

Abstract
Alterations to blood-brain barrier (BBB) adhesion molecules and junctional integrity during neuroinflammation can promote central nervous system (CNS) pathology. The chemokine CCL2 is elevated during CNS inflammation and is associated with endothelial dysfunction. The effects of CCL2 on endothelial adherens junctions (AJ) have not been defined. We demonstrate that CCL2 transiently induces Src-dependent disruption of human brain microvascular endothelial AJ. β-catenin is phosphorylated and traffics from the AJ to PECAM-1, where it is sequestered at the membrane. PECAM-1 is also tyrosine phosphorylated, an event associated with recruitment of the phosphatase SHP-2 to PECAM-1, β-catenin release from PECAM-1, and reassociation of β-catenin with the AJ. Surface localization of PECAM-1 is increased in response to CCL2. This may enable the endothelium to sustain CCL2-induced alterations in AJ and facilitate recruitment of leukocytes into the CNS. Our novel findings provide a mechanism for CCL2-mediated disruption of endothelial junctions that may contribute to BBB dysfunction and increased leukocyte recruitment in neuroinflammatory diseases.
Keywords: VE-cadherin, β-catenin, PECAM-1, CD31, MCP-1, endothelial cells, neuroinflammation
The interendothelial space is a dynamic compartment that regulates the movement of molecules and cells across vascular beds. Adhesion proteins protrude into this space and form complexes with adhesion proteins on adjacent endothelial cells, creating a functional barrier. There are many adhesion complexes that contribute to endothelial junctional properties including PECAM-1/CD31 (platelet endothelial cell adhesion molecule), tight junctions, and adherens junctions (AJ). These proteins are expressed at high levels on the endothelium of the blood-brain barrier (BBB) and contribute to its selectivity and “tightness” (1–3). The BBB responds to neuroinflammation by remodeling junctional complexes. Intracellular signaling mediates this response and results in upregulation of adhesion molecules (4), dynamic reorganization of junctional complexes (5), and endothelial cell retraction (6). The resulting BBB disruption increases leukocyte transmigration, impairs central nervous system (CNS) metabolic homeostasis, and facilitates entry of pathogens into the CNS, all of which contribute to neurologic compromise (7–10).
The chemokine CCL2/monocyte chemoattractant protein-1 is associated with endothelial dysfunction (11). CCL2 is elevated in the CNS and cerebrospinal fluid of individuals with neuroinflammatory conditions characterized by BBB disruption and leukocyte CNS infiltration (11). Understanding the mechanisms underlying CCL2-mediated BBB dysregulation is critical to developing effective therapeutics. Many studies of the effects of CCL2 on brain microvascular endothelium have been performed in the mouse and examined tight junction proteins. While tight junctions are important regulators of BBB permeability, few studies have addressed the role of AJ in CCL2-mediated endothelial dysfunction. To characterize further the mechanisms mediating CCL2-induced changes to human brain microvascular endothelium, we examined the in vitro effects of CCL2 on adherens junction proteins.
We hypothesized that the altered endothelial barrier properties that occur in response to CCL2 are attributable to dysregulation of multiple adhesion molecules. Adherens junctions, in particular, play a primary role in contact inhibition (12) and in maintaining the mechanical strength and stability of endothelial cell contacts (13). In the vasculature, these structures are formed by homodimers of VE-cadherin (vascular-endothelial cadherin) (14), a transmembrane protein that is linked to the filamentous actin cytoskeleton by catenins (15). VE-cadherin directly binds β-catenin, and β-catenin is anchored to actin through association with α-catenin/α-actinin complexes (16). Several stimuli induce Src kinase-dependent phosphorylation of VE-cadherin and β-catenin, leading to loss of the AJ complex and its link to the cytoskeleton (17–19).
We now demonstrate that CCL2 induces tyrosine-phosphorylation of VE-cadherin and β-catenin, causing transient dissociation of AJ proteins. This is associated with recruitment of β-catenin to PECAM-1 and its subsequent sequestration at the membrane. PECAM-1 is a transmembrane protein that participates in leukocyte adhesion through its extracellular domain (20), intracellular signaling through its cytoplasmic tail (21), and regulates BBB permeability (1). We show that PECAM-1 is tyrosine-phosphorylated in response to CCL2. This enables recruitment of the phosphatase SHP-2 (Src homology 2 domain-containing protein phosphatase) to PECAM-1, release of β-catenin, and reassociation of β-catenin at the AJ. Thus, PECAM-1 sequestration of β-catenin is transient, allowing reformation of AJ complexes. We also demonstrate that PECAM-1 expression and localization at the cell surface is increased in response to CCL2. We propose that this increases CCL2-mediated signaling between PECAM-1 and the AJ as well as increases the haptotactic gradient for leukocytes, thereby facilitating leukocyte transendothelial migration. Our findings identify a potential mechanism for the BBB dysregulation and increased leukocyte entry into the CNS that is seen in neuroinflammatory conditions associated with elevated CCL2.
Materials and Methods
Cell Culture and Treatment
Primary human brain microvascular endothelial cells (HBMVEC; Applied Cell Biology Research Institute, Kirkland, WA) and immortalized primary human brain microvascular endothelial cells (hCMEC/D3) were used in experiments with consistent and similar results. We and others previously demonstrated that the BBB properties of confluent HBMVEC (22,23)(also refer to the company web site for the complete characterization, http://www.cell-systems.com/cert/51/140/Products/Products/CSC-Primary-Human-Cells/Human-Microvascular-Endothelial-Cells) and hCMEC/D3 are comparable (24–27). We also showed that both cell types have similar responses to CCL2 treatment, in transient opening of the barrier, and in changes in AJ. Cells were grown in M199 media (Gibco, Grand Island, NY) supplemented with 20% heat-inactivated newborn calf serum (Gibco, Grand Island, NY), 5% heat-inactivated human serum type AB (Lonza, Walkersville, MD), 1% penicillin-streptomycin (Gibco, Grand Island, NY), 0.8% L-glutamine (Gibco, Grand Island, NY), 0.1% heparin (Sigma, St. Louis, MO), 0.1% ascorbic acid (Sigma, St. Louis, MO), 0.25% endothelial cell growth supplement (Sigma, St. Louis, MO), and 0.06% bovine brain extract (Clonetics, Walkersville, MD). Experiments were conducted using cell passages four through eight and characterization of each cell type for EC specific markers, TJP and expression of BBB markers was performed. Prior to experimentation, all plates of cells were examined under phase contrast light microscopy to eliminate subconfluent and overconfluent cells or plates with dying cells. When selecting plates for use as a control or treatment condition at each time point, care was taken to match cell density.
Recombinant human CCL2/MCP-1 was obtained from R&D Systems (Minneapolis, MN) and was resuspended in 0.1% BSA in PBS. CCL2 was added directly to cell culture media to a final concentration of 200 ng/mL. We also used 50, 100, 300 and 500 ng/ml of CCL2 and obtained similar results. Src Kinase Inhibitor I (4-[4′ -Phenoxyanilino]-6,7-dimethoxyquinazoline, Calbiochem, Gibbstown, NJ) and RS102895, a CCR2 inhibitor, were resuspended in DMSO (Sigma, St. Louis, MO) and was added to media to a final concentration of 10 and 20 µM, respectively. Src Inhibitor I binds potently and reversibly to both the ATP-binding domain and peptide-binding domain of Src kinase family members. Inhibition of c-Src occurs at a 2 fold lower concentration than inhibition of other Src kinase family members, and at 7–680 fold lower concentration than other intracellular kinases (Calbiochem, Gibbstown, NJ).
Live Cell Imaging
Primary HBMVEC were grown to confluence on 0.2% gelatin (Fisher Scientific, Pittsburgh, PA)-coated 35 mm MatTek dishes (Ashland, MA). Prior to time-lapse imaging, cells were incubated in warm Dulbecco’s modified Eagle’s medium/DMEM (Gibco, Grand Island, NY) supplemented with 25 mM HEPES (USB Corporation, Cleveland, OH) to minimize pH changes and were then mounted on the stage of a semi-motorized Axio-Observer Zeiss microscope. Images were obtained at 1 minute intervals using a 40X oil lens and the Axiovision Rel 4.7 program.
Immunofluorescence
Primary HBMVEC were grown to confluence on 0.2 % gelatin (Fisher Scientific, Pittsburgh, PA)-coated 35 mm MatTek dishes (Ashland, MA). Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 1 hour and were permeabilized with 0.1% Triton X-100 (Sigma, St. Louis, MO). Blocking was performed for 1 hour with 5 mM EDTA (Gibco, Grand Island, NY), 1% fish gelatin (Sigma, St. Louis, MO), 1% essentially immunoglobulin-free BSA (Sigma, St. Louis, MO), 1% heat-inactivated human serum type AB (Lonza, Walkersville, MD), and 1% goat serum (Vector, Burlingame, CA) in deionized water. Cells were incubated in primary antibody overnight at 4°C and in secondary antibody for one hour at room temperature. Primary antibodies were used at a 1:200 dilution: p-Y-576/p-Y-577-FAK (Cell Signaling, Boston, MA); FAK, actin, PECAM-1, β-catenin (Santa Cruz, Santa Cruz, CA); vimentin and CCR2 (Abcam, Cambridge, MA). Isotype- and species-matched controls were also used at a 1:200 dilution and include normal goat IgG, normal rabbit IgG (Santa Cruz, Santa Cruz, CA), and goat anti-mouse IgG1 (Abcam, Cambridge, MA; Thermo Fisher Scientific, Rockford, IL). Nuclei were stained with DAPI (ProLong Gold antifade reagent, Invitrogen, Eugene, OR). Flurochrome-conjugated secondary antibodies were used at a 1:50 dilution and include rabbit anti-goat-FITC, sheep anti-mouse-Cy3, goat anti-mouse-FITC (Sigma, St. Louis, MO), and donkey anti-rabbit-Cy5 (Millipore, Billerica, MA). Cells were imaged at 40× magnification using a Leica AOBS laser scanning confocal microscope. Antibody specificity was verified using isotype- or species-matched control antibodies. Non-specific background immunofluorescence was minimal in all experiments.
Immunoprecipitation and Western Blot
Immortalized primary HBMVEC (hCMEC/D3) were used to minimize variability between experimental replicates in phosphorylation and immunoprecipitation studies. Monolayers of hCMEC/D3 were grown to confluence on 0.2 % gelatin (Fisher Scientific, Pittsburgh, PA)-coated 100 mm tissue culture dishes. Cells were lysed with 100 µL of RIPA buffer (Cell Signaling, Boston, MA) supplemented with Halt phosphatase inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL) and protease inhibitor cocktail (Roche, Indianapolis, IN). Total cellular protein concentration was determined by the Bio-Rad (Hercules, CA) protein assay. 140 µg of cellular lysate was used in immunoprecipitation studies. Lysate was brought to a total volume of 200 µL with lysis buffer and was pre-cleared with 20 µL protein G-conjugated agarose beads (Pierce, Rockford, IL). Lysate was incubated with 2 µg of primary antibody or an isotype-matched or species-matched control antibody overnight at 4°C with gentle rocking. Primary antibody was captured with 10 µL of protein G-conjugated agarose beads. Lysate was washed 5 times with lysis buffer. After fifth wash, buffer was aspirated from beads. The beads were resuspended in 7.5 µL deionized water and 7.5 µL 5x loading buffer (300 mM Tris-HCl, pH 6.8–8.0; 10% w/v SDS; 12.5% beta-mercaptoethanol; 50% glycerol; 0.1% bromophenol blue in deionized water). Prior to SDS-PAGE, immunoprecipitated protein was eluted from beads by boiling for 5 minutes. 15 µL of total immunoprecipitate was evaluated by Western blot on either 7.5% or 4–15% gradient Tris-HCl gels (Bio-Rad, Hercules, CA). Specificity of immunoprecipitation was confirmed for each immunoprecipitating antibody by demonstrating a lack of non-specific binding to proteins of interest by an isotype- or species-matched control antibody.
In Western blot of non-immunoprecipitated proteins, 40 µg of total cell lysate were evaluated. Protein was transferred to Protran nitrocellulose membrane (Whatman, Dassel, Germany) and blocked in 1X TBS/Casein blocker (Bio-Rad, Hercules, CA). Blots were incubated in primary antibody at 4°C overnight with gentle rocking. Following a 1 hour wash in 0.1% Tween/TBS, blots were incubated in HRP-conjugated secondary antibody at room temperature for 1 hour. Blots were again washed for 1 hour and protein expression was visualized using Western Lightning Chemiluminescence Reagent (PerkinElmer, Boston, MA). Radiographic films were developed to a non-saturated exposure. Absence of significant protein degradation was demonstrated by discrete bands on Western blot at expected molecular weights.
Antibodies used for immunoprecipitation and Western blot include: VE-cadherin, p-Y654-β catenin, β-catenin, α-actinin, PECAM-1, SHP-2, FAK, Fyn, c-Yes, normal goat IgG, normal rabbit IgG (Santa Cruz, Santa Cruz, CA); p-Y100, c-Src, p-Y-576/p-Y-577-FAK (Cell Signaling, Boston, MA); CCR2, p-Y658-VE-cadherin, p-Y654-beta catenin, goat anti-mouse IgG1 (Abcam, Cambridge, MA); and α-tubulin (Sigma, St. Louis, MO). HRP-conjugated secondary antibodies used to visualize protein on immunoblot include donkey anti-goat, goat anti-rabbit, and horse anti-mouse (Cell Signaling, Boston, MA). Precision Plus Kaleidoscope protein standard was obtained from Bio-Rad (Hercules, CA). Positive control lysates were used at a protein concentration of 40 µg and were obtained from Santa Cruz (Santa Cruz, CA): A-431 for adherens junction proteins, PECAM-1, SHP-2, and CCR2; A-431 + Calyculin A for tyrosine phosphorylated VE-cadherin; ECV304 + pervanadate for tyrosine phosphorylated PECAM-1; and NIH/3T3 + anisomycin for tyrosine phosphorylated FAK.
Subcellular Fractionation
Protein from cytoplasmic, membrane, nuclear, and cytoskeletal fractions was obtained from confluent monolayers of primary HBMVEC grown on 0.2 % gelatin (Fisher Scientific, Pittsburgh, PA)-coated 100 mm tissue culture dishes using the ProteoExtract Subcellular Proteome Extraction Kit from Calbiochem (Gibbstown, NJ) according to manufacturer’s instructions. Briefly, growth medium was removed from cells and monolayers were washed with ice-cold wash buffer. Proteins from different cellular compartments were then sequentially extracted using proprietary buffers. Extraction buffer I solubilized cytosolic proteins, buffer II released proteins from membrane and membrane-bound organelles, buffer III extracted nuclear proteins, and buffer IV solubilized proteins of the cytoskeleton. Buffers I-III were kept on ice and protein extraction was performed at 4°C. Extraction of the cytoskeletal proteins with buffer IV was performed at room temperature. Protease inhibitor cocktail was added to each buffer and nuclease was added to buffer III.
The Bio-Rad (Hercules, CA) protein assay was used to determine total protein content of each lysate obtained from subcellular fractionation. 10 µg of protein were evaluated by SDS-PAGE on 4–15% gradient Tris-HCl gels (Bio-Rad, Hercules, CA). Protein expression was evaluated in each cellular fraction by Western blot. VE-cadherin, β-catenin, and PECAM-1 (Santa Cruz, Santa Cruz, CA) expression in each cellular faction was normalized to a compartment-specific loading control: HSP 70 (Santa Cruz, Santa Cruz, CA) for the cytoplasmic fraction, cytochrome P450 reductase (Santa Cruz, Santa Cruz, CA) for the membrane fraction, histone H1 (Santa Cruz, Santa Cruz, CA) for the nuclear compartment, and α-tubulin (Sigma, St. Louis, MO) for cytoskeletal localization.
