

Abstract
Objective
These studies were performed to determine the role of CCL21 and its corresponding receptor CCR7 in the pathogenesis of Rheumatoid Arthritis (RA).
Methods
Histological studies were performed to compare the expression of CCR7 and CCL21 in RA synovial tissues. Next the role of CCL21 and/or CCR7 in angiogenesis was examined employing in vitro chemotaxis, tube formation and in vivo matrigel plug assays. Finally the mechanism by which CCL21 mediates angiogenesis was determined by Western blot analysis, endothelial chemotaxis and tube formation.
Results
In this study, we document that CCR7 and CCL21 colocolize in VWF+ cells where their expression is elevated in RA synovial tissue. Hence the ability to induce angiogenesis was examined for CCR7 ligands, CCL19 and CCL21. CCL21, but not CCL19, at concentrations present in the RA joint, induces human microvascular endothelial cell (HMVEC) migration that is mediated through CCR7 ligation. Further, suppression of the PI3K pathway markedly reduces CCL21-induced HMVEC chemotaxis and tube formation, however suppression of ERK and JNK pathways has no effect on these processes. Neutralization of either CCL21 in RA synovial fluids or CCR7 on HMVECs significantly reduces the induction of HMVEC migration and/or tube formation by RA synovial fluid. We further demonstrate that CCL21 is angiogenic, by showing its ability to promote blood vessel growth in matrigel plugs in vivo at concentrations present in RA joint.
Conclusion
These observations identify a novel function for CCL21 as an angiogenic mediator in RA, supporting CCL21/CCR7 as a therapeutic target in RA.
Keywords: CCL21, CCR7, RA synovial fluid, angiogenesis and migration
Rheumatoid arthritis (RA) is a chronic systemic disorder characterized by the development through angiogenesis of new capillaries, which results in the infiltration of inflammatory cells leading to synovial hyperplasia and progressive bone destruction. Angiogenesis is dependent on endothelial cell activation, migration and proliferation, and inhibition of angiogenesis may provide a novel therapeutic approach in RA.
CCL21 and CCL19 are two CCR7 binding chemokines that play an important role by modulating the circulation of T cells and dendritic cells within lymphoid and peripheral organs (1). Consistently, CCR7 deficient mice show defective migration of lymphocytes and dendritic cells into the T-cell zones (2).
Previous studies have demonstrated that CCL21 is expressed by lymph node endothelial cells and functions as an endothelial cell growth factor (3). Expression of CCL21 is associated with formation of tertiary lymphoid tissue, a process called lymphoid neogenesis (4). Recent studies using antigen-induced arthritis in CCR7 deficient mice showed lessened arthritis. However due to abnormalities in lymphoid architecture detected in CCR7-/- mice it is unclear whether amelioration of arthritis is due to these structural issues (5). Nevertheless, a role for CCR7 and/or CCL21 in RA angiogenesis is undefined.
In a recent study we demonstrated that like CCR7, CCL21 expression is elevated on RA synovial tissue lining macrophages and fibroblasts as well as in sublining endothelial cells. Macrophages from RA synovial fluid express higher levels of CCL21 compared to RA and normal (NL) peripheral blood in vitro differentiated macrophages (6).
Ligation of CCL21 in RA fibroblasts and macrophages induced production of proangiogenic factors such as VEGF, Ang-1 and IL-8, suggesting that CCL21 plays an indirect role in RA angiogensis (6). In contrast, others have shown that CCL19 activated RA synovial tissue fibroblasts produce VEGF while this effect was not noted with CCL21 stimulation (7). These observations are consistent with the association of CCR7 expression with hypoxia, a process that is essential for initiation of angiogenesis (8). It was shown that Hypoxia Inducible Factors (HIF) 1α and 2α are responsible for upregulating CCR7 levels and inhibition of CCR7 and/or ERK1/2 signaling pathway significantly suppresses hypoxia induced cell migration and invasion, hence supporting the role of CCR7 in angiogenesis (8).
