

Abstract
To illustrate the impact developmental biology and genetics have already had on the clinical management of the million infants born worldwide each year with CHD, we have chosen three stories which have had particular relevance for pediatric cardiologists, cardiothoracic surgeons, cardiac anesthesiologists, and cardiac nurses. First, we show how Margaret Kirby’s finding of the unexpected contribution of an ectodermal cell population – the cranial neural crest – to the aortic arch arteries and arterial pole of the embryonic avian heart provided a key impetus to the field of cardiovascular patterning. Recognition that a majority of patients affected by the neurocristopathy DiGeorge syndrome have a chromosome 22q11 deletion, have also spurred tremendous efforts to characterize the molecular mechanisms contributing to this pathology, assigning a major role to the transcription factor Tbx1. Second, synthesizing the work of the last two decades by many laboratories on a wide gamut of metazoans (invertebrates, tunicates, agnathans, teleosts, lungfish, amphibians, and amniotes), we review the >20 major modifications and additions to the ancient circulatory arrangement composed solely of a unicameral (one-chambered), contractile myocardial tube and a short proximal aorta. Two changes will be discussed in detail – the interposition of a second cardiac chamber in the circulation and the septation of the cardiac ventricle. By comparing the developmental genetic data of several model organisms, we can better understand the origin of the various components of the multicameral (multi-chambered) heart seen in humans. Third, Martina Brueckner’s discovery that a faulty axonemal dynein was responsible for the phenotype of the iv/iv mouse (the first mammalian model of human heterotaxy) focused attention on the biology of cilia. We discuss how even the care of the complex cardiac and non-cardiac anomalies seen in heterotaxy syndrome, which have long seemed impervious to advancements in surgical and medical intensive care (Jacobs et al., 2011), may yet yield to strategies grounded in a better understanding of the cilium. The fact that all cardiac defects seen in patients with full-blown heterotaxy can also be seen in patients without obvious laterality defects hints at important roles for ciliary function not only in left-right axis specification but also in cardiovascular morphogenesis. These three developmental biology stories illustrate how the remaining unexplained mortality and morbidity of congenital heart disease can be solved.
Keywords: “neural crest”, “right ventricle”, “DiGeorge Syndrome”, “heterotaxy syndrome”, cilium, “congenital heart disease”
Introduction
In the 50 years since cardiothoracic surgeons began to correct congenital heart disease (CHD), which constitutes by far the most common fatal birth defect in the human newborn, early postoperative survival has improved from <5% to 95% in the USA (Welke et al., 2009). This dramatic turnaround in the fate of the 1 in 300 infants born with lethal cardiovascular developmental anomalies is one of the great successes of modern medicine (Norwood, 2010). However, there has been little additional progress in early survival since 1999, and long-term (20-year) outcomes have been unexpectedly poor for some subgroups, such as children with “functional single ventricle” - underdevelopment of one ventricular chamber (Khairy et al., 2008). As recently as 2008, the number of deaths from CHD was still double those from all forms of childhood cancer combined (http://www.heart.org/idc/groups/heart-public/@wcm/@sop/@smd/documents/downloadable/ucm_319830.pdf; http://www.cancer.org/acs/groups/content/@nho/documents/document/2008cafffinalsec uredpdf.pdf).
The available evidence suggests that the main reasons for the improvement plateau as well as the surprisingly high attrition among CHD patients over the long-term are: (a) the failure to understand the underlying basis for the CHD in most cases, including the presence of coexisting non-cardiac anomalies, (b) the fragmentary nature of current clinical databases (Welke et al., 2009; Williams, 2010) hampering our ability to correlate genotype with phenotype, and (c) the lack of a rigorous understanding of the performance characteristics of Francis Fontan’s operation for “single ventricle” disorders. As last year was the 40th anniversary of Fontan’s original report (Fontan and Baudet, 1971) describing a way to redirect the systemic venous flow directly into the pulmonary arteries while confining the pulmonary venous flow to the systemic arterial side of the circulation, the surgical treatment of univentricular disorders has been the subject of several recent reviews (Kreutzer et al., 2010; Chin et al., 2010) and will not be covered here.
The proportion of CHD cases likely to be genetic in etiology is at least one third (Formigari et al., 2009) and possibly far higher. Furthermore, in the case of conotruncal disorders occurring in DiGeorge syndrome, we already know that extracardiac anomalies play a major role in the early postoperative course (Ziolkowska et al., 2008; Shen et al., 2011) as well as in long-term outcome (Carotti et al., 2010). Hence, even with the anticipated availability of affordable whole genome sequencing within the next decade, characterizing the full phenotypic spectrum accompanying each genetic alteration will be necessary to optimize the short and long term care of individuals affected by CHD. Since the combination of full-body high-resolution imaging and serum proteomic profiling will remain prohibitively expensive over that time period, a first step is to better understand genotype-phenotype correlations in model organisms. To illustrate the impact developmental biology and genetics have already had on the clinical management of the million infants born worldwide each year with CHD, we have chosen three stories which have had particular relevance for pediatric cardiologists, cardiothoracic surgeons, cardiac anesthesiologists, and cardiac nurses.
First, we show how Margaret Kirby’s finding of the unexpected contribution of an ectodermal cell population – the cranial neural crest – to the aortic arch arteries and arterial pole of the embryonic avian heart provided a key impetus to the field of cardiovascular patterning. Recognition that a majority of patients affected by the neurocristopathy DiGeorge syndrome have a chromosome 22q11 deletion, have also spurred tremendous efforts to characterize the molecular mechanisms contributing to this pathology, assigning a major role to the transcription factor Tbx1. Second, synthesizing the work of the last two decades by many laboratories on a wide gamut of metazoans (invertebrates, tunicates, agnathans, teleosts, lungfish, amphibians, and amniotes), we review the >20 major modifications and additions to the ancient circulatory arrangement composed solely of a unicameral (one-chambered), contractile myocardial tube and a short proximal aorta. Two changes will be discussed in detail – the interposition of a second cardiac chamber in the circulation and the septation of the cardiac ventricle. By comparing the developmental genetic data of several model organisms, we can better understand the origin of the various components of the multicameral (multi-chambered) heart seen in humans. Third, Martina Brueckner’s discovery (Supp et al., 1997) that a faulty axonemal dynein was responsible for the phenotype of the iv/iv mouse (the first mammalian model of human heterotaxy) focused attention on the biology of cilia. We discuss how even the care of the complex cardiac and non-cardiac anomalies seen in heterotaxy syndrome, which have long seemed impervious to advancements in surgical and medical intensive care (Jacobs et al., 2011), may yet yield to strategies grounded in a better understanding of the cilium. The fact that all cardiac defects seen in patients with full-blown heterotaxy can also be seen in patients without obvious laterality defects hints at important roles for ciliary function not only in left-right axis specification but also in cardiovascular morphogenesis. These three developmental biology stories illustrate how the remaining unexplained mortality and morbidity of congenital heart disease can be solved.
On the origin of the cardiac neural crest
Almost 30 years ago, Margaret Kirby and her colleagues published the first evidence linking neural crest (NC) and heart development (Kirby et al., 1983). In this seminal work, sixteen chick embryos were subjected to a bilateral ablation of the NC over somites 1 to 3, a region then referred to as the occipital NC. Among these embryos, fifteen developed aorticopulmonary septal defects, fourteen of which were classified as persistent truncus arteriosus (Kirby et al., 1983), a phenotype well known to occur in humans. This finding was supported by quail-chick chimeras experiments, demonstrating the specific contribution of grafted quail NC cells to the septum of the host chick outflow tract and to the tunica media of the aorta and pulmonary trunk (Kirby et al., 1983). This work was the starting point of the ongoing quest to understand the specific requirements of the cardiac NC in cardiovascular development and its role in the pathogenesis of CHD.
The fate-map of the mammalian cardiac NC took much longer to describe due to the lack of appropriate markers to follow the cardiac NC, and the difficulty of manipulating embryonic tissues in the developing mouse. In mammals, the cardiac NC was first mapped using a transgenic line in which Lac-Z expression was driven by a 6.5kb upstream genomic fragment of the connexin 43 gene (Lo et al., 1997), which is transiently expressed in NC cells and their derivatives (Waldo et al., 1999). Three years later, Cre-Lox technology was used to lineage labeled NC cells in Wnt1-Cre and Pax3-Cre mice (Jiang et al., 2000; Li et al., 2000). From these studies in chick and mouse embryos, it is now well established that the cardiac NC represents a subdivision of the cranial NC, originating from the dorsal neural tube between the middle of the otic placode to the caudal border of somite 4, corresponding to rhombomeres 6–8 of the hindbrain (Fig. 1A; reviewed in Brown and Baldwin, 2006; Snider et al., 2007; Hutson and Kirby, 2007). Cardiac NC cells migrate through pharyngeal arches 3, 4 and 6, and contribute to the pharyngeal glands, thymus, thyroid and parathyroid (Le Lievre and Le Douarin, 1975; Bockman and Kirby, 1984). They also form the smooth muscle layer of the great arteries and play an important role in the remodeling of the five pairs of bilaterally symmetric pharyngeal arch arteries (numbered 1, 2, 3, 4, and 6; the fifth pharyngeal arch arteries are absent in amniotes) that eventually develop into the ascending aorta, proximal subclavian, carotid, and pulmonary arteries as well as the ductus arteriosus (Waldo et al., 1996; Jiang et al., 2000). A subset of cardiac NC cells migrate into the outflow tract cushions of the heart, where they form the spiral aorticopulmonary septum, which divides the truncus into the aorta and pulmonary arteries, thereby separating the systemic and pulmonary circulations at the arterial pole (Kirby et al., 1983; Jiang et al., 2000). A recent study suggests that the NC cells populating the outflow tract cushions also promote semilunar valve leaflet remodeling later in gestation (Jain et al., 2011). The parasympathetic innervation to the heart is also partially derived from the cardiac NC (Kirby and Stewart, 1983; Pietri et al., 2003). Finally, a number of lineage studies have proposed that the mammalian cardiac NC contributes to the myocardium and epicardium, however these observations remain controversial (reviewed in Stoller and Epstein, 2005).
Figure 1. Cardiac NC migration and its contribution to the cardiovascular system.
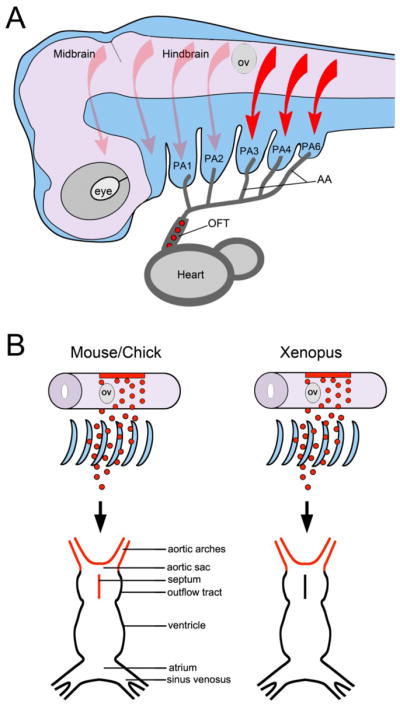
(A) In the mammalian embryo, NC cells delaminate form the dorsal neural tube and migrate in a stereotypical pattern. Based on their origin in the hindbrain (arrows) they populate individual pharyngeal arches (PA). The cardiac NC (red arrows) arises from a region posterior to the otic vesicle (OV) and migrates through pharyngeal arches 3, 4 and 6, along the aortic arch arteries (AA) and into the outflow tract (OFT) of the heart (red circles) to form the aorticopulmonary septum. There are no fifth pharyngeal arch arteries in amniotes. Lateral view, anterior to left, dorsal to top. (B) In chick and mouse the cardiac NC contributes to aortic arch arteries and forms the aorticopulmonary septum of the outflow tract. In Xenopus, cardiac NC cells populate the aortic sac and arch arteries, but do not colonize the outflow tract septum.
Several heart defects are associated with cardiac NC ablation in birds and mouse embryos; however, conotruncal dysmorphologies are the most frequent (Kirby et al., 1983; 1985; Porras and Brown, 2008). They include persistent truncus arteriosus and tetralogy of Fallot (the abnormal positioning, or “overriding”, of the aorta over a large ventricular septal defect permitting persistent communication between the two ventricular chambers) (reviewed in Creazzo et al., 1998). These defects are also present in a number of mouse mutants with cardiac NC deficiencies, with one of the best-studied examples, the Pax3 mouse mutant Splotch (Conway et al., 1997; Li et al, 1999; Epstein et al., 2000). Pax3 is normally expressed in NC progenitors at the dorsal aspect of the neural tube. In the absence of Pax3 function, the NC progenitors pool fails to expand and to complete its migration resulting in scarce cardiac NC derivatives (Conway et al., 1997; Conway et al., 2000). Homozygous Splotch mice die in utero with persistent truncus arteriosus and pharyngeal arch patterning defects (Conway et al., 1997; Epstein et al., 2000).
Like the rest of the NC, cardiac NC cells migrate long distances to reach their final location and are extremely sensitive to influences from adjacent tissues. For that reason, deletion of genes involved in NC migration and patterning can also result in similar cardiac defects in a cell-autonomous or non-cell-autonomous manner. This is the case, for example, with the secreted ligand Semaphorin 3C (Sema3C), an important regulator of cardiac NC cell migration into the outflow tract. Sema3C is not expressed by cardiac NC cells but is expressed by the outflow tract myocardium where it acts as a chemoattractant for the cardiac NC. Targeted deletion of Sema3C in mice causes interrupted aortic arch and persistent truncus arteriosus (Feiner et al., 2001). The cardiac NC cells express the semaphorin receptor, PlexinA2, and PlexinA2 mutant mouse embryos also display persistent truncus arteriosus and interrupted aortic arch (Brown et al., 2001).
