were subcutaneously injected into the NOD/SCID mice. Groups of mice were inoculated with SP or non-SP cells at 1 × 106, 1 × 105, 1 × 104 and 1 × 103 (six mice per group). The mice were killed 44 d after tumor cell injection.
Detection of Cell Surface Expression of ABCG2 and ABCB1 by Flow Cytometer
SP cells were collected and washed three times with an isotonic PBS buffer (supplemented with 0.5% bovine serum albumin [BSA]). For ABCG2 expression analysis, APC-conjugated anti-human Bcrp1/ABCG2 (R&D Systems, Minneapolis, MN, USA) reagent were mixed with 25 μL of Fc-blocked cells (1 × 106 cells). After incubating for 45 min at 4°C, the cells were washed twice with PBS buffer (supplemented with 0.5% BSA) and re-suspended in 400 μL PBS buffer for flow cytometric analysis. Isotype control samples were treated in an identical manner with allophycocyanin (APC)-labeled mouse immunoglobin G2b (IgG2b) antibody. For ABCB1 flow cytometric analysis, 1 × 106 cells (100 μL) were incubated at 4°C for 30 min with 10 μL of CD243-PE conjugated antibody (Beckman Coulter, Fullerton, CA, USA), cells were then washed and resuspended in PBS. Isotype control samples were treated with mouse IgG2a antibody in parallel. Tests and controls were analyzed with a flow cytometer.
Apoptosis Assay
Cells were seeded onto a six-well plate at a density of about 2.0 × 105 cells/well. After treatment with different concentrations of axitinib in the presence of 0.2 μmol/L topotecan or mitoxantrone for 48 h, both floating and attached cells were collected and washed with ice-cold PBS twice. Cells were resuspended in 100 μL of 1 × binding buffer, and the Alexa Fluoro 488 annexin V (5 μL) and propidium iodide (PI) (1 μL) were added before incubation at room temperature for 15 min. After the incubation period, we added 400 μL 1× binding buffer, mixed gently and analyzed via FACS.
Doxorubicin and Rhodamine 123 Accumulation
The effect of axitinib on the intracellular accumulation of Dox and rhodamine 123 was performed as previously described (41). Briefly, the cells were treated with axitinib of various concentration or vehicle at 37°C for 3 h. Subsequently, 10 μmol/L Dox or 5 μmol/L rhodamine 123 was added and the incubation was continued for an additional 3 h or 0.5 h, respectively. The cells were then collected, centrifuged and washed three times with cold PBS. Finally, the cells were analyzed with flow cytometric analysis (Cytomics FC500, Beckman Coulter). FTC was used as a control inhibitor of ABCG2 in S1 and S1-M1-80 cells (42).
ATPase Assay of ABCG2
The vanadate-sensitive ATPase activity of ABCG2 in cell membrane prepared from High-Five insect cells (cat. no. 453270) was measured by using the BD Gentest ATPase assay kit (both cells and kit from BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions. Briefly, the ATPase reaction was initiated by the addition of 12 mmol/L Mg-ATP into a total reaction mixture of 60 μL. After an incubation at 37°C for 10 min, the reactions were terminated by the addition of 30 μL of 10% sodium dodecyl sulfate (SDS) solution. The liberation of inorganic phosphate was detected by its absorbance at 800 nm and quantified by comparing the absorbance to a phosphate standard curve.
Western Blot Analysis
To determine whether axitinib affects the expression of ABCG2, the cells were incubated with 0.25, 0.5, 1.0 and 2.0 μmol/L axitinib for 48 h or with 1.0 μmol/L axitinib for 24, 48 and 72 h. To test whether axitinib blocks AKT or ERK1/2 phosphorylation, the cells were incubated with different concentrations of axitinib (0.25–2.0 μmol/L) for different periods of time (6–24 h). After drug treatment, cells were lysed after washing two times with ice cold PBS. Equal amounts of protein were resolved by SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Then, the protein–antibody complexes were visualized by the enhanced Phototope TM-HRP (horseradish peroxidase) detection kit (Cell Signaling) and exposed to a Kodak medical x-ray processor (Kodak, Rochester, NY, USA) (31).
Reverse Transcription– and Quantitative Real-Time–Polymerase Chain Reaction
After drug treatment, total cellular RNA was isolated by use of a Trizol Reagent RNA extraction kit according to the manufacturer’s instructions (Molecular Research Center, Cincinnati, OH, USA). The first strand of cDNA was synthesized by oligo dT primers with reverse transcriptase (Promega Corp., Madison, WI, USA). Oligonucleotide primers for ABCG2 and GAPDH were synthesized commercially (Invitrogen, Beijing, China). polymerase chain reaction (PCR) primers were 5′-TGG CTG TCA TGG CTT CAG TA-3′ (forward) and 5′-GCC ACG TGA TTC TTC CAC AA-3′ (reverse) for ABCG2 and 5′-CTT TGG TAT CGT GGA AGG A-3′ (forward) and 5′-CAC CCT GTT GCT GTA GCC-3′ (reverse) for GAPDH. Using the GeneAmp PCR system 9700 (PE Applied Biosystems, Norwalk, CT, USA), reactions were carried out at 94°C for 2 min for initial denaturation, and then at 94°C for 30 s, 58°C for 30 s and 72°C for 1 min. After 35 cycles of amplification, additional extensions were carried out at 72°C for 10 min. Products were resolved and examined by 1.5% agarose gel electrophoresis. The expected length of PCR products for ABCG2 is 235 bp and that for GAPDH is 475 bp.
Real-time PCR was performed with an ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster City, CA, USA). Reactions were carried out at least in triplicate repeats in two independent experiments. The geometric mean of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene was used as an internal control to normalize the variability in expression levels. All procedures were carried out according to the instructions. The forward primer for ABCG2 was 5′ CAC CTT ATT GGC CTC AGG AA-3′, the reverse primer was 5′ CCT GCT TGG AAG GCT CTA TG-3′. The forward primer for GAPDH was 5′ GAG TCA ACG GAT TTG GTC GT-3′, the reverse primer was 5′ GAT CTC GCT CCT GGA AGA TG-3′.
Statistical Analysis
All experiments were repeated at least thrice and the differences were determined by using the Student t test. The statistical significance was determined at P < 0.05.
All supplementary materials are available online at www.molmed.org.