In this study we show that expression of CCL21 and CCR7 in RA blood vessels is comparable and demonstrates a linear correlation. Additionally, cells in the RA synovial tissue lining including RA fibroblasts and macrophages activated with CCL21 produce potent proangiogenic factors (6) hence the direct role of CCL21 in RA angiogenesis was evaluated. Our results demonstrate that CCL21-induced HMVEC chemotaxis and tube formation are mediated by CCR7 ligation and activation of the PI3K pathway. Further we demonstrate that CCL21 enhances formation of blood vessels in vivo through recruitment of endothelial cells as well as endothelial progenitor cells (EPCs) in concentrations available in RA synovial fluid and tissue. Interestingly we show that factors in RA synovial fluid can greatly increase endothelial CCL21 expression making fluids an important source for CCR7+ cell attraction. Finally, we demonstrate that RA synovial fluid-mediated endothelial migration and/or tube formation is significantly reduced by CCL21 and/or CCR7 neutralization. In short our data suggest that therapy directed against CCR7 ligation may reduce leukocyte migration into the diseased joint by inhibiting angiogenesis in RA.
MATERIALS AND METHODS
Antibodies and immunohistochemistry
The studies were approved by the Institutional Review Board, and all donors gave informed written consent. RA synovial tissues were recruited from the practices of orthopedic surgeons and samples were de-identified. RA and NL synovial tissues were formalin fixed, paraffin embedded and sectioned. Synovial tissues were immunoperoxidase-stained using Vector ABC Kits (Vector Laboratories) with diaminobenzidine (DAB) as a chromogen. Slides were deparaffinized in xylene for 20 min followed by rehydration by transfer through graded alcohols. Antigens were unmasked by incubating slides in boiling citrate buffer for 15 min, followed by type II trypsin digestion for 30 min at 37°C. Nonspecific binding of avidin and biotin was blocked using an avidin/biotin blocking kit (Vector Laboratories). Tissues were incubated with antibodies to human CCR7 (1:500; R & D Systems, Minneapolis, MN), CCL21 (1:67; R&D Systems), LYVE-1 (1:25; R&D Systems), VWF (1:1000; Dako Carpinteria, CA) or IgG (Beckman Coulter). For immunohistochemistry performed in Figs. 1A, F and G slides were counterstained with Harris hematoxylin and treated with lithium carbonate for bluing. For CCL21+ VWF+ studies performed in Fig. 1E, Texas red labeled anti-goat (1:200; Abcam Cambridge, MA) was employed to visualize CCL21 staining and FITC-conjugated anti-rabbit (1:250; Abcam Cambridge, MA) was used to visualize VWF immunostaining in RA synovial tissues. Slides were evaluated by two blinded observers (A.M.M. and M.V.V.) (9-12). Tissue sections were scored for lining and endothelial staining (on a 0-5 scale) (6, 13, 14). CCR7 and CCL21 positive blood vessel staining was scored (0-5 scale) in 12 HPFs (blood vessels were identified by morphology in serial sections of CCR7 and CCL21 stained slides). CCR7 and CCL21 positive lymphatic vessel staining was scored in 15 HPFs (lymphatic vessels were identified by LYVE-1+ staining in serial sections of CCR7 and CCL21 stained slides). The correlation of the receptor and ligand expression on blood vessels was determined by regression analysis.
Figure 1. CCR7 and CCL21 are colocalized on RA synovial tissue (ST) endothelial cells.
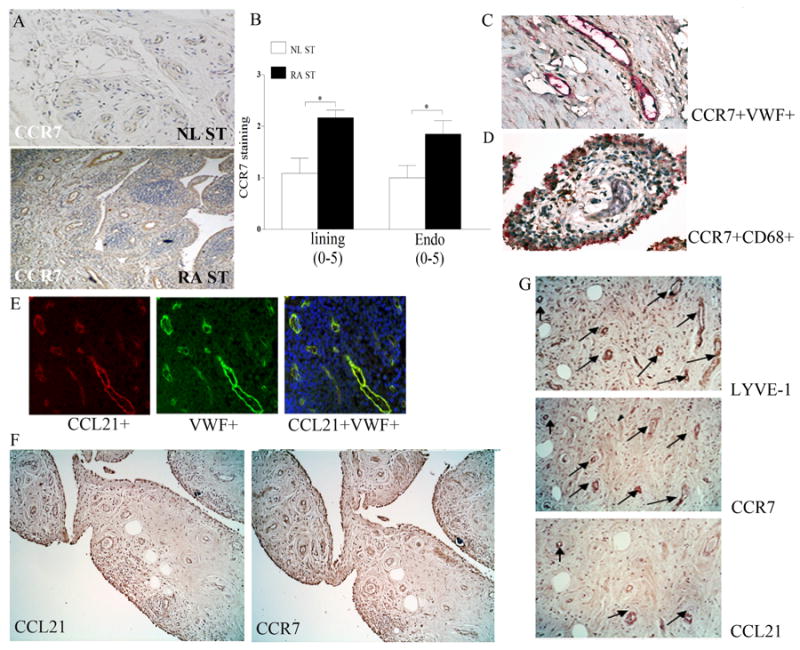