Over the last 10 years, a fairly long list of molecules acting in a cell-autonomous or non-cell-autonomous fashion during cardiac NC development has been associated with cardiovascular defects in mouse mutants. These cardiac anomalies can be the result of altered migration, proliferation, survival, or differentiation of cardiac NC progenitors (reviewed in Brown and Baldwin, 2006; Hutson and Kirby, 2007; Snider et al., 2007; Scholl and Kirby, 2009; Nelms and Labosky, 2010). These studies point to a very complex network of genes regulating the development of this cell population and its function in the morphogenesis of the cardiovascular system.
Cardiac NC and the clinical features of 22q11 deletion syndrome
Approximately 10% of patients with conotruncal defects carry a hemizygous microdeletion of Chromosome 22q11 (Lammer et al., 2009). More than half of patients with DiGeorge syndrome (DGS), a condition in which several derivatives of the cardiac NC are affected, have a chromosome 22q11 deletion (Greenberg, 1993). Clinical manifestations include heart defects, craniofacial dysmorphology with or without cleft palate, immunodeficiency and hypocalcemia due to thymus and parathyroid hypoplasia, respectively. Up to one-third of all individuals carrying the 22q11.2 deletion develop schizophrenia or schizoaffective disorder (Murphy, 2002; Karayiorgou, 2010). Similar deletions of the 22q11 region were found in patients with Velo-cardio-facial syndrome (VCFS) and conotruncal anomaly face syndrome (CAFS) (reviewed in Shprintzen, 2008), two other syndromes that share most of the phenotypic attributes of DGS, suggesting a common etiology. DGS, VCFS, and CAFS are now grouped together as the 22q11 deletion syndrome (reviewed in Yamagishi and Srivastava, 2003; Kobrinsky and Sullivan, 2007) with an estimated incidence of 1:4000 live births.
The range of defects seen in the 75% of 22q11 deletion syndrome patients with CHD include, (i) aberrant right subclavian artery (Fig. 2) in patients with left aortic arch; (ii) interruption of the aortic arch type B (Fig. 2), due to abnormal development of the left fourth pharyngeal arch artery, (iii) persistent truncus arteriosus usually accompanied by a large ventricular septal defect (reviewed in Momma, 2010), (iv) tetralogy of Fallot, and (v) tetralogy of Fallot with pulmonary atresia, characterized by malalignment of the great arteries over the ventricular chambers. Importantly, 22q11 deletion is not only a predictor of early postoperative hypocalcemia (Shen et al., 2011), but it is also an independent risk factor for late mortality in tetralogy of Fallot with pulmonary atresia (Carotti et al., 2010).
Figure 2. The two commonest cardiovascular phenotypes seen in 22q11 deletion syndrome are aberrant right subclavian artery and interrupted aortic arch type B.

Left Panel: Volume-rendered image from gadolinium-enhanced 3D magnetic resonance angiography shows interruption of the left fourth aortic arch artery between the left common carotid artery (LCCA) and the left subclavian artery (LSA), a phenotype called type B interrupted aortic arch by clinicians. The right ventricle (RV) ejects blood into the main pulmonary artery (MPA). Without surgical reconstruction of the left fourth aortic arch, viability of the lower body is dependent on continued patency of the ductus arteriosus which allows blood in the MPA to supply the descending aorta, mitigating the effect of the arch interruption. The right subclavian artery in this patient is “aberrant” (ARSA) since it arises from the descending aorta, distal to both the ductus-descending aorta junction and the origin of the LSA. In the normal patient, the first brachiocephalic artery to arise from the aorta is an innominate artery which bifurcates into a RCCA and a RSA. A = ascending aorta; PDA = patent ductus arteriosus; RA = right atrium; RCCA = right common carotid artery. Used with permission from Frank et al., 2010. Right Panel: Suprasternal echocardiographic identification of interrupted aortic arch Type B. Upper left: Frontal (coronal) view showing the takeoffs of the innominate artery (InnA) and LCCA. No connection is seen with the distal aorta. Upper right: Slightly more dorsal (posterior) frontal view showing the MPA, large left-sided PDA, and left-sided upper descending aorta (DescAo). The ascending aorta (AscAo) is not in continuity with the descending aorta. Lower left: Left oblique view showing the LCCA takeoff and no discernible aortic arch. Lower right: sagittal view showing the origin of the left subclavian artery (LSCA) from the DescAo. InnV= innominate vein; l=left; LPA=left pulmonary artery; p, l = posterior and leftward; s=superior; SVC=superior vena cava. Other abbreviations as in Left Panel.
Around 90% of 22q11 deletion syndrome patients have a deleted region of ~3.0 Mb, which contains 60 genes, while the remaining patients have a nested deletion of ~1.5 Mb spanning a region of 35 genes (reviewed in Karayiorgou, 2010). Candidate genes to explain the phenotypes observed in hemizygous individuals include HIRA, encoding a WD40 repeat protein involved in chromatin remodeling (Wilming et al., 1997; Farrell et al., 1999), UFD1L, encoding a factor involved in ubiquitin-mediated protein processing (Pizzuti et al., 1997) and CRKL, encoding an adaptor protein that is implicated in the response to growth factor and focal adhesion signaling (Guris et al., 2001). The strongest candidate, however, is the transcription factor, TBX1 (Chieffo et al., 1997). Since the 22q11.2 deletion is the commonest microdeletion syndrome encountered in humans (Botto et al., 2003), efforts to identify isolated TBX1 deletions or TBX1 point mutations in DGS patients without the large 1.5 Mb or 3.0 Mb 22q11 deletions were initially unsuccessful. In 2003, three TBX1 point mutations were finally identified in a cohort of DGS patients in Japan (Yagi et al., 2003). These mutations were associated with the expected CHD, providing a strong link between TBX1 function, cardiovascular development and the etiology of DGS.
Homozygous Tbx1 mutant mouse embryos have outflow tract septation defects and all the features of a severe DGS phenotype (Jerome and Papaioannou, 2001; Vitelli et al., 2002a). Haploinsufficiency of Tbx1 does not account for all the clinical manifestations associated with 22q11 deletion syndrome; although these animals have aortic arch defects (Lindsay et al., 2001; Merscher et al., 2001), the craniofacial component of the disease is rarely observed. By contrast, human patients with hemizygosity for 22q11 manifest both outflow tract and aortic arch defects as well as craniofacial malformations, suggesting inter-species and tissue-specific differences in Tbx1 dosage sensitivity. It is also important to mention that the severity of the malformations in 22q11 deletion syndrome patients is highly variable, suggesting that other genetic and epigenetic modifiers are involved in the presentation of the disease.
The role of Tbx1 in 22q11 deletion syndrome
Tbx1 is a transcriptional activator that belongs to the T-box gene family, a class of genes implicated in several aspects of cardiac development (reviewed in Greulich et al., 2011). Because Tbx1 is not expressed by premigratory cardiac NC cells or by migrating NC cells as they populate the pharyngeal arches, it is important to emphasize that the defects of NC-derived tissues in 22q11 deletion syndrome are occurring in a non-cell autonomous fashion. Instead, Tbx1 is expressed in all other components of the pharyngeal arch ectoderm, endoderm and mesoderm (Lindsay et al., 2001; Jerome and Papaioannou, 2001; Merscher et al., 2001), as well as in the anterior-most region of the cardiogenic mesoderm, the precursor of the outflow tract (Xu et al., 2004). This expression pattern appears to be well conserved across species (Garg et al., 2001; Piotrowski et al., 2003; Ataliotis et al., 2005; Showell et al., 2006).
The tissue requirements for Tbx1 have been analyzed using a variety of transgenic Cre lines and a conditional Tbx1 allele to mediate tissue-specific inactivation of Tbx1 in components of the pharyngeal arches and in the cardiogenic mesoderm (reviewed in Scambler, 2010). Pharyngeal mesoderm-specific deletion of Tbx1 using Mesp1-Cre resulted in mouse mutants resembling Tbx1-nulls, with severe pharyngeal patterning and cardiovascular defects suggesting a fundamental role for Tbx1 in this tissue (Zhang et al., 2006). However, reactivation of Tbx1 expression in the mesoderm of the mutants corrected the outflow tract septation defect but not the aortic arch phenotype (Zhang et al., 2006), suggesting that the aortic arch defects might be secondary to the pharyngeal segmentation defect. Conditional inactivation of Tbx1 in the cardiogenic mesoderm with Nkx2.5-Cre recapitulates the outflow tract septation defect, but not the aortic arch patterning phenotype of Tbx1 homozygote mutants (Xu et al., 2004). Therefore, it has been proposed that Tbx1 is primarily required in the anterior-most region of the cardiogenic mesoderm to regulate the growth and morphogenesis of the outflow tract, which is a prerequisite for its proper colonization by the cardiac NC (Xu et al., 2004).
A number of studies have reported a genetic link between Fibroblast growth factor 8 (Fgf8), expressed in the pharyngeal endoderm, and the 22q11 phenotype (Abu-Issa et al., 2002; Frank et al., 2002; Vitelli et al., 2002b). Mouse mutants for Fgf8 exhibit the craniofacial and cardiovascular defects typically seen in 22q11 deletion syndrome patients (Frank et al., 2002). Fgf8 expression is downregulated in the pharyngeal endoderm of Tbx1 mutants, and double Tbx1-Fgf8 heterozygote mutants have a higher incidence of aortic arch defects than either single heterozygote mutant (Vitelli et al., 2002b). These observations are consistent with a model in which Tbx1 regulates Fgf8 in the pharyngeal endoderm, which then signals to the adjacent cardiac NC cells to presumably promote their proliferation and/or survival (Abu-Issa et al., 2002; Zhang et al., 2006).
A more recent study has further refined this regulatory cascade providing novel information linking Tbx1 and Fgf8 (Guo et al., 2011). Double homozygote mutant mice for the transcription factor Six1 and its coactivator Eya1 exhibit a phenotype similar to Tbx1 and Fgf8 null embryos, including all the major craniofacial and cardiovascular defects seen in these mutants. Six1/Eya1 double null mutants show reduced Fgf8 expression in the pharyngeal ectoderm and endoderm and in the cardiogenic mesoderm, lineages in which Six1 and Fgf8 are normally co-expressed. Altered cell proliferation and survival in all these tissues appear to be the cause of the cardiovascular phenotypes in Six1/Eya1 mutants (Guo et al., 2011), similar to what was reported for Tbx1 and Fgf8 mouse mutants (Abu-Issa et al., 2002; Zhang et al., 2006). Using chromatin immunoprecipitation, the authors also demonstrate that the Six1/Eya1 transcription complex directly regulates Fgf8 gene expression in vivo (Guo et al., 2011). Overall this study provides strong genetic evidence that a regulatory pathway consisting of Tbx1 → Six1/Eya1 → Fgf8 is critical for the morphogenesis of cardiac and craniofacial structures. This suggests that mutations in SIX1 and EYA1 may contribute to the pathogenesis of 22q11 deletion-like syndromes (Guo et al., 2001), in addition to the well-documented association of SIX1 and EYA1 with branchio-oto-renal syndrome (reviewed in Kochhar et al., 2007).
The analysis of mouse models has provided invaluable information on the roles of TBX1 in the pathogenesis of 22q11 deletion syndrome. While Tbx1 does not directly regulate cardiac NC development, Tbx1 plays a critical role in multiple tissues with which the cardiac NC interacts during its migration, and as such, Tbx1 has a major impact on the ultimate fate of these cells. Efforts in recent years have been focused on characterization of the targets and pathways regulated by Tbx1 and upstream regulators of Tbx1 expression. This knowledge will be critical to understand the molecular mechanisms controlling the deployment of the cardiac NC and the conotruncal defects associated with 22q11 deletion syndrome. This is the starting point for understanding the risk factors for this class of human CHD.
Considerations on the evolution of the cardiac NC
While the cardiac NC has a critical function in outflow tract septation and in remodeling the aortic arch arteries in birds and mammals, its role in lower vertebrates is not as well understood. Zebrafish do not have separate systemic and pulmonary circulation and lack an outflow tract septum, but still possess a cardiac NC. Therefore, the existence of a cardiac NC population predated the need for a separated pulmonary circulation. In zebrafish, the cardiac NC arises from a much broader region of the hindbrain, anterior to the otic vesicle, and has been reported to contribute myocardial cells to all regions of the developing heart, including the outflow tract, atrium and ventricle (Sato and Yost, 2003; Sato et al., 2006; Li et al., 2003). With a single ventricular chamber, the separation of the pulmocutaneous and systemic blood is incomplete in amphibians, however, a spiral septum provides some separation at the level of the outflow tract (de Graaf, 1957; Farmer, 1999). A recent study using Xenopus laevis indicates, however, that the cardiac NC is not required for outflow tract septation. In this organism the spiral septum is exclusively derived from the anterior-most region of the cardiogenic mesoderm (Lee and Saint-Jeannet, 2011). In Xenopus, cardiac NC cells populate the aortic sac and arch arteries but halt their migration before entering the ouflow tract cushions.
How are these differences in the deployment of cardiac NC across species explained? With the transition from an aquatic to a terrestrial life came numerous changes in the cardio-respiratory system, including the complete separation of the pulmonary and systemic circulation. One can speculate that to provide species with the evolutionary advantage of a fully divided circulation, the NC was recruited into the outflow tract septum to complete the separation of the blood at the arterial pole of the heart. Since Xenopus is mostly aquatic, it may represent an exception among amphibians; therefore, it would be interesting to analyze the migration pattern of the cardiac NC in amphibian species that have a predominantly terrestrial lifestyle. If pulmonary respiration has been the evolutionary pressure to redirect migration of the cardiac NC into the outflow tract to complete the separation of the systemic and pulmonary circulations at the arterial pole, one would expect to find cardiac NC cells infiltrating the outflow tract of terrestrial amphibians. The lungfish (Protopterus) is one of the few fish species that possess true lungs. Unlike most amphibians, lungfish have a more complete separation of oxygenated and deoxygenated blood through its cardiac cavities, with a partially septated ventricle and an outflow tract lined with two ridges fused at the distal end providing an incomplete division of the outflow tract (Icardo et al., 2005b). This is an organism in which the fate of the cardiac NC has not been investigated, however, its study may provide important clues on the underlying processes driving the evolution of this cell population.
