

Abstract
Mutations affecting mobile domains of antithrombin induce conformational instability resulting in protein polymerization that associates with a severe clinical phenotype, probably by an unknown gain of function. By homology with other conformational diseases, we speculated that these variants might infect wild-type (WT) monomers reducing the anticoagulant capacity. Infective polymerization of WT polymers and different P1 mutants (p.R425del, p.R425C and p.R425H) were evaluated by using native gels and radiolabeled WT monomers and functional assays. Human embryonic kidney cells expressing the Epstein-Barr nuclear antigen 1 (HEK-EBNA) cells expressing inducible (p.R425del) or two novel constitutive (p.F271S and p.M370T) conformational variants were used to evaluate intracellular and secreted antithrombin under mild stress (pH 6.5 and 39°C for 5 h). We demonstrated the conformational sensitivity of antithrombin London (p.R425del) to form polymers under mild heating. Under these conditions purified antithrombin London recruited WT monomers into growing polymers, reducing the anticoagulant activity. This process was also observed in the plasma of patients with p.R425del, p.R425C and p.R425H mutations. Under moderate stress, coexpression of WT and conformational variants in HEK-EBNA cells increased the intracellular retention of antithrombin and the formation of disulfide-linked polymers, which correlated with impaired secretion and reduction of anticoagulant activity in the medium. Therefore, mutations inducing conformational instability in antithrombin allow its polymerization with the subsequent loss of function, which under stress could sequestrate WT monomers, resulting in a new prothrombotic gain of function, particularly relevant for intracellular antithrombin. The in vitro results suggest a temporal and severe plasma antithrombin deficiency that may contribute to the development of the thrombotic event and to the clinical severity of these mutations.
INTRODUCTION
The broad ranges of procoagulant serine proteases that are inhibited by anti-thrombin, together with its strong and efficient mechanism of inhibition, explain the embryonic lethality observed in knock-out mice and the high risk of thrombosis associated with the antithrombin heterozygous deficiency (1,2). Nevertheless, the risk and clinical severity of thrombosis reported in patients with antithrombin deficiency is heterogeneous. Thus, type II deficiencies, characterized by a heterogeneous population of mutant and wild-type (WT) antithrombin in plasma, usually have a milder thrombotic phenotype requiring the coexistence of additional risk factors, mainly when the heparin binding site is affected (3). Unfortunately, there are some type II antithrombin deficiencies with a very severe thrombotic phenotype. Interestingly, most individuals with this phenotype carry conformational mutations (4–6). These mutations have also been identified in other members of the serpin superfamily and affect predominantly mobile domains of the molecule (7). As a consequence of the high energy required for keeping the native metastable conformation of serpins, which is essential for the inhibitory activity of these molecules (7), these mutations cause aberrant conformational transitions, mainly resulting in a unique hyperstable ordered protein–protein linkage (polymerization) (8,9). These ordered polymers that are retained within the cell and result in protein overload, plasma deficiency and ultimately cell death are associated with a variety of diseases, called serpinopathies (10).
The aberrant β-strand linkages that underlie the serpinopathies have been used as a paradigm for the wider category of “conformational diseases” (11), which include amyloid, Alzheimer’s and Parkinson diseases. The mechanism of in vivo serpin polymerization is still under discussion (12), although recent data obtained with the use of antitrypsin as a model suggest that domain swapping is the molecular mechanism of polymerization (13). This mechanism is also involved in the aggregation of prion proteins that lead to spongiform encephalopathies, other conformational diseases (14). Interestingly, prion proteins are infectious molecules composed of the abnormal disease-causing isoform prion protein (PrP)Sc, which induces conformational conversion of the host-encoded normal cellular prion protein PrPC to PrPSc (15).
These data and the demonstration that the initiating step in serpin polymerization induced by denaturing conditions occurs when two molecules with coincidentally perturbed conformations link to form an initial dimer with two active interfaces (a donor and an acceptor), which acts as an infective seed (16), encouraged us to propose that conformational mutants that form unstable monomers of antithrombin could behave as an infective seed that recruits WT monomers into the growing polymers under mild stress. This new potential gain of function could contribute to explain the thus far unknown mechanism associated with the severity of conformational mutations of antithrombin (6). In this investigation, we evaluated this hypothesis by studying the infectivity of inducible or constitutive polymers caused by conformational mutations identified in patients with antithrombin deficiency.
MATERIALS AND METHODS
Patient and Family Studies
We studied six different antithrombin mutants detected in unrelated patients with venous or arterial thrombosis associated with antithrombin deficiency (Supplementary Table S1) from a selected investigation of self-referred families with thrombophilia. Patients and relatives were fully informed of the aim of this study, which was performed according to the Declaration of Helsinki, as amended in Edinburgh in 2000, and the study participants gave written informed consent for genetic analysis.
Functional and Genetic Analyses
Anti-FXa activity, antigen levels and heparin affinity of antithrombin from plasma of patient and family members, and polymerase chain reaction amplification and sequencing of the SERPINC1 gene were performed essentially as reported (17). Thrombophilic tests included quantification of free protein S and protein C activity, detection of an-tiphospholipid antibodies and genotyping of FV Leiden and prothrombin G20210A polymorphisms.
Additional anti-FXa activity assays were performed under particular conditions by heating plasma of four patients with P1 mutations and two controls at 37°C or 42°C for 3 d. The activity was also measured in plasma of six controls supplemented or not with 12.5 μmol/L WT antithrombin polymers generated by severe heating (60°C for 10 min) and incubated for up to 9 d at 42°C.
Protein Purification and Electrophoretic and Functional Evaluation of Antithrombin London (p.R425del)
Antithrombin from plasma of healthy subjects and patients carrying the p.R425del mutation (c.1272_1274delCCG) or antithrombin London (from now on also referred to as Δ393) was purified by heparin affinity chromatography on 1 mL HiTrap Heparin columns (GE Healthcare, Barcelona, Spain), using an ÄKTA Purifier (GE Healthcare) in 100 mmol/L Tris-HCl and 10 mmol/L citric acid, in a gradient from 0.15 to 2 mol/L NaCl. Fractions with antithrombin were applied to a HiTrap Q column (GE Healthcare). Finally, proteins were eluted in three different peaks and desalted over 5 mL HiTrap Desalting columns (GE Healthcare) and stored at −70°C.