Normal (NL) and and RA ST (A) were stained with anti-human CCR7 (R&D Systems) (original magnification × 200). B. Positive immunostaining was scored on a 0-5 scale and ST lining and endothelial immunostaining are shown as mean ±SEM, n=12. * represents p <0.05. RA synovial tissues were stained for CCR7 (brown staining) and Von willebrand factor (VWF) (red staining) (C) or for CCR7 (brown staining) and CD68 (red staining) (original magnification × 400) (D) in order to distinguish endothelial cells or macrophages that express CCR7. E. Colocalization of CCL21 on VWF+ cells was examined when sections were stained with Texas red labeled anti-goat CCL21 (red) or FITC conjugated anti-rabbit VWF (green staining) or staining overlay (yellow) (original magnification × 400). F. RA serial sections immunostained with anti-CCL21 and anti-CCR7 (original magnification × 200), n=12. G. RA serial sections were immunostained with anti-LYVE-1, anti-CCR7 or anti-CCL21 (original magnification × 400) and CCR7, CCL21 staining were read in 15 LYVE-1+ fields (3 fields in 5 different RA synovial tissues).
To localize CCR7 to macrophages or endothelial cells in RA synovial tissues, slides were deparaffinized and the antigen was unmasked by Proteinase K digestion buffer (Dako) for 10 min. Using an Invision G2 kit (Dako) tissues were stained with anti-CCR7 (1:25 dilution, R&D Systems) employing DAB (brown staining) as a chromogen. Thereafter tissues were blocked and stained with anti-VWF (1:1000, Dako) or anti-CD68 (1:100, Dako) employing Fast red (red staining) as a chromogen following manufacturers’ instruction (Dako).
Cell isolation, culture and procedures
NL peripheral blood (PB) mononuclear cells were isolated by Histopaque gradient centrifugation (Sigma-aldrich, St. Louis, MO). Monocytes were isolated from NL PB employing a negative selection kit (StemCell Technologies, Vancouver, Canada) according to the manufacturers’ instructions (6, 13). Monocytes were subsequently differentiated to macrophages by culturing in RPMI containing 20% FBS for 7 days. Synovial tissue fibroblasts were isolated from fresh RA synovial tissues by mincing and digestion in a solution of dispase, collagenase, and DNase and used between passages 3 and 9 (15). Macrophages or RA fibroblasts were untreated or treated with LPS (Sigma, 10 ng/ml), TNF-α (R&D Systems, 10 ng/ml), IL-1β (R&D Systems, 10 ng/ml), IL-17 (R&D Systems, 50 ng/ml) and RA synovial fluid (1:4). Cells were harvested after 6 h and the CCR7 mRNA levels were quantified by real-time RT-PCR (6, 13).
Since the number of endothelial cells obtained from RA synovial digest was very restricted and were easily overgrown by RA synovial tissue fibroblasts we compared the expression of CCR7 in a number of primary endothelial cell types in order to select one as a surrogate for RA endothelial cells. Skin and lung HMVECs (Lonza, Walkersville, Maryland) and human umbilical vein endothelial cells (HUVEC) (Lonza) were cultured in endothelial growth medium EGM-2MV. In a different experiment, skin HMVECs were either untreated or treated with IL-17 (R&D Systems, 50 ng/ml) and RA synovial fluid (1:4) for 6h. Subsequently cells were harvested and mRNA expression for CCR7 or CCL21 was quantified by real-time RT-PCR.
Quantification of CCR7 or CCL21 expression in different cell types
RNA was extracted using Trizol (Invitrogen, Carlsbad, CA) and reverse transcription and real-time RT-PCR were performed to determine CCR7 and/or CCL21 expression levels as previously described (16-18). Relative gene expression was determined by ΔΔCt method, and results were expressed as fold increase above conditions indicated in the figure legends. To determine levels of CCR7 (1:1000; R&D Systems) in lung and skin HMVECs as well as HUVECs, Western blot analysis was performed on cell lysates from each cell type and equal loading was determined by actin blotting (1:3000; Sigma).