Biotinylation
Confluent monolayers of primary HBMVEC grown on 0.2% gelatin (Fisher Scientific, Pittsburgh, PA)-coated 100 mm tissue culture dishes were washed three times with ice-cold PBS (pH 8.0). Cells were incubated in 2 mM Sulfo-NHS-biotin (Pierce, Rockford, IL) for 30 minutes at 4°C followed by 3 washes with ice cold 100 mM glycine in PBS to quench the reaction. Cells were lysed with 150 µL RIPA lysis buffer (Cell Signaling, Boston, MA) supplemented with Halt phosphatase inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL) and protease inhibitor cocktail (Roche, Indianapolis, IN) and were incubated for 1 hour on ice. Lysate was centrifuged at 14,000 rpm for 20 minutes at 4°C. Total protein content of supernatant was evaluated by the Bio-Rad (Hercules, CA) protein assay. 300 µg of total protein was brought to a final volume of 500 µL with lysis buffer and was incubated with 100 µL streptavidin-conjugated agarose beads (Invitrogen, Eugene, OR) overnight at 4°C. Lysate was again centrifuged at 14,000 rpm for 5 minutes at 4°C. Non-biotinylated supernatant was collected and kept on ice while protein-bound beads were washed 5 times with ice cold lysis buffer. After fifth wash, beads were aspirated dry and resuspended with 50 µL 1x Laemmli sample buffer (Bio-Rad, Hercules, CA). Biotinylated protein was separated from beads by boiling for 5 minutes and 30 µL was loaded on a 7.5% Tris-HCl gel (Bio-Rad, Hercules, CA). An equal volume of the non-biotinylated protein fraction was also loaded on the gel as a control. Expression of PECAM-1 and VE-cadherin (Santa Cruz, Santa Cruz, CA) were analyzed by Western blot. Restricted expression of α-tubulin (Sigma, St. Louis, MO) to the non-biotinylated fraction served as an internal control.
ELISA
Media was removed from confluent cultures of primary HBMVEC after treatment with CCL2 or 0.1% BSA/PBS. Media from 5 culture dishes (15 mL total) was concentrated to 250 µL using a Centriplus-YM50 centrifugal filter device (Millipore, Bellerica, MA) according to manufacturer’s instructions. 15 mL of complete M199 was also concentrated to the same volume to control for soluble protein present in the sera component of the supplemented media. ELISA was used to determine soluble VE-cadherin and soluble PECAM-1 shed from cells in response to CCL2- or vehicle-treatment and present in media. ELISA was performed according to manufacturer’s instructions. The limit of detection using the Human VE-cadherin/CD144 ELISA (PromoKine, Heidelberg, Germany) and ZyQuik sPECAM-1 ELISA Kit (Zymed, Carlsbad, CA) are 0.16 ng/mL and 0.48 ng/mL, respectively.
Blood brain barrier (BBB) model using endothelial cells
To examine whether CCL2 induced endothelial disruption that results in changes in permeability, we used BMVEC cultured on a gelatin-coated, 3 µm pore-size tissue culture insert (Falcon, BD, Franklin lakes, NJ) as we previously described, but without the human astrocytes (28–32). Confluent cells were treated with CCL2, SrcI, a CCR2 inhibitor, or vehicle and subsequent changes were evaluated by assessing permeability of the BMVEC monolayer to albumin conjugated to Evans blue dye as previously described (28). We did not detect any significant change in permeability with any treatment. This is consistent with our previous data (33) showing that CCL2 together with an additional insult changes permeability.
Densitometry and Data Analysis
When evaluating protein phosphorylation, optical density (OD) of phosphorylated protein was compared to OD of non-phosphorylated band after normalization to α-tubulin. Immunoblots were stripped (Restore Plus Western Stripping Buffer, Pierce, Rockford, IL) of primary antibody to phosphoprotein and re-blocked prior to re-probing with antibody specific to non-phosphorylated form of protein. Loss of signal from phospho-specific antibody was confirmed prior to re-probing. In immunoprecipitation studies, OD of immunoprecipitated proteins was compared to OD of protein used for immunoprecipitation. In every co-immunoprecipitation experiment, each protein in the complex was sequentially used as immunoprecipitating protein with consistent results. To evaluate relative expression of protein in subcellular fractionation studies, OD of protein of interest was evaluated against loading control specific to each fraction. Similarly, to evaluate total cellular expression of PECAM-1, OD of protein of interest was compared to OD of α-tubulin. Densitometry was performed using Un-Scan-It gel analysis software (Silk Scientific, Orem, Utah). Densitometric analysis is reported as fold change and compares relative protein expression in CCL2-treated cells to vehicle-treated cells of same time point. To control for potential cell growth or death between time points, a control plate of cells was also used at the same time point as the experimental treatment. Thus, protein from CCL2-treated cells was compared to vehicle-treated cells at the same time point. This controlled for differences in protein expression seen between control plates at different time points. Statistics were obtained using a single factor ANOVA. Results were considered significant when p < 0.05. All data were reproducible in at least three independent experiments.
Results
CCL2 disrupts junctional stability of human brain microvascular endothelial cells in a Src-dependent manner
CCL2 is elevated in many neuroinflammatory conditions in which there is increased leukocyte recruitment and BBB disruption (34–38). The role of CCL2 in BBB disruption and leukocyte transmigration has been established both in vitro and in vivo (39). In particular, CCL2-mediated disruption of mouse endothelial tight junctions has been well described (39–41). However, few studies have evaluated the mechanisms by which CCL2-alters AJ at the BBB in human endothelium. We therefore examined the in vitro effect of CCL2 on the junctional integrity of primary human brain microvascular endothelial cells (HBMVEC) and of a human brain endothelial cell line (hCMEC/D3) (26).
We first confirmed the expression of CCR2, the receptor for CCL2 (42), on the endothelial cells (EC) used in these studies by both Western blot and immunofluorescence (Figure 1). CCR2 expression was similar in HBMVEC and hCMEC/D3. Two CCR2 isoforms are produced by alternate mRNA splicing (42): CCR2A (45 kDa) and CCR2B (42 kDa). Each has similar affinity for CCL2 (43). The antibody used in our studies does not distinguish between the CCR2 isoforms. As a result, multiple bands are seen on Western blot.
Figure 1. CCR2 is expressed by primary HBMVEC and hCMEC/D3 cells.
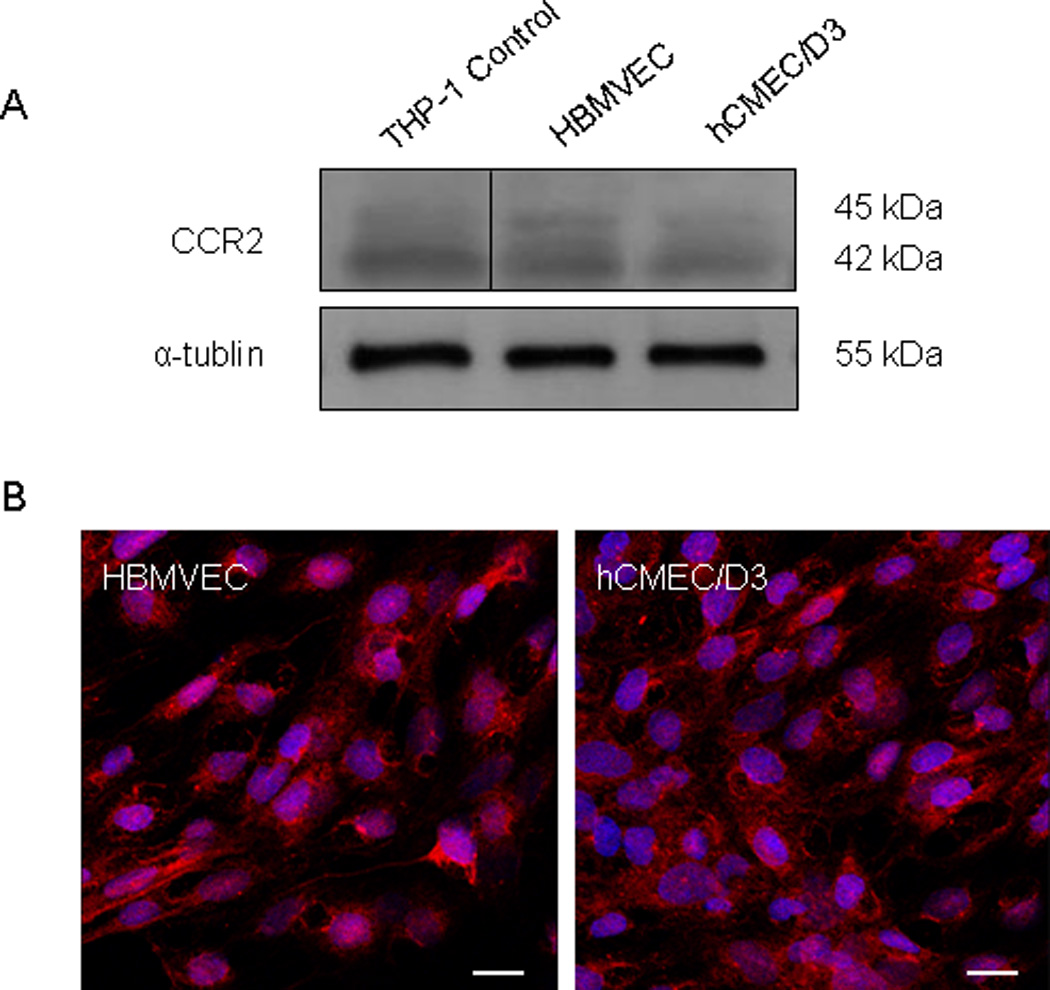
Expression of CCR2, the receptor for CCL2, was evaluated in the brain microvascular endothelial cells used in these studies. (A) 40 µg of total cellular protein from primary human brain microvascular endothelial cells (HBMVEC) and from the HBMVEC cell line hCMEC/D3 were evaluated by Western blot. An equal protein concentration from the THP-1 cell line was used as a positive control for CCR2. Two bands at 45 and 42 kDa represent splice variants of CCR2, CCR2A and CCR2B, respectively. (B) CCR2 expression was also evaluated by immunofluorescence and confocal microscopy. CCR2 (Cy3-red) is expressed in both cell types. Cell nuclei were stained with DAPI (blue). Bar = 18.75 µm.
The expression of CCR2 on cultured BMVEC and its functional interaction with CCL2 has been established (44, 45). CCL2 present in the perivascular space binds abluminal CCR2, is internalized, transcytosed, and displayed on luminal proteoglycans to create a chemokine gradient (46, 47). This enables leukocyte recruitment to sites of inflammation. When leukocytes interact with CCL2 on the vascular luminal surface, they become activated and release cytokines and chemokines, including CCL2, that facilitate their transmigration by opening junctional complexes (48). We therefore treated EC with CCL2 on the luminal surface to model CCL2 signaling during the immune response to CNS inflammation.
We treated confluent monolayers of primary HBMVEC with either 200 ng/mL CCL2 or an equal volume of 0.1% BSA in PBS, the CCL2 diluent, for 30 minutes and performed time-lapse imaging. This concentration is consistent with our previous studies in which 100–500 ng/mL CCL2 promoted leukocyte transmigration, and may represent local concentrations at sites of neuroinflammation (33). Others also found 200 ng/mL CCL2 to induce cellular chemotaxis (49, 50). Disruption of mouse BMVEC was demonstrated at 100 ng/mL CCL2 (51, 52). In our studies with human BMVEC, 200 ng/mL CCL2 was the optimal concentration at which we detected transient opening of the monolayer of BMVEC by microscopy, although we also detected similar changes in preliminary studies with 100 ng/ml of CCL2. Figure 2 demonstrates that CCL2 causes marked disruption of the EC monolayer as compared to vehicle-treated cells. Still images taken from the live-cell imaging series show gap formation between EC in response to CCL2 (Figure 2D, E and F) that are not seen when HBMVEC are treated with diluent (Figure 2A, B and C). Videos from which these still images for all of the conditions were taken are included as supplemental material (see supplemental material). As EC retract, they maintain contact through cellular processes, and then recoil such that monolayer integrity is only transiently open. This disruption of endothelial junctional integrity was observed over 30 minutes in the presence of CCL2 only. Diluent-treated cells did not retract or demonstrate visual evidence of disruption to monolayer integrity.
Figure 2. CCL2 modulates junctional stability of HBMVEC by promoting Src and CCR2-dependent cell retraction and transient loss of monolayer confluence.
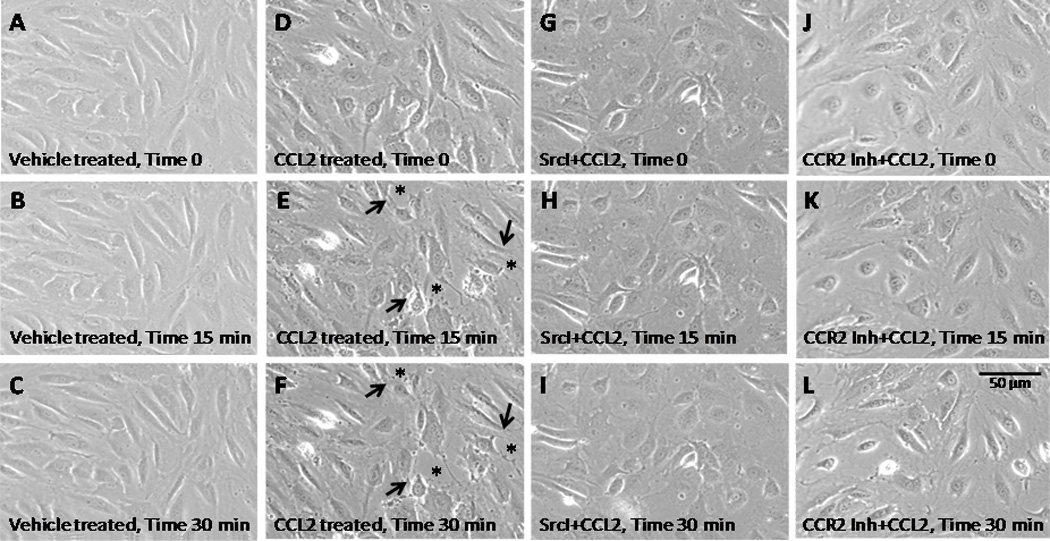
Confluent primary HBMVEC were pre-treated with 10 µM Src inhibitor, 20 µM CCR2 inhibitor (CCR2 Inh, RS102895) or DMSO (diluent) for 2 hours followed by treatment with 200 ng/mL CCL2 or diluent for 30 minutes. Time-lapse images were obtained every minute during the 30 minute CCL2 or diluent treatment. Figure 2 represents still images captured at the start of time-lapse imaging (A, D, G and J), after 15 min (B, E, H and K) and after 30 minutes (C, F, I and L). Images are representative of 3 independent experiments that yielded consistent results. CCL2 induces gaps in the EC monolayer (D, asterisks) that are not seen before CCL2 treatment (C) or in control diluent treated cells (B). This is abrogated by pre-treatment with Src inhibitor (H and I) or with CCR2 inhibitor (K and L). Cell retraction did not occur after diluents, Src or CCR2 inhibitor treatment without CCL2 treatment.