RESULTS
Axitinib Particularly Reversed ABCG2-Mediated MDR In Vitro
To examine the reversal activity of axitinib in ABCB1-, ABCG2-, ABCC1-, ABCC4- and LRP-mediated MDR in cancer cells, we first examined the cytotoxic activity of axitinib in different cell lines via MTT assay. As shown in Supplementary Figure S1, more than 90% of cells survived when treated with axitinib alone up to 1.0 μmol/L in S1, S1-M1-80, KB, KBv200, SW1573, SW1573/2R120, NIH3T3 and NIH3T3/MRP4-2 cells, and up to 0.4 μmol/L in HL60 and HL60/ADR cells. Therefore, we used 1.0 or 0.4 μmol/L (in HL60 and HL60/ADR cells) axitinib to perform an MDR reversal assay in vitro. As shown in Table 1, the S1-M1-80 cells were significantly resistant to topotecan and mitoxantrone: the IC50 values were 12.790 ± 0.241 and 14.668 ± 0.636 μmol/L in S1-M1-80 cells, respectively, which were 51- and 76-fold higher than that in S1 cells (0.250 ± 0.014 and 0.193 ± 0.021 μmol/L, respectively). Thus, overexpression of ABCG2 resulted in a significant resistance to topotecan and mitoxantrone. Axitinib strongly enhanced the cytotoxicity of topotecan and mitoxantrone in S1-M1-80 cells in a dose-dependent manner, but had no such effect on drug-sensitive parent S1 cells. At the presence of 1.0 μmol/L axitinib, the IC50 values of topotecan and mitoxantrone in S1-M1-80 cells reduced from 12.79 ± 0.241 to 3.020 ± 0.242 μmol/L and from 14.668 ± 0.636 to 2.904 ± 0.482 μmol/L, respectively, representing a 4.11-and 5.05-fold drug sensitization. In addition, axitinib almost entirely reversed the resistance to mitoxantrone in ABCG2-482-G2 and ABCG2-482-T7 cells, which were transfected with ABCG2 (Table 2). However, axitinib did not alter the cytotoxicity of non-ABCG2 substrate (cisplatin) in MDR cells or their parental sensitive cells. All cell drug-resistance models with overexpression of ABCG2 showed no detectable expression of other MDR proteins (Supplementary Figure S2). These results suggest that axitinib is an effective modulator in restoring the sensitivity of resistant cells to anticancer drugs in vitro.
Table 1.
Effect of axitinib on reversing ABCG2-mediated drug resistance.a
| IC50 ± SD, μmol/L (Fold reversal) | ||||
|---|---|---|---|---|
|
|
||||
| Compounds | S1 | S1-M1-80(ABCG2) | ||
| Topetecan | 0.250 ± 0.014 | (1.00) | 12.79 ± 0.241 | (1.00) |
| +0.25 μmol/L Axitinib | 0.249 ± 0.020 | (1.01) | 6.692 ± 0.504b | (1.85) |
| +0.5 μmol/L Axitinib | 0.255 ± 0.017 | (0.98) | 4.964 ± 0.320b | (2.47) |
| +1.0 μmol/L Axitinib | 0.199 ± 0.006 | (1.26) | 3.020 ± 0.242b | (4.11) |
| +0.5 μmol/L FTC | 0.244 ± 0.013 | (1.02) | 1.195 ± 0.108b | (10.7) |
| Mitoxantrone | 0.193 ± 0.021 | (1.00) | 14.668 ± 0.636 | (1.00) |
| +0.25 μmol/L Axitinib | 0.194 ± 0.057 | (0.99) | 7.992 ± 0.675b | (1.84) |
| +0.5 μmol/L Axitinib | 0.201 ± 0.011 | (0.96) | 4.140 ± 0.301b | (3.54) |
| +1.0 μmol/L Axitinib | 0.185 ± 0.011 | (1.12) | 2.904 ± 0.482b | (5.05) |
| +0.5 μmol/L FTC | 0.188 ± 0.011 | (1.03) | 1.272 ± 0.056b | (11.5) |
| Cisplatin | 14.877 ± 1.126 | (1.00) | 14.496 ± 1.112 | (1.00) |
| +1.0 μmol/L Axitinib | 14.206 ± 1.460 | (1.05) | 14.360 ± 1.812 | (1.01) |
|
| ||||
| KB | KBv200(ABCB1) | |||
|
| ||||
| Doxorubicin | 0.024 ± 0.002 | (1.00) | 2.204 ± 0.108 | (1.00) |
| +0.25 μmol/L Axitinib | 0.024 ± 0.002 | (1.00) | 2.022 ± 0.185 | (1.08) |
| +0.5 μmol/L Axitinib | 0.025 ± 0.001 | (0.96) | 2.153 ± 0.146 | (1.02) |
| +1.0 μmol/L Axitinib | 0.015 ± 0.004c | (1.63) | 1.250 ± 0.215c | (1.76) |
| +10 μmol/L Verapamil | 0.024 ± 0.003 | (1.00) | 0.109 ± 0.008b | (20.2) |
| Cispatin | 1.552 ± 0.121 | (1.00) | 2.364 ± 0.241 | (1.00) |
| +1.0 μmol/L Axitinib | 1.514 ± 0.117 | (1.03) | 2.412 ± 0.225 | (0.98) |
|
| ||||
| HL60 | HL60/ADR(ABCC1) | |||
|
| ||||
| Doxorubicin | 0.055 ± 0.011 | (1.00) | 5.725 ± 0.203 | (1.00) |
| +0.1 μmol/L Axitinib | 0.054 ± 0.006 | (1.02) | 5.719 ± 0.591 | (1.00) |
| +0.2 μmol/L Axitinib | 0.056 ± 0.006 | (0.98) | 5.759 ± 0.659 | (0.99) |
| +0.4 μmol/L Axitinib | 0.049 ± 0.005 | (1.13) | 5.476 ± 0.384 | (1.04) |
| +40 μmol/L MK571 | 0.054 ± 0.005 | (1.02) | 1.108 ± 0.182b | (5.17) |
| Cisplatin | 1.456 ± 0.187 | (1.00) | 1.658 ± 0.118 | (1.00) |
| +0.4 μmol/L Axitinib | 1.516 ± 0.125 | (0.96) | 1.524 ± 0.175 | (1.08) |
|
| ||||
| SW1573 | SW1573/2R120(LRP) | |||
|
| ||||
| Doxorubicin | 0.108 ± 0.012 | (1.00) | 1.266 ± 0.121 | (1.00) |
| +0.25 μmol/L Axitinib | 0.096 ± 0.004 | (1.12) | 1.214 ± 0.174 | (1.04) |
| +0.5 μmol/L Axitinib | 0.098 ± 0.004 | (1.10) | 1.236 ± 0.122 | (1.02) |
| +1.0 μmol/L Axitinib | 0.094 ± 0.006 | (1.14) | 1.138 ± 0.088 | (1.11) |
| Cisplatin | 2.473 ± 0.167 | (1.00) | 9.659 ± 1.073 | (1.00) |
| +1.0 μmol/L Axitinib | 2.790 ± 0.147 | (0.89) | 9.180 ± 0.881 | (1.05) |
|
| ||||
| NIH3T3 | NIH3T3/MRP4-2(ABCC4) | |||
|
| ||||
| 6-MP | 0.298 ± 0.050 | (1.00) | 11.852 ± 1.564 | (1.00) |
| +0.25 μmol/L Axitinib | 0.285 ± 0.034 | (1.04) | 11.145 ± 1.048 | (1.06) |
| +0.5 μmol/L Axitinib | 0.318 ± 0.045 | (0.94) | 10.709 ± 1.637 | (1.11) |
| +1.0 μmol/L Axitinib | 0.265 ± 0.011 | (1.12) | 12.319 ± 1.704 | (0.96) |
| Cisplatin | 6.140 ± 0.728 | (1.00) | 5.668 ± 0.690 | (1.00) |
| +1.0 μmol/L Axitinib | 5.033 ± 0.296 | (1.21) | 5.842 ± 0.691 | (0.97) |
Cell survival was determined by MTT assays as described in Materials and Methods. Data are the mean ± standard deviation (SD) of at least three independent experiments performed in triplicate. The fold reversal of MDR (values given in parentheses) was calculated by dividing the IC50 for cells with the anticancer drugs in the absence of axitinib by that obtained in the presence of axitinib.