The fate and the migration pattern of the cardiac NC appears to vary considerably among species, from a broad colonization of all regions of the heart including cardiomyocytes in zebrafish (Sato and Yost, 2003; Sato et al., 2006; Li et al., 2003), to a much more constrained migration that stops short of entering the outflow tract in Xenopus (Lee and Saint-Jeannet, 2011), to a narrowly targeted migration into the outflow tract cushions in chick and mouse (Kirby et al., 1983; Jiang et al., 2000) (Fig. 1B). Defining the factors that regulate the migration of cardiac NC cells once they exit the pharyngeal arches would be especially critical to understand these differences. Because Xenopus cardiac NC cells stop their migration before entering the outflow tract, Xenopus represents an excellent system in which to test factors that may promote the migration of these cells into the outflow tract cushions. Identification of such factors may provide important insights into the CHD resulting from abnormal deployment of the cardiac NC as seen in 22q11 deletion syndromes.
As indicated earlier the cardiac NC makes an important contribution to the autonomic innervation of the heart. The autonomic nervous system consists of two major divisions: the sympathetic and parasympathetic systems. The sympathetic system is organized in two bilateral chains of ganglia running parallel to the spinal cord. The parasympathetic system is lying in or near the innervated organs. In gnathostomes the autonomic innervation of the heart is almost entirely derived from the NC (reviewed in Hildreth et al, 2009). The sympathetic division comes from the trunk NC (Kirby et al., 1983; Jiang et al., 2000), while the parasympathetic system arises from the cardiac NC (Kirby and Stewart, 1983; Pietri et al., 2003), with additional contribution from the nodose placode providing sensory neurons to the distal ganglion of cranial nerve X or vagus nerve (D’Amico-Martel and Noden, 1983). The jawless vertebrates (agnathans), lamprey and hagfish, constitute the most basal vertebrate group, bearing many characteristics of the ancestral vertebrate. They possess NC cells and NC derivatives (McCauley and Bronner-Fraser 2003; Ota et al., 2007) but completely lack the sympathetic ganglia chains (Nicol, 1952; Haming et al., 2011). The lamprey is the first group of chordates with a vagal innervation of the heart, which is anatomically similar to the modern fish two-chambered heart. This innervation, however, differs from the rest of the vertebrates by being excitatory rather than inhibitory. The inhibitory action of the vagus nerve appeared with cartilagenous fishes, and the opposing excitatory sympathetic innervation evolved later with the emergence of bony fishes (reviewed in Burnstock 1969; Wang, 2011). Interestingly, unlike higher vertebrates, the vagal innervation of the heart in the lamprey is entirely placode derived (McCauley and Bronner-Fraser 2003), suggesting that the contribution of the cardiac NC to the parasympathetic division of the autonomic nervous system is a derived character in vertebrate evolution, which has presumably evolved in parallel with the specialization of the heart.
The rise of evolutionary developmental biology
While the chick (Gallus gallus) remained the dominant model system for the study of embryonic heart development for more than 10 years after Kirby’s pioneering experiment, a variety of other organisms have drawn investigators’ attention since the early 1990s. Although invertebrates like Drosophila melanogaster (fruit fly) and Ciona intestinalis (sea squirt) and vertebrates like Danio rerio (zebrafish) and Mus musculus (mouse) have provided the bulk of the insights, lungfish species and amphibians (both anuran and urodele) have contributed in important ways as well. A more extensive history of the evolutionary approach to cardiac developmental biology, with particular focus on the development of the vasculature and the epicardium, was contributed by Pérez-Pomares et al. (2009). Nothing better illustrates the power of using a broad spectrum of animals to unravel the likely genetic and genomic drivers of a morphologically complex organ like the cardiovascular system than the saga of the transcription factor Islet-1 (Isl1).
First identified as a protein which bound to a cis-regulatory element of the rat insulin I gene (Karlsson et al., 1990), Isl1 was rapidly recognized as playing a crucial role in motoneuron development. However, 8 years ago, Cai et al. (2003) reported that Isl1 was expressed in pharyngeal mesoderm and additionally required for proper heart development in the mouse embryo. Isl1 persists postnatally in the outflow tract, right ventricle (RV), and right atrium of the mouse, and the Isl1 null embryo dies at mid-gestation with a missing outflow tract and RV. Based on these observations, it was proposed that an Isl1-positive group of cells in the pharyngeal mesoderm contributing to the arterial pole of the heart evolved in amniotes specifically to give rise to the cardiac segments “associated with the pulmonary circulation” of air-breathing organisms, such as the RV (Olson, 2006). This Isl1-positive group of cells was given the name “the second heart field” (SHF) to distinguish them from progenitors of the left ventricle (LV) which were called “first heart field” (FHF). However, Isl1 expression was also identified in the left atrium in the mouse postnatally as well as in a cardiac pluripotent cell population in both mouse and human (Bu et al., 2009). Of great interest to the field of regenerative medicine, the latter finding also argued for a broader and earlier embryonic function for the Isl1 gene than just the fashioning of the outflow tract, RV, and right atrium. We would predict that not only should Isl1 be among the earliest cardiac markers in the mouse embryo, but also that examination of Isl1 in lower vertebrates and even invertebrates should reveal a required role for Isl1 in cardiac progenitors.
Isl1 has indeed been identified in the precardiac mesoderm of mouse embryos (Cai et al., 2003; Prall et al., 2007). More importantly, analysis of the expression of Isl1 homologs and their loss-of-function phenotypes in Xenopus laevis (Brade et al., 2007), zebrafish (de Pater et al., 2009), lamprey (Kokubo et al., 2010), and the fruit fly (Tao et al., 2007) confirms that Isl1’s role in the heart does not appear to be evolutionarily new. Since none of these animals has a septated ventricle, and since the fly has only one cardiac chamber, the evolutionarily ancient function of Isl1 is in fact pancardiac, not “pulmonary circulation”-related.
In the zebrafish, in which atrial and ventricular precursors are situated near the margin in a ventral-to-dorsal configuration at 40% epiboly, just prior to the onset of gastrulation, fate mapping analysis at 44 hours post fertilization (hpf) reveals pharyngeal mesoderm progenitors at the dorsal-most extent of the ventricular “field” (Keegan et al., 2004) (Fig 3). Cells in this region of the blastula were also found to contribute to the outflow tract at 72 hpf by other investigators (Hami et al., 2011). By immunohistochemistry, Isl1 was observed in cardiac progenitors and pharyngeal mesoderm at 16 hpf as well as in pharyngeal mesoderm adjacent to the arterial pole at 24, 36, and 48, but not 72 hpf, and labeling of pharyngeal mesoderm with the lipophilic dye DiI allowed the tracking of descendants into the outflow tract at 48 and 72 hpf (Hami et al., 2011). By exploiting a photoconvertible fluorescent marker, Lazic and Scott (2011) showed that while the majority of addition of myocardial cells to the arterial pole occurred before 36 hpf, later addition of myocardial cells to the heart did occur and that the addition was exclusively to the arterial pole (Fig. 3). This late-added population of progenitors is Mef2cb-positive (Lazic and Scott, 2011) and Ltbp3-positive (Zhou et al., 2011). These recent papers suggest both an early role for Isl1 as well as evolutionary conservation of late-added myocardial cells at the arterial pole.
Figure 3. Stages of cardiac development in the zebrafish embryo.

(A) Lateral view. At 5 h post fertilization (hpf), the blastula (white) covers approximately 50% of the large yolk cell (yellow). Cardiac progenitor cells are located bilaterally in the lateral marginal zone. Atrial progenitor cells (pink) are located more ventrally (v) than the ventricle progenitor cells (light blue) and pharyngeal mesoderm progenitors (red circles). During gastrulation, the cardiac progenitor cells move dorsally (d) towards the midline to end up in the anterior lateral plate mesoderm by the 12-somite stage (B; B–F are dorsal views). Pharyngeal mesoderm progenitors remain rostral to this region. (C) The endocardial cells (light green) are the first to migrate towards the midline, with the myocardial cells following slightly later. When the bilateral heart fields fuse at the midline, they form a shallow cardiac cone with the endocardial cells in the center, ventricular myocytes at the circumference and atrial myocytes at the periphery (D). Proliferation transforms the cardiac cone into a cardiac tube. The endocardium forms the inner lining of the myocardial tube. (E) By 28 hpf, the elongating heart tube has reoriented from the dorsal-ventral axis to the rostal-caudal axis, with the venous pole swinging to the left while the arterial pole remains fixed at the midline. New cardiomyocytes are added to the arterial pole from the pharyngeal/cranial mesoderm (red). At 36 hpf, cardiac looping is well underway, and the constriction at the position of the AV canal is obvious (F). The heart tube continuous to loop and forms an S-shaped loop (G, ventral view). Ellipsoid extracardiac pro-epicardial cells (brown) are located near the AV canal (yellow), from where they start to cover the myocardium with an epicardial layer. The pacemaker is present in the inner curvature of the atrium near the venous pole (dark green). This figure is modified from Bakkers (2011) and used with permission.
Gessert and Kuhl (2009) showed robust Isl1 expression in the cardiac progenitors at stage 12/13 in Xenopus laevis (late gastrula/ early neurula; roughly analogous to 15 hpf in zebrafish), continuing through stages 15/16 (neurula; similar to 18 hpf in zebrafish), 20 (end of neurulation; roughly equivalent to 22 hpf in zebrafish), and 24 (early tailbud; similar to 26 hpf in zebrafish). It then turns off at stage 24 in Tbx5-positive cells starting terminal differentiation but persists in a domain rostral to the Nkx2.5-positive cells; this rostral domain eventually contributes to the outflow tract.
In addition, a re-examination of the SHF and FHF in the mouse embryo suggests less disparity in Isl1 expression than previously believed. Although cells descended from cardiovascular progenitors are heritably and irreversibly marked by recombination of Cre-activated reporter genes (Soriano, 1999), different floxed loci exhibit differential susceptibility to Cre recombination (Novak et al., 2000; Vooijs et al., 2001). A Gata4-based reporter, Gata4flap, that was more susceptible to Cre recombination than a Rosa26-based reporter (Zhou et al., 2008), was recombined by Isl1Cre and Nkx2-5Cre in a substantially broader domain than previously reported using standard Cre-activated reporters (Ma et al., 2008). The expanded Isl1 and Nkx2-5 cardiac fate maps were remarkably similar, including in both cases extensive contributions to cardiomyocyte, endocardial, and smooth muscle lineages in all four cardiac chambers (Ma et al., 2008). Since Isl1 is expressed in both left and right heart precursors, it does not qualitatively distinguish SHF and FHF progenitors (Ma et al., 2008), a point also emphasized by Pérez-Pomares et al. (2009). Recently, Isl1 derivatives in the heart were also shown to be of neural crest origin (Engleka et al., 2012).
Hence, one potentially unifying model for vertebrates is that there is a common progenitor population in which all cells initially express Isl1, which is extinguished only in those cells starting to terminally differentiate while it remains active for a longer time period in those cells added to the arterial and venous poles of the heart. For chordates, the bHLH transcription factor Mesp1 is upstream of Isl1 and is the earliest marker of multipotent cardiovascular progenitors, descendants of which account for all components of the heart but a small portion of the ventricular conduction system and the outflow cushions (reviewed in Bondue and Blanpain, 2010). The outflow tract and the RV derive from a population of head mesoderm which also gives rise to craniofacial muscles (Lescroart et al., 2010; reviewed in Sambasivan et al., 2011). Even though the concept of two heart fields – one an add-on by air-breathing organisms and one evolutionarily more ancient – has proved to be erroneous, the underlying genetic and genomic basis for the evolution of form in the heart remains a fundamental question for the field of cardiovascular development.
By examining a range of organisms – invertebrates, urochordates, agnathans, teleosts, lungfish, amphibians, and amniotes – we can see at least twenty stepwise, major modifications to the ancient circulatory arrangement, seen in Drosophila. The dorsal vessel of the fly is composed of myocardial cells surrounded by nephrocytes (pericardial cells) (Weaver et al., 2009). Subsequent modifications are seen in the Table. Of these twenty morphological modifications, the two we will discuss in detail – the interposition of a second cardiac chamber between the time of tunicates and the appearance of agnathans; and the post-amphibian sculpting of two equal-sized compartments within the ventricular chamber by making one muscular trabecula taller than all others – have been genetically dissected in recent years. The new understanding of how changes in gene regulation can drive morphological change bears directly on complex human congenital heart disorders. While atrial aplasia has never been reported in humans and atrial hypoplasia is rare, ventricular hypoplasia occurs in >1000 newborns annually in the US with most cases involving an unequal partitioning of the ventricular chamber, such that the LV is much smaller than normal while the chamber to the right of the ventricular septum appears much less affected (Fyler et al., 1980).
The emergence of a two-chambered pump
In the model tunicate Ciona intestinalis, the heart arises from the B7.5 blastomere pair. Ci-Mesp, the sole ortholog of the vertebrate Mesp genes, drives the expression of Ets1/2 in the cells of this pair of blastomeres and all their descendants. (Hirano and Nishida 1997; Davidson and Levine 2003). Following two cell divisions, the rostral B7.5 lineages normally migrate anteriorly to form the heart, and the caudal B7.5 lineages migrate posteriorly to form muscle at the base of the tail (Fig. 4). Targeted expression of a constitutively activated form of Ets1/2, EtsVp16, causes both lineages to migrate into the head transforming the proximal tail muscle lineage into supernumerary heart cells, and nine percent of the juveniles contain beating hearts with two distinct myocardial compartments within a single pericardium (Davidson et al., 2006). Most dramatically, the two compartments are connected and function to drive blood flow efficiently through the juvenile body cavity (Fig. 4E–G). The independence of the two compartments is proved by periodic bouts of asynchronous beating (Davidson et al., 2006). This phenotype is distinct from cardia bifida, in that the two compartments are contained in a single pericardium and constitute a single tube (Davidson et al., 2006). This is an elegant illustration of how a change in the regulation of a transcription factor, rather than a change in its protein-coding region, could underlie a key transition in the emergence of dual-chambered hearts in a basal vertebrate such as the lamprey, namely the recruitment of additional heart precursor cells. All extant vertebrate species including man have hearts with at least two chambers connected in series.