Purity and separation of proteins were checked by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), performed in 10% (w/v) poly-acrylamide and nondenaturing PAGE in the presence and absence of 6 mol/L urea, essentially performed as indicated elsewhere (17,18). SDS-PAGE under nonreducing conditions was run to detect disulfide-linked dimers (19). After separation, proteins were transblotted onto a polyvinylidene difluoride membrane. Antithrombin was immunostained with rabbit anti-human antithrombin polyclonal antibody (Sigma-Aldrich, Madrid, Spain), followed by donkey anti-rabbit IgG–horseradish peroxidase conjugate (GE Healthcare), with detection via an ECL kit (Amersham Biosciences, Piscataway, NJ, USA).
Purified antithrombin at a final concentration of 100 nmol/L was incubated with 10 nmol/L thrombin, and activity was quantified by measuring its inhibitory activity by using S-2238, as a chromogenic substrate (Chromogenix, IZASA, Madrid, Spain). Samples were previously incubated with or without unfractionated heparin (ROVI, Madrid, Spain) (1 nmol/L or 1 μmol/L) at room temperature for 30 min. Heparin effective concentration was calculated by stoichiometric titration against a solution of known antithrombin concentration. The absorbance of the reaction was measured at a wavelength of 405 nm by using a plate reader in a continuous method (Synergy HT, Biotek, Bedfordshire, UK) for up to 30 min. Additionally, formation of a thrombin–antithrombin complex was evaluated by incubation of 129 nmol/L antithrombin with 10 nmol/L thrombin and incubated for 15 min at 37°C. The reaction was carried out with and without previous incubation of antithrombin with 1 nmol/L heparin for 30 min. These samples were analyzed by SDS-PAGE as indicated previously.
Matrix-Assisted Laser Desorption Ionization/Time of Flight–Mass Spectrometry Analysis
A solution of 3,5-dimethoxy-4- hydroxycinnamic acid (10 g/L) in acetonitrile/water/trifluoroacetic acid (50:50:0.1 by vol.) was chosen for protein analysis. Experiments were carried out on a Voyager-DE™ STR Biospectrometry workstation (Applied Biosystems, Madrid, Spain), equipped with an N2 laser (337 nm). Recorded data were processed with Data Explorer™ software (Applied Biosystems).
Antithrombin samples were also digested in 100 mmol/L NH4HCO3 (pH 7.8) containing trypsin (ratio enzyme/substrate 1:50) at 37°C for 16 h. Peptide mixtures from in situ digestion of proteins were desalted in a GELoader tip packed with 0.5 μL POROS-10 R2 (PerSeptive Biosystem, East Lyme, CT, USA) slurry.
Calorimetry Measurements
The heat capacity Cp of samples was recorded over a temperature range of 10°C–130°C by using differential scanning calorimetry (VP-DSC, MicroCal, LLC, Northampton, MA, USA), which consisted of two fixed cells, a reference cell and a sample cell. Prior to all the measurements, the buffer and protein solutions were degassed. The volume of the calorimetric cells was 0.5 mL and the protein concentration used was 0.2 mg/mL. The measurements were carried out with a microcalorimeter at a scan rate of 60°C/h with WT and antithrombin London, purified from plasma. Data were analyzed by using the ORIGIN DSC software that was provided by MicroCal Inc.
Circular Dichroism
Changes of protein secondary structure with temperature in WT and antithrombin London were also measured by monitoring a circular dichroism signal at 222 nm with a spectrapolarimeter (Applied Photophysic, Surrey, UK) with a temperature control and N2 stream system. One scan was completed within 1.21 min. Samples (0.1 mg/mL) were assessed in a 20 mmol/L sodium phosphate buffer, pH 7.4, containing 0.1 mol/L NaCl, 0.1 mmol/L EDTA and 0.1% polyethylene glycol 8000. The temperature within the cuvette was maintained by a computer-controlled water bath connected to a peltier and monitored by a sensor directly located in the cuvette. Data were fitted to a two-state protein-unfolding model as previously described for other serpins (20).
Infectivity of P1 Mutants and Polymers of WT Antithrombin
Monomers of WT antithrombin were radiolabeled with I−125, following the procedure previously described (17). The native conformation of the radiolabeled protein was checked, employing native PAGE analysis in the presence and absence of 6 mol/L urea. The latent component was less than 5% of total protein and no polymers were detected (data not shown). Purified WT monomers, heat- induced polymers, and antithrombin Δ393 (2.5 μmol/L) were incubated with equimolecular amounts of I−125-radiolabeled antithrombin at 42°C for 3 d. Similarly, citrate plasma of patients with P1 mutations, and control plasma supplemented or not with 12.5 μmol/L polymers of WT antithrombin generated by heating were incubated with 2.5 μmol/L of I−125-radiolabeled antithrombin at 42°C for up to 3 d. Samples were run in native PAGE, fixed and dried, and radioactivity was detected with x-ray films, essentially as described (17).