P < 0.01;
P < 0.05 versus the values obtained in the absence of inhibitor.
Table 2.
Effect of axitinib on reversing ABCG2-mediated MDR in transfected cell lines.a
| IC50 ± SD (μmol/L) (fold reversal) | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| Compounds | HEK293/pcDNA3.1 | ABCG2-G482-G2 | ABCG2-482-T7 | |||
| Mitoxantrones | 0.029 ± 0.0015 | (1.00) | 1.176 ± 0.241 | (1.00) | 1.195 ± 0.301 | (1.00) |
| +0.25 μmol/L Axitinib | 0.031 ± 0.0024 | (0.94) | 0.488 ± 0.085b | (2.41) | 0.338 ± 0.028b | (3.54) |
| +0.5 μmol/L Axitinib | 0.041 ± 0.0029 | (0.71) | 0.247 ± 0.076b | (4.76) | 0.206 ± 0.013b | (5.80) |
| +1.0 μmol/L Axitinib | 0.042 ± 0.0033 | (0.70) | 0.079 ± 0.007b | (14.9) | 0.076 ± 0.005b | (15.7) |
| +0.5 μmol/L FTC | 0.040 ± 0.0011 | (0.73) | 0.138 ± 0.005b | (8.52) | 0.096 ± 0.003b | (12.4) |
| Cisplatin | 4.069 ± 0.258 | (1.00) | 5.141 ± 0.197 | (1.00) | 4.324 ± 0.176b | (1.00) |
| +1.0 μmol/L Axitinib | 5.449 ± 0.269 | (0.75) | 4.512 ± 0.224 | (1.14) | 5.078 ± 0.209b | (0.85) |
Cell survival was determined by MTT assays as described in Materials and Methods. Data are the mean ± standard deviation (SD) of at least three independent experiments performed in triplicate. The fold reversal of MDR (values given in parentheses) was calculated by dividing the IC50 for cells with the anticancer drugs in the absence of axitinib by that obtained in the presence of axitinib.
P < 0.01 versus the values obtained in the absence of inhibitor.
Moreover, to investigate whether axitinib had the potential ability to reverse MDR mediated by other ABC transporters, we examined the modulator activity of axitinib on ABCB1/P-gp-, ABCC1/MRP1-, ABCC4/MRP4-, and LRP-mediated drug resistance in cancer cells. As illustrated in Table 1, axitinib displayed no modulating activity in KBv200, HL60/ADR, NIH3T3/MRP4-2 and SW1573/2R120 cell lines. Taken together, our data suggest that axitinib probably specifically reverses ABCG2-mediated MDR.
Axitinib Reversed ABCG2-Mediated MDR In Vivo
An established S1-M1-80 cell xenograft model in nude mice was used to evaluate the efficacy of axitinib to reverse the resistance to topotecan in vivo. Topotecan and axitinib had minimal inhibitory activity toward S1-M1-80 tumors when administrated alone. Surprisingly, the antitumor activity of topotecan was significantly enhanced when it was administered in combination with axitinib (Figure 1, P < 0.05 comparing topotecan to topotecan plus axitinib). The weight of tumors excised from mice were 0.704 ± 0.418, 0.651 ± 0.280, 0.555 ± 0.344 and 0.224 ± 0.097 g for saline, topotecan, axitinib and combination groups, respectively. The inhibition rate in the combination group was 68.2%. No significant body weight loss or treatment-related deaths occurred during the study, indicating that axitinib effectually enhanced the antitumor activity of topotecan without causing additional toxicity. The S1 cell xenograft model in nude mice was established to examine the effect of axitinib on the parental sensitive cells. As shown in Supplementary Figure S3, after treatment of the S1 cell xenograft model in the same way as the S1-M1-80 tumor model, compared with animals treated with saline or axitinib alone, both topotecan and the combination of axitinib with topotecan produced significant inhibition of tumor growth. S1 cells remained sensitive to topotecan and there was no significant difference in tumor size between topotecan and the combination group.
Figure 1.