Src family kinases regulate endothelial permeability through cytoskeletal contraction (53), focal adhesion remodeling (54), and tight junction and adherens junction disassembly (55–57). Thus, we examined the role of Src in CCL2-mediated disruption of endothelial junctional integrity. Confluent monolayers of primary HBMVEC were pre-treated with 10 µM Src Inhibitor I (4-[4′ -Phenoxyanilino]-6,7-dimethoxyquinazoline), or an equal volume of DMSO (diluent), for 2 hours prior to CCL2 exposure. Pre-treatment with Src inhibitor abrogated the effects of CCL2 on monolayer integrity (Fig. 2G, H and I). These findings indicate that CCL2-mediated modulation of HBMVEC junctional integrity is a Src-dependent process.
To demonstrate that CCR2, the receptor for CCL2, is involved in the opening of the monolayer of HBMVEC, we used video time lapse as described above. Pretreatment with RS102895, a CCR2 inhibitor (20 µM), as described and calibrated (58), was used to examine whether this compound blocked the effects of CCL2. RS102895 blocked completely the effects of CCL2 on monolayer integrity (Fig. 2J, K and L). These findings indicate that CCL2-mediated modulation of HBMVEC integrity is CCR2 dependent.
CCL2 mediates Src-dependent FAK activation, which correlates with HBMVEC stress fiber and interendothelial gap formation
Focal adhesion kinase (FAK) and c-Src form a signaling complex that regulates contact between EC and the extracellular matrix (59). At adhesive interfaces called focal adhesions, integrins anchor cells to the basal lamina by binding to matrix proteins (60). During cell migration, focal adhesions are remodeled such that cells are able to move relative to the extracellular matrix (61). Association of FAK with c-Src and subsequent activation of both kinases is required for focal adhesion remodeling (54, 62). We examined the tyrosine phosphorylation of FAK in response to CCL2 at sites known to be phosphorylated by c-Src, as an additional mechanism of CCL2-mediated and Src-dependent EC motility.
Immortalized HBMVEC were used to examine protein phosphorylation to minimize variability between experimental replicates and to enable use of many experimental plates of cells at a low passage number. Monolayers of hCMEC/D3 were grown to confluence on a collagen matrix and were treated with either 200 ng/mL CCL2 or diluent. Cells were lysed and whole-cell lysate was evaluated by Western blot (Figure 3A). Antibody specific to c-Src-phosphorylated tyrosine residues 576/577 on FAK was used as a marker of FAK activation (62). Blots were first evaluated with p-Y-576/p-Y-577-FAK antibody, followed by stripping, and re-probing with antibody to total FAK. Optical densities of tyrosine-phosphorylated FAK bands were evaluated relative to un-phosphorylated FAK and are reported as fold change relative to control at the same time point (Figure 3B). CCL2-treated cells were compared to vehicle-treated cells at each time point to control internally for cell growth or death between time points. Tyrosine phosphorylation of FAK is increased in response to CCL2 relative to control cells at 15 and 30 minutes. By 45 minutes, FAK activation is the same as control (Figure 3A and 3B). This corresponds with the time-lapse imaging sequence in which cell retraction and EC gap formation is evident after 30 minutes of CCL2 exposure.
Figure 3. CCL2 promotes Src-dependent FAK activation and stress fiber formation in retracting EC.
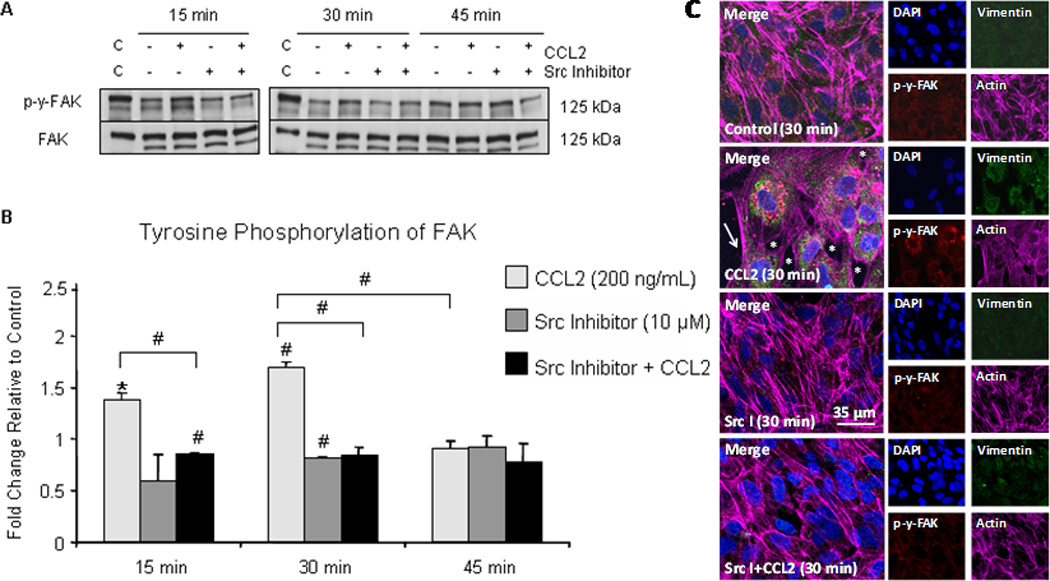
(A) hCMEC/D3 were grown to confluence on gelatin-coated tissue culture dishes and pre-treated with 10 µM Src inhibitor or DMSO for 2 hours followed by treatment with 200 ng/mL CCL2 or diluent. Cells were lysed and 40 µg of total protein was analyzed by Western blot. Blots were first evaluated with p-Y-576/p-Y-577-FAK antibody, followed by stripping of phospho-specific antibody and re-probing with antibody to total FAK. Figure 3A is a representative blot of 3 independent experiments. C = control lysate. (B) Optical density (OD) of tyrosine phosphorylated FAK was evaluated relative to OD of non-phosphorylated FAK. Densitometry is reported as fold change in tyrosine phosphorylated FAK in response to CCL2 relative to control at same time point. n=3. (#) p < 0.01. FAK activation, as evident by phosphorylation, is increased at 15 minutes and 30 minutes and is reduced with Src inhibitor pre-treatment. By 45 minutes, the level of FAK activation is equal to control. (C) Primary HBMVEC were grown to confluence on gelatin-coated MatTek dishes. Following Src inhibitor pre-treatment, cells were treated with 200 ng/mL CCL2 or diluent. Cells were fixed, permeabilized, and immunofluorescently stained with DAPI (blue)- nucleus, Cy3 (red)- p-y-FAK, FITC (green)- vimentin, and Cy5 (magenta)- actin. Cells treated with CCL2 demonstrate increased p-y-FAK staining in retracting cells relative to control cells. Interendothelial gaps in monolayer (asterisk) and EC stress fiber formation (arrow) are evident. Pre-treatment with Src inhibitor abrogates FAK phosphorylation and formation of interendothelial gaps and EC stress fibers.
To demonstrate Src-dependent FAK activation in response to CCL2, confluent hCMEC/D3 monolayers were pre-treated with 10 µM Src inhibitor or an equal volume of DMSO for 2 hours prior to chemokine treatment. CCL2 or diluent was then added to the culture medium. Cell lysates were obtained and evaluated by Western blot. Phosphorylation of FAK at known c-Src sites after 15 and 30 minutes of CCL2 exposure was abrogated by pre-treatment with Src inhibitor (Figure 3A and 3B), demonstrating further the role of c-Src in CCL2-mediated disruption of endothelial barrier integrity.
We also demonstrated FAK activation to be associated with interendothelial gap and cellular stress fiber formation in EC monolayers (Figure 3C). Primary HBMVEC were grown to confluence on collagen-coated MatTek dishes and were pre-treated for 2 hours with either 10 µM Src inhibitor or DMSO. Cells were then treated with 200 ng/mL CCL2 or diluent. After 30 minutes, cells were washed, fixed, and permeabilized. Cell nuclei (DAPI-blue), vimentin (FITC-green), α-actin (Cy3-red), and p-Y-576/p-Y-577-FAK (Cy5-magenta) were immunofluorescently labeled and visualized with confocal microscopy (Figure 3C). There is minimal phosphorylation of FAK at c-Src sites in control cells (DMSO + 0.1% BSA/PBS). In contrast, in CCL2-treated cells, FAK phosphorylation is increased in association with formation of interendothelial gaps (Figure 3C, asterisk), cellular stress fibers, and prominent perinuclear vimentin rings. Actin staining outlines the cortical actin ring and also demonstrates stress fiber formation in retracting cells (Figure 3C, arrow). Vimentin is prominently organized in perinuclear rings in response to CCL2, consistent with internal force transduction from focal adhesions through intermediate filaments (63). Pretreatment with Src inhibitor reduces FAK phosphorylation and formation of interendothelial gaps and cellular stress fibers in response to CCL2 (Figure 3C). These findings support a role for Src-dependent CCL2 signaling in focal adhesion remodeling, cell motility, and disruption of EC junctional integrity.
CCL2 mediates Src-dependent tyrosine phosphorylation of VE-cadherin and β-catenin
Adherens junctions are dynamic structures in EC that are rapidly remodeled in response to external stimuli (64). Src family kinases regulate AJ complexes (55), with tyrosine phosphorylation of VE-cadherin and β-catenin associated with AJ disassembly and increased EC permeability (17, 57). Since we found CCL2-mediated, Src-dependent disruption of EC junctions, we examined phosphorylation of VE-cadherin and β-catenin in response to CCL2.
During monolayer formation, AJ proteins are constitutively phosphorylated (65). However once contact inhibition is established and EC become confluent, tyrosine phosphorylation of adhesive complexes is reduced (65). Therefore, we grew monolayers of hCMEC/D3 to confluence and pre-treated them with 10 µM Src inhibitor or DMSO for 2 hours followed by exposure to either 200 ng/mL CCL2 or diluent. Whole-cell lysates were obtained and evaluated by Western blot (Figure 4A and 4C). Antibodies to Y658 of VE-cadherin and Y654 of β-catenin, sites of c-Src phosphorylation (66, 67), were used to evaluate phosphorylation. Blots were stripped and re-probed with antibody to non-phosphorylated VE-cadherin and β-catenin. Densitometry was performed in which phosphorylated protein was compared to its non-phosphorylated form, and these values were compared between CCL2-treated and control-treated cells. Results are reported as fold change relative to control (Figure 4B and 4D). In response to CCL2, tyrosine phosphorylation of both VE-cadherin and β-catenin at c-Src-specific sites is increased at 30 minutes relative to control and relative to the 15 and 45 minute time points (Figure 4). Pretreatment with Src inhibitor abrogates this phosphorylation. These findings indicate that CCL2 mediates disruption of AJ complexes, and are consistent with the live-cell imaging and confocal microscopy in which EC retraction and interendothelial gap formation are evident after 30 minutes of CCL2 treatment.
Figure 4. Adherens junction proteins are tyrosine phosphorylated after 30 minutes of CCL2 treatment.
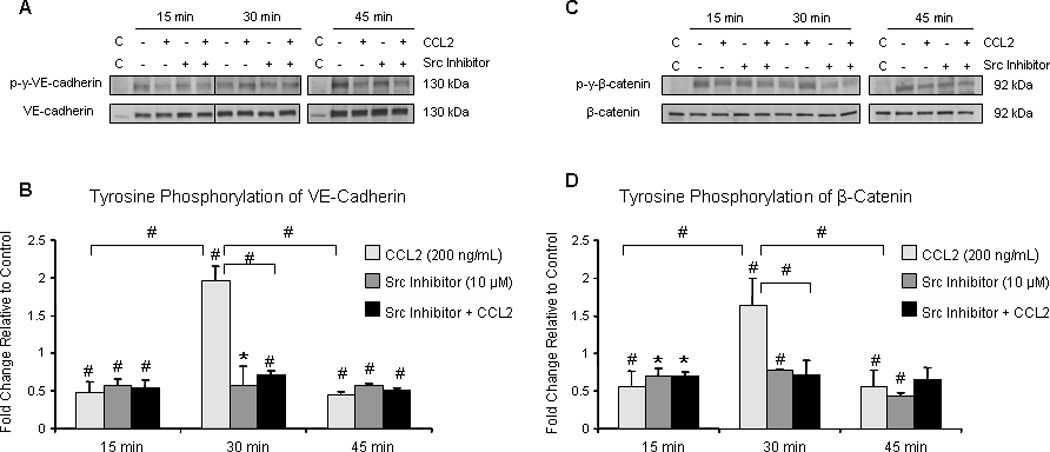
Confluent hCMEC/D3 were pre-treated with 10 µM Src inhibitor or DMSO for 2 hours followed by treatment with 200 ng/mL CCL2 or diluent. Cells were lysed and 40 µg of total protein were analyzed by Western blot. Blots were probed with phospho-specific antibody, stripped, and re-probed with antibody specific to non-phosphorylated protein. Optical density of phosphorylated protein was compared to OD of non-phosphorylated form. Densitometry is reported as fold change in protein phosphorylation in response to CCL2 relative to control at same time point. An intervening time point was removed from image and is represented by line in blot. Blots are representative of 3 independent experiments. C = control lysate. n=3. (*) p < 0.05. (#) p < 0.01. Tyrosine phosphorylation of VE-cadherin (A and B) and β-catenin (C and D) is increased at 30 minutes, relative to control and to the 15 and 45 minute time points. Src inhibitor pre-treatment abrogates VE-cadherin and β-catenin phosphorylation.
CCL2 induces adherens junction disassembly
Tyrosine phosphorylation of AJ proteins regulates the dynamic state and functional integrity of the interendothelial junction (68). We found VE-cadherin and β-catenin to be tyrosine-phosphorylated in response to 30 minutes of CCL2 treatment. Therefore, we examined the association of proteins at the AJ complex. Cultures of confluent hCMEC/D3 were treated with either 200 ng/mL CCL2 or diluent. Cells were lysed and AJ proteins were immunoprecipitated and evaluated by Western blot (Figure 5A). Optical densities (OD) of co-immunoprecipitating proteins were evaluated relative to OD of the protein used for immunoprecipitation. Protein association in response to CCL2 was compared to control cells treated with vehicle for the same time period and is reported as fold change (Figure 5B). The data are consistent regardless of whether antibody to VE-cadherin, β-catenin, or α-actinin is used as the immunoprecipitating protein. Species- or isotype-matched control antibodies were also used for immunoprecipitation to ensure that proteins were not non-specifically bound by antibody (data not shown). CCL2 induces a loss of association between VE-cadherin, β-catenin, and α-actinin at 30 minutes relative to control cells and relative to the 15 and 45 minute time points (Figure 5A and 5B). This is in agreement with increased tyrosine phosphorylation of VE-cadherin and β-catenin after 30 minutes of CCL2 exposure and suggests disassembly of the AJ.
Figure 5. CCL2 promotes disassembly of the AJ.