Recombinant Expression of WT and Antithrombin Mutants
Analysis of intracellular and secreted antithrombin under moderate stress conditions
Recombinants of antithrombin were constructed on the β-glycoform p.S169A (from now on referred to as S137A) antithrombin background to reduce glycosylation heterogeneity and to facilitate purification. Site-directed mutagenesis of the pCEP4-S137A/antithrombin plasmid was performed with the Stratagene QuikChange Site-Directed Mutagenesis kit and the appropriate primers for the following mutations: Δ393, p.F271S (from now on referred to as F239S) and p.M370T (from now on referred to as M338T). Human embryonic kidney cells expressing the Epstein-Barr nuclear antigen 1 (HEK-EBNA) were grown in DMEM with GlutaMAX-I medium (Invitrogen, Barcelona, Spain) supplemented with 5% fetal bovine serum (Sigma-Aldrich) to 60% confluence at 37°C and 5% CO2 in a humidified incubator. Transfection was performed by addition of pCEP4-S137A WT antithrombin and plasmids encoding for the three antithrombin mutants (200 μg/mL) independently or cotransfected (adding the same final concentration of DNA) and preincubated for 30 min in serum-free OptiMEM culture medium with Lipofectamine LTX reagent (Invitrogen) according to the manufacturer’s protocol. Twenty-four hours after transfection the cells were washed with phosphate-buffered saline (PBS) and exchanged into CD-CHO medium (Invitrogen) supplemented with 4 mmol/L l-glutamine and 0.25 mg/mL Geneticin (Invitrogen). Cells were grown at different conditions (at 37°C and 39°C, and at pH 6.5, 7.5 and 8.5) for 5 h. After that, culture medium was collected and cells were harvested for analysis. Transfected cells were then lysed with 500 μL of lysis buffer (10 mmol/L Tris-HCl, 0.5 mmol/L dithiothreitol, 0.035% SDS, 1 mmol/L EGTA [ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid], 50 mmol/L sodium fluoride, 50 μmol/L sodium orthovanadate, 5 mmol/L benza-midine and 20 mmol/L phenylmethyl-sulphonyl fluoride) and stored at −70°C. Bradford assays (Bio Rad, Madrid, Spain) were carried out to determine the protein concentration. Intracellular antithrombin was evaluated by Western blotting, essentially as indicated above, running 40 μg of cell lysates in 10% SDS-PAGE under reducing and nonreducing conditions. Moreover, we also performed immunofluorescence to study intracellular antithrombin, essentially as previously reported (21). Briefly, HEK-EBNA cells were fixed in 4% (w/v) paraformalde-hyde diluted in PBS buffer, pH 7.4, for 20 min at room temperature and then washed with PBS and permeabilized with 0.1% saponin, 0.2% gelatin and 0.02% azide (3 × 5 min). All subsequent incubations and washes contained 0.1% saponin, 0.2% gelatin and 0.02% azide in PBS buffer. Anti-antithrombin antibody was used at 1:500 and incubated for 1 h at room temperature. We carried out indirect immunofluorescence using the appropriate fluorescein-conjugated goat anti-rabbit antibody. Fluorescence was analyzed on a Leica 6000B microscope with its associated software (Leica Microsystems, Barcelona, Spain).
Secreted antithrombin was evaluated by Western blotting, and by measuring the anti-FXa activity, as previously described.
All supplementary materials are available online at www.molmed.org.
RESULTS
Purification and Functional Effect of Antithrombin London (Δ393): Loss of Anticoagulant and Procoagulant Gain of Function
Genetic analysis revealed heterozygous deletion of the P1 residue (R393) responsible for the variant antithrombin London in two unrelated Spanish families with a type II antithrombin deficiency without the heparin-binding defect.
Purification of antithrombin London from plasma of carriers of Δ393 mutation by heparin affinity chromatography was facilitated by the increased heparin affinity associated with the deletion of P1 (22). Thus, the variant eluted at a higher NaCl concentration (1.8 mol/L) than α (1 mol/L) and β (1.5 mol/L) WT iso-forms. Moreover, the deletion produced a variant with faster electrophoretic mobility in native gels, consistent with deletion of the positively charged R393, which helped in the identification of the mutant molecule (Supplementary Figure S1). Fractions containing the high heparin-affinity variant antithrombin peak were pooled, concentrated and dialyzed into equilibration buffer (50 mmol/L Tris-HCl, pH 7.4). SDS-PAGE of the purified protein revealed more than 95% of purity (Supplementary Figure S1). Proteomic analysis by matrix-assisted laser desorption ionization/time of flight–mass spectrometry confirmed the deletion of P1 in the purified variant (data not shown).
Functional chromogenic assays performed with purified antithrombin London confirmed that this variant had no inhibitory activity, because it did not inhibit thrombin in the presence or absence of heparin. Moreover, equimolecular amounts (100 nmol/L) of antithrombin London significantly impaired the inhibition of thrombin by WT molecules only under subsaturating amounts of heparin (1 nmol/L) (Supplementary Figure S2). This effect was more severe for the αWT isoform (19-fold reduction of its activity) than for the βWT isoform (4-fold reduction), and it was not observed with saturating heparin (1 μmol/L) (Supplementary Figure S2). Consistent with this result, the formation of thrombin–antithrombin complexes was reduced in the presence of antithrombin London (Supplementary Figure S2).
Conformational Instability of Antithrombin London
Antithrombin London (Δ393) was predominantly secreted as monomer to the plasma of patients carrying this mutation (Figure 1A). Recombinant expression of antithrombin London showed similar results, with secretion rates comparable to WT antithrombin (data not shown). However, our data supported a conformational instability for the variant lacking the P1 residue. Thus, incubation of plasma-purified antithrombin London at 37°C and 42°C for 3 d generated polymers, while these conditions slightly affected the conformation of the WT molecule (Figure 1A). Calorimetry assay was also performed to identify the melting temperature (Tm) of antithrombin London. However, the profile obtained was compatible with an already polymerized protein, perhaps owing to the ultrafiltration steps required for an optimal concentration of the protein (Supplementary Figures S3A–C), because the monomeric conformation was not altered after freezing and thawing cycles or any of the purification steps. Indeed, this was confirmed by gel filtration chromatography (Supplementary Figure S3C), in which no monomer was detected. To validate the conformational instability of antithrom-bin London, circular dichroism assay was performed with recombinant molecules, because we had no further variant purified from plasma of patients. As shown in Supplementary Figure S3D, antithrombin London was significantly more unstable (Tm = 43.54°C ± 1.34°C) than WT protein (Tm = 57.36°C ± 0.14°C), which explained its high propensity to polymerize.
Figure 1.

Conformationally unstable antithrombin mutants. (A) 6 μmol/L WT and London (Δ393) antithrombin purified from plasma were incubated at 37 °C and 42°C for 3 d. Proteins were separated by nondenaturing electrophoresis and identified by western blot. Polymers were generated by heating WT antithrombin 10 min at 60°C. (B) Intracellular disulfide-linked polymers produced by constitutively conformational unstable recombinant antithrombin mutants. Intracellular lysate from cells transfected with WT plasmid was also run as a control. Disulfide-linked polymers were detected by SDS under nonreducing (SDS-nr) and reducing (SDS-r) conditions and Western blot.