HMVEC chemotaxis
To examine chemotaxis, skin HMVECs (1.25×104 cells) were placed in a 48-well Boyden chemotaxis chamber (NeuroProbe, Cabin John, MD) (19, 20). Chambers were inverted and incubated at 37°C for 2h, allowing cell attachment. Chambers were reinverted, and PBS, positive control VEGF (10 ng/ml; R&D Systems), CCL19 or CCL21 at varying concentrations from 0.001 to 100 ng/ml (R&D Systems) was added and chambers were incubated for 2h at 37°C. The number of migrating cells was counted and the data represent an average of three high power × 40 fields/well, averaged for each triplicate. To test the specificity of CCL21-induced HMVEC migration (1, 10 and 50 ng/ml), HMVECs were blocked by anti-CCR7 antibody or IgG control (10μg/ml for 1hr in 37°C) (R&D Systems) (18, 21). To define which signaling pathway(s) mediated CCL21-induced HMVEC chemotaxis, HMVECs were incubated with inhibitors to PI3K (LY294002; 1 or 5 μM), ERK (PD98059; 1 or 5 μM), JNK (SP600125; 1 or 5 μM) or DMSO for 2h in the Boyden chamber with CCL21 (10 ng/ml). To determine the role of CCL21 and/or CCR7 in RA synovial fluid-mediated endothelial migration, HMVEC chemotaxis induced by RA synovial fluids was examined following incubation of fluids (1:20) with control IgG or anti-CCL21 antibody (10 μg/ml; R&D Systems) (18, 21) or treatment of HMVECs with antibodies to CCR7 (10 μg/ml) or IgG control (18, 21).
Characterization of CCL21 signaling pathways in HMVECs
HMVECs were untreated or treated with CCL21 (10 ng/ml) for 5 to 65 min. Cell lysates were examined by Western blot analysis, as previously described (16, 18, 22). Blots were probed with phospho (p)-AKT, pERK and pJNK (Cell Signaling; 1:1000) and/or with AKT, ERK and JNK (Cell Signaling; 1:3000) overnight.
HMVEC tube formation assay
To perform the matrigel tube formation assay, BD Matrigel Matrix (BD Pharmingen, 50 μl) was polymerized for 30 min at 37°C in a 96-well plate. To examine which signaling pathways contribute to CCL21-mediated HMVEC tube formation, HMVECs (4×105 cells/ml) were incubated with inhibitors (1 or 5 μM) to PI3K (LY294002), ERK (PD98059), JNK (SP600125) or DMSO for 45 minutes at 37°C prior to adding to polymerized matrigel. CCL21 (10ng/ml) was then added to the wells and the plate was incubated for 16h at 37°C. To determine that CCR7 plays a role in RA synovial fluid-induced HMVEC tube formation, HMVECs were incubated with antibody to CCR7 or IgG for 45 minutes at 37°C. Cells were then added to polymerized matrigel, RA synovial fluid (1:20) was added to the wells, and incubated for 16h at 37°C. Each condition was performed in triplicate, FGF (20 ng/ml) and PBS were used as positive or negative control. Thereafter, tube formation was quantified using calcein fluorescent dye (BD Pharmingen) according to the manufacturers’ instructions. Subsequently, the number of branch points/tubes was quantified as previously described (20, 21, 23).
Matrigel plug assay in vivo
To examine the effect of CCL21 on angiogenesis in vivo, we used a matrigel plug assay. C57BL/6 mice were injected subcutaneously with 500 μl matrigel containing PBS or bFGF (100 ng) as negative or positive control and mouse CCL21 (4 μg) served as the experimental condition. After 10 days, mice were sacrificed; matrigel plugs were removed and analyzed for vascularity. For hemoglobin measurement, plugs were weighed and homogenized and a serial dilution of methemoglobin was prepared for quantification purposes (21, 23). Fifty microliters of supernatants or standards were added to a plate in duplicate and 50 μl tetramethylbenzidine were added to each sample. The plate was allowed to develop at room temperature, and absorbance was read. To calculate hemoglobin concentrations, the values (g/dl) were normalized to the weights of the plugs (g) (21, 23). CCL21 levels were also quantified from day 10 matrigel plug homogenates employing ELISA (R&D systems).