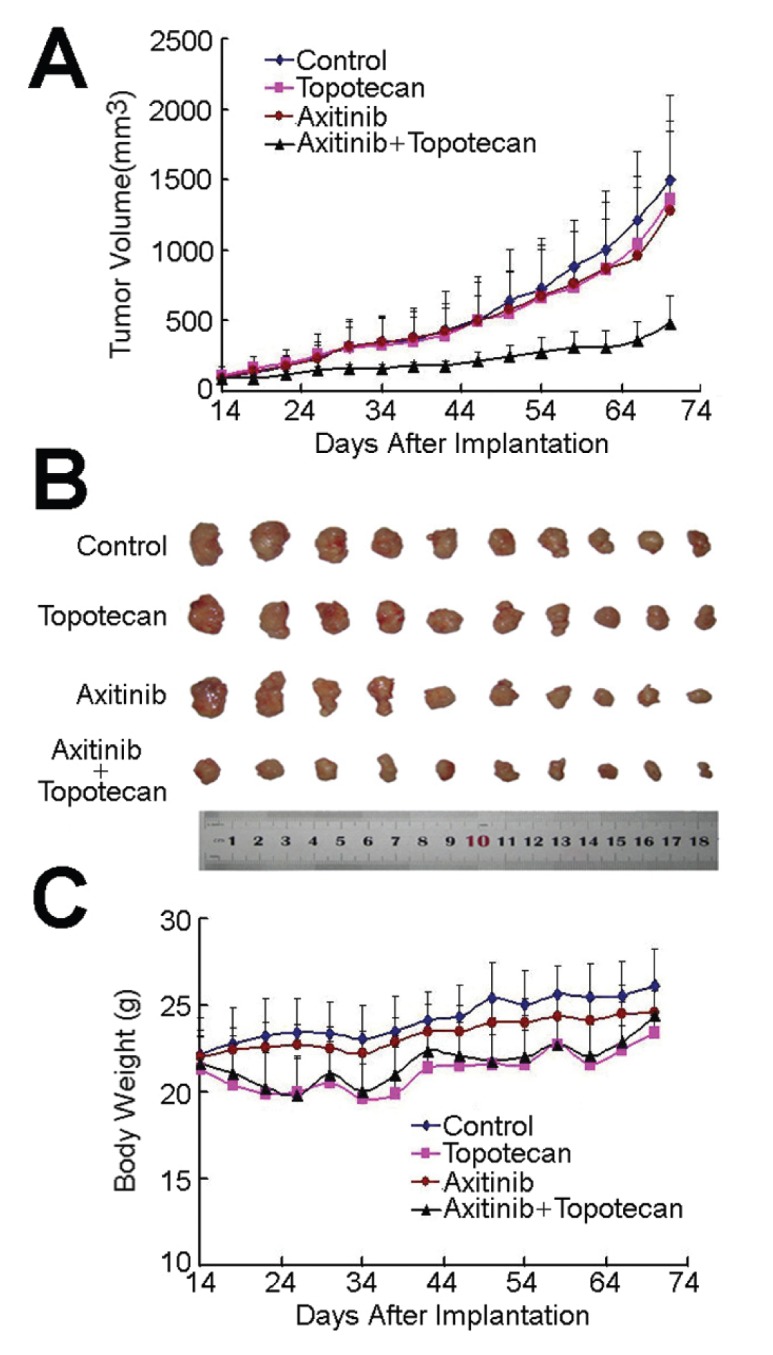
Potentiation of the antitumor effects of topotecan by axitinib in an S1-M1-80 cell xenograft model in athymic nude mice. (A) Changes in tumor volume with time. Each point represents the mean ± standard deviation (SD) of tumor volumes from 10 mice in the group. (B) Tumor size. The picture was taken on the 70th d after implantation. (C) Changes in body weight with time after tumor cell inoculation. Each point represents the mean ± SD of body weight from 10 mice in the group. The various treatments were as follows: (a) saline (every 4 d × 9, IP); (b) topotecan (every 4 d × 9, IP, 3 mg/kg); (c) axitinib (every 4 d × 9, PO, 25 mg/kg); (d) topotecan (every 4 d × 9, IP, 3 mg/kg) and axitinib (every 4 d × 9, PO, 25 mg/kg) (axitinib was given 1 h before topotecan administration). The whole administration process was divided into three cycles with a 10-d drug-free recovery period between every two cycles.
Axitinib Targeted to SP Cells and Enhanced the Efficacy of Chemotherapeutic Drugs in SP Cells
We examined the existence of SP cells in A549 cells by Hoechst 33342 staining to generate a Hoechst blue-red profile. The SP gate was defined as the diminished region in the presence of FTC, which blocked the activity of Hoechst 33342 dye transporter. A549 cells contained about 5.06% SP cells, which decreased significantly in the presence of FTC (Figure 2A).
Figure 2.

Axitinib targeted to SP cells and enhanced the efficacy of topotecan and mitoxantrone in the inhibition of proliferation and induction of apoptosis. (A) The A549 cells were stained with Hoechst 33342 as described in Materials and Methods. Gated on forward and side scatter to exclude debris, Hoechst red versus Hoechst blue (R2) was used to sort SP cells. (B) The cell surface expression of ABCG2 and ABCB1. (C, D) Induction of 50% cell death in SP and non-SP cells by topotecan, mitoxantrone and axitinib. Growth inhibition was determined by the MTT assay according to the protocol described in Materials and Methods. (E, F) Sorted SP and non-SP cells treated with toptecan, mitoxantrone and axitinib in the indicated concentrations for 48 h, respectively. Apoptosis was analyzed by flow cytometry as the percentage of cells labeled by annexin V and propidium iodide. All of these experiments were repeated at least thrice, and a representative experiment is shown. Columns, means of triplicate determinations; *P < 0.05; **P < 0.01, compared with topotecan or mitoxantrone treatment.
To test whether SP cells isolated in our study were enriched for tumorigenic cells, we examined the tumor formation rate of the SP and non-SP cells in a xenograft model. Our results showed that the SP cells gave rise to tumors with 1 × 104 cells (one of six animals), whereas at least 1 × 106 non-SP cells were needed to form a tumor. At the same injection dose (1 × 106 cells), the tumor generated by the SP cells (389 mm3) is 3.6-fold larger in volume than that of the non-SP cells (107 mm3) (Supplementary Figure S4).
We next analyzed the cell surface expression of ABCG2 and ABCB1. The SP cells showed much higher expression of ABCG2 than the non-SP cells. The A549 cells also showed a low expression of ABCG2. All of the A549 cell subsets showed no expression of ABCB1 (Figure 2B). Then we tested whether axitinib could enhance the cytotoxic effect of chemotherapeutics. As shown in Figure 2C, the SP cells exhibited higher resistance to chemotherapeutic drugs than non-SP cells. It is interesting to note that axitinib significantly enhanced the sensitivity of SP cells to topotecan and mitoxantrone in a dose-dependent manner, but had no such effect on non-SP cells (Figures 2C, D). Axitinib had no effect on the apoptosis induced by topotecan and mitoxantrone in non-SP cells, but it drastically enhanced the apoptosis of SP cells (Figures 2E, F). Quantitative analysis of the apoptotic SP cells showed that axitinib induced an increase of the percentage of apoptotic cells in a dose-dependent manner: topotecan from 5.6% ± 0.9% to 12.2% ± 1.1 % and mitoxantrone from 7.5% ± 1.1% to 14.4% ± 1.3% (Supplementary Figure S5). The results of the apoptosis assay reveal that axitinib can target to SP cells and increase the cell apoptosis induced by topotecan and mitoxantrone.