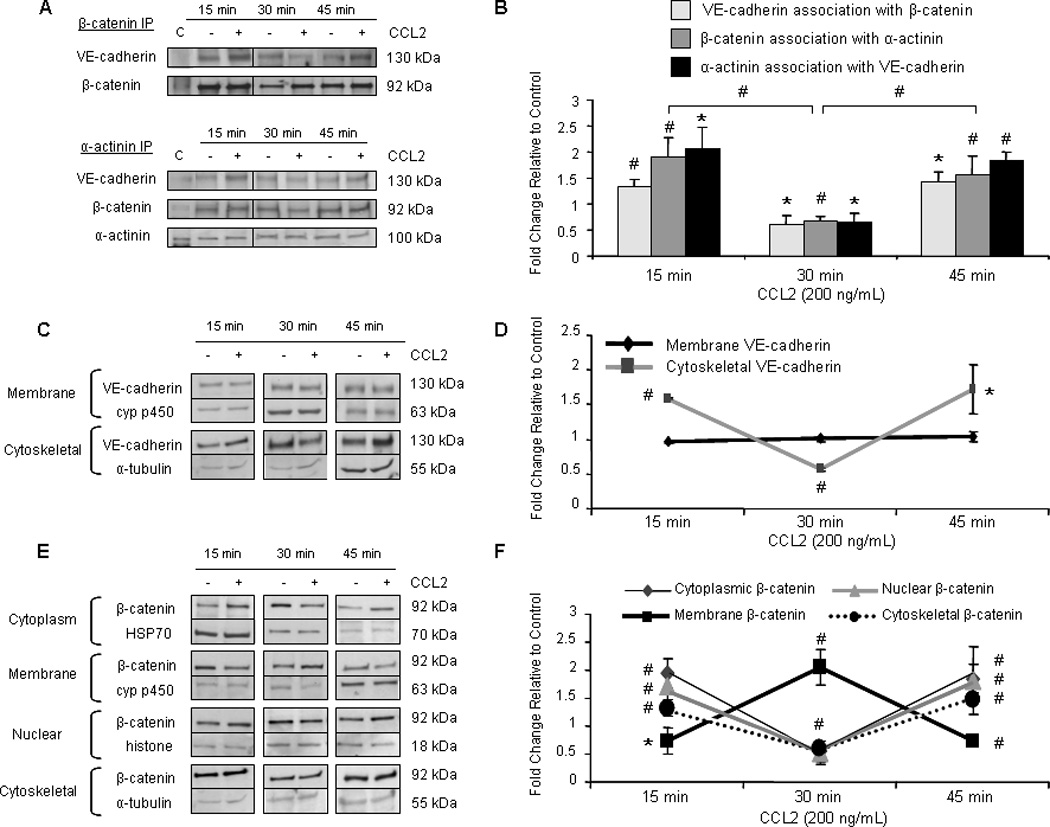
(A) Confluent hCMEC/D3 were treated with 200 ng/mL CCL2 or diluent. Cells were lysed and 140 µg of total protein were immunoprecipitated and evaluated by Western blot. Optical density of pulled-down protein was compared to OD of immunoprecipitating protein. Protein association in response to CCL2 was compared to control cells at the same time point and is reported as fold change. Findings are consistent when VE-cadherin, β-catenin, or α-actinin is used to pull-down other components of the AJ. Blots are representative of 3 independent experiments. An intervening time point was removed from the image and is represented by a dividing line in the blot. C = control lysate. Association of VE-cadherin with β-catenin and α-actinin is reduced after 30 minutes of CCL2 treatment, indicating loss of AJ integrity. Adherens junction disassembly corresponds to VE-cadherin and β-catenin phosphorylation. (C–F) Cytoplasmic, membrane, nuclear, and cytoskeletal fractions were isolated using different buffers from CCL2- or vehicle-treated primary HBMVEC. 10 µg of total protein from each fraction was evaluated by Western blot. VE-cadherin was quantifiable in only the membrane and cytoskeletal fractions. Optical density of VE-cadherin and β-catenin were compared to OD of loading control specific to each fraction. Changes in protein localization in response to CCL2 were compared to control cells at the same time point and are reported as fold change. n=3. (*) p < 0.05, (#) p < 0.01. (C–D) After 30 minutes of CCL2 treatment, association of VE-cadherin with the cytoskeleton is reduced. This corresponds with VE-cadherin phosphorylation and decreased association with β-catenin and α-actinin. (E–F) In response to 30 minutes of CCL2 treatment, β-catenin localization to the cytoplasm, nucleus, and cytoskeleton is reduced, while its localization in the membrane is increased. This corresponds with β-catenin phosphorylation and AJ disassembly and indicates that β-catenin is being sequestered at the membrane away from other cellular compartments in response to 30 minutes of CCL2 treatment.
The cellular localization of VE-cadherin and β-catenin upon AJ disassembly was next examined. After exposure to 200 ng/mL CCL2 or diluent, confluent monolayers of primary HBMVEC were lysed with sequential buffers to fractionate the cellular lysate into cytoplasmic, membrane, nuclear, and cytoskeletal components. VE-cadherin and β-catenin were evaluated in each fraction by Western blot (Figure 5C and 5E). Optical densities of AJ proteins were evaluated relative to loading control specific to each fraction. CCL2-mediated changes in VE-cadherin and β-catenin in each fraction were compared to vehicle-treated control cells and are reported as fold change (Figure 5D and 5F). Quantifiable levels of VE-cadherin were detected in the membrane and cytoskeletal compartments (Figure 5C). CCL2 did not alter the amount of VE-cadherin in the membrane compartment. However, concordant with its tyrosine phosphorylation, VE-cadherin distribution to the cytoskeletal fraction after 30 minutes of CCL2 treatment was reduced when compared to control and to the 15 and 45 minute time points (Figure 5C and 5D). This suggests dissociation of VE-cadherin from filamentous actin and AJ disassembly.
The antibodies used to detect VE-cadherin are specific to the cytoplasmic domain. However, the extracellular domain of VE-cadherin can be shed proteolytically (69). To confirm that CCL2-mediated AJ disassembly is due to VE-cadherin tyrosine phosphorylation and subsequent dissociation from β-catenin and the actin cytoskeleton and not due to cleavage of the VE-cadherin ectodomain, soluble VE-cadherin (sVE-cadherin) in the culture media of primary HBMVEC was assayed by ELISA. Soluble VE-cadherin levels were so low as to be at the limit of detection of the assay, despite concentration of the culture media. There was no difference in the amount of VE-cadherin shed from HBMVEC cultures treated with 200 ng/mL CCL2 or with diluent at any time point assayed (data not shown). The serum component of the media also had undetectable levels of sVE-cadherin. Thus, CCL2 modulates junctional integrity by altering the cellular localization of AJ proteins and their association in multi-protein complexes, but not by inducing VE-cadherin shedding.
β-catenin was detected and quantifiable in all four compartments isolated by subcellular fractionation (Figure 5E). In response to 30 minutes of CCL2 treatment, significant reductions in β-catenin partitioning to the cytoplasmic, nuclear, and cytoskeletal compartments occurred relative to control and relative to the 15 and 45 minute time points (Figure 5E and 5F). This coincides with a significant increase in localization of β-catenin at the membrane. These findings indicate that when CCL2 induces β-catenin tyrosine phosphorylation, β-catenin not only dissociates from the AJ complex, but is also mobilized from other cellular compartments to the membrane, where it is sequestered.
CCL2 mediates PECAM-1 sequestration of β-catenin
PECAM-1 binds tyrosine-phosphorylated β-catenin and regulates its phosphorylation and cellular localization (70). We therefore evaluated association of β-catenin with PECAM-1 in response to CCL2 as a mechanism for its increased membrane localization. Confluent monolayers of hCMEC/D3 were treated with either 200 ng/mL CCL2 or diluent. Cells were lysed and PECAM-1 or β-catenin was immunoprecipitated from total cell lysate. Western blot was used to evaluate complex formation between these two proteins (Figure 6). Data were consistent when either PECAM-1 or β-catenin was used as immunoprecipitant. Species-matched control antibodies were also used to ensure that non-specific binding did not occur (data not shown). In response to 30 minutes of CCL2 treatment, coincident with β-catenin tyrosine phosphorylation, there is increased association between PECAM-1 and β-catenin relative to control and to the 15 and 45 minute time points (Figure 6A and 6B).
Figure 6. PECAM-1 sequesters β-catenin at the membrane in response to CCL2.
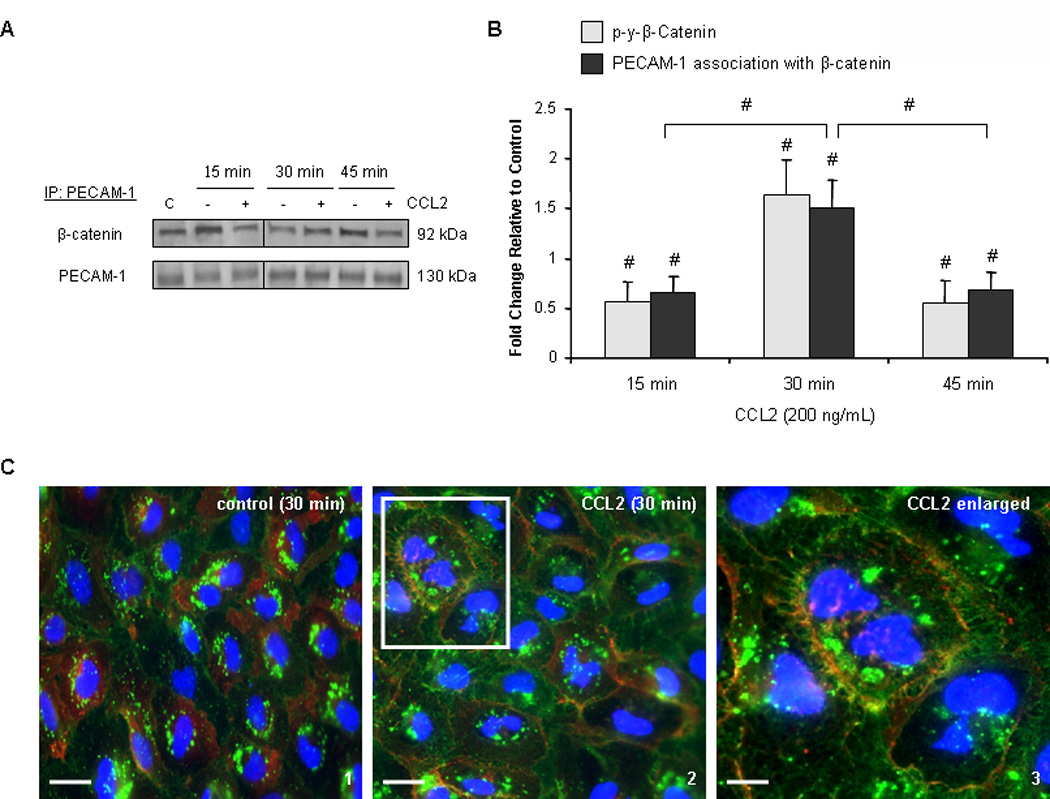
(A–B) 140 µg of total protein were immunoprecipitated from hCMEC/D3 treated with 200 ng/mL CCL2 or diluent. Results were consistent when either PECAM-1 or β-catenin was used in pull-down assay. Blots are representative of 3 independent experiments. An intervening time point was removed from the image and is represented by a dividing line in the blot. Optical densities were compared to OD of immunoprecipitating protein. Densitometry is represented as fold change in CCL2-treated cells relative to control cells. C= control lysate. n=3. (#) p <0.01. β-catenin is found in increased association with PECAM-1 in response to CCL2 relative to control cells and relative to the 15 and 45 minute time points. β-catenin/PECAM-1 association is coincident with β-catenin phosphorylation and dissociation from the adherens junction. (C) Immunocytochemistry was performed on primary HBMVEC treated with 200 ng/mL CCL2 or diluent for 30 minutes. Cells were fixed and permeabilized. β-catenin was stained with Cy3 (red) and PECAM-1 with FITC (green). The third panel is increased magnification of the area outlined in the second panel. After 30 minutes of CCL2 treatment, β-catenin is sequestered at the membrane by PECAM-1.
To confirm these findings, PECAM-1 and β-catenin in confluent primary HBMVEC monolayers were labeled with fluorescent antibodies and evaluated for their cellular localization with confocal microscopy. In control cells treated with diluent, β-catenin localizes to both the membrane and cytoplasm (Figure 6C, panel 1). However after 30 minutes of CCL2 treatment, cytoplasmic β-catenin is recruited to the plasma membrane (Figure 6C, panel 2) and to the cytoplasmic domain of PECAM-1 (Figure 6C, panel 3). This supports the subcellular fractionation and co-immunoprecipitation findings in which β-catenin partitioning to the membrane compartment is increased in response to CCL2. We conclude that in response to CCL2, β-catenin is tyrosine phosphorylated, dissociates from the AJ, and is sequestered at the membrane by PECAM-1.
PECAM-1 has variable affinity for β-catenin and γ-catenin and has altered cellular signaling depending on whether its ectodomain has been cleaved from the cell surface (71). The antibody used to detect PECAM-1 is specific to the cytoplasmic domain. We therefore examined whether the ectodomain of PECAM-1 is shed in response to CCL2. Media from confluent cultures of primary HBMVEC treated with either 200 ng/mL CCL2 or diluent was concentrated 60 fold and evaluated by ELISA. There was minimal, if any, soluble PECAM-1 detected in the media. There was no difference in quantity of PECAM-1 shed from endothelial cells in response to CCL2 (data not shown). This indicates that CCL2 does not induce PECAM-1 shedding from HBMVEC. Thus, the sequestration of β-catenin by PECAM-1 is specific to full-length PECAM-1 and is not a consequence of altered PECAM-1 signaling properties.
CCL2 increases Src-dependent tyrosine phosphorylation of PECAM-1, which facilitates the rapid reconstitution of the adherens junction
The time-lapse imaging studies demonstrate a transient disruption of junctional integrity in response to CCL2 in which endothelial cells retract and recoil. This is supported by the immunoprecipitation and subcellular fractionation studies where β-catenin dissociation from the AJ and recruitment to PECAM-1 at the plasma membrane is significantly increased after 30 minutes of CCL2 treatment, but rapidly reduced by 45 minutes. Thus, a mechanism must exist to liberate β-catenin from PECAM-1 such that it can traffic back to VE-cadherin and reform the AJ. The cytoplasmic domain of PECAM-1 contains two immunoreceptor tyrosine-based inhibitory motif (ITIM) domains (72) which, when phosphorylated by c-Src, can recruit a variety of SH2 domain-containing proteins, including protein phosphatases (73, 74). This provides a mechanism for dephosphorylating β-catenin and freeing it from its PECAM-1 anchor (75). We therefore examined PECAM-1 phosphorylation in response to CCL2.
Confluent monolayers of hCMEC/D3 were pre-treated with 10 µM Src inhibitor followed by treatment with either 200 ng/mL CCL2 or diluent. Cells were lysed and PECAM-1 was immunoprecipitated with a pan-phosphotyrosine antibody. Immunoblots were probed with antibody to PECAM-1. Cells treated with CCL2 or Src inhibitor were compared to vehicle-treated cells at the same time point as an internal control. Differences in PECAM-1 phosphorylation are reported as fold change relative to control. Figure 7A and 7B demonstrate that in response to 30 minutes of CCL2 treatment, tyrosine phosphorylation of PECAM-1 is increased relative to control cells and to the 15 and 45 minute time points. PECAM-1 phosphorylation is Src-dependent, as pre-treatment with Src inhibitor reduces its phosphorylation.
Figure 7. SHP-2 binds tyrosine phosphorylated PECAM-1 after 30 minutes of CCL2 treatment, allowing β-catenin to be dephosphorylated and released from PECAM-1 to reassemble the AJ.