Figure 4. A single change in the regulation of the transcription factor Ets1/2, the Ciona homolog of Ets1, is sufficient to convert a unicameral (one-chambered) heart to a bicameral (two-chambered) heart.

(A) Summary of the gene network controlling heart specification in Ciona. Mesp drives expression of Ets1/2 in all descendants of the B7.5 blastomeres. (B) FGF signaling activates Ets1/2 in the rostral daughters, leading to the expression of FoxF and ultimately to differentiation into heart myocytes. The caudal daughter cells become muscle at the base of the tail. (C) Diagram illustrating a model of chordate heart evolution suggested by the results of experimental manipulation of Ets1/2 activation in Ciona embryos. (D) Control Mesp-GFP transgenic juveniles have a unicameral (one-chambered) heart (h). (E) In Mesp-EtsVp16 transgenic juveniles, expansion of Ets1/2 induction within a broad heart field leads to a fate switch in the caudal B7.5 descendants and to the emergence of two hearts (denoted by 1 and 2). The two hearts are connected to each other, since blood cells (in this still frame, one such blood cell can be seen exiting one chamber) can be easily be tracked visually. In basal vertebrates such as agnathans, this two-heart arrangement could then have been further patterned and modified to form one heart with two distinct chambers, an atrium and a ventricle with an atrio-ventricular valve (C). This composite figure is modified from a figure and still frames from two supplementary movies provided in Davidson et al. (2006) and is used with permission.
Ventricular septation
The evolutionary record of another transcription factor, Tbx5, argues further that it is the more dorsal, morphologic left ventricle (LV), and not the ventral, morphologic RV, which is the more recent modification to the heart. In humans and other mammals, Tbx5 expression correlates with the formation of the ventricular septum (Fig. 5A–D) - high in the left ventricle and low in the right, with a sharp boundary of expression exactly at the location where the septum forms (Koshiba-Takeuchi et al., 2009). The Tbx5 null mouse embryo has a severely hypoplastic LV (Bruneau et al., 2001). During early development in the turtle, an animal with only one ventricle, Tbx5 is expressed throughout its lone ventricular chamber. To prove that the level of Tbx5 is causal of ventricular septum formation rather than merely correlative, the Bruneau lab genetically engineered mice to express Tbx5 at a moderate level throughout the developing heart, as in turtles. Offspring from these mice had only a single ventricle. By mimicking the turtle pattern of Tbx5, these investigators had created mouse hearts, which now resembled turtle hearts (Koshiba-Takeuchi et al., 2009). Therefore, a sharp line delineating an area of high expression of Tbx5 is critical to induce the formation of a ventricular septum, a precursor for the fashioning of a separate, specialized ventricular compartment (Fig. 5). For clinicians who have long judged chambers by their gross anatomical landmarks (e.g., assessment of whether apical trabecular pattern is fine or coarse), a modern, molecular definition of the morphologic LV is that it is the persistently Tbx5-positive and Isl1-negative chamber of the heart.
Figure 5. A sharp spatial gradient of Tbx5 is necessary and sufficient to septate the ventricle.

(A) In the mouse, although the T-box transcription factor Tbx5 is expressed in the cardiac crescent at embryonic day 8.0 (E8.0), it is rapidly extinguished by E8.5 except in the sinus venosus, atrium (a), and the portion of the ventricular chamber destined to be the left ventricle (v) (B). The embryo in Panel C is from slightly later on E8.5 than the embryo in Panel B. (D) E11.5 embryo. (E) In evolution, the emergence of a separated left ventricle correlates with the gradual spatial restriction of the expression of Tbx5. The infant shown with haploinsufficiency for TBX5 (Holt-Oram Syndrome) has muscular ventricular septal defects, holes in the trabecular septum. la=left atrium; lv=left ventricle; ot=outflow tract; ra=right atrium; rv=right ventricle. Panels A through D are from Bruneau et al. (1999) and are reproduced with permission. Panel E is modified from Zina Deretsky, National Science Foundation, after Benoit Bruneau, Gladstone Institute of Cardiovascular Disease (http://www.nsf.gov/news/news_images.jsp?cntn_id1/4115520&org1/4IOS).
For the last 40 years, controversy has raged over whether an infant born with hypoplasia of the LV could ever be expected to have a normal lifespan and quality of life if the surgically reconstructed circulatory pattern required a dependence on the RV. Although patients with transposition of the great arteries (TGA) and large ventricular septal defect (VSD) treated with atrial inversion and VSD closure did experience ongoing attrition unrelated to sinus node dysfunction, the evidence that patients with TGA alone suffered RV dysfunction has not been persuasive (reviewed in Chin et al., 2010). Moreover, although several analyses of Fontan survivors have identified having a morphological RV as a risk factor for poor long-term outcome, other reports disagree (reviewed in Chin et al., 2010).
In addition, it is instructive to examine the question: Are there long-lived univentricular organisms, with high-pressure circulations, which predate the transition to air-breathing and emergence of the LV? Sharks, including the dogfish shark and the whale shark, can live to 100 years (Mattingly et al., 2004; Bradshaw et al., 2007). The systolic blood pressure of shortfin mako sharks, even anesthetized, is 70 mmHg (Lai et al., 1997). They are entirely dependent on a single ventricle. The fiber arrangement and intramyocardial connective tissue architecture is remarkably similar to that of mammals (Sanchez-Quintana et al., 1996), as is their cardiac myosin heavy chain (Franco et al., 2002). By scanning electron microscopy and diffusion tensor magnetic resonance imaging, there is remarkably little difference in the fiber arrangement between the right and left ventricles of the human heart (Fernandez-Teran and Hurle, 1982; Rohmer et al., 2006).
If one defines the mammalian RV as the persistently Isl1-positive chamber, then the RV is really the ancient chamber. An Isl-positive cardiac chamber has existed at least as early as Drosophila melanogaster, in which the islet homolog (tailup) is absolutely required for heart development (Tao et al., 2007). The appearance of the LV or, more rigorously speaking, the two-ventricle configuration does not occur until a change in the spatial expression pattern of Tbx5 occurs (Koshiba-Takeuchi et al., 2009), namely the new imposition of a late, sharp-edged gradient in Tbx5 such that the left part of the ventricular mass has high Tbx5 while the right part of the ventricular mass has low Tbx5. So the mammalian LV, the “Isl1-negative, persistently Tbx5-positive” chamber, is clearly a more recent evolutionary occurrence. There is no obvious Tbx5 homolog in Drosophila, the closest being omb (optomotor blind), and omb is not expressed in the dorsal vessel (Drosophila heart) (Poeck et al., 1993).
Hence, although the LV clearly has outstanding performance characteristics (including inflow and outflow valves which are superior to those of the RV), the RV underwent 250 million years of refinement (the evolutionary distance between fly and fish) before air-breathing animals even arose. The RV is not “an afterthought”, added only to handle a second, low-pressure vascular bed, namely the lungs. It is the sole ventricular chamber for all fish species, amphibians, lizards, and turtles. The RV should not be assumed a priori to be incapable of sustaining the systemic circulation. If the morphologic RV in any individual human patient appears to perform poorly, the cause (whether genetic or acquired) is likely to be specific to that patient rather than a systematic inherent “RV design flaw”.
Finally, whether the emergence of a second atrium in anuran amphibians occurred by: (a) a change in the spatial expression of a single transcription factor which was sufficient to add a second, in-series cardiac chamber as in Ciona (Davidson et al., 2006), (b) a change in the spatial gradient of a transcription factor accentuating a ridge within a chamber as shown by Bruneau in the ventricle of the mouse (Koshiba-Takeuchi et al., 2009), or (c) via a Fgf-mediated reapportionment of fates of marginal progenitor cells at the blastula margin as recently proposed by Simões, based on work in zebrafish (Simões et al., 2011), is still an open question. However, the evolutionarily intermediate atrial septation steps evident in lungfish species (Icardo et al., 2005a) and urodele amphibians (Putnam and Parkerson, 1985) may favor Bruneau’s mechanism (Koshiba-Takeuchi et al., 2009).
Many forms of congenital heart disease may be ciliopathies
Another important question in cardiovascular developmental biology is how the vertebrate heart acquires its chirality. Although most organs are chiral, derived from the Greek kheir meaning “hand”, in that they are not identical to, and cannot be superimposed onto, their mirror image, positioning of the heart tube with respect to the midline and its subsequent bending (looping) constitute the first morphological evidence of left-right asymmetry in the vertebrate embryo.
Although human lateralization disorders have been recognized for at least two centuries (Martin, 1826), only in the last two decades have genetic alterations responsible for their occurrence been identified. In 1997 Martina Brueckner showed that the locus of the iv mutation in mouse (the first mammalian model of incomplete lateralization, or heterotaxy, derived from the Greek heteros meaning “other” and taxis, meaning “order”) encoded an axonemal dynein Lrd (Supp et al., 1997), for which the human homolog is DNAH11/DNAHC11. Since 40% of iv homozygotes have cardiovascular phenotypes resembling those seen in human heterotaxy patients (Icardo and Sanchez de Vega, 1991; Seo et al., 1992), the fact that lrd expression at embryonic day 7.5 was confined to the few hundred ciliated cells of the ventral surface of the node, the fluid-filled pit-shaped structure at the rostral end of the primitive streak, quickly focused investigators’ attention on the cilium.
The cilium is an evolutionarily conserved ancient cellular organelle
Cilia are complex organelles composed of at least 600 different proteins (Pazour et al., 2005) organized around a microtubule based projection called the axoneme. While cilia can be motile or nonmotile, most nondividing cells harbor a single nonmotile primary cilium (Fig. 6; Pazour, 2004). The biological importance of the cilia is indicated by their high degree of conservation despite nearly 2 billion years of evolution. For example, motile cilia in the green alga Chlamydomonas reinhardtii and humans are nearly identical at the ultrastructural level, and the majority of the ciliary proteins identified in Chlamydomonas have clear homologues in humans (Pazour et al., 2005; 2006). The microtubules of the ciliary axoneme are templated from the basal body, a structure derived from the centriole, also a highly conserved organelle (Fig. 6). While the centriole is best known for its roles in mitosis and in organizing the interphase cytoskeleton, it is also postulated to be an important control center of the cell, integrating signals that regulate morphology, migration and proliferation (Satir and Christensen, 2007; Doxsey, 2001).
Figure 6. Mechanism of ciliogenesis.

(A). Schematic diagram showing the transport of proteins into and out of the cilia via intraflagellar transport (IFT) particles mediated by kinesin-II and dynein, respectively. Note the cilium is built on a basal body template that is the mother centriole, with ciliogenesis also requiring BBS gene products. Diagram provided courtesy of Dr. Gregory Pazour (Pazour, 2004). (B). Cross sectional view of motile, tracheal airway cilium, showing the 9+2 arrangement of microtubule doublets (each doublet consisting of an A component with 13 protofilaments and a B component of 10 protofilaments) surrounding a central pair of singlets.
(C, D). Mks1 null mutant fibroblast cells exhibit a primary cilia defect.
Mks1 encodes a protein localized to the centrosome and basal body. In wild-type (WT) mouse embryonic fibroblasts (C), ciliary axoneme is delineated by antibody to acetylated tubulin (red), with the tip and base stained by antibody to IFT140 (green). In contrast, Mks1 null mutant (D) fibroblasts showed IFT140 (green) staining but no obvious cilium can be detected.
Given the complexity and high degree of evolutionary conservation of the cilia, it is perhaps not surprising that cilia assembly is mediated by an elaborate and highly conserved intraflagellar transport (IFT) system which carries ciliary component precursors from sites of synthesis in the cytoplasm to sites of assembly in the cilium (Fig. 6A). IFT transport also plays an essential role in the removal and turnover of proteins in the cilia (Rosenbaum and Witman, 2002; Scholey, 2003). In addition, recent studies show Bardet-Biedl syndrome (BBS) multiprotein complexes also play a role in ciliary assembly and turnover, possibly in transporting membrane proteins to the cilium (Nachury et al. 2007; Berbari et al., 2008), and to the proteasome (Gerdes et al. 2007).
The ciliary axoneme provides the scaffold for motor proteins required for ciliary motion, and also a multitude of other proteins required for sensory transduction of signals detected by the cilia. This includes ciliary membrane localized receptors, such as Patched and Smoothened involved in Hedgehog signaling, a pathway indispensible in cardiovascular development and cardiac septation (Goddeeris et al., 2008; Hoffman et al., 2009), as well as the polycystins which are involved in mechanosensory signal transduction and flow sensing, such as in endothelial cells and renal tubules (Nauli et al. 2003; 2006; Praetorius and Spring, 2001; 2005). Significantly, some of the proteins found in the cilia can traffic to the nucleus and alter transcriptional regulation. For example, the Gli3 transcription factor that mediates Shh signaling is sequestered in the cilia (Haycraft et al., 2005; Huangfu and Anderson, 2005). In fact, the pivotal role that the cilium plays in vertebrate Shh signaling (reviewed in Ingham et al., 2011) may well explain why the most prominent cardiovascular phenotypes reported so far involve not only the chirality of the venous pole but also septation of most portions of the heart - the atria (Hoffmann et al., 2009), the atrioventricular canal (Goddeeris et al., 2008), and the arterial pole (e.g., subpulmonary stenosis associated with double-outlet right ventricle, transposition of the great arteries, or tetralogy of Fallot; subaortic stenosis, sometimes associated with double-outlet right ventricle) (Hoffmann et al., 2009).
Polycystin-1, a protein associated with polycystic kidney disease, is localized to cilia and can form a complex with p100-Stat6 via its cytoplasmic tail. Upon changes in fluid flow, cleavage of the C-terminal cytoplasmic tail allows Stat6 to translocate to the nucleus (Low et al., 2006). The activity of other Stats such as Stat3 requires membrane- anchored polycystin-1 (Talbot et al., 2011). These observations suggest that sensory cilia integrate extracellular signals with changes in transcriptional regulation.