Axitinib Inhibited the Function of ABCG2-Mediated Transport
The results above indicated that axitinib could enhance the sensitivity of MDR cancer cells to certain ABCG2 substrate anticancer drugs. To ascertain the potential mechanisms, we examined the effect of axitinib on the accumulation of Dox and Rho 123 in cells overexpressing ABCG2. In the absence of axitinib, the intracellular levels of Dox and Rho 123 were very low in MDR cells, whereas axitinib significantly increased the intracellular accumulation of Dox and Rho 123 in a dose-dependent manner (Figure 3). The fluorescence index of Dox in the presence of 0.25, 0.5 and 1.0 μmol/L of axitinib was increased by 1.28-, 1.67- and 2.16-fold in S1-M1-80 cells, respectively (Figures 3A, C). As shown in Figures 3B, D, axitinib at 0.25, 0.5 and 1.0 μmol/L increased the intracellular accumulation of Rho 123 by 1.38-, 1.81-and 2.91-fold in S1-M1-80 cells, respectively. However, axitinib did not alter the intracellular accumulation of Dox and Rho 123 in the parental sensitive S1 cells. Taken together, these results suggest that axitinib significantly inhibits ABCG2- mediated transport function.
Figure 3.

Effect of axitinib on the intracellular accumulation of Dox and Rho123 in S1 and S1-M1-80 cells. The accumulation of Dox (A) and Rho 123 (B) was measured by flow cytometric analysis after cells were preincubated with or without axitinib and FTC for 3 h at 37°C and then incubated with 10 μmol/L Dox or 5 μmol/L Rho123 for another 3 h or 0.5 h at 37°C, respectively, as described in Materials and Methods. (A) Intracellular accumulation of Dox and Rho123 was not significant affected by axitinib in S1 cells. (B) The accumulation of Dox and Rho123 was increased in a dose-dependent manner in S1-M1-80 cells. (C, D) Dox and Rho123 levels were expressed as fold change in fluorescence intensity relative to control MDR cells. They are calculated by dividing the fluorescence intensity of each sample with that of MDR cells treated with Dox and Rho123. Columns, means of triplicate determinations; bars, standard deviation. **P < 0.01 versus the MDR control group. Independent experiments were performed at least three times, and a representative experiment is shown.
Drug efflux function of ABCG2 is associated with ATP hydrolysis that is stimulated in the presence of its substrates. To assess the effect of axitinib on the ATPase activity of ABCG2, we measured ABCG2-mediated ATP hydrolysis using a range of concentrations of axitinib under conditions in which the activity of other major membrane ATPases was suppressed by sodium vanadate. As shown in Figure 4, axitinib stimulated the ATPase activity of ABCG2 in a concentration-dependent manner. A maximum ABCG2 ATPase activity of 63.4 ± 2.8 nmol Pi/min per mg protein was attained in the presence of a low concentration of axitinib (~400 nmol/L) (inset of Figure 4). At a higher concentration of axitinib, a drop in the stimulated ABCG2 ATPase activity was observed. The data suggested that axitinib may be a substrate of ABCG2.
Figure 4.
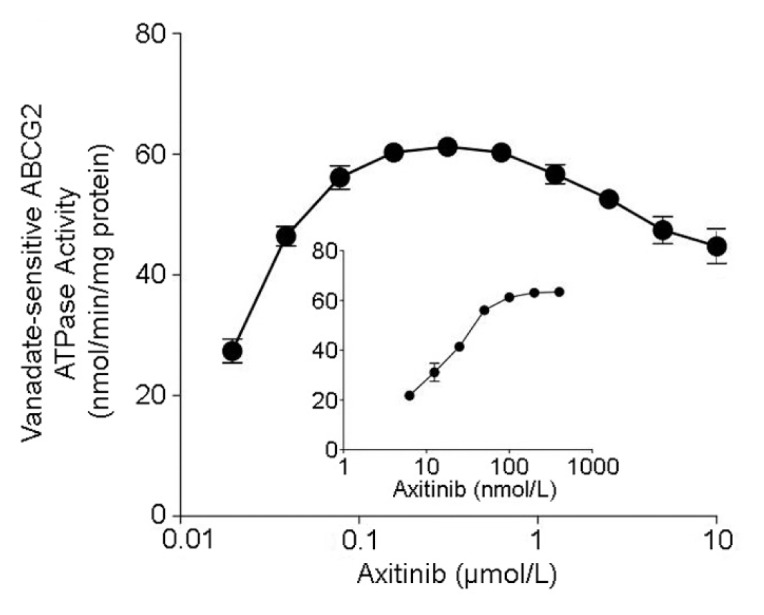
Effect of axitinib on the ATPase activity of ABCG2. The vanadate-sensitive ATPase activity of ABCG2 was determined at different concentrations of axitinib. The inset shows the effect of lower concentrations of axitinib on ABCG2 ATPase activity. ATP hydrolysis was monitored by measuring the amount of inorganic phosphate released using a colorimetric assay.
Axitinib Did Not Alter the Expression Level of ABCG2 at the mRNA or Protein Level
The reversal of ABCG2-mediated MDR can be achieved by either inhibiting ABCG2 function or lowering ABCG2 expression. Therefore, we determined the effect of axitinib on the expression of ABCG2 at the mRNA and protein levels. S1-M1-80 cells were incubated with axitinib at 1.0 μmol/L for 24, 48 and 72 h, or at 0.25, 0.5 1.0 and 2.0 μmol/L for 48 h. Our results indicated that axitinib did not significantly alter the protein or mRNA expression level of ABCG2 in S1-M1-80 cells (Figure 5). These data suggest that axitinib most likely exerts its MDR-reversal activity via direct inhibition of ABCG2-mediated efflux, rather than downregulation of its expression.
Figure 5.

Effect of axitinib on the expression of ABCG2 in MDR cells at the protein (A) and mRNA (B, C) levels. S1-M1-80 cells were treated with axitinib at 1.0 μmol/L for 12, 24 and 48 h, or at 0.25, 0.5, 1.0 and 2.0 μmol/L for 48 h, respectively. The change at mRNA level was determined by reverse transcription–PCR (B) and quantitative real-time PCR (C) as described in Materials and Methods. Independent experiments were performed at least three times and results from a representative experiment are shown.