Histology slides from different treatment groups were examined by H&E or Masson’s trichrome staining and scored by two blinded observers (A.M.M. and M.V.V.) on a 0-4 scale (14, 21). To determine whether matrigel plugs containing CCL21 were capable of recruiting EPCs, matrigels from all three treatment groups were stained with anti-VEGFR2 (1:25; R&D systems) antibody which is a known marker for EPC staining. Slides were processed and immunoperoxidase-stained using Vector Elite ABC Kits with DAB as a chromogen as described above in the immunohistochemistry description with the exception that the antigen did not require unmasking.
Statistical Analysis
The data were analyzed using 2-tailed Student’s t tests for paired and unpaired samples. P values < 0.05 were considered significant.
RESULTS
CCR7 and CCL21 colocalize in RA synovial tissue sublining endothelial cells
RA and NL synovial tissues were stained with anti-CCR7 antibody in order to characterize their expression pattern in RA patients compared to NL individuals. We found that CCR7 expression was significantly increased in RA synovial tissue lining and endothelial cells compared to NL synovial tissue (Figs. 1A-B). We next validated that CCR7 is expressed in RA synovial tissue macrophages in the lining and endothelial cells in the sublining by colocalizing CCR7 expression on CD68+ and VWF+ cells (Figs. 1C-D). Since the focus of this study was to determine RA angiogenesis mediated by CCL21 ligation to CCR7, studies were performed to colocalize CCL21 and CCR7 expression in VWF+ cells. We show that CCL21 was positively stained in blood vessels of all 12 RA synovial tissues examined (1.75 ± 0.22; supplementary material) (Fig. 1E) and its expression levels are comparable to and closely correlate with CCR7 immunostaining on blood vessels from the same tissues (1.8 ± 0.26; supplementary material) (R2=0.4; p=0.02) (Fig. 1F). Given that CCL21 and CCR7 are involved in lymphoid neogenesis, RA synovial tissue serial sections were stained with anti-CCL21, anti-CCR7 or anti-LYVE-1 antibodies to distinguish whether CCL21 and/or CCR7 were expressed in lymphatic vessels. We found that in RA joints, 8 of the 15 LYVE-1+ fields did not positively stain for CCL21 (0.43 ± 0.12) while CCR7 was present in all LYVE-1+ fields (1.5 ± 0.09) (Fig. 1G), hence, expression of CCL21 and CCR7 on lymphatic vessels were uncorrelated (R2=0.009; p=0.7). These results support the role of CCL21 and CCR7 in RA angiogenesis.
RA synovial fluids modulate expression of CCR7 in macrophages and RA fibroblasts and levels of CCL21 and CCR7 in endothelial cells. Additionally skin HMVECs may function as a surrogate for RA endothelial cells because of elevated levels of CCR7 on skin compared to lung HMVECs
Since expression levels of CCR7 and CCL21 were elevated in RA synovial tissue lining as well as sublining endothelial cells, we asked which factors may affect their expression in macrophages, fibroblasts or endothelial cells. We demonstrate that in macrophages stimulation with LPS, IL-1β and RA synovial fluid (Fig. 2A) and in RA fibroblasts activation with IL-17 and RA synovial fluid can significantly increase CCR7 expression levels (Fig. 2B). Interestingly, in endothelial cells expression levels of CCR7 and CCL21 were modulated by IL-17 and RA synovial fluid stimulation (Fig. 2C) perhaps explaining the relationship between the expression of this receptor and its ligand on RA blood vessels. Because RA synovial fluid was the common factor capable of driving the expression of CCR7 and/or CCL21 in all examined cell types, the role of RA synovial fluid is highlighted in CCL21 production and attraction of CCR7+ cells. Although RA synovial tissue endothelial cells express CCR7, obtaining sufficient quantities of RA endothelial cells from synovial tissues was not possible and these cells were not commercially available; therefore, expression of this receptor was compared among endothelial primary cell types. We found that the levels of CCR7 expression were comparable in skin HMVECs and HUVECs which were both greater than levels expressed in lung HMVECs (Figs. 2D-E). Therefore skin HMVECs were used as surrogates for RA endothelial cells.
Figure 2. Expression of CCR7 and CCL21 are modulated by IL-17 or RA synovial fluid (SF) in HMVECs and skin HMVECs can be used as surrogates for RA endothelial cells because of their elevated levels of CCR7.