Identification and Conformational Effect of Other Antithrombin Mutants
For this study we also selected four additional unrelated thrombophilic Spanish families with antithrombin deficiency. Supplementary Table S1 shows the functional and antigen levels of plasma antithrombin, as well as the clinical data of these families. Genetic analysis revealed missense heterozygous mutations in all cases. Two families carried mutations affecting the P1 residue (c.1274G > A and c.1273C > T; p.R425H and p.R425C, from now also referred to as R393H and R393C, respectively) associated with type II deficiency without the heparin-binding defect. We also selected two novel conformational mutations (c.1109T > C, M338T; and c.812T > C, F239S) on the basis of their consequences. M338T carriers had disulfide-linked dimers in plasma identical to those described previously by our group in patients with a P80S mutation (19). Additionally, recombinant expression of the M338T variant also revealed disulfide-linked dimers in the culture medium and increased intracellular retention associated with intracellular disulfide-linked polymers (Figure 1B). The F239S variant was almost not secreted to the plasma, and similarly, its expression in HEK-EBNA cells resulted in a very mild secretion to the culture medium. This variant also formed intracellular disulfide-linked polymers (Figure 1B). Therefore, the recombinant variants seemed to fulfill the features of the variants detected in the plasma of the patients.
Infectivity of WT Polymers and Δ393 Monomers under Moderate Stress
To detect the incorporation of WT antithrombin monomers to polymers we used a new method, which also validated the infectivity of WT polymers. To do so, we incubated at 24°C or 42°C equimolecular amounts of 125I-radiolabeled WT monomers with WT monomers or heat-induced polymers, and ran the obtained products on a native PAGE. With the detection of radioactivity in the polymers we could conclude that radiolabeled monomers were being recruited into growing polymers, identifying the particle that incorporates the WT monomer (dimer, trimer, and so on). We point out that this method is applicable to biological samples, supplementing plasma with 125I-radiolabeled WT monomers.
No radioactive polymers were detected when we incubated 125I-radiolabeled WT monomers in the absence of WT polymers, or if the incubations with WT polymers were performed at 24°C (data not shown). However, at 42°C in the presence of WT polymers, we detected incorporation of radiolabeled antithrombin in trimers and higher species but not in dimers (Figure 2A). Interestingly, when we incubated 125I-radiolabeled WT at 42°C with a monomer of antithrombin London purified from two patients, radioactivity was also detected in dimers (Figure 2B). In agreement with this result, 42°C incubation of plasma of patients with the Δ393 mutation, supplemented with traces of 125I-radiolabeled WT antithrombin, also revealed radioactivity in dimers and trimers (Figure 2C). Similar results were obtained with plasma of patients carrying other P1 mutations, but not when using plasma from healthy subjects (Figure 2C and Supplementary Figure S4).
Figure 2.

Infectivity of antithrombin polymers and P1 variants. (A) Immunostaining and autoradiography of 2.5 μmol/L 125I-radiolabeled WT antithrombin added to WT polymers. B) Autoradiography of 2.5 μmol/L 125I-radiolabeled WT antithrombin added to 2.5 μmol/L purified antithrombin London (Δ393) after 3 d incubated at 42°C. C) Polymerization of 2.5 μmol/L 125I-radiolabeled WT antithrombin added to 1μL control plasma and 1μL plasma of carriers of P1 mutants heated at 42°C up to 3 d. Samples were run in native gels. Arrows indicate monomer (M), dimer (D) and trimer (T).
Functional Consequences of Antithrombin Infectivity in Plasma
This infectivity might be a prothrombotic gain of function if it affects the anticoagulant activity of WT antithrombin. Thus, the loss of anticoagulant function was exacerbated in the plasma of patients with the P1 mutation compared with that of the controls when incubated at 42°C for up to 3 d (Table 1). To verify this effect, we determined the anticoagulant activity of control plasma supplemented with 12.5 μmol/L of WT antithrombin polymers and incubated at 42°C for up to 9 d. The presence of high levels of antithrombin polymers mildly but significantly increased the loss of anticoagulant function induced by moderate heating (Figure 3).
Table 1.
Anti-FXa activity in plasma of patients carrying P1 mutations and controls after 3 d of incubations at 37°C and 42°C.a
| Anti-FXa activity, %b | |||
|---|---|---|---|
|
|
|||
| 37°C | 42°C | Activity reduction, %c | |
| Control | 100.0 ± 5.1 | 73.5 ± 3.7 | 26.5 |
| Δ393 | 47.8 ± 2.3 | 28.4 ± 2.2 | 40.6 |
| R393H | 54.8 ± 1.8 | 21.3 ± 3.5 | 61.1 |
| R393C | 50.5 ± 1.1 | 25.3 ± 4.1 | 49.9 |
Data are mean ± SD of three independent measurements.
Percentage of the activity observed in a pool of 100 healthy subjects.
Reduction of activity induced by heating at 42°C and considering the value observed at 37°C for control plasma as 100%.
Figure 3.
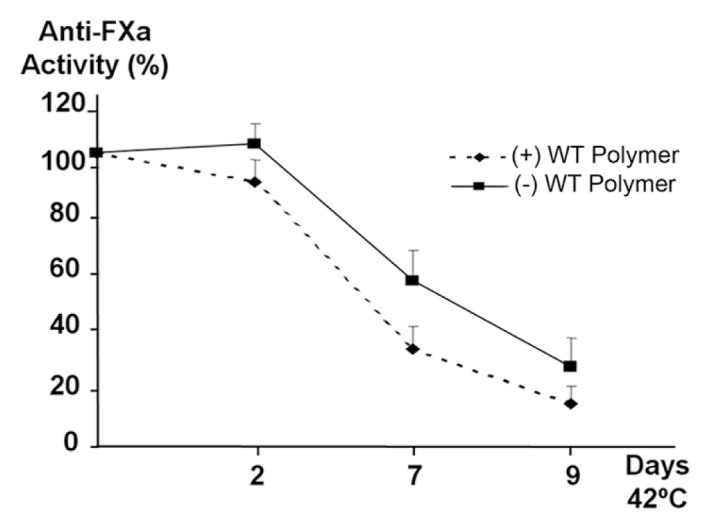
Anti-FXa activity of control plasma incubated with antithrombin polymers. Control plasma was incubated at 42°C up to 9 d in the presence and absence of 12.5 μmol/L WT antithrombin polymers. The graphic shows the average from 3 independent experiments, represented as % of the basal activity ± standard deviation.