Axitinib Did Not Block the Phosphorylation of AKT and ERK1/2 at MDR Reversal Concentrations
Accumulating studies have shown that the inhibition of AKT and ERK1/2 pathways may decrease the resistance to antineoplastic drugs in cancer cells (43,44). To determine whether the concentration of axitinib used in our experiments attenuated cell survival signaling pathways, we measured the change of total and phosphorylated forms of AKT and ERK1/2 in S1 and S1-M1-80 cells. As shown in Figure 6, axitinib did not alter the total or phosphorylated forms of AKT and ERK1/2 in S1 (Figure 6A) and S1-M1-80 cells (Figure 6B). This suggests that the MDR reversal effect of axitinib in S1-M1-80 cells is independent of the blockade of AKT and ERK1/2 signal transduction pathway.
Figure 6.

Effect of axitinib on the blockade of AKT and ERK1/2 phosphorylation. S1 and S1-M1-80 were treated with axitinib at 1.0 μmol/L for 0, 3, 6 and 12 h (lanes 1–4, respectively), or at 0, 0.25, 0.5, 1.0 and 2.0 μmol/L for 24 h (lanes 5–9, respectively), and lane 10 was the positive control (cells were treated with lapatinib at 10 μmol/L). An equal amount of protein was loaded for Western blot analysis as described in Materials and Methods. The experiments were repeated at least three times independently, and a representative experiment is shown.
DISCUSSION
The cancer stem cell hypothesis suggests that the formation and growth of tumors are driven by rare cancer stem cells, and increasing evidence also indicates that cancer stem cells play an important role in tumor initiation, progression and metastasis, as well as chemoresistance (45–47). Isolation and observation of CSCs have been achieved through selecting the SP cells, the subset of cells capable of effluxing the DNA- intercalating dye Hoechst 33342. SP cells have been identified in both human primary tumors and human cancer cell lines of several tissue origins, including thyroid, ovary, breast, glial cells and hepatic oval cells, and in all these cases the SP cells exhibit features of CSCs. Recent strong evidence has shown that cancer stemlike phenotypes are often correlated with expression and function of ABCG2, which may be responsible for their drug resistance phenotype (48–50). Elevated expression of ABCG2 has been observed in a number of cancer stem cells isolated from retinoblastoma, pancreas, liver and lung. In addition, ABCG2 and CD133, a widely identified CSC marker, are coexpressed in melanoma and pancreatic carcinoma. These data suggest that ABCG2 is a promising molecular marker for identification of CSCs in tumors. New therapeutic strategies targeting ABCG2-positive CSCs may effectively eliminate CSCs and overcome current chemotherapeutic limitations.
Axitinib is an oral small-molecule inhibitor of VEGFR-1, -2 and -3; PDGFR and c-KIT TKs. Further studies demonstrated that axitinib alone produced remarkable antitumor efficacy associated with antiangiogenesis effects across pre-clinical models regardless of the RTK expression profile in tumor cells. Clinical trials with axitinib are showing promising antitumor activity against advanced renal cell carcinoma (7), thyroid cancer (8) and non–small cell lung cancer (6). In combination studies, additive or synergistic enhancement of TKIs and response to chemotherapeutic agents alone was observed when axitinib was combined with docetaxel, carboplatin and gemcitabine. Importantly, combining axitinib with docetaxel generated marked suppression of disease progression compared with docetaxel alone in a docetaxel-resistant Lewis lung carcinoma model (51–55). More studies are underway to provide deeper insight into how axitinib and chemotherapeutic agents can be best used for maximal activity in animal models.
In the present study, we examined the effect of axitinib on enhancing chemo-therapeutic efficacy in SP cells and the ability of axitinib to reverse MDR in drug-resistant cell lines. Our data showed that axitinib enhanced the chemotherapeutic sensitivity of topotecan and mitoxantrone and increased apoptosis induced by the two drugs in SP cells. In addition, nontoxic concentrations of axitinib produced a 4.11-fold topotecan sensitization and a 5.05-fold mitoxantrone sensitization in S1-M1-80 cells, but had no such effect in the drug-sensitive parent S1 cells, indicating that the sensitization of the resistant cells by axitinib was attributable to its specific effect on ABCG2.
To determine whether the favorable effects of axitinib in vitro can be extended to an in vivo paradigm, we have examined the effect of axitinib on enhancing the antitumor activity of topotecan in S1-M1-80 cell xenograft model in mice. Consistent with the in vitro results, our data indicated that axitinib in combination with topotecan resulted in markedly enhanced antitumor activity of topotecan in this ABCG2-overexpressing tumor xenograft model and did not increase the toxic side effects (Figure 1 and Supplementary Figure S3).
To investigate the mechanisms of reversal of ABCG2-mediated MDR by axitinib, ABCG2 expression and transport activity were examined. Consistent with the overexpression and therefore higher transport function of ABCG2, S1-M1-80 cells had lower intracellular accumulation of Dox and rhodamine 123 than S1 cells (Figure 3 and Supplementary Figure S2). Axitinib treatment significantly increased the accumulation of Dox and rhodamine 123 in a dose-dependent manner but had no effect in the parent S1 cells. We also found that axitinib stimulated the ATPase activity of ABCG2 in a concentration-dependent manner (Figure 4), indicating that axitinib may directly interacts with the drug-substrate binding site on ABCG2.
As shown in Supplementary Figure S4, SP cells that are isolated by their ability to efflux Hoechst 33342 dye were enriched in tumor-initiating capability compared with non-SP cells. We also found that axitinib enhanced the cytotoxicity of topotecan and mitoxantrone in SP cells in vitro. Kataoka et al. have reported that treatment of SP cells with dofequidar reversed the drug resistance of xenografted SP cells in vivo just as it did in vitro (56). Because the SP cells isolated in our study did overexpress ABCG2 (Figure 2B), we can conclude that the in vitro effects of axitinib on SP cells can be extended to an in vivo pardigm as efficient as dofequidar. Thus it could be used in conjunction with other conventional anticancer drugs to eradicate the cancer stem cells.
Taken together, these data strongly indicated that axitinib can inhibit the transport function of ABCG2, thereby increasing the intracellular concentration of its substrate chemotherapeutic drugs. It is possible that the downregulation of ABCG2 expression may potentiate the reversal effect of axitinib on ABCG2- mediated MDR. However, axitinib treatment did not change the expression of ABCG2 at both mRNA and protein levels (Figure 5). We thus proposed that the MDR reversal effect of axitinib was due to the inhibition of efflux function of ABCG2 as revealed in the drug accumulation assay.