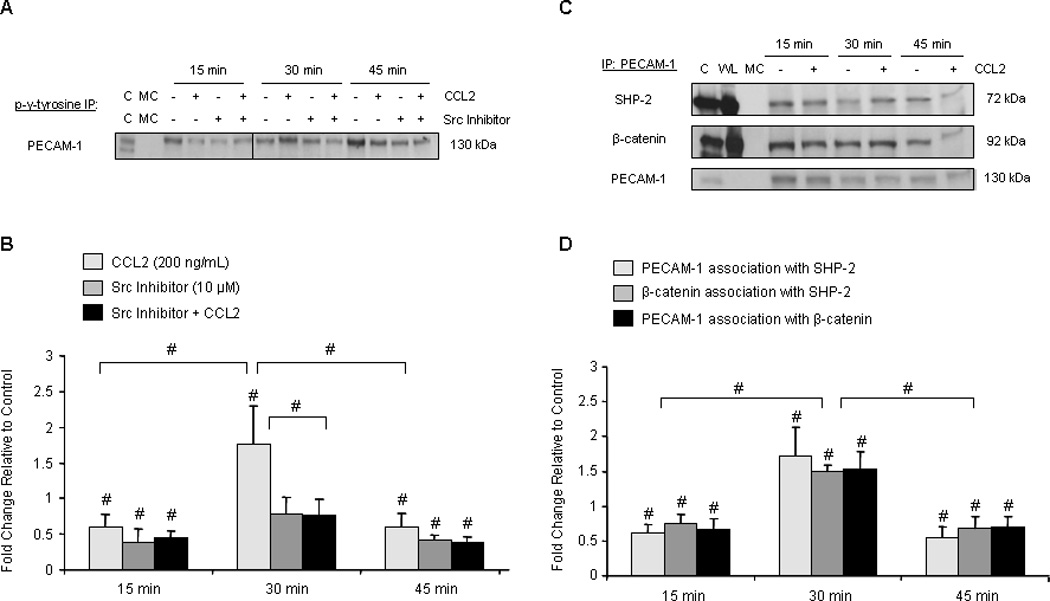
140 µg of total protein were immunoprecipitated from hCMEC/D3 treated with 200 ng/mL CCL2 or diluent and evaluated by Western blot. Optical density of immunoprecipitated protein was normalized to OD of protein used as immunoprecipitant. Densitometry represents fold change in response to CCL2 treatment relative to control cells. Blots are representative of 3 independent experiments. C = control lysate. MC = immunoprecipitation with species- or isotype-matched control antibody. WL = whole cell lysate (not immunoprecipitated). n=3. (#) p < 0.01. (A–B) Cells were pre-treated with 10 µM Src inhibitor or DMSO for 2 hours prior to CCL2 treatment. Results were consistent when antibody to phosphotyrosine or PECAM-1 was used for immunoprecipitation. An intervening time point was removed from image and is represented as a line in the blot. CCL2 increases tyrosine-phosphorylation of PECAM-1 at 30 minutes, which is abrogated by pre-treatment with Src inhibitor. (C–D) PECAM-1 tyrosine phosphorylation corresponds with increased recruitment of SHP-2 and PECAM-1/SHP-2/β-catenin complex formation. Results were consistent when PECAM-1, β-catenin, or SHP-2 was used for immunoprecipitation.
We further demonstrated c-Src-mediated tyrosine phosphorylation of PECAM-1 by comparing PECAM-1/c-Src association to the association of PECAM-1 with the other Src family members. PECAM-1 was immunoprecipitated from CCL2- or diluent-treated hCMEC/D3 monolayers and its association with c-Src, fyn, and c-Yes was evaluated by Western blot. Results were similar when Src kinase family member was used to immunoprecipitate PECAM-1. c-Src was specifically found in increased association with PECAM-1 after 30 minutes of CCL2 treatment as compared to control cells, the 15 and 45 minute time points, and relative to the association of PECAM-1 with Fyn and c-Yes (Figure 8A and B). No significant changes in interaction between PECAM-1 and Fyn (Figure 8 c and D) or c-Yes kinase (Figure 8 E and F) were observed. Collectively, our data indicate that CCL2 induces c-Src-dependent tyrosine phosphorylation of PECAM-1 after 30 minutes.
Figure 8. CCL2 preferentially promotes association of c-Src with PECAM-1 relative to other Src family kinases.
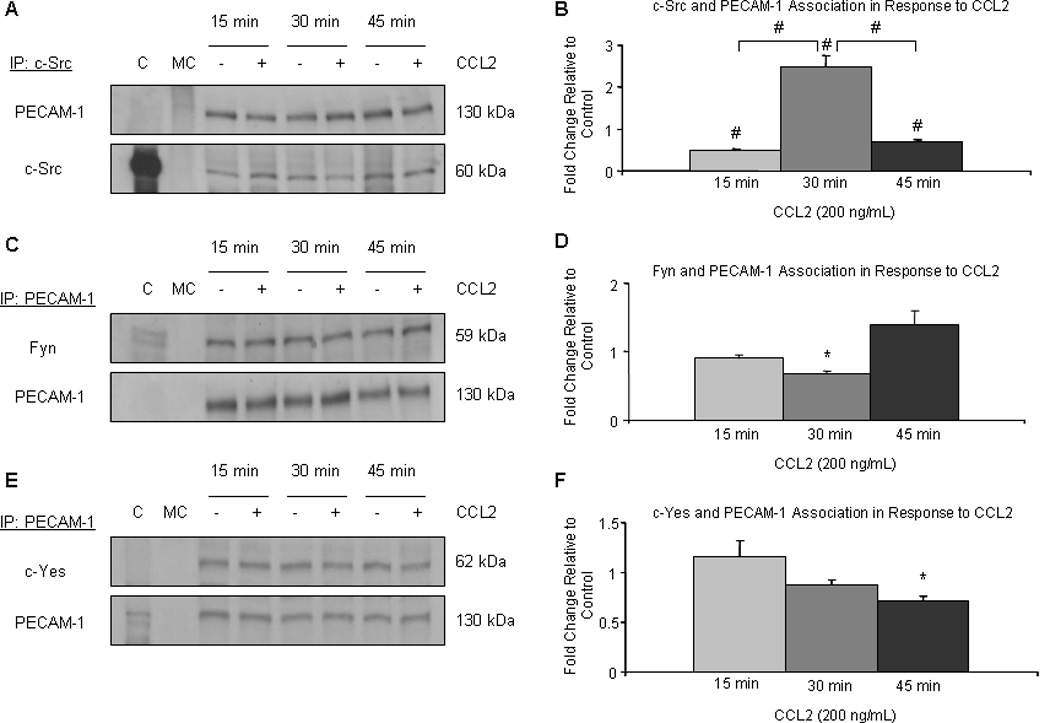
Confluent cultures of hCMEC/D3 were treated with 200 ng/mL CCL2 or diluent. 140 µg of total protein were immunoprecipitated and evaluated by Western blot. Results were consistent when either PECAM-1 or Src family member were used as immunoprecipitating protein. Optical density of immunoprecipitated protein was normalized to OD of protein used as immunoprecipitant. Densitometry represents fold change relative to control cells. Blots are representative of 3 independent experiments. (*) p < 0.05, (#) p < 0.01. CCL2 increases the association of c-Src with PECAM-1 at 30 minutes, compared to the 15 and 45 minute time points and compared to control. This is consistent with an increase in tyrosine phosphorylation of PECAM-1 after 30 minutes of CCL2 treatment. Fyn and c-Yes were not found in increased association with PECAM-1 after 30 minutes of CCL2 treatment.
The protein phosphatase SHP-2 is recruited to PECAM-1 in response to PECAM-1 tyrosine phosphorylation (76), where it is able to interact with β-catenin (75). Thus, we examined the association of SHP-2 with PECAM-1 and β-catenin during CCL2-mediated AJ disassembly. Immunoprecipitation of these three proteins with each other was examined in confluent monolayers of hCMEC/D3 treated with 200 ng/mL CCL2 or diluent. Results were consistent regardless of which protein was used as immunoprecipitant. Optical density of immunoprecipitated protein was normalized to OD of protein used as immunoprecipitant. Association of proteins in response to CCL2 treatment was compared to vehicle-treated cells at same time point and is reported as fold change.
Relative to control-treated cells and to the 15 and 45 minute time points, association between PECAM-1, SHP-2, and β-catenin is increased after 30 minutes of CCL2 treatment (Figure 7C and 7D). This corresponds with tyrosine phosphorylation of β-catenin (Figure 4C and 4D), a protein modification that enables its binding to PECAM-1 (70). It also corresponds with tyrosine phosphorylation of PECAM-1 (Figure 7A and 7B), which creates an SH2 binding domain at each of its two ITIM domains (76). As a result, SHP-2 is recruited to PECAM-1, interacts with β-catenin, and dephosphorylates it (75). Consistent with the subcellular fractionation data (Figure 5E and 5F), β-catenin then dissociates from PECAM-1, trafficking back to the AJ by 45 minutes. This suggests that PECAM-1 serves as a temporary sink for β-catenin and that c-Src-dependent tyrosine phosphorylation of PECAM-1 regulates β-catenin trafficking in response to CCL2.
CCL2 increases surface and whole cell expression of PECAM-1
We found that PECAM-1 sequesters β-catenin at the membrane during CCL2-mediated AJ disassembly and endothelial disruption. PECAM-1 is an important regulator of the BBB (1). Endothelial cells contain approximately 1 million PECAM-1 molecules at their cell surface (77), with most molecules localizing to intercellular contacts (78). Therefore, we evaluated CCL2-mediated alterations in PECAM-1 cellular localization and expression.
Subcellular fractionation of confluent monolayers of primary HBMVEC treated with either 200 ng/mL CCL2 or diluent demonstrates that PECAM-1 is enriched in the membrane in response to CCL2 (Figure 9A and 9B). After 15 minutes of CCL2 treatment, cytoplasmic and membrane PECAM-1 levels are elevated relative to control, suggesting increased protein stability and trafficking to the membrane. After 30 minutes, PECAM-1 enrichment at the membrane is maintained in response to CCL2 while cytosolic levels are decreased. This suggests decreased stability of cytosolic PECAM-1. However no degradation products were evident by Western blot, as PECAM-1 continued to be identified as a single molecular weight band. After 45 minutes of CCL2 treatment, PECAM-1 enrichment at the membrane relative to vehicle-treated cells is maintained, while cytoplasmic levels are near baseline. These data demonstrate a CCL2-mediated increase in PECAM-1 membrane localization, consistent with an enhanced ability to sequester β-catenin.
Figure 9. PECAM-1 surface localization is increased in response to CCL2.
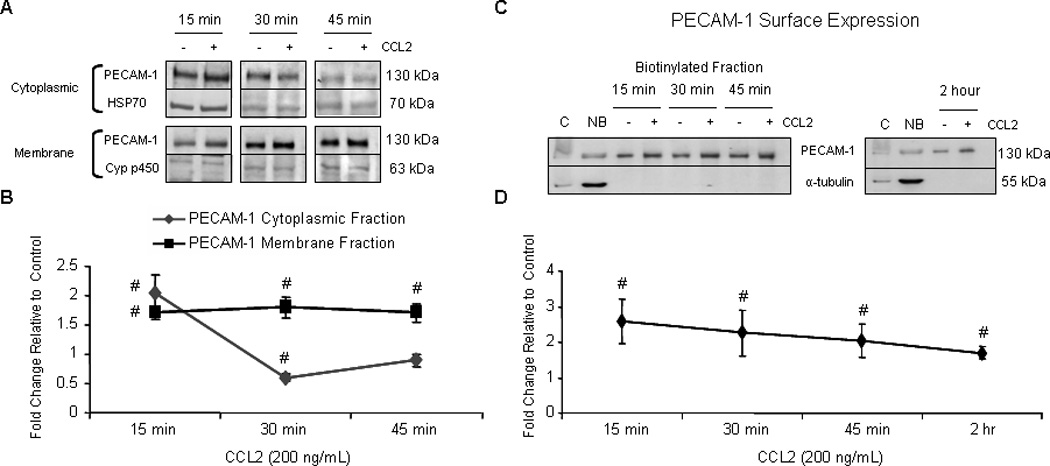
Confluent primary HBMVEC were exposed to 200 ng/mL CCL2 or diluent. (A and B) Cytoplasmic, membrane, nuclear, and cytoskeletal fractions were isolated using different buffer systems. 10 µg of total protein from each fraction was evaluated by Western blot. PECAM-1 was quantifiable in cytoplasmic and membrane fractions. Optical density of PECAM-1 was compared to OD of loading control specific to each fraction. Densitometry is reported as fold change in CCL2-treated cells relative to control cells at the same time point. Blots are representative of 3 independent experiments. (#) p < 0.01. After 15, 30, and 45 minutes of CCL2 treatment, PECAM-1 localization to the membrane compartment is increased. (C–D) After treatment with either CCL2 or diluent, cell surface proteins were biotinylated, immunoprecipitated with streptavidin conjugated to agarose beads, and evaluated by Western blot. Optical densities of PECAM-1 from CCL2-treated cells were compared to vehicle-treated cells from same time point. Densitometry is reported as fold change in CCL2-treated cells relative to control. Immunoreactivity of non-biotinylated, but not biotinylated fraction, to α-tubulin demonstrates that only surface proteins were biotinylated. C = control lysate. NB = non-biotinylated fraction. n=3. (#) p < 0.01. CCL2 induces PECAM-1 mobilization to the cell surface by 15 minutes and is maintained for at least 2 hours.
To determine whether PECAM-1 is localizing specifically to the cell surface as compared to other membrane compartments in response to CCL2, biotinylation was performed. Surface proteins from CCL2- or vehicle-treated confluent monolayers of primary HBMVEC were biotinylated and evaluated for PECAM-1 by Western blot. Surface PECAM-1 expression in response to CCL2 was compared to vehicle treated cells and is reported as fold change. Absence of α-tubulin expression in biotinylated fractions serves as an internal control. Consistent with subcellular fractionation data, surface PECAM-1 is increased in response to CCL2 (Figure 9C and 9D). PECAM-1 enrichment at the plasma membrane is maintained after 2 hours of CCL2 treatment.
To confirm these findings, we immunofluorescently labeled PECAM-1 in confluent primary HBMVEC cultures. Consistent with the subcellular fractionation and biotinylation studies, PECAM-1 membrane immunoreactivity (FITC-green) was increased in response to CCL2 at 30 minutes (Figure 6C) and at 2 hours (Figure 10A). PECAM-1 also had prominent perinuclear localization, suggestive of synthesis and processing in the endoplasmic reticulum-golgi secretory pathway. However, differential perinuclear localization in response to CCL2 was not observed.
Figure 10. CCL2 increases total cellular PECAM-1.