Conformational Mutations and Secretion of WT Antithrombin under Mild Stress Conditions
High concentrations of WT antithrombin polymers (12.5 μmol/L) and long incubations at 42°C slightly impaired anti-coagulant activity. Obviously, these are not pathological conditions, which suggests that the infectivity of plasma WT molecules induced by antithrombin mutants might have no physiopathological relevance. However, we speculated that this infectivity process could be more relevant at an intracellular level. To test this hypothesis, we performed experiments with a cellular model of recombinant expression of antithrombin, cotransfecting WT antithrombin and different conformational mutants, evaluating both the intracellular and secreted antithrombin. Cotransfection experiments were designed to simulate the heterozygous state observed in patients. Interestingly, pathological temperatures (39°C) and pH (8.5 and 6.5) during short periods of time (5 h) reduced secretion of antithrombin in cells lines coexpressing with antithrombin London (Figure 4).
Figure 4.

Antithrombin secretion and retention under stress conditions. HEK-EBNA cells were transfected with 200 ng of WT/WT plasmid or cotransfected with 100 ng of WT plasmid and 100 ng of Δ393 plasmid (WT/London) and then treated at different pHs (A) and temperatures (B). Culture medium from cells were collected after treatment and analyzed by SDS-PAGE under reducing conditions by western blot with an antibody to antithrombin. Intracellular antithrombin was detected by immunofluorescence.
In addition, we further evaluated the consequences of cotransfection with constitutive conformational mutations: M338T and F239S. The experiment was undertaken at pH 6.5 because it provoked the most potent impairment on extracellular secretion (Figure 4). Moreover, in this experiment, the final amount of WT plasmid transfected was the same in the control as in those cells cotransfected with mutant plasmids. Similarly to that found for the London variant, cotransfection of M338T, F239S increased the intracellular retention of antithrombin (Figure 5A). Interestingly, for all mutants, the increased retention of antithrombin associated to the stress was accompanied by an augmentation of disulfide-linked polymers (Figure 5B), even for antithrombin London. Moreover, the anti-FXa activity of supernatants was significantly reduced in cells cotransfected with conformational mutants (50% ± 5%, 26.7% ± 4.1%, and 39.3% ± 2.5%, for Δ393, F239S and M338T mutations, respectively, compared with the activity observed in control cells: 100%).
Figure 5.
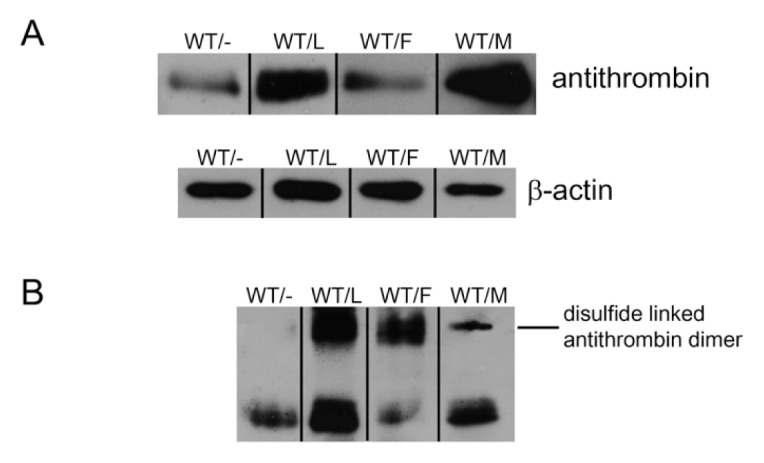
Intracellular antithrombin in HEK-EBNA cells transfected with conformational unstable mutants under moderate stress. Cells were transfected with WT plasmid alone (WT/−) or in combination with Δ393 (WT/L), F239S (WT/F) and M338T (WT/mol/L). Cell lysates after treatment for 5 h at pH 6.5 were run in SDS-PAGE under reducing (A) and nonreducing conditions (B) and antithrombin was detected by Western blotting. β-Actin was used as loading control.
DISCUSSION
Mutations associated with loss of function are involved in a high number of different diseases due to a reduced or abolished protein function. In contrast, gain of function mutations, which are much less common, confer an abnormal activity on a protein, which can also associate with the development of pathologies (23). The superfamily of serpins, crucial for many systems, displays particular features: high homology, structural flexibility, and target specificity, that supports the presence of both loss-of-function and gain-of-function mutations associated with different disorders. Similarly, most of the mutations affecting serpins belong to the loss-of-function type. Different mechanisms have been identified for these mutations, such as generation of stop codons, or significant modification of key functional residues or domains (3). Some loss-of-function mutations are specific for serpins and other conformationally sensitive proteins, because they induce or promote a transition of the protein conformation rendering inactive molecules that, in some cases, can also be toxic (10). Few gain-of-function mutations have been described in serpins. The best example is antitrypsin Pittsburg (24), for which the change affecting the P1 residue (p.M382R or M358R) greatly diminishes the anti-elastase activity of α1-antitrypsin but markedly affects antithrombin activity, increasing the risk of bleeding (24). In this study report, we present the data obtained with plasma and recombinant antithrombin London (Δ393), which is associated with loss- and gain-of-function mutations, both inducing the loss of anticoagulant capacity (22).
In addition, we describe a new prothrombotic gain of function for this mutation, the infectivity of WT molecules, which can be extrapolated to other antithrombin mutations, and potentially other serpins and molecules involved in conformational diseases.