Cilia in development and human diseases
With rapid advances and insight into ciliary biology over the past decade, the cilium has gone from relative obscurity to being recognized as a key player both in vertebrate development and in human diseases. It has become apparent that defects in cilia underlie a large number of important human diseases collectively known as “ciliopathies”. Some ciliopathies such as Leber’s congenital amaurosis (LCA) type 6, characterized by both childhood-onset profound retinal dysfunction and nystagmus (involuntary eye movement) and attributed to mutations in RPGRIP1, cause defects in only one organ. Other ciliopathies like the Bardet-Biedl, Alström, Meckel-Gruber, Joubert’s, Orofaciodigital type 1, Senior-Løken, Jeune asphyxiating thoracic dystrophy, and short rib-polydactyly syndromes cause widespread disease with defects in a variety of organs. The anomalies caused by each of the syndromes vary, but cystic kidney disease, polydactyly, and laterality defects are commonly observed. Obesity, mental retardation, encephalocele, retinal degeneration, lung hypoplasia, deafness, and skeletal and facial abnormalities such as short-limb dwarfism and cleft palate, are more variably observed (Cardenas-Rodriguez and Badano, 2009; Baker and Beales, 2009). The defective genes in the syndromic ciliopathies tend to encode proteins required for basic functions of ciliary assembly and structure, such as basal body and ciliary axonemal proteins, as well as IFT and BBS proteins required for ciliogenesis.
Another class of syndromes known as primary ciliary dyskinesia (PCD), formerly known as Kartagener’s syndrome, is caused by mutations in genes required to produce ciliary force and consequently affect only motile cilia. These syndromes result in highly-penetrant laterality defects, male infertility due to immotile sperm, hydrocephaly, and recurrent respiratory infections (Satir and Christensen, 2007). Patients with PCD develop sinusitis, bronchitis, pneumonia, and eventually bronchiectasis (destruction of bronchial smooth muscle and elastic tissue resulting in dilated airways) due to mucociliary clearance defects resulting from immotile or dyskinetic cilia in the airway. Approximately 50% of PCD patients have the complete mirror-image reversal of visceral organ situs throughout the body, or situs inversus totalis. This reflects the dual requirement for motile cilia in both the specification of left-right patterning during embryonic development (Hirokawa et al., 2006) and in postnatal airway mucociliary clearance.
Virtually all motile cilia possess a central bundle of microtubules, called the axoneme, in which nine outer microtubule doublets (each doublet consisting of an A component with 13 protofilaments and a B component of 10 protofilaments) surround a central pair of singlet microtubules (Fig. 6B). Because ventral node cilia in the mouse embryo are missing the central doublet (i.e., have a “9+0” configuration of microtubule doublets seen in sensory cilia, rather than the “9+2” configuration typically seen in motile cilia), they were originally not believed to be motile. However, closer scrutiny showed that they do in fact move, despite their 9+0 arrangement of microtubules. In fact, uniquely among cilia, they rotate at 600 rpm (Okada et al., 1999). Whereas iv heterozygotes had cilia that rotated at 600 rpm, iv homozygotes had immotile cilia (Okada et al., 1999). Half of mouse embryos missing either Kif3a or Kif3b, components of the kinesin-2 molecular motor which, like dynein, is responsible for IFT along microtubules within cilia, had inverted heart chirality (Nonaka et al., 1998; Marszalek et al., 1999). Inspection of ventral node cells of Kif3b and Kif3a nulls revealed either sporadic, very short cilia or absent cilia (Nonaka et al., 1998; Marszalek et al., 1999).
Since it was hypothesized that a left-right asymmetric cascade of signaling molecules transmitting positional information from the midline node to lateral regions of the embryo was the way chirality was established during gastrulation, it remained to be explained how rotatory ciliary action could produce a consistently leftward (rather than a vortical) flow of signals within the node pit. In a remarkable series of experiments, the Hirokawa lab proved that a posterior tilt in the orientation of the cilia, as well as a difference in the viscous drag at the fluid surface compared with the base of the pit, is necessary and sufficient to cause the fluid within the node to move unidirectionally to the left (reviewed in Hirokawa et al., 2009). In one model, this flow sweeps signaling molecules or sheathed lipidic particles (termed “nodal vesicular” parcels) containing signaling molecules to the left side of the node (Tanaka et al., 2005). In another, termed the “two-cilia hypothesis”, left-right specification further requires calcium signaling, transduced by polycystin-2 and the Pkd1-related protein Pkd1l1 (Field et al., 2011) in nonmotile sensory cilia localized in cells at the node periphery (McGrath and Brueckner, 2003). Finally, activation of a cascade of left-determining genes in the lateral plate mesoderm (the evolutionarily conserved nodal-Pitx2 pathway) completes the establishment of the left-right body axis (Shiratori and Hamada, 2006; Raya and Belmonte, 2006; Shen, 2007). Mutations in genes encoding proteins in the basal body, ciliary axoneme, or in BBS and IFT proteins have all been shown to cause laterality defects.
Identification of cilia defects in human heterotaxy
More recently, cilia mutations implicated in PCD has been shown to play a role not only in situs inversus totalis but also in heterotaxy, the randomization of left-right patterning, in both human and mouse (Kennedy et al.,2007; Tan et al., 2007). As heterotaxy and PCD are both rare phenotypes with an incidence of 1/10,000 to 15,000, this coincidental finding is unlikely to have occurred purely by chance. However, given the inherent genetic heterogeneity of the human population, it was not possible to ascertain whether the heterotaxy was elicited by the PCD-causing genetic lesion or some other lesions in the genetic background of the patients (Kennedy et al.,2007). Using inbred mutant mouse models, we were able to definitively answer this question. Thus, mice homozygous for a mutation in Dnahc5, a motor dynein gene whose human homolog DNAH5 is commonly mutated in patients with PCD (with or without situs inversus totalis), exhibited a 40% incidence of heterotaxy (with 60% being situs solitus or situs inversus) (Fig. 7C). As the PCD mouse model is inbred and thus all animals are genetically identical except for the disease-causing mutation, we can conclude a single mutation can cause both PCD and also heterotaxy. It is significant to note in the Dnahc5 mutant mouse embryos, cilia are retained in the embryonic node, but they are entirely immotile (Fig. 8A, B).
Figure 7. Situs anomalies in Dnahc5 mouse mutants.

(A) Situs solitus with levocardia. The right lung (R) has 4 lobes, and the left lung (L) has 1. Arrow indicates direction of heart (H) apex. The stomach (S) is on the left. (B) Situs inversus totalis with dextrocardia. Note 4 left (labeled 1, 2, 3, and 4) and 1 right (R1) lung lobes, with stomach on the right. (C and D) Heterotaxy with levocardia. Note right aortic arch (RAA), 1 lung lobe on each side (R1 and L1), and stomach on the right (C). Reprinted from Tan et al., 2007.
Figure 8. Nodal cilia in two mutant mouse models with laterality defects.

In the early mouse embryo, cells in the embryonic node have a single motile cilia (A, C). In the Dnahc5 PCD mutant mouse model, cilia in the embryonic node are retained, but the cilia are immotile (B). Homozygous Dnahc5 mutants can exhibit situs solitus, situs inversus or heterotaxy. In comparison, in the Meckel-Gruber syndrome (MKS) mouse model harboring a mutation in Mks1, cells at the node do not have cilia (D, with magnified views in F–H, as compared to magnified view of cilia in wild-type node in E). Mks1 mutants exhibit heterotaxy exclusively. Panels A, B are reprinted from Tan et al., 2007; Panels C–H are reprinted from Cui et al., 2010.
PCD mutant mice exhibiting heterotaxy all died prenatally or neonatally from complex structural heart defects. In contrast, mutants with situs solitus or situs inversus totalis were viable postnatally with no CHD. Clinically, half of PCD patients with heterotaxy also were found to have CHD such as double-outlet right ventricle, common atrioventricular canal, and systemic venous anomalies. Together, these observations suggest cilia play essential roles not only in left-right patterning but also in cardiovascular morphogenesis. They suggest the dynamic events that pattern left-right visceral organ situs are inextricably intertwined with the developmental processes that pattern left-right asymmetries of the heart. Overall, these observations suggest PCD patients and their families should be routinely screened for heterotaxy and for structural heart defects.
The observation that Dnahc5 mutant mice with immotile cilia at the embryonic node are able to establish a consistent left-right body axis more than 50% of the time - either as normal situs solitus or mirror-image situs inversus totalis - suggests that motile cilia function is not absolutely required for breaking symmetry and specifying the left-right body axis. Since mouse mutants with no nodal cilia at all typically exhibit only heterotaxy, such as in the Mks1 mutant mouse model of Meckel-Gruber syndrome (Fig. 8C–H) (Cui et al, 2010), this would argue the normal function of motile cilia at the node may involve an additional, non-motility-related function that may play a primary role in breaking symmetry and specifying the left-right body axis. It should be noted animals such as Xenopus laevis (reviewed in Vandenberg and Levin, 2010), chick (Gros et al., 2009), and pig (Gros et al., 2009) appear to use non-cilia-dependent strategies for breaking symmetry during development. It is interesting to note that Mks1 null mutant fibroblast cells also showed defect in assembly of the primary cilia (Fig. 6D), but this is not observed in all cell types, suggesting possible differences in functional redundancies of proteins required for ciliogenesis.
Ciliary dysfunction affects the management and outcome of congenital heart disease in patients with heterotaxy
Human heterotaxy is usually associated with complex CHD, and such patients must undergo extensive cardiac surgical reconstruction. A number of studies have shown heterotaxy patients have unexplained worse outcomes, with higher postsurgical morbidity and mortality, even after adjusting for surgical complexity. For example, respiratory complications are common and often severe in heterotaxy patients (Swisher et al., 2009). Since sinusitis and bronchiectasis were included with situs inversus totalis in the original Kartagener’s triad (now renamed as PCD), could the ciliary dysfunction in heterotaxy patients also contribute to pre- and post-operative respiratory complications leading to worse outcomes? In particular, it is unclear whether mutations that disrupt nodal cilia function underlying the laterality defects in heterotaxy patients also cause PCD-like mucociliary clearance defects. A small pilot study indeed showed 42% of CHD patients with heterotaxy had ciliary dysfunction similar to that observed in PCD patients (Nakhleh et al., 2012). Screening only by looking for normal-appearing axonemes is insufficient since DNAH11/DNAHC11/Lrd mutated cilia appear normal, even while demonstrating abnormal beating behavior on high-speed video microscopy (Schwabe et al., 2008). These findings suggest the possibility that undiagnosed ciliary dysfunction overlapping with that of PCD may contribute to respiratory complications and worse outcomes in heterotaxy patients (Swisher et al., 2011). While replication of these findings in a larger cohort together with outcome studies are needed to validate the potential mechanistic link between ciliary dysfunction and respiratory complications in CHD patients with heterotaxy, these findings show the potential value of insights into disease mechanism for improving patient care.
Patients with LCA type 10 are reported to have abnormal airway ciliary motility together with a clinical history of recurrent inflammatory diseases of the airway (Papon et al., 2010). These findings suggest increased respiratory disease associated with motile ciliary dysfunction and may have relevance for other ciliopathies. As the disease in LCA patients arises from a defect in the primary cilium (of photoreceptor cells), the connection to respiratory disease may be much broader, extending to disorders involving either motile or nonmotile cilia.
Summary
Although the progress in surgical reconstructive techniques in a half-century commonly called the Golden Age of Cardiology has been astounding, most of the remaining improvements in the care of patients with congenital heart disease will depend on an understanding of the developmental biology and genetics of the metazoan cardiovascular system. Even though these latter fields have blossomed only in the last 30 years, they have already yielded considerable insights relevant to clinicians, as exemplified by the above vignettes on outflow tract development, “single ventricle” disorders, and ciliopathies with or without heterotaxy. Over the next 30 years, progressively more powerful tools and databases will become available to the biomedical community. In addition, the fields of stem cell biology and regenerative medicine may well contribute to a second Golden Age of Cardiology.
Table.
Chordate phylogeny and twenty major modifications to the ancient circulatory arrangement seen in Drosophila melanogaster.
| ||
|---|---|---|
| Organism | Modifications | References |
| tunicates | Addition of a vasculature | Davidson, 2007 |
| agnathans | Appearance of a separate endocardial cell layer | Hatta, 1897 |
| Appearance of desmosome as a myocyte linker | Grosskurth et al., 2008 | |
| Interposition of a second cardiac chamber | Kokubo et al., 2010 | |
| the sculpting of a valve between those two chambers providing for unidirectional circulation | Tahara et al., 1988 | |
| the emergence of the epicardium (visceral pericardium) from the pronephric external glomerulus | Pombal et al., 2008; reviewed in Pérez- Pomares et al., 2009 | |
| the appearance of two myocardial zones (trabecular and compact | ||
| development of neural crest cells required for normal assembly of the ventral aorta | Newth, 1956 | |
| teleosts | the appearance of coronary circulation | Tota et al., 1983 |
| chiral, rather than ventral-dorsal, looping of the heart tube | Senior, 1909 | |
| the eventual reliance on the gut-derived swimbladder as not only a buoyancy organ but also as a respiratory organ | Graham, 1997 | |
| lungfishes | complete co-option of the swimbladder as a lung communicating with the systemic circulation by means of a pulmonary vein passing across the roof of the sinus venosus to the atria divided only at their caudalmost extent | Icardo et al., 2005a |
| emergence of outflow tract ridges | (Icardo et al., 2005b | |
| appearance of pulmonary arteries as branches from the sixth aortic arch arteries bilaterally | Johansen and Hanson, 1968 | |
| amphibians | septation of the pharynx to yield a trachea and bronchi | |
| complete separation of the pulmonary venous return and the systemic venous return | ||
| various degrees of atrial septation | Putnam and Parkerson, 1985 | |
| amniotes | progressive separation of the streams at the arterial pole | |
| “walling off” of the left ventricle (LV) | Koshiba-Takeuchi et al., 2009 | |
| decrease in the LV’s trabecular/compact zone ratio with increasing workloads | Eliason et al., 2011 | |
22q11 hemizygosity: aberrant right subclavian artery and interrupted aortic arch type B.