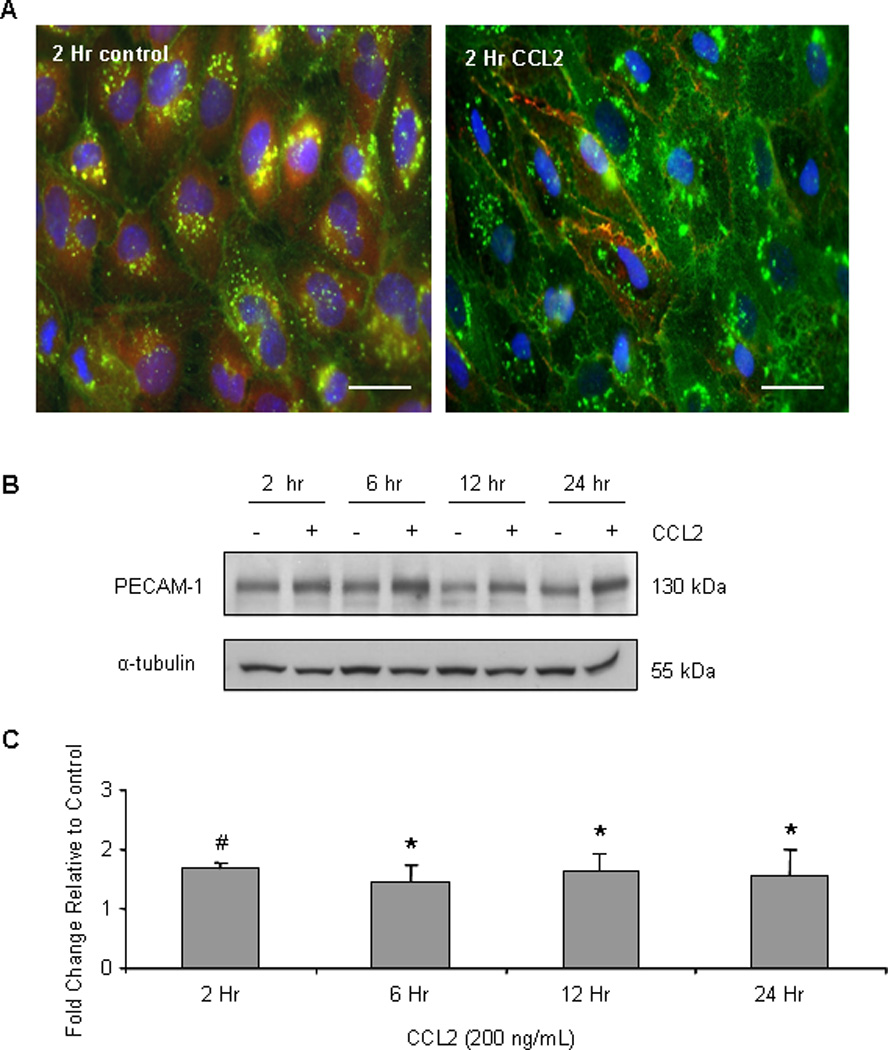
Primary HBMVEC were treated with 200 ng/mL CCL2 or diluent. (A) Cells were fixed and permeabilized. Immunofluorescence was used to visualize PECAM-1 distribution and abundance (FITC- green). Nuclei were stained with DAPI (blue) and β-catenin was stained with Cy3 (red). After 2 hours of CCL2 exposure, whole cell expression of PECAM-1 is increased. (B–C) 40 µg of protein was evaluated by Western blot. Optical density of PECAM-1 was normalized to the OD of α-tubulin. Densitometry represents fold change in PECAM-1 expression in response to CCL2 relative to control cells at same time point. n=6. (*) p < 0.05, (#) p < 0.01. CCL2 increases total cellular PECAM-1 from 2–24 hours.
We also detected increased whole-cell PECAM-1 expression by immunofluorescence after 2 hours of CCL2 treatment (Figure 10A). To examine this further, we evaluated the effects of prolonged CCL2 treatment on PECAM-1 expression by Western blot. Confluent monolayers of primary HBMVEC were treated with 200 ng/mL CCL2 or diluent. After normalization to α-tubulin, PECAM-1 expression in response to CCL2 was compared to control cells of the same time point. Figures 10B and 10C demonstrate that CCL2 increases whole-cell PECAM-1 by 1.5–1.8 fold from 2 to 24 hours. There are approximately 1 million PECAM-1 molecules expressed at the junctional surface of an endothelial cell (77). This 1.5 fold or greater increase in PECAM-1 expression may have substantial effects on barrier integrity and leukocyte diapedesis. Collectively, our data indicate that in addition to disruption of the EC junctional integrity, CCL2 mobilizes PECAM-1 to the cell surface and increases the total PECAM-1 content in cells, thereby facilitating PECAM-1 sequestration of β-catenin during AJ disassembly.
CCL2 treatment does not result in detectable permeability of HBMVEC, despite the transient opening of the monolayer
To determine whether changes in adherens junctions in HBMVEC induced by CCL2 treatment result in permeability of the barrier, permeability was measured by albumin conjugated to Evans blue dye or by TEER. We cultured primary human BMVEC on 3 µm tissue culture inserts placed within a 24 well tissue culture dish. Cells were tested after 3 days in culture to allow them to reach confluence and impermeability. Inserts with confluent BMVEC were impermeable to albumin conjugated to Evans blue dye (data not shown) and showed resistance as measured by TEER. Treatment with 200 ng/ml or 500 ng/ml of CCL2 was performed for 30 and 45 min. Permeability was assayed using albumin conjugated to Evans blue dye as described in the Materials and Methods or by TEER. None of the treatments altered the permeability of the HBMVEC despite the transient opening of the barrier detected by microscopy (data not illustrated). Thus, our data indicate that despite the transient retraction of the HBMVEC described above, these changes are not sufficient to result in BBB permeability. In agreement, our previous published data demonstrated that CCL2 alone did not disrupt our human BBB model of endothelial cells cocultured with human astrocytes (33). However, in the presence of HIV infected leukocytes and CCL2, BBB integrity was compromised (33), while when there was no CCL2 or no HIV infection of leukocytes, there was no barrier disruption. Thus, while CCL2 is necessary for increased barrier permeability, additional factors, for example HIV infection, must be required for detectable disruption of the barrier.
Discussion
Blood-brain barrier disruption and leukocyte infiltration are common sequelae of neuroinflammation and are seen in meningitis (79, 80), HIV-associated neurocognitive disorder (81, 82), Alzheimer disease (83, 84), multiple sclerosis (85, 86), neuropsychiatric lupus (87, 88), and ischemic stroke (89, 90). CNS CCL2 is elevated in all of these conditions, and often corresponds with severity of disease (34, 37, 38, 91–93). CCL2 is a potent chemoattractant for monocytes (94) and is associated with EC dysfunction (39). Our results using human brain microvascular endothelial cells indicated that CCL2 treatment alone did not disrupt the barrier sufficiently to change BBB permeability. However, we propose that CCL2 “primes” the barrier to disruption by additional insults, including infectious agents such as HIV (28, 33). Its role in tight junction disruption has been established (40, 95); however, its effects on AJ and PECAM-1, two important regulators of BBB integrity (1, 96), have not been extensively examined. This study demonstrates a novel mechanism by which the EC junctional compartment may be disrupted during CCL2-associated neuroinflammation. We show for the first time an exquisite regulation between the AJ and PECAM-1 in response to CCL2, potentially altering BBB integrity. We also demonstrate that CCL2 increases the cell surface localization of PECAM-1, a response that may facilitate leukocyte entry into the CNS. Understanding the mechanisms of CCL2-mediated BBB disruption and leukocyte CNS recruitment is important for designing therapeutics directed at reducing neuroinflammation. Our data identify potential treatment targets that have clinical relevance for neuroinflammatory disease.
We demonstrate that CCL2 destabilizes AJ complexes. In response to CCL2, HBMVEC retract and form transient interendothelial gaps in confluent monolayers. This is associated with endothelial FAK activation and stress fiber formation, indicating focal adhesion remodeling. CCL2 induces tyrosine phosphorylation of VE-cadherin and β-catenin after 30 minutes, and this corresponds with loss of association of VE-cadherin and β-catenin with each other, with their α-actinin link to actin, and with their localization to the cytoskeleton. Upon dissociation from the AJ, tyrosine-phosphorylated β-catenin is sequestered at the plasma membrane by PECAM-1. Thus, we propose that this is an important regulatory mechanism to minimize β-catenin transcriptional activity and activation of cellular proliferation genes during its liberation from the AJ.
We find CCL2-mediated AJ disruption and β-catenin association with PECAM-1 to be transient. Concurrent with β-catenin phosphorylation and sequestration by PECAM-1, PECAM-1 is tyrosine-phosphorylated after 30 minutes of CCL2 treatment. This creates a binding site for proteins containing an SH2 domain (73). Upon PECAM-1 tyrosine phosphorylation, SHP-2 is recruited to PECAM-1. SHP-2 interacts with PECAM-1-bound tyrosine-phosphorylated β-catenin and likely dephosphorylates it, as we find a reduction in association of β-catenin with PECAM-1 after β-catenin/SHP-2/PECAM-1 complex formation. Thus, β-catenin is released from PECAM-1 and re-associates with VE-cadherin, resulting in reconstitution of the AJ. This is supported by increased partitioning of β-catenin to the cytoskeletal, cytoplasmic, and nuclear compartments after 45 minutes of CCL2 treatment relative to the 30 minute time point. Collectively, our data suggest a response by the BBB to CCL2 in which AJ are transiently disrupted as β-catenin shuttles back-and-forth between the AJ and PECAM-1 (Figure 11). Fluxes in EC permeability may not only disrupt the metabolic homeostasis of the CNS, but may also permit the entry of inflammatory and neurotoxic substances, pathogens, and leukocytes through the BBB. Thus, CCL2-induced AJ disruption could contribute to CNS pathology in neuroinflammatory disease.
Figure 11. CCL2 induces shuttling of β-catenin between PECAM-1 and the AJ in HBMVEC such that the AJ and monolayer integrity are transiently disrupted.
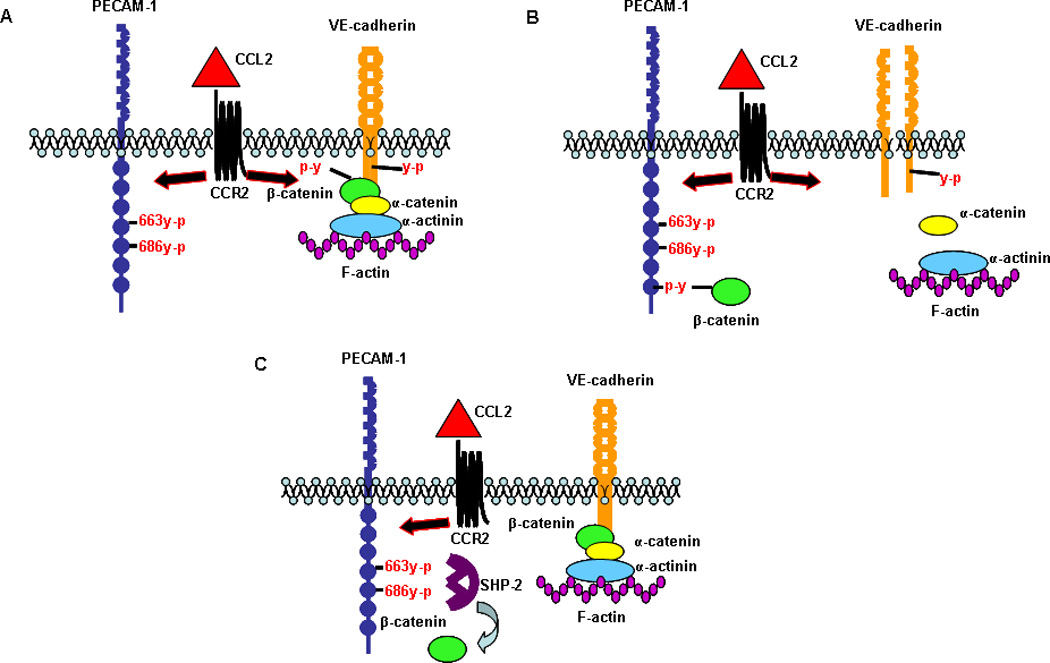
(A) CCL2 induces tyrosine phosphorylation of VE-cadherin, β-catenin, and PECAM-1 after 30 minutes of exposure. (B) VE-cadherin and β-catenin tyrosine phosphorylation result in loss of AJ integrity. Tyrosine phosphorylated β-catenin binds to PECAM-1 and is sequestered at the plasma membrane. (C) Tyrosine phosphorylation of PECAM-1 in response to CCL2 creates a docking site for the protein phosphatase SHP-2. Binding of SHP-2 to PECAM-1 promotes dephosphorylation of β-catenin by SHP-2 and release of β-catenin from PECAM-1. β-catenin reassociates with VE-cadherin and reforms the AJ. Thus, CCL2 mediates a transient disruption of the AJ through the temporary sequestration of β-catenin by PECAM-1 at the plasma membrane.
The mechanisms regulating the trafficking of β-catenin during CCL2-mediated BBB disruption are currently being examined in our laboratory. Ubiquitination regulates both the cellular localization and abundance of proteins (97). Glycogen synthase kinase 3 beta (GSK-3β) facilitates serine/threonine phosphorylation of β-catenin (98), thereby targeting β-catenin for ubiquitination (99). Akt is an upstream regulator of GSK-3β (75). Tyrosine-phosphorylated PECAM-1 recruits and activates phosphatidylinositol-3 kinase, an activator of Akt (100). Thus, by inducing PECAM-1 tyrosine phosphorylation, CCL2 may regulate GSK-3β -mediated ubiquitination of β-catenin, thereby altering its cellular localization.
Adherens junction disruption in response to CCL2 is potentially attenuated by PECAM-1 recruitment to the cell membrane. We find cell surface PECAM-1 to increase after CCL2 treatment from 15 minutes to 2 hours. This would increase the opportunity for β-catenin to be sequestered at the membrane away from the AJ and other cellular compartments in response to CCL2. The CCL2-mediated increase in total cellular PECAM-1 beginning at 2 hours and lasting for at least 24 hours may also sustain CCL2-mediated signaling between PECAM-1 and AJ for prolonged periods. The endothelium may respond to high CCL2 levels by increasing PECAM-1 and its surface localization to provide a temporary sink for β-catenin at the membrane so the AJ might be rapidly reconstituted. Upregulation of endothelial PECAM-1 expression and membrane localization may be a compensatory mechanism for CCL2-induced alterations in AJ integrity. This would be consistent with upregulation of CNS endothelial PECAM-1 in response to cerebral ischemia in rodents (101) and an earlier disease onset in experimentally induced autoimmune encephalitis in PECAM-1-deficient mice (1). Thus, by facilitating reconstitution of the CCL2-disrupted AJ, PECAM-1 may be protective during neuroinflammation.
Elevated PECAM-1 expression and cell surface localization also have implications for leukocyte transendothelial migration. Interactions between EC and leukocyte PECAM-1 facilitate leukocyte diapedesis through the interendothelial space (102). Disruption of these interactions by blocking antibody or peptide results in reduced leukocyte accumulation at sites of inflammation (103). Leukocyte transendothelial migration can occur over a time frame of 30 minutes to 24 hours (33, 102, 104). Thus, a CCL2-mediated increase in PECAM-1 expression and surface localization may enhance leukocyte-EC interactions and facilitate leukocyte entry into the CNS.
Our data support a role for c-Src in CCL2-mediated disruption of EC junctional integrity. CCL2-induced cellular retraction and interendothelial gap formation is inhibited by pre-treatment with Src inhibitor. Although the Src inhibitor has activity against multiple Src family members, CCL2-mediated FAK activation is specifically inhibited in our studies. The antibody used to assay FAK activation is specific to the tyrosine residue identified as a site of c-Src phosphorylation (62). Src inhibition also reduces CCL2-induced phosphorylation of VE-cadherin and β-catenin at c-Src-specific sites. PECAM-1 is tyrosine phosphorylated by c-Src (74) and our studies demonstrate reduced tyrosine phosphorylation in response to CCL2 with Src inhibitor pre-treatment. We also find increased association of c-Src with PECAM-1 after 30 minutes of CCL2 treatment, a time point associated with increased PECAM-1 tyrosine phosphorylation. Fyn and c-Yes, the other two Src family members that are constitutively found in EC (105), do not immunoprecipitate with PECAM-1 at increased levels after 30 minutes of CCL2 treatment. Collectively, our data indicate that c-Src mediates CCL2-induced signaling to PECAM-1 and the AJ and associated remodeling of the EC junctional space. This suggests that c-Src is a potential target for therapeutic intervention in neuroinflammatory disease. Small molecule Src inhibitors such as Dasatinib, AZD-0530, and SKI-606, are currently in phase II clinical trials for the treatment of cancer in part because Src-mediated vascular permeability facilitates tumor extravasation and metastasis (106). Dasatinib, in particular, has been shown to cross the BBB and have efficacy against CNS-penetrating leukemia (107). Thus, the potential exists for targeting Src-mediated EC dysfunction in neuroinflammatory disease.