Deletion of the P1 residue of the reactive center loop abolishes inhibitory activity but enhances heparin affinity (22). A similar effect has also been reported when R393 is changed to His, Pro, Leu, Thr (25,26) and Cys residues (27), or when R393 is citrullinated (28). All these mutations and citrullination share a common consequence: the rupture of the salt bridge established between E237 and P1. The rupture of this interaction causes a partial activation of the molecule that also has structural consequences on the heparin-binding domain, resulting in significantly higher heparin affinity (26). The consequences are particularly significant for antithrombin London, because the tension produced as a consequence of the shortening in the reactive center loop by deletion of P1 causes additional changes in the structural orientation of the residues of the heparin-binding site (22). The coexistence in plasma of similar amounts of antithrombin London and WT together with the limiting amounts of endothelial cell surface heparan sulfate molecules that contain the pentasac-charide sequence (around 1%) (29,30) explains that the inactive variant with high heparin affinity impairs (19-fold) the activation of WT antithrombin by competing with the normal molecule for the binding of heparin. Moreover, our results might also help to clarify the development of arterial thrombosis in carriers of this mutation. Indeed, we here demonstrate that the anticoagulant activity of the βiso-form is also impaired (4-fold) by the presence of the London variant (Supplementary Figure S2). The βisoform, with higher heparin affinity than the main βisoform (31), has been suggested to be the main entity responsible for the control of thrombin at the endothelial surface and to play a relevant role in arterial thrombosis (32,33). Importantly, all these results encourage the development of new diagnostic methods of antithrombin deficiency by use of subsaturating concentrations of heparin and shorter incubation times to detect this gain of function associated with a more severe clinical phenotype.
In addition, we here demonstrate that antithrombin London, like other P1 mutants, also has increased sensitivity to polymerize, because the energy required to transform the native form to polymers in WT antithrombin is significantly reduced. Thus, mild stress (42°C) allows the formation of mutant polymers (Figure 1). The partially activated conformation of these mutants might significantly facilitate the domain swapping required for polymerization (12,13). However, activated antithrombin does not tend to polymerize. Therefore, P1 mutants should become polymerization nucleating units because they are unstable and may unfold at lower temperatures than WT antithrombin (Supplementary Figure S3D). Indeed, we have recently reported that citrullination of antithrombin also caused the polymerization of the molecule (34). The identification of an increased sensitivity of P1 mutants to polymerize could also indicate the presence of a new prothrombotic gain of function. Elegant studies have suggested that ser-pin dimers (the study was performed with antithrombin and α1-antitrypsin) contain an active interface that allows progressive intermolecular β-linkages with resultant oligomeric extension and propagated polymerization, describing how the active and infective interface of oligomers is inherently toxic (16,35). This property, called “infectivity” encouraged us to study the potential deleterious effects on WT antithrombin of antithrombin London. Our results indicate that mutant polymers present in even small amounts in plasma of patients with antithrombin London and other P1 mutations are infective under moderate stress conditions, incorporating WT monomers into the growing polymers (Figure 2). The stress is necessary to induce at least a minor modification in WT molecules to provide active donor or acceptor ends (12,16). Moreover, the identification of radiolabeled dimers suggests that not only the dimer is infective, but also the mutant monomer. Thus, under mild stress (42°C in our experiments) antithrombin London monomers might polymerize, more probably by interacting with another mutant molecule, but also by forming a dimer with a WT monomer (Figure 2). Similarly, infectivity has been described for other β-structured conformational diseases such as prion diseases (36) and Alzheimer’s disease (37).
Although this infectivity impairs the anticoagulant capacity of plasma (Figure 3 and Table 1), long incubations with relatively high temperatures and concentrations of infective molecules are required to make evident this effect in secreted and folded WT antithrombin, which are nonphysiological conditions. Therefore, infectivity in plasma would never have pathological consequences, and this also explains the absence of polymers in the plasma of patients carrying these mutations. However, the circumstances are different inside the cell, with increased concentrations of antithrombin and the presence of intermediate folded protein that might facilitate polymerization and intracellular retention (38). Indeed, intracellular antithrombin seems to be more sensitive to conformational changes than plasma antithrombin (21,39,40). Accordingly, our cell models indicate that the presence of not only antithrombin London (a stress-inducible variant), but also other novel constitutive conformational mutations (M338T, F239S), significantly reduce the secretion of WT antithrombin and increase the amount of disulfide-linked polymers, leading to higher intracellular accumulation of antithrombin under moderate stress conditions (Figures 4 and 5). It has been recently demonstrated for α1-antitrypsin (13) that, during folding, the instability of the protein caused by different conformational mutations provokes the domain swapping of the C-terminal strands of the molecule, in which the C-terminal disulfide bond is included. Thus, this domain swapping between monomers allows a disulfide bridge between different monomers, rendering disulfide linked polymers. M338T and F239S seem to be conformational mutations because they associate with few amounts of disulfide-linked dimers in plasma, which are potentially the main oligomers that can be secreted. The stress-inducible mutant antithrombin London is also able to polymerize by forming disulfide intermolecular linkages intracellularly. This polymerization might be explained by the existence of intermediate folding states in the endoplasmic reticulum during protein synthesis, which can follow a more stable conformation under these conditions, remaining accumulated inside the cells or being driven to the proteasome (41–43). Antithrombin has three disulfide bridges that should be formed early during folding. Then, mutations that affect the early folding steps would tend to disrupt intramolecular disulfide bridging and promote intermolecular associations. This seems to be the case for the two mutants at residues 239 and 338, located at s4C and helix I, respectively. By attaining some homology with the polymerization of neuroserpin (42) and α1-antitrypsin (13,43), both mutations could affect the previous step to achieve native conformation, in which the s1C-s4B-s5B region cannot efficiently associate with the already folded N-terminal region. The P1 mutations might slow down the last folding step, when all disulfide bridges are already formed (13,43). Thus, the mutation does not reduce secretion as much as seen with the constitutive mutants, and some protein ends up in the plasma. However, these mutants are still sensitive and can return to the last folding step and turn polymerizable. Thus, it is tentative to speculate that the P1-E237 salt bridge interaction might play a role in accelerating folding and stabilizing native antithrombin conformation.
Therefore, in plasma samples from carriers of these mutations only those small polymer species composed of two or three monomers can be detected, because they may escape and be secreted into the plasma but they are indeed reflecting a secretion defect due to intracellular retention of the polymers (44). Whether or not these effects may be mediated by infectivity of mutant molecules, the final consequence is an acquired exacerbated deficiency of antithrombin in plasma (<50% of functional antithrombin) that might contribute to the thrombotic event. Actually, it is common for the onset of thrombosis in patients with conformational mutations to be associated with stress-inducing conditions such as infection or inflammation (4,6,19,38,41,44).