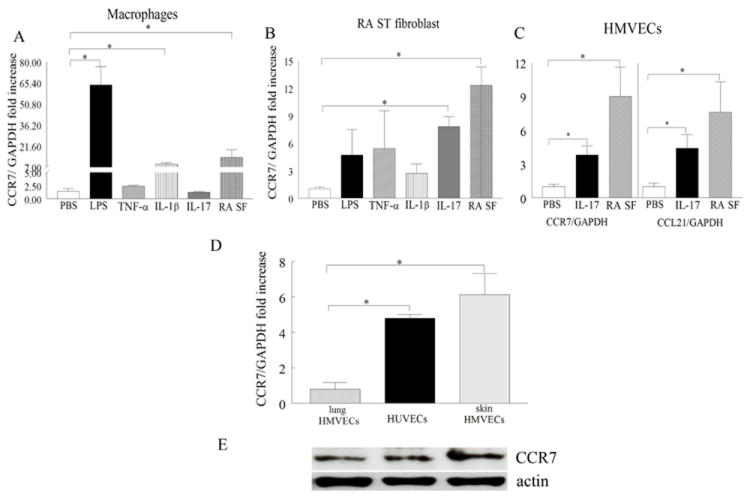
A. Cells were untreated (PBS) or treated with LPS (10 ng/ml), TNF-α (10 ng/ml), IL-1β (10 ng/ml), IL-17 (50 ng/ml) or RA SF (1:4) for 6h and expression of CCR7 was determined in PB in vitro differentiated macrophages (A) or RA fibroblasts (B) by real-time RT-PCR (n=5-10). C. HMVECs were either untreated or treated with IL-17 (50 ng/ml) or RA SF (1:4) for 6h and expression of CCR7 and CCL21 were determined by real-time RT-PCR (n=4-5). The data are shown as fold increase above untreated cells (A-C) and are normalized to GAPDH. D. CCR7 mRNA levels were determined in lung HMVECs, HUVECs and skin HMVEC and results were expressed as fold increase above levels detected in lung HMVECs, n=3-6. Values are the mean ± SE. * represents p <0.05. E. CCR7 (1:1000; o/n) protein levels were determined employing Western blot analysis on lung HMVECs, HUVECs and skin HMVECs lysates (75 μg) and equal loading was determined by actin blotting (1:3000).
CCL21 but not CCL19 induces endothelial (HMVEC) migration
Next, experiments were performed to determine whether CCR7-corresponding ligands were involved in HMVEC (skin) chemotaxis and/or tube formation. We found that while CCL21 was chemotactic for HMVECs at concentrations ranging from 1 to 100 ng/ml (p<0.05; Fig. 3B), CCL19 did not induce HMVEC migration (Fig. 3A). The mean concentration of CCL21 in the RA synovial fluids (n=74) and tissues (n=11) analyzed was 519 ± 38 pg/ml (up to 3.4 ng/ml) and 824 ± 104 pg/mg (up to 1.6 ng/mg) respectively (6), concentrations that were highly chemotactic for HMVEC migration. Further, incubation of HMVECs with neutralizing antibody to CCR7 suppressed CCL21-induced HMVEC migration suggesting that the chemotactic effect was due to CCR7 ligation (Fig. 3C). These results suggest that CCL21 can mediate HMVEC migration at concentrations present in the RA joint via CCR7 ligation.
Figure 3. CCL21 induces HMVEC migration through binding to CCR7.
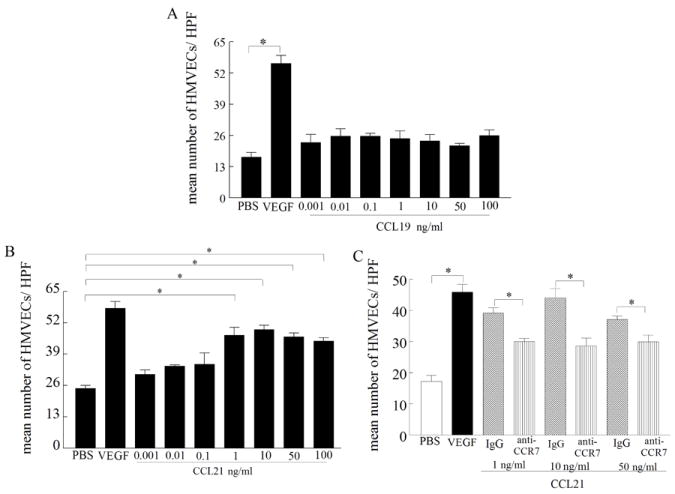
Dose-response curve of CCL19 (A) and CCL21-induced (B) HMVEC chemotaxis was performed in a Boyden chemotaxis chamber with varying concentrations of the mentioned chemokines, n=3. C. CCL21-induced HMVEC chemotaxis (1, 10 and 50 ng/ml) was suppressed by neutralization of CCR7 (10 μg/ml) by anti-CCR7 antibody but not by IgG control (10 μg/ml 1h preincubation with cells in 37°C) for 2h, n=3. Values are the mean ± SE. * represents p <0.05.