One change in the regulation of Ciona Ets1/2 is sufficient to convert a unicameral heart to a bicameral one.
A sharp spatial gradient of Tbx5 is necessary and sufficient to septate the ventricle.
Cilia mutations cause both heterotaxy and situs inversus totalis.
Ciliary dysfunction affects the outcome of congenital heart disease in patients with heterotaxy.
Acknowledgments
We are grateful to Drs. Patricia Labosky, Karl Degenhardt, and Lazaros Kochilas, as well as Ms. Allison Williams for comments on the manuscript. This work was supported by grants from the Children’s Heart Foundation and the Children’s Hospital of Philadelphia Cardiac Center to AJC and by the National Institutes of Health to J-P S-J (R01-DE014212) and CWL (U01-HL098180).
Footnotes
The corresponding author attests that no undisclosed authors contributed to the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- Ataliotis P, Ivins S, Mohun TJ, Scambler PJ. XTbx1 is a transcriptional activator involved in head and pharyngeal arch development in Xenopus laevis. Dev Dyn. 2005;232:979–991. doi: 10.1002/dvdy.20276. [DOI] [PubMed] [Google Scholar]
- Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- Bakkers J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc Res. 2011;91:279–288. doi: 10.1093/cvr/cvr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol Biol Cell. 2008;19:1540–1547. doi: 10.1091/mbc.E07-09-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockman DE, Kirby ML. Dependence of thymus development on derivatives of the neural crest. Science. 1984;223:498–500. doi: 10.1126/science.6606851. [DOI] [PubMed] [Google Scholar]
- Bondue A, Blanpain C. Mesp1. A key regulator of cardiovascular lineage commitment. Circulation Research. 2010;107:1414–1422. doi: 10.1161/CIRCRESAHA.110.227058. [DOI] [PubMed] [Google Scholar]
- Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O’Leary LA, Wong L-Y, Elixson EM, Mahle WT, Campbell RM. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- Brade T, Gessert S, Kühl M, Pandur P. The amphibian second heart field: Xenopus islet-1 is required for cardiovascular development. Dev Biol. 2007;311:297–310. doi: 10.1016/j.ydbio.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Bradshaw CJA, Mollet HF, Meekan MG. Inferring population trends for the world’s largest fish from mark-recapture estimates of survival. J Anim Ecol. 2007;76:480–489. doi: 10.1111/j.1365-2656.2006.01201.x. [DOI] [PubMed] [Google Scholar]
- Brown CB, Feiner L, Lu MM, Li J, Ma XK, Webber AL, Jia L, Raper JA, Epstein JA. PlexinA2 and semaphorin signaling during cardiac neural crest development. Development. 2001;128:3071–3080. doi: 10.1242/dev.128.16.3071. [DOI] [PubMed] [Google Scholar]
- Brown CB, Baldwin HS. Neural crest contribution to the cardiovascular system. Adv Exp Med Biol. 2006;589:134–154. doi: 10.1007/978-0-387-46954-6_8. [DOI] [PubMed] [Google Scholar]
- Bruneau BG, Logan M, Davis N, Levi T, Tabin CJ, Seidman JG, Seidman CE. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram Syndrome. Dev Biol. 1999;211:100–108. doi: 10.1006/dbio.1999.9298. [DOI] [PubMed] [Google Scholar]
- Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. A murine model of Holt-Oram Syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–721. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, Roberts DJ, Huang PL, Domian IJ, Chien KR. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature. 2009;460:113–7. doi: 10.1038/nature08191. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Evolution of the autonomic innervation of visceral and cardiovascular systems in vertebrates. Pharmcol Rev. 1969;21:247–324. [PubMed] [Google Scholar]
- Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 Identifies a Cardiac Progenitor Population that Proliferates Prior to Differentiation and Contributes a Majority of Cells to the Heart. Developmental Cell. 2003;25:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas-Rodriguez M, Badano JL. Ciliary biology: understanding the cellular and genetic basis of human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:263–280. doi: 10.1002/ajmg.c.30227. [DOI] [PubMed] [Google Scholar]
- Carotti A, Albanese SB, Filippelli S, Rava L, Guccione P, Pongiglione P, DiDonato RM. Determinants of outcome after surgical treatment of pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries. J Thorac Cardiovasc Surg. 2010;140:1092–1103. doi: 10.1016/j.jtcvs.2010.07.087. [DOI] [PubMed] [Google Scholar]
- Chieffo C, Garvey N, Gong W, Roe B, Zhang G, Silver L, Emanuel BS, Budarf ML. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics. 1997;43:267–277. doi: 10.1006/geno.1997.4829. [DOI] [PubMed] [Google Scholar]
- Chin AJ, Whitehead KK, Watrous RL. Insights After 40 Years of the Fontan Operation. World Journal for Pediatric and Congenital Heart Surgery. 2010;1:328–343. doi: 10.1177/2150135110379623. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Henderson DJ, Copp AJ. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development. 1997;124:505–514. doi: 10.1242/dev.124.2.505. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Bundy J, Chen J, Dickman E, Rogers R, Will BM. Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovasc Res. 2000;47:314–328. doi: 10.1016/s0008-6363(00)00098-5. [DOI] [PubMed] [Google Scholar]
- Creazzo TL, Godt RE, Leatherbury L, Conway S, Kirby ML. Role of cardiac neural crest cells in cardiovascular development. Annu Rev Physiol. 1998;60:267–286. doi: 10.1146/annurev.physiol.60.1.267. [DOI] [PubMed] [Google Scholar]
- Cui C, Chatterjee B, Francis D, Yu Q, SanAgustin JT, Francis R, Tansey T, Henry C, Wang B, Lemley B, Pazour GJ, Lo CW. Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel-Gruber Syndrome. Disease Mechanisms and Models. 2010;4:43–56. doi: 10.1242/dmm.006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’amico-Martel A, Noden DM. Contributions of placodal and neural crest cells to avian cranial peripheral ganglia. Am J Anat. 1983;166:445–468. doi: 10.1002/aja.1001660406. [DOI] [PubMed] [Google Scholar]
- Davidson B, Levine M. Evolutionary origins of the vertebrate heart: Specification of the cardiac lineage in Ciona intestinalis. Proc Natl Acad Sci. 2003;100:11469–11473. doi: 10.1073/pnas.1634991100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson B, Shi W, Beh J, Christiaen L, Levine M. FGF signaling delineates the cardiac progenitor field in the simple chordate, Ciona intestinalis. Genes Dev. 2006;20:2728–2738. doi: 10.1101/gad.1467706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson B. Ciona intestinalis as a model for cardiac development. Semin Cell Dev Biol. 2007;18:16–26. doi: 10.1016/j.semcdb.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf AR. Investigations into the distribution of blood in the heart and aortic arches of Xenopus laevis. J Exp Biol. 1957;34:143–172. [Google Scholar]
- de Pater E, Clijsters L, Marques SR, Lin YF, Garavito-Aguilar ZV, Yelon D, Bakkers J. Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development. 2009;136:1633–1641. doi: 10.1242/dev.030924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxsey S. Re-evaluating centrosome function. Nat Rev Mol Cell Biol. 2001;2:688–698. doi: 10.1038/35089575. [DOI] [PubMed] [Google Scholar]
- Eliason EJ, Clark TD, Hague MJ, Hanson LM, Gallagher ZS, Jeffries KM, Gale MK, Patterson DA, Hinch SG, Farrell AP. Differences in thermal tolerance among sockeye salmon populations. Science. 2011;332:109–112. doi: 10.1126/science.1199158. [DOI] [PubMed] [Google Scholar]
- Engleka KA, Manderfield LJ, Brust RD, Li L, Cohen A, Dymecki SM, Epstein JA. Islet1 derivatives in the heart are of both neural crest and second heart field origin. Circ Res. 2012;110:922–926. doi: 10.1161/CIRCRESAHA.112.266510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein JA, Li J, Lang D, Chen F, Brown CB, Jin FZ, Lu MM, Thomas M, Liu E, Wessels A, Lo CW. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127:1869–1878. doi: 10.1242/dev.127.9.1869. [DOI] [PubMed] [Google Scholar]
- Farmer CG. Evolution of the vertebrate cardio-pulmonary system. Ann Rev Physiol. 1999;61:573–592. doi: 10.1146/annurev.physiol.61.1.573. [DOI] [PubMed] [Google Scholar]
- Farrell MJ, Stadt H, Wallis KT, Scambler P, Hixon RL, Wolfe R, Leatherbury L, Kirby ML. HIRA, a DiGeorge syndrome candidate gene, is required for cardiac outflow tract septation. Circ Res. 1999;84:127–135. doi: 10.1161/01.res.84.2.127. [DOI] [PubMed] [Google Scholar]
- Feiner L, Webber AL, Brown CB, Lu MM, Jia L, Feinstein P, Mombaerts P, Epstein JA, Raper JA. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development. 2001;128:3061–3070. doi: 10.1242/dev.128.16.3061. [DOI] [PubMed] [Google Scholar]
- Fernandez-Teran MA, Hurle JM. Myocardial fiber architecture of the human heart ventricles. Anat Rec. 1982;204:137–147. doi: 10.1002/ar.1092040207. [DOI] [PubMed] [Google Scholar]
- Field S, Riley KL, Grimes DT, Hilton H, Simon M, Powles-Glover N, Siggers P, Bogani D, Greenfield A, Norris DP. Pkd1/1 establishes left-right asymmetry and physically interacts with Pkd2. Development. 2011;138:1131–1142. doi: 10.1242/dev.058149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontan F, Baudet E. Surgical repair of tricuspid atresia. Thorax. 1971;26:240–248. doi: 10.1136/thx.26.3.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formigari R, Michielon G, Digilio MC, Piacentini G, Carotti A, Giardini A, Di Donato RM, Marino B. Genetic syndromes and congenital heart defects: how is surgical management affected? Eur J Cardiothorac Surg. 2009;35:606–614. doi: 10.1016/j.ejcts.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Franco D, Gallego A, Habets PEMH, Sans-Coma V, Moorman AFM. Species-specific differences of myosin content in the developing cardiac chambers of fish, birds, and mammals. The Anatomical Record. 2002;268:27–37. doi: 10.1002/ar.10126. [DOI] [PubMed] [Google Scholar]
- Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–4603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank L, Dillman JR, Parish V, Mueller GC, Kazerooni EA, Bell A, Attili AK. Cardiovascular MR imaging of conotruncal anomalies. RadioGraphics. 2010;30:1069–1094. doi: 10.1148/rg.304095158. [DOI] [PubMed] [Google Scholar]
- Fyler DC, Buckley LP, Hellenbrand WE, Cohn HE. Report of the New England Regional Infant Cardiac Program. Pediatrics. 1980;65:375–461. [PubMed] [Google Scholar]
- Garg V, Yamagishi C, Hu T, Kathiriya IS, Yamagishi H, Srivastava D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev Biol. 2001;235:62–73. doi: 10.1006/dbio.2001.0283. [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, Badano JL, Katsanis N. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet. 39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- Gessert S, Kühl M. Comparative gene expression analysis and fate mapping studies suggest an early segregation of cardiogenic lineages in Xenopus laevis. Dev Biol. 2009;334:395–408. doi: 10.1016/j.ydbio.2009.07.037. [DOI] [PubMed] [Google Scholar]
- Goddeeris MM, Rho S, Petiet A, Davenport CK, Johnson GA, Meyers EN, Klingensmith J. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development. 2008;135:1887–1895. doi: 10.1242/dev.016147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JB. Air-breathing fishes evolution, diversity, and adaptation. Academic Press; San Diego: 1997. p. 299. [Google Scholar]
- Greenberg F. DiGeorge syndrome: an historical review of clinical and cytogenetic features. J Med Genet. 1993;30:803–806. doi: 10.1136/jmg.30.10.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich F, Rudat C, Kispert A. Mechanisms of T-box gene function in the developing heart. Cardiovasc Res. 2011;91:212–222. doi: 10.1093/cvr/cvr112. [DOI] [PubMed] [Google Scholar]
- Gros J, Feistel K, Viebahn C, Blum M, Tabin CJ. Cell movements at Hensen’s node establish left/right asymmetric gene expression in the chick. Science. 2009;324:941–944. doi: 10.1126/science.1172478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskurth SE, Bhattacharya D, Wang Q, Lin JJ-C. Emergence of Xin Demarcates a Key Innovation in Heart Evolution. PLoS ONE. 2008;3(8):e2857. doi: 10.1371/journal.pone.0002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Sun Y, Zhou B, Adam RM, Li X, Pu WT, Morrow BE, Moon A, Li X. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J Clin Invest. 2011;121:1585–1595. doi: 10.1172/JCI44630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–298. doi: 10.1038/85855. [DOI] [PubMed] [Google Scholar]
- Hami D, Grimes AC, Tsai H-J, Kirby ML. Zebrafish cardiac development requires a conserved secondary heart field. Development. 2011;138:2389–2398. doi: 10.1242/dev.061473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta S. Contribution to the morphology of cyclostomata. I On the formation of the heart in Petromyzon. J Coll Sci Imp Univ Tokyo. 1897;10:225–237. [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildreth V, Anderson RH, Henderson DJ. Autonomic innervation of the developing heart: origins and function. Clin Anat. 2009;22:36–46. doi: 10.1002/ca.20695. [DOI] [PubMed] [Google Scholar]