CONCLUSION
In summary, our results sustain the clinical severity of conformational antithrombin mutations and the development of vein thrombosis under certain conditions of stress, such as hyperthermia or metabolic acidosis. In addition to the loss of function, and specific gain of functions such as the higher heparin affinity of P1 mutants, unstable mutant monomers of antithrombin could initiate protein polymerization under mild stress that sequestrate WT functional monomers. We suggest that this process could be a new prothrombotic gain of function, more relevant for intracellular antithrombin, which results in a temporal but very severe plasma deficiency that may contribute to the development of the thrombotic event.
Supplemental Data
ACKNOWLEDGMENTS
The authors thank Lorena Velázquez and José Padilla for their excellent technical assistance. Authors also thank Ale-jandro Torrecillas from Centro de Apoyo a la Investigación y Desarrollo [CAID] (Murcia, Spain) for his help. I Martínez-Martínez and C Martínez are researchers from Fundación para la Formación e Investigación Sanitarias. J Navarro- Fernández holds postdoctoral contract Sara Borrell from ISCIII. A Miñano and ME de la Morena-Barrio are holders of a predoctoral research grant from ISCIII and S Águila is the holder of an FPI grant from Ministerio de Ciencia y Tecnología. This work was supported by SAF2009-08993 (MCYT & FEDER), RETICS RECAVA RD06/0014/0039 (ISCIII & FEDER), Fundación Séneca (04515/GERM/06) and Fundación Mutua Madrileña.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Abildgaard U. Antithrombin—early prophecies and present challenges. Thromb Haemost. 2007;98:97–104. [PubMed] [Google Scholar]
- 2.Quinsey NS, Greedy AL, Bottomley SP, Whisstock JC, Pike RN. Antithrombin: in control of coagulation. Int J Biochem Cell Biol. 2004;36:386–9. doi: 10.1016/s1357-2725(03)00244-9. [DOI] [PubMed] [Google Scholar]
- 3.Lane DA, et al. Antithrombin mutation database: 2nd 1997 update. For the Plasma Coagulation Inhibitors Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1997;77:197–211. [PubMed] [Google Scholar]
- 4.Corral J, Vicente V, Carrell RW. Thrombosis as a conformational disease. Haematologica. 2005;90:238–46. [PubMed] [Google Scholar]
- 5.Carrell RW, Huntington JA. How serpins change their fold for better and for worse. Biochem Soc Symp. 2003;70:163–78. doi: 10.1042/bss0700163. [DOI] [PubMed] [Google Scholar]
- 6.Carrell RW, Huntington JA, Mushunje A, Zhou A. The conformational basis of thrombosis. Thromb Haemost. 2001;86:14–22. [PubMed] [Google Scholar]
- 7.Whisstock JC, Bottomley SP. Molecular gymnastics: serpin structure, folding and misfolding. Curr Opin Struct Biol. 2006;16:761–8. doi: 10.1016/j.sbi.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Gooptu B, Lomas DA. Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu Rev Biochem. 2009;78:147–76. doi: 10.1146/annurev.biochem.78.082107.133320. [DOI] [PubMed] [Google Scholar]
- 9.Gooptu B, Lomas DA. Polymers and inflammation: disease mechanisms of the serpinopathies. J Exp Med. 2008;205:1529–34. doi: 10.1084/jem.20072080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roussel BD, et al. Unraveling the twists and turns of the serpinopathies. FEBS J. 2011;278:3859–67. doi: 10.1111/j.1742-4658.2011.08201.x. [DOI] [PubMed] [Google Scholar]
- 11.Carrell RW, Lomas DA. Conformational diseases. Lancet. 1997;350:134–8. doi: 10.1016/S0140-6736(97)02073-4. [DOI] [PubMed] [Google Scholar]
- 12.Yamasaki M, Li W, Johnson DJ, Huntington JA. Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature. 2008;455:1255–8. doi: 10.1038/nature07394. [DOI] [PubMed] [Google Scholar]
- 13.Yamasaki M, Sendall TJ, Pearce MC, Whisstock JC, Huntington JA. Molecular basis of α1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO Rep. 2011;12:1011–7. doi: 10.1038/embor.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Zhao YP. Donut-shaped fingerprint in homologous polypeptide relationships—a topological feature related to pathogenic structural changes in conformational disease. J Theor Biol. 2009;258:294–301. doi: 10.1016/j.jtbi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev. 2009;89:1105–52. doi: 10.1152/physrev.00006.2009. [DOI] [PubMed] [Google Scholar]
- 16.Zhou A, Carrell RW. Dimers initiate and propagate serine protease inhibitor polymerization. J Mol Biol. 2008;375:36–42. doi: 10.1016/j.jmb.2007.10.055. [DOI] [PubMed] [Google Scholar]
- 17.Corral J, et al. Latent and polymeric antithrombin: clearance and potential thrombotic risk. Exp Biol Med (Maywood) 2007;232:219–26. [PubMed] [Google Scholar]
- 18.Mushunje A, Evans G, Brennan SO, Carrell RW, Zhou A. Latent antithrombin and its detection, formation and turnover in the circulation. J Thromb Haemost. 2004;2:2170–7. doi: 10.1111/j.1538-7836.2004.01047.x. [DOI] [PubMed] [Google Scholar]
- 19.Corral J, et al. Mutations in the shutter region of antithrombin result in formation of disulfide-linked dimers and severe venous thrombosis. J Thromb Haemost. 2004;2:931–9. doi: 10.1111/j.1538-7836.2004.00749.x. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence DA, Olson ST, Palaniappan S, Ginsburg D. Engineering plasminogen activator inhibitor 1 mutants with increased functional stability. Biochemistry. 1994;33:3643–8. doi: 10.1021/bi00178a022. [DOI] [PubMed] [Google Scholar]