Our data demonstrate some of the exquisitely controlled mechanisms by which CCL2 regulates the transient opening of the adherens junctions between brain endothelial cells. We propose that this transient opening of the junctions that does not result in detectable changes in BBB permeability necessary for transmigration of leukocytes into the brain. However, additional factors are required for significant transmigration above baseline. In pathological conditions, such as HIV infection, both factors, CCL2 and HIV infection, are necessary to result in disruption and increased transmigration of HIV infected leukocytes across the BBB, as we described previously (33). Our previous data also demonstrated that in the case of HIV infection of leukocytes, some of the adhesion molecules regulated by CCL2 are shed into the extracellular space and we propose that these soluble molecules also compete for the endothelial-endothelial interaction resulting in destabilization of the BBB (108, 109). It is possible that for other CNS pathologies characterized by elevated CCL2, such as multiple sclerosis, additional mediators likely act in concert with CCL2 (e.g. autoreactive T cells or other cytokines) to disrupt the barrier integrity. Thus, our current data provide a demonstration of the complex cellular and molecular mechanisms that facilitate the transient movement of adherents junction proteins in response to CCL2 and indicate future areas of investigation to examine how pathological conditions such as HIV infection, affect this highly regulated BBB integrity.
In summary, we provide the first demonstration in human cerebral EC of (1) CCL2-mediated disruption to the VE-cadherin AJ, (2) CCL2-induced signaling between PECAM-1 and the AJ, and (3) CCL2-associated increases in PECAM-1 expression and cell surface localization. These in vitro findings suggest BBB dysregulation during neuroinflammation. Specifically, our data provide a potential mechanism for loss of BBB integrity and increased CNS leukocyte transendothelial migration in response to CCL2. These findings have important implications for understanding and treating neuroinflammation and suggest that inhibition of Src signaling is a potential therapeutic target in the treatment of neuroinflammatory disease.
Supplementary Material
Acknowledgments
We would like to thank the Analytical Imaging Facility at AECOM for microscopy support, as well as the CFAR Pathology and Immunology core for assistance with the live cell imaging.
Grant Numbers and Sources of Support: This work was supported by NIH grants MH075679 and DA025567 (J.W.B.), MH076679 and MH096625 (E.A.E.), by grant AI-051519 from the NIH Centers for AIDS Research and by AI071326 pre-doctoral fellowship (T.K.R).
Abbreviations
- AJ
adherens junction
- BBB
blood-brain barrier
- CCL2
chemokine [C-C motif] ligand 2
- CCR2
chemokine [C-C motif] receptor 2
- CNS
central nervous system
- EC
endothelial cells
- FAK
focal adhesion kinase
- GSK-3β
Glycogen synthase kinase 3 beta
- HBMVEC
human brain microvascular endothelial cells
- ITIM
immunoreceptor tyrosine-based inhibition motif
- OD
optical densities
- PECAM-1
platelet endothelial cell adhesion molecule
- SHP-2
Src homology 2 domain-containing protein phosphatase
- VE-cadherin
vascular-endothelial cadherin
References
- 1.Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, et al. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest. 2002 Feb;109(3):383–392. doi: 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Navratil E, Couvelard A, Rey A, Henin D, Scoazec JY. Expression of cell adhesion molecules by microvascular endothelial cells in the cortical and subcortical regions of the normal human brain: an immunohistochemical analysis. Neuropathol Appl Neurobiol. 1997 Feb;23(1):68–80. [PubMed] [Google Scholar]
- 3.Petty MA, Lo EH. Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Prog Neurobiol. 2002 Dec;68(5):311–323. doi: 10.1016/s0301-0082(02)00128-4. [DOI] [PubMed] [Google Scholar]
- 4.Williams AJ, Wei HH, Dave JR, Tortella FC. Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J Neuroinflammation. 2007;4:17. doi: 10.1186/1742-2094-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez JI, Teale JM. Evidence for differential changes of junctional complex proteins in murine neurocysticercosis dependent upon CNS vasculature. Brain Res. 2007 Sep 12;1169:98–111. doi: 10.1016/j.brainres.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haorah J, Heilman D, Knipe B, Chrastil J, Leibhart J, Ghorpade A, et al. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol Clin Exp Res. 2005 Jun;29(6):999–1009. doi: 10.1097/01.alc.0000166944.79914.0a. [DOI] [PubMed] [Google Scholar]
- 7.Nottet HS. Interactions between macrophages and brain microvascular endothelial cells: role in pathogenesis of HIV-1 infection and blood - brain barrier function. J Neurovirol. 1999 Dec;5(6):659–669. doi: 10.3109/13550289909021294. [DOI] [PubMed] [Google Scholar]
- 8.Ifergan I, Wosik K, Cayrol R, Kebir H, Auger C, Bernard M, et al. Statins reduce human blood-brain barrier permeability and restrict leukocyte migration: relevance to multiple sclerosis. Ann Neurol. 2006 Jul;60(1):45–55. doi: 10.1002/ana.20875. [DOI] [PubMed] [Google Scholar]
- 9.Weiss N, Miller F, Cazaubon S, Couraud PO. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta. 2009 Apr;1788(4):842–857. doi: 10.1016/j.bbamem.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 10.Wang P, Dai J, Bai F, Kong KF, Wong SJ, Montgomery RR, et al. Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J Virol. 2008 Sep;82(18):8978–8985. doi: 10.1128/JVI.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Morris A, Sunkara M, Layne J, Toborek M, Hennig B. Epigallocatechin-gallate stimulates NF-E2-related factor and heme oxygenase-1 via caveolin-1 displacement. J Nutr Biochem. 2012 Feb;23(2):163–168. doi: 10.1016/j.jnutbio.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caveda L, Martin-Padura I, Navarro P, Breviario F, Corada M, Gulino D, et al. Inhibition of cultured cell growth by vascular endothelial cadherin (cadherin-5/VE-cadherin) J Clin Invest. 1996 Aug 15;98(4):886–893. doi: 10.1172/JCI118870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999 Aug 17;96(17):9815–9820. doi: 10.1073/pnas.96.17.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bibert S, Jaquinod M, Concord E, Ebel C, Hewat E, Vanbelle C, et al. Synergy between extracellular modules of vascular endothelial cadherin promotes homotypic hexameric interactions. J Biol Chem. 2002 Apr 12;277(15):12790–12801. doi: 10.1074/jbc.M111597200. [DOI] [PubMed] [Google Scholar]
- 15.Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B, et al. The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin) J Cell Biol. 1995 Apr;129(1):203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knudsen KA, Soler AP, Johnson KR, Wheelock MJ. Interaction of alpha-actinin with the cadherin/catenin cell-cell adhesion complex via alpha-catenin. J Cell Biol. 1995 Jul;130(1):67–77. doi: 10.1083/jcb.130.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci. 1998 Jul;111(Pt 13):1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 18.Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol. 1999 Oct;19(10):2286–2297. doi: 10.1161/01.atv.19.10.2286. [DOI] [PubMed] [Google Scholar]
- 19.van Wetering S, van Buul JD, Quik S, Mul FP, Anthony EC, ten Klooster JP, et al. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J Cell Sci. 2002 May 1;115(Pt 9):1837–1846. doi: 10.1242/jcs.115.9.1837. [DOI] [PubMed] [Google Scholar]
- 20.Liao F, Huynh HK, Eiroa A, Greene T, Polizzi E, Muller WA. Migration of monocytes across endothelium and passage through extracellular matrix involve separate molecular domains of PECAM-1. J Exp Med. 1995 Nov 1;182(5):1337–1343. doi: 10.1084/jem.182.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji G, O'Brien CD, Feldman M, Manevich Y, Lim P, Sun J, et al. PECAM-1 (CD31) regulates a hydrogen peroxide-activated nonselective cation channel in endothelial cells. J Cell Biol. 2002 Apr 1;157(1):173–184. doi: 10.1083/jcb.200110056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park M, Hennig B, Toborek M. Methamphetamine alters occludin expression via NADPH oxidase-induced oxidative insult and intact caveolae. J Cell Mol Med. 2012 Feb;16(2):362–375. doi: 10.1111/j.1582-4934.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams DW, Eugenin EA, Calderon TM, Berman JW. Monocyte maturation, HIV susceptibility, and transmigration across the blood brain barrier are critical in HIV neuropathogenesis. J Leukoc Biol. 2012 Mar;91(3):401–415. doi: 10.1189/jlb.0811394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mkrtchyan H, Scheler S, Klein I, Fahr A, Couraud PO, Romero IA, et al. Molecular cytogenetic characterization of the human cerebral microvessel endothelial cell line hCMEC/D3. Cytogenet Genome Res. [Research Support, Non-U.S. Gov't] 2009;126(4):313–317. doi: 10.1159/000253080. [DOI] [PubMed] [Google Scholar]
- 25.Vu K, Weksler B, Romero I, Couraud PO, Gelli A. Immortalized human brain endothelial cell line HCMEC/D3 as a model of the blood-brain barrier facilitates in vitro studies of central nervous system infection by Cryptococcus neoformans. Eukaryot Cell. [Research Support, N.I.H. Extramural] 2009 Nov;8(11):1803–1807. doi: 10.1128/EC.00240-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weksler BB, Subileau EA, Perriere N, Charneau P, Holloway K, Leveque M, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005 Nov;19(13):1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 27.Cucullo L, Couraud PO, Weksler B, Romero IA, Hossain M, Rapp E, et al. Immortalized human brain endothelial cells and flow-based vascular modeling: a marriage of convenience for rational neurovascular studies. J Cereb Blood Flow Metab. [Research Support N.I.H., Extramural Research Support, Non-U.S. Gov't] 2008 Feb;28(2):312–328. doi: 10.1038/sj.jcbfm.9600525. [DOI] [PubMed] [Google Scholar]
- 28.Eugenin EA, Berman JW. Chemokine-dependent mechanisms of leukocyte trafficking across a model of the blood-brain barrier. Methods. 2003 Apr;29(4):351–361. doi: 10.1016/s1046-2023(02)00359-6. [DOI] [PubMed] [Google Scholar]
- 29.Hurwitz AA, Berman JW, Lyman WD. The role of the blood-brain barrier in HIV infection of the central nervous system. Adv Neuroimmunol. 1994;4(3):249–256. doi: 10.1016/s0960-5428(06)80263-9. [DOI] [PubMed] [Google Scholar]
- 30.Hurwitz AA, Berman JW, Rashbaum WK, Lyman WD. Human fetal astrocytes induce the expression of blood-brain barrier specific proteins by autologous endothelial cells. Brain Res. 1993;625(2):238–243. doi: 10.1016/0006-8993(93)91064-y. [DOI] [PubMed] [Google Scholar]
- 31.Weiss JM, Downie SA, Lyman WD, Berman JW. Astrocyte-derived monocyte-chemoattractant protein-1 directs the transmigration of leukocytes across a model of the human blood-brain barrier. J Immunol. 1998 Dec 15;161(12):6896–6903. [PubMed] [Google Scholar]
- 32.Eugenin EA, Berman JW. Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J Neurosci. 2007 Nov 21;27(47):12844–12850. doi: 10.1523/JNEUROSCI.4154-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006 Jan 25;26(4):1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE. Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol. 1998 Nov;44(5):831–835. doi: 10.1002/ana.410440521. [DOI] [PubMed] [Google Scholar]
- 35.Spanaus KS, Nadal D, Pfister HW, Seebach J, Widmer U, Frei K, et al. C-X-C and C-C chemokines are expressed in the cerebrospinal fluid in bacterial meningitis and mediate chemotactic activity on peripheral blood-derived polymorphonuclear and mononuclear cells in vitro. J Immunol. 1997 Feb 15;158(4):1956–1964. [PubMed] [Google Scholar]
- 36.Dogan RN, Elhofy A, Karpus WJ. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF- and iNOS-expressing macrophages and myeloid dendritic cells. J Immunol. 2008 Jun 1;180(11):7376–7384. doi: 10.4049/jimmunol.180.11.7376. [DOI] [PubMed] [Google Scholar]
- 37.Galimberti D, Schoonenboom N, Scarpini E, Scheltens P. Chemokines in serum and cerebrospinal fluid of Alzheimer's disease patients. Ann Neurol. 2003 Apr;53(4):547–548. doi: 10.1002/ana.10531. [DOI] [PubMed] [Google Scholar]
- 38.Iikuni N, Okamoto H, Yoshio T, Sato E, Kamitsuji S, Iwamoto T, et al. Raised monocyte chemotactic protein-1 (MCP-1)/CCL2 in cerebrospinal fluid of patients with neuropsychiatric lupus. Ann Rheum Dis. 2006 Feb;65(2):253–256. doi: 10.1136/ard.2005.041640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, et al. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab. 2005 May;25(5):593–606. doi: 10.1038/sj.jcbfm.9600055. [DOI] [PubMed] [Google Scholar]
- 40.Song L, Pachter JS. Monocyte chemoattractant protein-1 alters expression of tight junction-associated proteins in brain microvascular endothelial cells. Microvasc Res. 2004 Jan;67(1):78–89. doi: 10.1016/j.mvr.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2006 Jun;26(6):797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- 42.Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A. 1994 Mar 29;91(7):2752–2756. doi: 10.1073/pnas.91.7.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu L, Tseng SC, Rollins BJ. Monocyte chemoattractant protein-1. Chem Immunol. 1999;72:7–29. doi: 10.1159/000058723. [DOI] [PubMed] [Google Scholar]
- 44.Dzenko KA, Andjelkovic AV, Kuziel WA, Pachter JS. The chemokine receptor CCR2 mediates the binding and internalization of monocyte chemoattractant protein-1 along brain microvessels. J Neurosci. 2001 Dec 1;21(23):9214–9223. doi: 10.1523/JNEUROSCI.21-23-09214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dzenko KA, Song L, Ge S, Kuziel WA, Pachter JS. CCR2 expression by brain microvascular endothelial cells is critical for macrophage transendothelial migration in response to CCL2. Microvasc Res. 2005 Jul;70(1–2):53–64. doi: 10.1016/j.mvr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Ge S, Song L, Serwanski DR, Kuziel WA, Pachter JS. Transcellular transport of CCL2 across brain microvascular endothelial cells. J Neurochem. 2008 Mar;104(5):1219–1232. doi: 10.1111/j.1471-4159.2007.05056.x. [DOI] [PubMed] [Google Scholar]
- 47.Andjelkovic AV, Pachter JS. Characterization of binding sites for chemokines MCP-1 and MIP-1alpha on human brain microvessels. J Neurochem. 2000 Nov;75(5):1898–1906. doi: 10.1046/j.1471-4159.2000.0751898.x. [DOI] [PubMed] [Google Scholar]