- Hirano T, Nishida H. Developmental fates of larval tissues after metamorphosis in ascidian Halocynthia roretzi I Origin of mesodermal tissues of the juvenile. Dev Biol. 1997;192:199–210. doi: 10.1006/dbio.1997.8772. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Tanaka K, Okada Y, Takeda S. Nodal flow and the generation of left-right asymmetry. Cell. 2006;125:33–45. doi: 10.1016/j.cell.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Tanaka Y, Okada Y. Left Right Determination: Involvement of Molecular Motor KIF3, Cilia, and Nodal Flow. Cold Spring Harb Perspect Biol 2009. 2009;1:a000802. doi: 10.1101/cshperspect.a000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AD, Peterson MA, Friedland-Little JM, Anderson SA, Moskowitz IP. Sonic hedgehog is required in the pulmonary endoderm for atrial septation. Development. 2009;136:1761–1770. doi: 10.1242/dev.034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and hedgehog responsiveness in the mouse. PNAS. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol. 2007;18:101–110. doi: 10.1016/j.semcdb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Icardo JM, Sanchez de Vega MJ. Spectrum of heart malformations in mice with situs solitus, situs inversus, and associated visceral heterotaxy. Circulation. 1991;84:2547–58. doi: 10.1161/01.cir.84.6.2547. [DOI] [PubMed] [Google Scholar]
- Icardo JM, Ojeda JL, Colvee E, Tota B, Wong WP, Ip YK. Heart inflow tract of the African lungfish Protopterus dolloi. J Morph. 2005a;263:30–38. doi: 10.1002/jmor.10286. [DOI] [PubMed] [Google Scholar]
- Icardo JM, Brunelli E, Perrotta I, Colvee E, Wong WP, Ip YK. Ventricle and outflow tract of the African lungfish Protopterus dolloi. J Morph. 2005b;265:43–51. doi: 10.1002/jmor.10340. [DOI] [PubMed] [Google Scholar]
- Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signaling across the metazoa. Nat Rev Genet. 2011;12:393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- Jacobs JP, Pasquali SK, Morales DLS, Jacobs ML, Mavroudis C, Chai PJ, Tchervenkov CI, Lacour-Gayet FG, Walters H, III, Quintessenza JA. Heterotaxy: Lessons Learned About Patterns of Practice and Outcomes From the Congenital Heart Surgery Database of the Society of Thoracic Surgeons. World Journal for Pediatric and Congenital Heart Surgery. 2011;2:278–286. doi: 10.1177/2150135110397670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Engleka KA, Rentschler SL, Manderfield LJ, Li L, Yuan L, Epstein JA. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J Clin Invest. 2011;121:422–430. doi: 10.1172/JCI44244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Johansen K, Hanson D. Functional anatomy of the hearts of lungfishes and amphibians. Am Zool. 1968;8:191–210. doi: 10.1093/icb/8.2.191. [DOI] [PubMed] [Google Scholar]
- Karayiorgou M, Simon TJ, Gogos JA. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nature Reviews Neuroscience. 2010;11:402–416. doi: 10.1038/nrn2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson O, Thor S, Norberg T, Ohlsson H, Edlund T. Insulin gene enhancer binding protein Isl-1 is a member of a novel class of proteins containing both a homeo-and a Cys His domain. Nature. 1990;344:879–882. doi: 10.1038/344879a0. [DOI] [PubMed] [Google Scholar]
- Keegan BR, Meyer D, Yelon D. Organization of cardiac chamber progenitors in the zebrafish blastula. Development. 2004;131:3081–3091. doi: 10.1242/dev.01185. [DOI] [PubMed] [Google Scholar]
- Kennedy MP, Omran H, Leigh MW, Dell S, Morgan L, Molina PL, Robinson BV, Minnix SK, Olbrich H, Severin T, Ahrens P, Lange L, Morillas HN, Noone PG, Zariwala MA, Knowles MR. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- Khairy P, Fernandes SM, Mayer JE, Jr, Triedman JK, Walsh EP, Lock JE, Landzberg MJ. Long-term survival, modes of death, and predictors of mortality in patients with Fontan surgery. Circulation. 2008;117:85–92. doi: 10.1161/CIRCULATIONAHA.107.738559. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Stewart DE. Neural crest origin of cardiac ganglion cells in the chick embryo: identification and extirpation. Dev Biol. 1983;97:433–443. doi: 10.1016/0012-1606(83)90100-8. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to aorticopulmonary septation. Science. 1983;220:1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Turnage KL, Hays BM. Characterization of conotruncal malformations following ablation of “cardiac” neural crest. Anat Rec. 1985;213:87–93. doi: 10.1002/ar.1092130112. [DOI] [PubMed] [Google Scholar]
- Kobrinsky LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- Kochhar A, Fischer SM, Kimberling WJ, Smith RJH. Branchio-oto-renal syndrome. Am J Med Genet. 2007;143A:1671–1678. doi: 10.1002/ajmg.a.31561. [DOI] [PubMed] [Google Scholar]
- Kokubo N, Matsuura M, Onimaru K, Tiecke E, Kuraku S, Kuratani S, Tanaka M. Mechanisms of heart development in the Japanese lamprey, Lethenteron japonicum. Evolution and Development. 2010;12:34–44. doi: 10.1111/j.1525-142X.2009.00389.x. [DOI] [PubMed] [Google Scholar]
- Koshiba-Takeuchi K, Mori AD, Kaynak BL, Cebra-Thomas J, Sukonnik T, Georges RO, Latham S, Beck L, Henkelman RM, Black BL, Olson EN, Wade J, Takeuchi JK, Nemer M, Gilbert SF, Bruneau BG. Reptilian heart development and the molecular basis of cardiac chamber evolution. Nature. 2009;461:95–98. doi: 10.1038/nature08324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzer GO, Schlichter AJ, Kreutzer C. The Fontan/Kreutzer Procedure At 40: An Operation for the Correction of Tricuspid Atresia. Seminars in thoracic and cardiovascular surgery. Pediatric cardiac surgery annual. 2010;13:84–90. doi: 10.1053/j.pcsu.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Kureshi S, Nakhleh N, Setton M, Giese R, Francis R, Chatterjee B, Dami I, Kuehl K, Olivier KN, Jonas R, Tian X, Leigh MW, Knowles MR, Leatherbury L, Lo CW. Nasal nitric oxide and ciliary function in patients with non-heterotaxy congenital heart disease. Am J Resp Crit Care Med. 2010;181:A3323. [Google Scholar]
- Lai NC, Korsmeyer KE, Katz S, Holts DB, Laughlin LM, Graham JB. Hemodynamics and Blood Properties of the Shortfin Mako Shark, Isurus oxyrinchus. Copeia. 1997;2:424–428. [Google Scholar]
- Lammer EJ, Chak JS, Iovannisci DM, Schultz K, Osoegawa K, Yang W, Carmichael SL, Shaw GM. Chromosomal abnormalities among children born with conotruncal cardiac defects. Birth Defects Research Part A: Clinical and Molecular Teratology. 2009;85:30–35. doi: 10.1002/bdra.20541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazic S, Scott IC. Mef2cb regulates late myocardial cell addition from a second heart field-like population of progenitors in zebrafish. Dev Biol. 2011;354:123–133. doi: 10.1016/j.ydbio.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Lee YH, Saint-Jeannet JP. Cardiac neural crest is dispensable for outflow tract septation in Xenopus. Development. 2011;138:2025–2034. doi: 10.1242/dev.061614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Lievre CS, Le Douarin NM. Mesenchymal derivatives of the neural crest. Analysis of chimaeric quail and chick embryos. J Embryol Exp Morphol. 1975;34:125–154. [PubMed] [Google Scholar]
- Lescroart F, Kelly RG, Le Garrec JF, Nicolas JF, Meilhac SM, Muckingham M. Clonal analysis reveals common lineage relationships between head muscles and second heart field derivatives in the mouse embryo. Development. 2010;137:3269–3279. doi: 10.1242/dev.050674. [DOI] [PubMed] [Google Scholar]
- Li J, Liu KC, Jin F, Lu MM, Epstein JA. Transgenic rescue of congenital heart disease and spina bifida in Splotch mice. Development. 1999;126:2495–2503. doi: 10.1242/dev.126.11.2495. [DOI] [PubMed] [Google Scholar]
- Li J, Chen F, Epstein JA. Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis. 2000;26:162–164. doi: 10.1002/(sici)1526-968x(200002)26:2<162::aid-gene21>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Li YX, Zdanowicz M, Young L, Kumiski D, Leatherbury L, Kirby ML. Cardiac neural crest in zebrafish embryos contributes to myocardial cell lineage and early heart function. Dev Dyn. 2003;226:540–550. doi: 10.1002/dvdy.10264. [DOI] [PubMed] [Google Scholar]
- Lindsay EA. Chromosome microdeletions: dissecting del22q11 syndrome. Nat Rev Genet. 2001;2:858–868. doi: 10.1038/35098574. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- Lo CW, Cohen MF, Huang GY, Lazatin BO, Patel N, Sullivan R, Pauken C, Park SM. Cx43 gap junction gene expression and gap junctional communication in mouse neural crest cells. Dev Genet. 1997;20:119–132. doi: 10.1002/(SICI)1520-6408(1997)20:2<119::AID-DVG5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, Kane ME, Obara T, Weimbs T. Poycystin-1, STAT6, and p100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell. 2006;10:57–69. doi: 10.1016/j.devcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Ma Q, Zhou B, Pu WT. Reassessment of Isl1 and Nkx2.5 cardiac fate maps using a Gata4-based reporter of Cre activity. Dev Biol. 2008;323:98–104. doi: 10.1016/j.ydbio.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LSB. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the Kif3A subunit of kinesin-II. PNAS. 1999;96:5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G. Observation d’une deviation organique de l’estomac, d’une anomalie dans la situation et dans le configuration du coeur et des vaisseaux qui en partent ou qui s’y rendant. Bull Soc Anat Paris. 1826;1:40–48. [Google Scholar]
- Mattingly C, Parton A, Dowell L, Rafferty J, Barnes D. Cell and Molecular Biology of Marine Elasmobranchs: Squalus acanthias and Raja erinacea. Zebrafish. 2004;1:111–120. doi: 10.1089/zeb.2004.1.111. [DOI] [PubMed] [Google Scholar]
- McCauley DW, Bronner-Fraser M. Neural crest contributions to the lamprey head. Development. 2003;130:2317–2327. doi: 10.1242/dev.00451. [DOI] [PubMed] [Google Scholar]
- McGrath J, Brueckner M. Cilia are at the heart of vertebrate left-right asymmetry. Curr Opin Genet Develop. 2003;13:385–392. doi: 10.1016/s0959-437x(03)00091-1. [DOI] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. TBX1 is responsible for cardiovascular defects in velo cardio facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Momma K. Cardiovascular Anomalies Associated With Chromosome 22q11.2 Deletion Syndrome. Am J Cardiology. 2010;105:1617–1624. doi: 10.1016/j.amjcard.2010.01.333. [DOI] [PubMed] [Google Scholar]
- Murphy KC. Schizophrenia and velo-cardio-facial syndrome. Lancet. 2002;359:426–430. doi: 10.1016/S0140-6736(02)07604-3. [DOI] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- Nakhleh N, Francis R, Giese RA, Tian X, Li Y, Zariwala MA, Yagi H, Khalifa O, Kureshi S, Chatterjee B, Sabol SL, Swisher M, Connelly PS, Daniels MP, Srinivasan A, Kuehl K, Kravitz N, Burns K, Sami I, Omran H, Barmada M, Olivier K, Chawla KK, Leigh M, Jonas R, Knowles M, Leatherbury L, Lo CW. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation. 2012;125:2232–42. doi: 10.1161/CIRCULATIONAHA.111.079780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, Harris PC, Ingber DE, Loghman-Adham M, Zhou J. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol. 2006;17:1015–1025. doi: 10.1681/ASN.2005080830. [DOI] [PubMed] [Google Scholar]
- Nelms BL, Labosky PA. Colloquium Series on Developmental Biology. 1. Vol. 1. Morgan & Claypool Publishers; 2010. Transcriptional control of neural crest development. [PubMed] [Google Scholar]
- Newth DR. On the neural crest of the lamprey embryo. J Embryol Exp Morph. 1956;4:358–375. [Google Scholar]
- Nicol JAC. Autonomic Nervous Systems in Lower Chordates. Biol Rev Cambridge Phil Soc. 1952;27:1–50. [Google Scholar]
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking Kif3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- Norwood WI. Our Roots, Our Future. World Journal for Pediatric and Congenital Heart Surgery. 2010;1:127–131. doi: 10.1177/2150135109360814. [DOI] [PubMed] [Google Scholar]
- Okada Y, Nonaka S, Tanaka Y, Saijoh Y, Hamada H, Hirokawa N. Abnormal nodal flow precedes situs inversus in iv and inv mutant mice. Mol Cell. 1999;4:459–468. doi: 10.1016/s1097-2765(00)80197-5. [DOI] [PubMed] [Google Scholar]
- Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota KG, Kuraku S, Kuratani S. Hagfish embryology with reference to the evolution of the neural crest. Nature. 2007;446:672–675. doi: 10.1038/nature05633. [DOI] [PubMed] [Google Scholar]
- Papon JF, Perrault I, Costa A, Louis B, Gerard X, Hanein S, Fares-Taie L, Gerber S, Defoort-Dhellemmes S, Vojtek AM, Kaplan J, Rozet JM, Escudier E. Abnormal respiratory cilia in non-syndromic Leber congenital amaurosis with CEP290 mutations. J Med Genet. 2010;47:829–834. doi: 10.1136/jmg.2010.077883. [DOI] [PubMed] [Google Scholar]
- Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol. 2004;15:2528–36. doi: 10.1097/01.ASN.0000141055.57643.E0. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Agrin N, Leszyk J, Witman GB. Proteomic analysis of a eukaryotic cilium. J Cell Biol. 2005;170:103–113. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Agrin N, Walker BK, Witman GB. Identification of predicted human outer dynein arm genes: candidates for primary ciliary dyskinesia genes. J Med Genet. 2006;43:62–73. doi: 10.1136/jmg.2005.033001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Pomares JM, González-Rosa JM, Muñoz-Chápuli R. Building the vertebrate heart an evolutionary approach to cardiac development. Int J Dev Biol. 2009;53:1427–1443. doi: 10.1387/ijdb.072409jp. [DOI] [PubMed] [Google Scholar]
- Pietri T, Eder O, Blanche M, Thiery JP, Dufour S. The human tissue plasminogen activator-Cre mouse: a new tool for targeting specifically neural crest cells and their derivatives in vivo. Dev Biol. 2003;259:176–187. doi: 10.1016/s0012-1606(03)00175-1. [DOI] [PubMed] [Google Scholar]
- Piotrowski T, Ahn DG, Schilling TF, Nair S, Ruvinsky I, Geisler R, Rauch GJ, Haffter P, Zon LI, Zhou Y, Foott H, Dawid IB, Ho RK. The zebrafish van gogh mutation disrupts tbx1, which is involved in the DiGeorge deletion syndrome in humans. Development. 2003;130:5043–5052. doi: 10.1242/dev.00704. [DOI] [PubMed] [Google Scholar]
- Pizzuti A, Novelli G, Ratti A, Amati F, Mari A, Calabrese G, Nicolis S, Silani V, Marino B, Scarlato G, Ottolenghi S, Dallapiccola B. UFD1L, a developmentally expressed ubiquitination gene, is deleted in CATCH 22 syndrome. Hum Mol Genet. 1997;6:259–265. doi: 10.1093/hmg/6.2.259. [DOI] [PubMed] [Google Scholar]
- Poeck B, Hofbauer A, Pflugfelder GO. Expression of the Drosophila optomotor-blind gene transcript in neuronal and glial cells of the developing nervous system. Development. 1993;117:1017–1029. doi: 10.1242/dev.117.3.1017. [DOI] [PubMed] [Google Scholar]
- Pombal MA, Carmona R, Megas M, Ruiz A, Pérez-Pomares JM, Muñoz-Chápuli R. Epicardial development in lamprey supports an evolutionary origin of the vertebrate epicardium from an ancestral pronephric external glomerulus. Evolution and Development. 2008;10:210–216. doi: 10.1111/j.1525-142X.2008.00228.x. [DOI] [PubMed] [Google Scholar]
- Porras D, Brown CB. Temporal-spatial ablation of neural crest in the mouse results in cardiovascular defects. Dev Dyn. 2008;237:153–162. doi: 10.1002/dvdy.21382. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. A physiological review of the primary cilium. Annu Rev Physiol. 2005;67:515–529. doi: 10.1146/annurev.physiol.67.040403.101353. [DOI] [PubMed] [Google Scholar]
- Prall OWJ, Menon MK, Solloway MJ, Watanabe Y, Zaffran S, Bajolle F, Biben C, McBride JJ, Robertson BR, Chaulet H, Stennard FA, Wise N, Schaft D, Wolstein O, Furtado MB, Shiratori H, Chien KR, Hamada H, Black BL, Saga Y, Robertson EJ, Buckingham ME, Harvey RP. An Nkx2–5/Bmp2/Smad1 Negative Feedback Loop Controls Heart Progenitor Specification and Proliferation. Cell. 2007;128:947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam JL, Parkerson JB. Anatomy of the heart of the amphibia. II Cryptobranchus alleganiensis. Herpetologica. 1985;41:287–298. [Google Scholar]
- Raya A, Belmonte JCI. Left-right asymmetry in the vertebrate embryo: from early information to higher-level integration. Nat Rev Genet. 2006;7:283–293. doi: 10.1038/nrg1830. [DOI] [PubMed] [Google Scholar]
- Rohmer D, Sitek A, Gullberg GT. Visualization of fiber structure in the left and right ventricle of the human heart. Lawrence Berkeley National Laboratory technical report LBNL-61064. 2006 doi: 10.2172/920253. [DOI] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Sambasivan R, Kuratani S, Tajbakhsh S. An eye on the head: the development and evolution of craniofacial muscles. Development. 2011;128:2401–2415. doi: 10.1242/dev.040972. [DOI] [PubMed] [Google Scholar]
- Sanchez-Quintana D, García-Martínez V, Climent V, Hurlé JM. Myocardial fiber and connective tissue architecture in the fish heart ventricle. J Exp Zool. 1996;275:112–124. doi: 10.1016/S0940-9602(11)80198-6. [DOI] [PubMed] [Google Scholar]
- Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- Sato M, Yost HJ. Cardiac neural crest contributes to cardiomyogenesis in zebrafish. Dev Biol. 2003;257:127–139. doi: 10.1016/s0012-1606(03)00037-x. [DOI] [PubMed] [Google Scholar]
- Sato M, Tsai HJ, Yost HJ. Semaphorin3D regulates invasion of cardiac neural crest cells into the primary heart field. Dev Biol. 2006;298:12–21. doi: 10.1016/j.ydbio.2006.05.033. [DOI] [PubMed] [Google Scholar]
- Scambler PJ. 22q11 Deletion Syndrome: A Role for TBX1 in Pharyngeal and Cardiovascular Development. Pediatr Cardiol. 2010;31:378–390. doi: 10.1007/s00246-009-9613-0. [DOI] [PubMed] [Google Scholar]
- Scholey JM. Intraflagellar transport. Annu Rev Cell Dev Biol. 2003;19:423–443. doi: 10.1146/annurev.cellbio.19.111401.091318. [DOI] [PubMed] [Google Scholar]
- Scholl AM, Kirby ML. Signals controlling neural crest contributions to the heart. Wiley Interdiscip Rev Syst Biol Med. 2009;1:220–227. doi: 10.1002/wsbm.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe GC, Hoffmann K, Loges NT, Birker D, Rossier C, de Santi MM, Olbrich H, Fliegauf M, Failly M, Liebers U, Collura M, Gaedicke G, Mundlos S, Wahn U, Blouin JL, Niggemann B, Omran H, Antonarakis SE, Bartoloni L. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mut. 2008;29:289–298. doi: 10.1002/humu.20656. [DOI] [PubMed] [Google Scholar]
- Seo JW, Brown NA, Ho SY, Anderson RH. Abnormal laterality and congenital cardiac anomalies. Relations of visceral and cardiac morphologies in the iv/iv mouse. Circulation. 1992;86:642–50. doi: 10.1161/01.cir.86.2.642. [DOI] [PubMed] [Google Scholar]
- Senior HD. The development of the heart in shad. (Alosa Sapadissima, Wilson) With a note on the classification of teleostean embryos from a morphological standpoint. Amer J Anat. 1909;9:211–262. [Google Scholar]
- Shen L, Gu H, Wang D, Yang, Xu Z, Jing H, Jiang Y, Ding Y, Hou H, Ge Z, Chen S, Mo X, Yi L. Influence of Chromosome 22q11.2 Microdeletion on Postoperative Calcium Level After Cardiac-Correction Surgery. Ped Cardiol. 2011;32:904–909. doi: 10.1007/s00246-011-0012-y. [DOI] [PubMed] [Google Scholar]
- Shen MM. Nodal signaling: developmental roles and regulation. Development. 2007;134:1023–1034. doi: 10.1242/dev.000166. [DOI] [PubMed] [Google Scholar]
- Shiratori H, Hamada H. The left-right axis in the mouse: from origin to morphology. Development. 2006;133:2095–2104. doi: 10.1242/dev.02384. [DOI] [PubMed] [Google Scholar]
- Showell C, Christine KS, Mandel EM, Conlon FL. Developmental Expression Patterns of Tbx1, Tbx2, Tbx5, and Tbx20 in Xenopus tropicalis. Dev Dyn. 2006;235:1623–1630. doi: 10.1002/dvdy.20714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shprintzen RJ. Velo-Cardio-Facial Syndrome: 30 Years of Study. Dev Disabil Res Rev. 2008;14:3–10. doi: 10.1002/ddrr.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões FC, Peterkin T, Patient R. Fgf differentially controls cross-antagonism between cardiac and hemangioblast regulators. Development. 2011;138:3235–3245. doi: 10.1242/dev.059634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P, Olaopa M, Firulli AB, Conway SJ. Cardiovascular development and the colonizing cardiac neural crest lineage. Scientific World Journal. 2007;7:1090–1113. doi: 10.1100/tsw.2007.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stoller JZ, Epstein JA. Cardiac neural crest. Semin Cell Dev Biol. 2005;16:704–715. doi: 10.1016/j.semcdb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Supp DM, Witte DP, Potter SS, Brueckner M. Mutation of an axonemal dynein affects left-right asymmetry in inversus viscerum mice. Nature. 1997;389:963–966. doi: 10.1038/40140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher MS, Jonas R, Tian X, Lee ES, Lo CW, Leatherbury L. Increased postoperative and respiratory complications in patients with congenital heart disease associated with heterotaxy. J Thorac Cardiovasc Surg. 2011;141:637–644. doi: 10.1016/j.jtcvs.2010.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara Y. Normal stages of development in the lamprey, Lampetra reissneri (Dybowski) Zool Sci. 1988;5:109–118. [Google Scholar]
- Talbot JJ, Shillingford JM, Vasanth S, Doerr N, Mukherjee S, Kinter MT, Watnick T, Weimbs T. Polycystin-1 regulates STAT activity by a dual mechanism. PNAS. 2011;108:7985–7990. doi: 10.1073/pnas.1103816108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SY, Rosenthal J, Zhao XQ, Francis RJ, Chatterjee B, Sabol SL, Linask KL, Bracero L, Connelly PS, Daniels MP, Yu Q, Omran H, Leatherbury L, Lo CW. Heterotaxy and complex structural defects in a mutant mouse model of primary ciliary dyskinesia. J Clin Invest. 2007;117:3742–3752. doi: 10.1172/JCI33284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Okada Y, Hirokawa N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left–right determination 2005. Nature. 2005;435:172–177. doi: 10.1038/nature03494. [DOI] [PubMed] [Google Scholar]
- Tao Y, Wang J, Tokusumi T, Gajewski K, Schulz RA. Requirement of the LIM Homeodomain Transcription Factor Tailup for Normal Heart and Hematopoietic Organ Formation in Drosophila melanogaster. Mol Cell Biol. 2007;27:3962–3969. doi: 10.1128/MCB.00093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tota B, Cimini V, Salvatore G, Zummo G. Comparative study of the arterial and lacunary systems of the ventricular myocardium of elasmobranch and teleost fishes. Am J Anat. 1983;167:15–32. doi: 10.1002/aja.1001670103. [DOI] [PubMed] [Google Scholar]
- Vandenberg LN, Levin M. Far from solved: a perspective on what we know about early mechanisms of left-right asymmetry. Dev Dyn. 2010;239:3131–3146. doi: 10.1002/dvdy.22450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002a;11:915–922. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. 2002b;129:4605–4611. doi: 10.1242/dev.129.19.4605. [DOI] [PubMed] [Google Scholar]
- Vooijs M, Jonkers J, Berns A. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep. 2001;2:292–297. doi: 10.1093/embo-reports/kve064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo KL, Kumiski D, Kirby ML. Cardiac neural crest is essential for the persistence rather than the formation of an arch artery. Dev Dyn. 1996;205:281–292. doi: 10.1002/(SICI)1097-0177(199603)205:3<281::AID-AJA8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Lo CW, Kirby ML. Connexin 43 expression reflects neural crest patterns during cardiovascular development. Dev Biol. 1999;208:307–323. doi: 10.1006/dbio.1999.9219. [DOI] [PubMed] [Google Scholar]
- Wang T. Evolution of the cardiovascular autonomic nervous system in vertebrates. In: Robertson D, Biaggioni I, Burnstock G, Low PA, Paton JFR, editors. Primer on the Autonomic Nervous System. Elsevier, Academic Press; 2011. pp. 669–674. [Google Scholar]
- Weavers H, Prieto-Sánchez S, Grawe F, Garcia-López A, Artero R, Wilsch-Bräuninger M, Ruiz-Gómez M, Skaer H, Denholm B. The insect nephrocyte is a podocyte-like cell with a filtration slit diaphragm. Nature. 2009;457:322–326. doi: 10.1038/nature07526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welke KF, Diggs BS, Karamlou T, Ungerleider RM. Comparison of pediatric cardiac surgical mortality rates from national administrative data to contemporary clinical standards. Ann Thorac Surg. 2009;87:216–222. doi: 10.1016/j.athoracsur.2008.10.032. [DOI] [PubMed] [Google Scholar]
- Wilming LG, Snoeren CA, van Rijswijk A, Grosveld F, Meijers C. The murine homologue of HIRA, a DiGeorge syndrome candidate gene, is expressed in embryonic structures affected in human CATCH22 patients. Hum Mol Genet. 1997;6:247–258. doi: 10.1093/hmg/6.2.247. [DOI] [PubMed] [Google Scholar]
- Williams WG. Uses and Limitations of Registry and Academic Databases. Seminars in Thoracic and Cardiovascular Surgery. Pediatric Cardiac Surgery Annual. 2010;13:66–70. doi: 10.1053/j.pcsu.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131:3217–27. doi: 10.1242/dev.01174. [DOI] [PubMed] [Google Scholar]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- Yamagishi H, Srivastava D. Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med. 2003;9:383–389. doi: 10.1016/s1471-4914(03)00141-2. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development. 2006;133:3587–3595. doi: 10.1242/dev.02539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, Von Gise A, Ikeda S, Chien KR, Pu WT. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454:109–113. doi: 10.1038/nature07060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cashman TJ, Nevis KR, Obregon P, Carney SA, Liu Y, Gu A, Mosimann C, Sondalle S, Peterson RE, Heideman W, Burns CE, Burns CG. Latent TGF-β binding protein 3 identifies a second heart field in zebrafish. Nature. 2011;474:645–648. doi: 10.1038/nature10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolkowska L, Kawalec W, Turska-Kmiec A, Krajewska-Walasek M, Brzezinska-Rajszys G, Daszkowska J, Maruszewski B, Burczynski P. Chromosome 22q11.2 microdeletion in children with conotruncal heart defects: frequency, associated cardiovascular anomalies, and outcome following cardiac surgery. Eur J Ped. 2008;167:1135–1140. doi: 10.1007/s00431-007-0645-2. [DOI] [PubMed] [Google Scholar]