Receptor TKs such as VEGFR, PDGFR and c-Kit play a key role in modulating cell proliferation, differentiation and survival by activating downstream signal molecules such as signal transducers and activators, PI3K/AKT and ERK1/2) (57). Aberrant activation of receptor TKs is believed to be associated with cancer growth, angiogenesis and metastasis. Moreover, several studies have revealed that activation of the PI3K/AKT and/or ERK pathways is associated with resistance to conventional chemotherapeutic drugs (58,59). Our data revealed that total and phosphorylation forms of AKT and ERK1/2 remained unchanged in S1 and S1-M1-80 cells after treatment with different concentrations of axitinib (Figure 6), indicating that blockade of AKT and ERK1/2 activation was not involved in the reversal of ABCG2-mediated MDR by axitinib. Compared with other ABCG2 inhibitors, axitinib is more potent and specific, which is ideal for future clinical studies. Nonetheless, as with other modulators it will be essential to evaluate the effect of the axitinib on the pharmacokinetic disposition of other antineoplastic drugs.
CONCLUSION
In conclusion, axitinib can enhance the efficacy of conventional chemotherapeutic drugs in SP cells and ABCG2-overexpressing MDR cells via directly inhibiting the drug transport function of ABCG2. Our results suggest that axitinib may be used in combination with conventional ABCG2 substrate chemotherapeutic drugs to overcome multidrug resistance in the clinic. It should be discussed that axitinib would be used both as an antineoplastic drug and as an MDR reversal agent in the future.
Supplemental Data
ACKNOWLEDGMENTS
We thank S Bates (National Cancer Institute, NIH) for the ABCG2-overexpressing and ABCG2 transfectant cell lines; X-Y Liu (Cancer Hospital of Beijing, Beijing, China) for the KB and KBv200 cell lines; EA Wiemer (Department of Medical Oncology, Erasmus Medical Center) for the SW1573 and W1573/2R120 cell lines; Z-S Chen (St John’s University) for the NIH3T3 and NIH3T3/MRP4-2 cell lines. The work was supported by grants from 973 Project Foundation no. 2012CB96700 (L Fu); National Natural Sciences Foundation of China (no. 81072669 and no. 81061160); Natural Sciences Foundation of Guangdong Province (no. 2008B030301331) and NSFC/RGC Joint Research Scheme 2010/11 (no. CUHK443/10).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Choueiri TK. Axitinib, a novel anti-angiogenic drug with promising activity in various solid tumors. Curr Opin Investig Drugs. 2008;9:658–71. [PubMed] [Google Scholar]
- 2.Kelly RJ, Rixe O. Axitinib—a selective inhibitor of the vascular endothelial growth factor (VEGF) receptor. Target Oncol. 2009;4:297–305. doi: 10.1007/s11523-009-0126-9. [DOI] [PubMed] [Google Scholar]
- 3.Kelly RJ, Rixe O. Axitinib (AG-013736) Recent Results Cancer Res. 2010;184:33–44. doi: 10.1007/978-3-642-01222-8_3. [DOI] [PubMed] [Google Scholar]
- 4.Sakurai T, Kudo M. Signaling pathways governing tumor angiogenesis. Oncology. 2011;81(Suppl 1):24–9. doi: 10.1159/000333256. [DOI] [PubMed] [Google Scholar]
- 5.Aftab DT, McDonald DM. MET and VEGF: synergistic targets in castration-resistant prostate cancer. Clin Transl Oncol. 2011;13:703–9. doi: 10.1007/s12094-011-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schiller JH, et al. Efficacy and safety of axitinib in patients with advanced non-small-cell lung cancer: results from a phase II study. J Clin Oncol. 2009;27:3836–41. doi: 10.1200/JCO.2008.20.8355. [DOI] [PubMed] [Google Scholar]
- 7.Rixe O, et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007;8:975–84. doi: 10.1016/S1470-2045(07)70285-1. [DOI] [PubMed] [Google Scholar]
- 8.Cohen EE, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol. 2008;26:4708–13. doi: 10.1200/JCO.2007.15.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13:1859–76. doi: 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- 10.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–66. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 11.Doyle LA, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyake K, et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59:8–13. [PubMed] [Google Scholar]
- 13.Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58:5337–9. [PubMed] [Google Scholar]
- 14.Bailey-Dell KJ, Hassel B, Doyle LA, Ross DD. Promoter characterization and genomic organization of the human breast cancer resistance protein (ATP-binding cassette transporter G2) gene. Biochim Biophys Acta. 2001;1520:234–41. doi: 10.1016/s0167-4781(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 15.Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–58. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 16.Honjo Y, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001;61:6635–9. [PubMed] [Google Scholar]
- 17.Allen JD, Jackson SC, Schinkel AH. A mutation hot spot in the Bcrp1 (Abcg2) multidrug transporter in mouse cell lines selected for Doxorubicin resistance. Cancer Res. 2002;62:2294–9. [PubMed] [Google Scholar]
- 18.Xu Q, Thompson JE, Carroll M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood. 2005;106:4261–8. doi: 10.1182/blood-2004-11-4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007;13:470–81. doi: 10.1016/j.molmed.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haraguchi N, et al. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells. 2006;24:506–13. doi: 10.1634/stemcells.2005-0282. [DOI] [PubMed] [Google Scholar]
- 21.Zhou S, et al. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci U S A. 2002;99:12339–44. doi: 10.1073/pnas.192276999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemos C, Jansen G, Peters GJ. Drug transporters: recent advances concerning BCRP and tyrosine kinase inhibitors. Br J Cancer. 2008;98:857–62. doi: 10.1038/sj.bjc.6604213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rabindran SK, et al. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998;58:5850–8. [PubMed] [Google Scholar]
- 24.Allen JD, et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 2002;1:417–25. [PubMed] [Google Scholar]
- 25.Hegedus T, et al. Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim Biophys Acta. 2002;1587:318–25. doi: 10.1016/s0925-4439(02)00095-9. [DOI] [PubMed] [Google Scholar]