- 21.Hernandez-Espinosa D, et al. L-asparaginase-induced antithrombin type I deficiency: implications for conformational diseases. Am J Pathol. 2006;169:142–53. doi: 10.2353/ajpath.2006.051238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raja SM, et al. Deletion of P1 arginine in a novel antithrombin variant (antithrombin London) abolishes inhibitory activity but enhances heparin affinity and is associated with early onset thrombosis. J Biol Chem. 2003;278:13688–95. doi: 10.1074/jbc.M300062200. [DOI] [PubMed] [Google Scholar]
- 23.Coleman WB, Tsongalis GJ. Molecular Pathology: The Molecular Basis of Human Disease. Academic Press; Burlington: 2009. pp. 365–394. [Google Scholar]
- 24.Hua B, Fan L, Liang Y, Zhao Y, Tuddenham EG. Alpha1-antitrypsin Pittsburgh in a family with bleeding tendency. Haematologica. 2009;94:881–4. doi: 10.3324/haematol.2008.004739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson DJ, Langdown J, Li W, Luis SA, Baglin TP, Huntington JA. Crystal structure of monomeric native antithrombin reveals a novel reactive center loop conformation. J Biol Chem. 2006;281:35478–86. doi: 10.1074/jbc.M607204200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chuang YJ, Swanson R, Raja SM, Bock SC, Olson ST. The antithrombin P1 residue is important for target proteinase specificity but not for heparin activation of the serpin. Characterization of P1 antithrombin variants with altered proteinase specificity but normal heparin activation. Biochemistry. 2001;40:6670–9. doi: 10.1021/bi002933d. [DOI] [PubMed] [Google Scholar]
- 27.Lane DA, et al. Antithrombin III North-wick Park: demonstration of an inactive high MW complex with increased affinity for heparin. Br J Haematol. 1987;65:451–6. doi: 10.1111/j.1365-2141.1987.tb04149.x. [DOI] [PubMed] [Google Scholar]
- 28.Pike RN, et al. Heparin-dependent modification of the reactive center arginine of antithrombin and consequent increase in heparin binding affinity. J Biol Chem. 1997;272:19652–5. doi: 10.1074/jbc.272.32.19652. [DOI] [PubMed] [Google Scholar]
- 29.Marcum JA, et al. Cloned bovine aortic endothelial cells synthesize anticoagulantly active heparan sulfate proteoglycan. J Biol Chem. 1986;261:7507–17. [PubMed] [Google Scholar]
- 30.Olson ST, Bjork I, Bock SC. Identification of critical molecular interactions mediating heparin activation of antithrombin: implications for the design of improved heparin anticoagulants. Trends Cardiovasc Med. 2002;12:198–205. doi: 10.1016/s1050-1738(02)00160-3. [DOI] [PubMed] [Google Scholar]
- 31.McCoy AJ, Pei XY, Skinner R, Abrahams JP, Carrell RW. Structure of beta-antithrombin and the effect of glycosylation on antithrombin’s heparin affinity and activity. J Mol Biol. 2003;326:823–33. doi: 10.1016/s0022-2836(02)01382-7. [DOI] [PubMed] [Google Scholar]
- 32.Swedenborg J. The mechanisms of action of alpha- and beta-isoforms of antithrombin. Blood Coagul. Fibrinolysis. 1998;9(Suppl3):S7–10. [PubMed] [Google Scholar]
- 33.Frebelius S, Isaksson S, Swedenborg J. Thrombin inhibition by antithrombin III on the subendothelium is explained by the isoform AT beta. Arterioscler Thromb Vasc Biol. 1996;16:1292–7. doi: 10.1161/01.atv.16.10.1292. [DOI] [PubMed] [Google Scholar]
- 34.Ordonez A, et al. Effect of citrullination on the function and conformation of antithrombin. FEBS J. 2009;276:6763–72. doi: 10.1111/j.1742-4658.2009.07391.x. [DOI] [PubMed] [Google Scholar]
- 35.Carrell RW, Mushunje A, Zhou A. Serpins show structural basis for oligomer toxicity and amyloid ubiquity. FEBS Lett. 2008;582:2537–41. doi: 10.1016/j.febslet.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sweeting B, Khan MQ, Chakrabartty A, Pai EF. Structural factors underlying the species barrier and susceptibility to infection in prion disease. Biochem Cell Biol. 2010;88:195–202. doi: 10.1139/o09-172. [DOI] [PubMed] [Google Scholar]
- 37.Eisele YS, et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330:980–2. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beauchamp NJ, et al. Antithrombins Wibble and Wobble (T85M/K): archetypal conformational diseases with in vivo latent-transition, thrombosis, and heparin activation. Blood. 1998;92:2696–706. [PubMed] [Google Scholar]
- 39.Hernandez-Espinosa D, et al. In vivo effects of hyperthermia on the functional and conformational characteristics of antithrombin. J Thromb Haemost. 2007;5:963–70. doi: 10.1111/j.1538-7836.2007.02479.x. [DOI] [PubMed] [Google Scholar]
- 40.Hernandez-Espinosa D, et al. Hyperglycaemia impairs antithrombin secretion: possible contribution to the thrombotic risk of diabetes. Thromb Res. 2009;124:483–9. doi: 10.1016/j.thromres.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 41.Whisstock JC, Bottomley SP. Structural biology: serpins’ mystery solved. Nature. 2008;455:1189–90. doi: 10.1038/4551189a. [DOI] [PubMed] [Google Scholar]
- 42.Huntington JA. Serpin structure, function and dysfunction. J. Thromb. Haemost. 2011;9(Suppl 1):26–34. doi: 10.1111/j.1538-7836.2011.04360.x. [DOI] [PubMed] [Google Scholar]
- 43.Takehara S, et al. Refolding and polymerization pathways of neuroserpin. J Mol Biol. 2010;403:751–62. doi: 10.1016/j.jmb.2010.07.047. [DOI] [PubMed] [Google Scholar]
- 44.Dolmer K, Gettins PG. How the serpin α1-proteinase inhibitor folds. J Biol Chem. 2012;287:12425–32. doi: 10.1074/jbc.M111.315465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yanada M, et al. Impact of antithrombin deficiency in thrombogenesis: lipopolysaccharide and stress-induced thrombus formation in heterozygous antithrombin-deficient mice. Blood. 2002;99:2455–8. doi: 10.1182/blood.v99.7.2455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.