- 48.Roberts TK, Buckner CM, Berman JW. Leukocyte transmigration across the blood-brain barrier: perspectives on neuroAIDS. Front Biosci. 15:478–536. doi: 10.2741/3631. [DOI] [PubMed] [Google Scholar]
- 49.Yamauchi LM, Aliberti JC, Baruffi MD, Portela RW, Rossi MA, Gazzinelli RT, et al. The binding of CCL2 to the surface of Trypanosoma cruzi induces chemo-attraction and morphogenesis. Microbes Infect. 2007 Jan;9(1):111–118. doi: 10.1016/j.micinf.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 50.Denney H, Clench MR, Woodroofe MN. Cleavage of chemokines CCL2 and CXCL10 by matrix metalloproteinases-2 and-9: implications for chemotaxis. Biochem Biophys Res Commun. 2009 May 1;382(2):341–347. doi: 10.1016/j.bbrc.2009.02.164. [DOI] [PubMed] [Google Scholar]
- 51.Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV. Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006 Mar 31;281(13):8379–8388. doi: 10.1074/jbc.M513122200. [DOI] [PubMed] [Google Scholar]
- 52.Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009 Jul 10;284(28):19053–19066. doi: 10.1074/jbc.M109.000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mucha DR, Myers CL, Schaeffer RC., Jr Endothelial contraction and monolayer hyperpermeability are regulated by Src kinase. Am J Physiol Heart Circ Physiol. 2003 Mar;284(3):H994–H1002. doi: 10.1152/ajpheart.00862.2002. [DOI] [PubMed] [Google Scholar]
- 54.Fincham VJ, Frame MC. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J. 1998 Jan 2;17(1):81–92. doi: 10.1093/emboj/17.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wallez Y, Cand F, Cruzalegui F, Wernstedt C, Souchelnytskyi S, Vilgrain I, et al. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene. 2007 Feb 15;26(7):1067–1077. doi: 10.1038/sj.onc.1209855. [DOI] [PubMed] [Google Scholar]
- 56.Groten T, Pierce AA, Huen AC, Schnaper HW. 17 beta-estradiol transiently disrupts adherens junctions in endothelial cells. FASEB J. 2005 Aug;19(10):1368–1370. doi: 10.1096/fj.04-2558fje. [DOI] [PubMed] [Google Scholar]
- 57.Tinsley JH, Ustinova EE, Xu W, Yuan SY. Src-dependent, neutrophil-mediated vascular hyperpermeability and beta-catenin modification. Am J Physiol Cell Physiol. 2002 Dec;283(6):C1745–C1751. doi: 10.1152/ajpcell.00230.2002. [DOI] [PubMed] [Google Scholar]
- 58.Zhu H, Zhao J, Zhu B, Collazo J, Gal J, Shi P, et al. EMMPRIN regulates cytoskeleton reorganization and cell adhesion in prostate cancer. Prostate. 2012 Jan;72(1):72–81. doi: 10.1002/pros.21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996 Oct;16(10):5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng YF, Kramer RH. Human microvascular endothelial cells express integrin-related complexes that mediate adhesion to the extracellular matrix. J Cell Physiol. 1989 May;139(2):275–286. doi: 10.1002/jcp.1041390209. [DOI] [PubMed] [Google Scholar]
- 61.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995 Oct 12;377(6549):539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 62.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995 Feb;15(2):954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Helmke BP, Goldman RD, Davies PF. Rapid displacement of vimentin intermediate filaments in living endothelial cells exposed to flow. Circ Res. 2000 Apr 14;86(7):745–752. doi: 10.1161/01.res.86.7.745. [DOI] [PubMed] [Google Scholar]
- 64.Angelini DJ, Hyun SW, Grigoryev DN, Garg P, Gong P, Singh IS, et al. TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am J Physiol Lung Cell Mol Physiol. 2006 Dec;291(6):L1232–L1245. doi: 10.1152/ajplung.00109.2006. [DOI] [PubMed] [Google Scholar]
- 65.Lampugnani MG, Corada M, Andriopoulou P, Esser S, Risau W, Dejana E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997 Sep;110(Pt 17):2065–2077. doi: 10.1242/jcs.110.17.2065. [DOI] [PubMed] [Google Scholar]
- 66.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005 Sep 9;280(36):31906–31912. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 67.Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999 Dec 17;274(51):36734–36740. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- 68.Bogatcheva NV, Garcia JG, Verin AD. Role of tyrosine kinase signaling in endothelial cell barrier regulation. Vascul Pharmacol. 2002 Nov;39(4–5):201–212. doi: 10.1016/s1537-1891(03)00009-0. [DOI] [PubMed] [Google Scholar]
- 69.Navaratna D, McGuire PG, Menicucci G, Das A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes. 2007 Sep;56(9):2380–2387. doi: 10.2337/db06-1694. [DOI] [PubMed] [Google Scholar]
- 70.Ilan N, Mahooti S, Rimm DL, Madri JA. PECAM-1 (CD31) functions as a reservoir for and a modulator of tyrosine-phosphorylated beta-catenin. J Cell Sci. 1999 Sep;112(Pt 18):3005–3014. doi: 10.1242/jcs.112.18.3005. [DOI] [PubMed] [Google Scholar]
- 71.Ilan N, Mohsenin A, Cheung L, Madri JA. PECAM-1 shedding during apoptosis generates a membrane-anchored truncated molecule with unique signaling characteristics. FASEB J. 2001 Feb;15(2):362–372. doi: 10.1096/fj.00-0372com. [DOI] [PubMed] [Google Scholar]
- 72.Newman DK, Hamilton C, Newman PJ. Inhibition of antigen-receptor signaling by Platelet Endothelial Cell Adhesion Molecule-1 (CD31) requires functional ITIMs, SHP-2, and p569(lck) Blood. 2001 Apr 15;97(8):2351–2357. doi: 10.1182/blood.v97.8.2351. [DOI] [PubMed] [Google Scholar]
- 73.Cao MY, Huber M, Beauchemin N, Famiglietti J, Albelda SM, Veillette A. Regulation of mouse PECAM-1 tyrosine phosphorylation by the Src and Csk families of protein-tyrosine kinases. J Biol Chem. 1998 Jun 19;273(25):15765–15772. doi: 10.1074/jbc.273.25.15765. [DOI] [PubMed] [Google Scholar]
- 74.Lu TT, Barreuther M, Davis S, Madri JA. Platelet endothelial cell adhesion molecule-1 is phosphorylatable by c-Src, binds Src-Src homology 2 domain, and exhibits immunoreceptor tyrosine-based activation motif-like properties. J Biol Chem. 1997 May 30;272(22):14442–14446. doi: 10.1074/jbc.272.22.14442. [DOI] [PubMed] [Google Scholar]
- 75.Biswas P, Canosa S, Schoenfeld D, Schoenfeld J, Li P, Cheas LC, et al. PECAM-1 affects GSK-3beta-mediated beta-catenin phosphorylation and degradation. Am J Pathol. 2006 Jul;169(1):314–324. doi: 10.2353/ajpath.2006.051112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jackson DE, Kupcho KR, Newman PJ. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of platelet/endothelial cell adhesion molecule-1 (PECAM-1) that are required for the cellular association and activation of the protein-tyrosine phosphatase, SHP-2. J Biol Chem. 1997 Oct 3;272(40):24868–24875. doi: 10.1074/jbc.272.40.24868. [DOI] [PubMed] [Google Scholar]
- 77.Newman PJ. The role of PECAM-1 in vascular cell biology. Ann N Y Acad Sci. 1994 Apr 18;714:165–174. doi: 10.1111/j.1749-6632.1994.tb12041.x. [DOI] [PubMed] [Google Scholar]
- 78.Muller WA, Ratti CM, McDonnell SL, Cohn ZA. A human endothelial cell-restricted, externally disposed plasmalemmal protein enriched in intercellular junctions. J Exp Med. 1989 Aug 1;170(2):399–414. doi: 10.1084/jem.170.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sharief MK, Ciardi M, Thompson EJ. Blood-brain barrier damage in patients with bacterial meningitis: association with tumor necrosis factor-alpha but not interleukin-1 beta. J Infect Dis. 1992 Aug;166(2):350–358. doi: 10.1093/infdis/166.2.350. [DOI] [PubMed] [Google Scholar]
- 80.Del Maschio A, De Luigi A, Martin-Padura I, Brockhaus M, Bartfai T, Fruscella P, et al. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM) J Exp Med. 1999 Nov 1;190(9):1351–1356. doi: 10.1084/jem.190.9.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986 Jun;19(6):525–535. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- 82.Petito CK, Cash KS. Blood-brain barrier abnormalities in the acquired immunodeficiency syndrome: immunohistochemical localization of serum proteins in postmortem brain. Ann Neurol. 1992 Nov;32(5):658–666. doi: 10.1002/ana.410320509. [DOI] [PubMed] [Google Scholar]
- 83.Zilka N, Stozicka Z, Kovac A, Pilipcinec E, Bugos O, Novak M. Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. J Neuroimmunol. 2009 Apr 30;209(1–2):16–25. doi: 10.1016/j.jneuroim.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 84.Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM, et al. Microvascular injury and blood-brain barrier leakage in Alzheimer's disease. Neurobiol Aging. 2007 Jul;28(7):977–986. doi: 10.1016/j.neurobiolaging.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 85.Gay D, Esiri M. Blood-brain barrier damage in acute multiple sclerosis plaques. An immunocytological study. Brain. 1991 Feb;114(Pt 1B):557–572. doi: 10.1093/brain/114.1.557. [DOI] [PubMed] [Google Scholar]
- 86.Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006 Mar 2;354(9):942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 87.Ellis SG, Verity MA. Central nervous system involvement in systemic lupus erythematosus: a review of neuropathologic findings in 57 cases, 1955--1977. Semin Arthritis Rheum. 1979 Feb;8(3):212–221. doi: 10.1016/s0049-0172(79)80009-8. [DOI] [PubMed] [Google Scholar]
- 88.Winfield JB, Shaw M, Silverman LM, Eisenberg RA, Wilson HA, 3rd, Koffler D. Intrathecal IgG synthesis and blood-brain barrier impairment in patients with systemic lupus erythematosus and central nervous system dysfunction. Am J Med. 1983 May;74(5):837–844. doi: 10.1016/0002-9343(83)91075-6. [DOI] [PubMed] [Google Scholar]
- 89.Neumann-Haefelin T, Kastrup A, de Crespigny A, Yenari MA, Ringer T, Sun GH, et al. Serial MRI after transient focal cerebral ischemia in rats: dynamics of tissue injury, blood-brain barrier damage, and edema formation. Stroke. 2000 Aug;31(8):1965–1972. doi: 10.1161/01.str.31.8.1965. discussion 72-3. [DOI] [PubMed] [Google Scholar]
- 90.Barone FC, Hillegass LM, Tzimas MN, Schmidt DB, Foley JJ, White RF, et al. Time-related changes in myeloperoxidase activity and leukotriene B4 receptor binding reflect leukocyte influx in cerebral focal stroke. Mol Chem Neuropathol. 1995 Jan;24(1):13–30. doi: 10.1007/BF03160109. [DOI] [PubMed] [Google Scholar]
- 91.McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol. 1998 Jun 1;86(1):20–29. doi: 10.1016/s0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- 92.Sprenger H, Rosler A, Tonn P, Braune HJ, Huffmann G, Gemsa D. Chemokines in the cerebrospinal fluid of patients with meningitis. Clin Immunol Immunopathol. 1996 Aug;80(2):155–161. doi: 10.1006/clin.1996.0109. [DOI] [PubMed] [Google Scholar]
- 93.Losy J, Zaremba J. Monocyte chemoattractant protein-1 is increased in the cerebrospinal fluid of patients with ischemic stroke. Stroke. 2001 Nov;32(11):2695–2696. doi: 10.1161/hs1101.097380. [DOI] [PubMed] [Google Scholar]
- 94.Bell MD, Taub DD, Perry VH. Overriding the brain's intrinsic resistance to leukocyte recruitment with intraparenchymal injections of recombinant chemokines. Neuroscience. 1996 Sep;74(1):283–292. doi: 10.1016/0306-4522(96)00083-8. [DOI] [PubMed] [Google Scholar]
- 95.Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. Potential role of MCP-1 in endothelial cell tight junction 'opening': signaling via Rho and Rho kinase. J Cell Sci. 2003 Nov 15;116(Pt 22):4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- 96.Yap AS, Brieher WM, Gumbiner BM. Molecular and functional analysis of cadherin-based adherens junctions. Annu Rev Cell Dev Biol. 1997;13:119–146. doi: 10.1146/annurev.cellbio.13.1.119. [DOI] [PubMed] [Google Scholar]
- 97.Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001 Mar;2(3):195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 98.Hagen T, Di Daniel E, Culbert AA, Reith AD. Expression and characterization of GSK-3 mutants and their effect on beta-catenin phosphorylation in intact cells. J Biol Chem. 2002 Jun 28;277(26):23330–23335. doi: 10.1074/jbc.M201364200. [DOI] [PubMed] [Google Scholar]
- 99.Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc Natl Acad Sci U S A. 1999 May 25;96(11):6273–6278. doi: 10.1073/pnas.96.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pellegatta F, Chierchia SL, Zocchi MR. Functional association of platelet endothelial cell adhesion molecule-1 and phosphoinositide 3-kinase in human neutrophils. J Biol Chem. 1998 Oct 23;273(43):27768–27771. doi: 10.1074/jbc.273.43.27768. [DOI] [PubMed] [Google Scholar]
- 101.Hwang IK, Kim DW, Yoo KY, Jung BK, Song JH, Jung JY, et al. Ischemia-induced changes of platelet endothelial cell adhesion molecule-1 in the hippocampal CA1 region in gerbils. Brain Res. 2005 Jun 28;1048(1–2):251–257. doi: 10.1016/j.brainres.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 102.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993 Aug 1;178(2):449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liao F, Ali J, Greene T, Muller WA. Soluble domain 1 of platelet-endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. J Exp Med. 1997 Apr 7;185(7):1349–1357. doi: 10.1084/jem.185.7.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Keuschnigg J, Henttinen T, Auvinen K, Karikoski M, Salmi M, Jalkanen S. The prototype endothelial marker PAL-E is a leukocyte trafficking molecule. Blood. 2009 Jul 9;114(2):478–484. doi: 10.1182/blood-2008-11-188763. [DOI] [PubMed] [Google Scholar]
- 105.Werdich XQ, Penn JS Src, Fyn and Yes play differential roles in VEGF-mediated endothelial cell events. Angiogenesis. 2005;8(4):315–326. doi: 10.1007/s10456-005-9021-x. [DOI] [PubMed] [Google Scholar]
- 106.Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004 Oct 25;167(2):223–229. doi: 10.1083/jcb.200408130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Porkka K, Koskenvesa P, Lundan T, Rimpilainen J, Mustjoki S, Smykla R, et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008 Aug 15;112(4):1005–1012. doi: 10.1182/blood-2008-02-140665. [DOI] [PubMed] [Google Scholar]
- 108.Zhong Y, Zhang B, Eum SY, Toborek M. HIV-1 Tat triggers nuclear localization of ZO-1 via Rho signaling and cAMP response element-binding protein activation. J Neurosci. 2012 Jan 4;32(1):143–150. doi: 10.1523/JNEUROSCI.4266-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sipos E, Chen L, Andras IE, Wrobel J, Zhang B, Pu H, et al. Proinflammatory Adhesion Molecules Facilitate Polychlorinated Biphenyl-Mediated Enhancement of Brain Metastasis Formation. Toxicol Sci. 2012 Jan 12; doi: 10.1093/toxsci/kfr349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.