- 26.Tiwari AK, et al. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol. 2009;78:153–61. doi: 10.1016/j.bcp.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Kitazaki T, et al. Gefitinib, an EGFR tyro-sine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer. 2005;49:337–43. doi: 10.1016/j.lungcan.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura Y, et al. Gefitinib (“Iressa,” ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, reverses breast cancer resistance protein/ABCG2-mediated drug resistance. Cancer Res. 2005;65:1541–6. doi: 10.1158/0008-5472.CAN-03-2417. [DOI] [PubMed] [Google Scholar]
- 29.Leggas M, et al. Gefitinib modulates the function of multiple ATP-binding cassette transporters in vivo. Cancer Res. 2006;66:4802–7. doi: 10.1158/0008-5472.CAN-05-2915. [DOI] [PubMed] [Google Scholar]
- 30.Dai CL, et al. Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett. 2009;279:74–83. doi: 10.1016/j.canlet.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 31.Dai CL, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–14. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Litman T, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2) J. Cell Sci. 2000;113(Pt 11):2011–21. doi: 10.1242/jcs.113.11.2011. [DOI] [PubMed] [Google Scholar]
- 33.Zhang JY, et al. Anthracenedione derivative 1403P-3 induces apoptosis in KB and KBv200 cells via reactive oxygen species-independent mitochondrial pathway and death receptor pathway. Cancer Biol Ther. 2007;6:1413–21. doi: 10.4161/cbt.6.9.4543. [DOI] [PubMed] [Google Scholar]
- 34.van Zon A, et al. Efflux kinetics and intracellular distribution of daunorubicin are not affected by major vault protein/lung resistance-related protein (vault) expression. Cancer Res. 2004;64:4887–92. doi: 10.1158/0008-5472.CAN-03-3891. [DOI] [PubMed] [Google Scholar]
- 35.Lee K, Klein-Szanto AJ, Kruh GD. Analysis of the MRP4 drug resistance profile in transfected NIH3T3 cells. J Natl Cancer Inst. 2000;92:1934–40. doi: 10.1093/jnci/92.23.1934. [DOI] [PubMed] [Google Scholar]
- 36.Robey RW, et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971–8. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan YY, et al. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Mol Pharm. 2011;8:1687–97. doi: 10.1021/mp2000499. [DOI] [PubMed] [Google Scholar]
- 38.Shi Z, et al. Reversal of MDR1/P-glycoprotein-mediated multidrug resistance by vector-based RNA interference in vitro and in vivo. Cancer Biol Ther. 2006;5:39–47. doi: 10.4161/cbt.5.1.2236. [DOI] [PubMed] [Google Scholar]
- 39.Chen LM, et al. Reversal of P-gp mediated multidrug resistance in-vitro and in-vivo by FG020318. J Pharm Pharmacol. 2004;56:1061–6. doi: 10.1211/0022357043879. [DOI] [PubMed] [Google Scholar]
- 40.Goodell MA, et al. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. 1997;3:1337–45. doi: 10.1038/nm1297-1337. [DOI] [PubMed] [Google Scholar]
- 41.Fu L, et al. Characterization of tetrandrine, a potent inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Chemother Pharmacol. 2004;53:349–56. doi: 10.1007/s00280-003-0742-5. [DOI] [PubMed] [Google Scholar]
- 42.Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res. 2000;60:47–50. [PubMed] [Google Scholar]
- 43.Gagnon V, Van Themsche C, Turner S, Leblanc V, Asselin E. Akt and XIAP regulate the sensitivity of human uterine cancer cells to cisplatin, doxorubicin and taxol. Apoptosis. 2008;13:259–71. doi: 10.1007/s10495-007-0165-6. [DOI] [PubMed] [Google Scholar]
- 44.Oh SY, et al. ERK activation by thymosin-beta-4 (TB4) overexpression induces paclitaxel-resistance. Exp Cell Res. 2006;312:1651–7. doi: 10.1016/j.yexcr.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 45.Yoon SK. The biology of cancer stem cells and its clinical implication in hepatocellular carcinoma. Gut Liver. 2012;6:29–40. doi: 10.5009/gnl.2012.6.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaiopoulos AG, Kostakis ID, Koutsilieris M, Pa-pavassiliou AG. Colorectal cancer stem cells. Stem Cells. 2012;30:363–71. doi: 10.1002/stem.1031. [DOI] [PubMed] [Google Scholar]
- 47.Gong X, Schwartz PH, Linskey ME, Bota DA. Neural stem/progenitors and glioma stem-like cells have differential sensitivity to chemotherapy. Neurology. 2011;76:1126–34. doi: 10.1212/WNL.0b013e318212a89f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding XW, Wu JH, Jiang CP. ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci. 2010;86:631–7. doi: 10.1016/j.lfs.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 49.Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67:4827–33. doi: 10.1158/0008-5472.CAN-06-3557. [DOI] [PubMed] [Google Scholar]
- 50.Balbuena J, et al. ABCG2 is required to control the sonic hedgehog pathway in side population cells with stem-like properties. Cytometry A. 2011;79:672–83. doi: 10.1002/cyto.a.21103. [DOI] [PubMed] [Google Scholar]
- 51.Rugo HS, et al. Randomized, placebo-controlled, double-blind, phase II study of axitinib plus docetaxel versus docetaxel plus placebo in patients with metastatic breast cancer. J Clin Oncol. 2011;29:2459–65. doi: 10.1200/JCO.2010.31.2975. [DOI] [PubMed] [Google Scholar]
- 52.Michael M, et al. Phase Ib study of CP-868,596, a PDGFR inhibitor, combined with docetaxel with or without axitinib, a VEGFR inhibitor. Br J Cancer. 2010;103:1554–61. doi: 10.1038/sj.bjc.6605941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kindler HL, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol. 2011;12:256–62. doi: 10.1016/S1470-2045(11)70004-3. [DOI] [PubMed] [Google Scholar]
- 54.Spano JP, et al. Efficacy of gemcitabine plus axitinib compared with gemcitabine alone in patients with advanced pancreatic cancer: an open-label randomised phase II study. Lancet. 2008;371:2101–8. doi: 10.1016/S0140-6736(08)60661-3. [DOI] [PubMed] [Google Scholar]
- 55.Hu-Lowe DD, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res. 2008;14:7272–83. doi: 10.1158/1078-0432.CCR-08-0652. [DOI] [PubMed] [Google Scholar]
- 56.Katayama R, et al. Dofequidar fumarate sensitizes cancer stem-like side population cells to chemotherapeutic drugs by inhibiting ABCG2/BCRP-mediated drug export. Cancer Sci. 2009;100:2060–8. doi: 10.1111/j.1349-7006.2009.01288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kessler T, Fehrmann F, Bieker R, Berdel WE, Mesters RM. Vascular endothelial growth factor and its receptor as drug targets in hematological malignancies. Curr Drug Targets. 2007;8:257–68. doi: 10.2174/138945007779940089. [DOI] [PubMed] [Google Scholar]
- 58.West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist Updat. 2002;5:234–48. doi: 10.1016/s1368-7646(02)00120-6. [DOI] [PubMed] [Google Scholar]
- 59.Knuefermann C, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22:3205–12. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.