CCL21 activates the PI3K, ERK and JNK pathways in HMVECs, however only inhibition of PI3K reduces CCL21-induced HMVEC chemotaxis and tube formation
To determine which signaling pathways in HMVECs are activated by CCL21, phosphorylation of the MAPK and AKT pathways was determined by immunoblot analysis. Our data demonstrate that CCL21 phosphorylates ERK and AKT1 as early as 35 min while JNK is activated later, at 65 min (Figs. 4A-C). To determine which signaling pathways mediate HMVEC migration, chemical inhibitors at concentrations of 1 or 5 μM were utilized, while 10 μM was toxic and resulted in cell death, as determined by trypan blue staining (data not shown). Inhibition of ERK and JNK was ineffective in suppressing CCL21-induced HMVEC chemotaxis, while inhibition of PI3K reduced (p<0.05) chemotaxis starting at 1 μM (Fig. 4D). Similarly, while inhibition of PI3K (starting at 1μM) decreased CCL21-mediated tube formation by 35-40% (p<0.05) (Figs. 4E-K), suppression of ERK and JNK had no effect on this process. These results suggest that CCL21-induced HMVEC chemotaxis and tube formation are mediated through the PI3K/AKT1 pathway.
Figure 4. CCL21-induced HMVEC migration and tube formation is suppressed by PI3K inhibition.
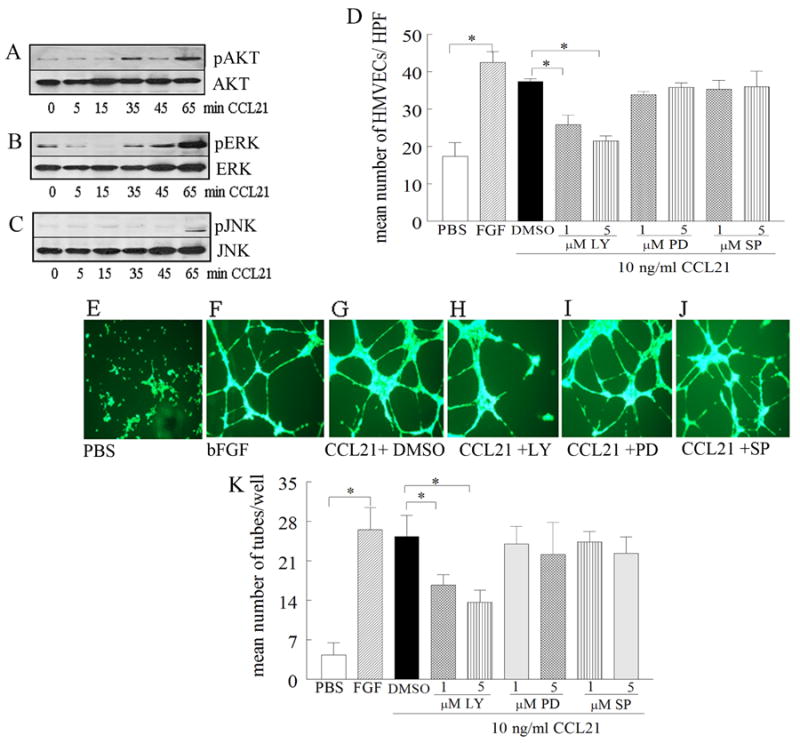
To determine the mechanism of CCL21 in HMVEC migration, cells were stimulated with CCL21 (10 ng/ml) for 0-65 minutes, and cell lysates were probed for pAKT (A), pERK (B), or pJNK (C) (n=3). D. To determine signaling pathways associated with CCL21 HMVEC migration, cells were treated with (1 or 5 μM) of the identified chemical inhibitors for PI3K (LY294002), ERK (PD98059) or JNK (SP600125) for 2h. To examine the mechanism of CCL21-induced tube formation, HMVECs were incubated with inhibitors (1 or 5 μM) to PI3K (LY294002), ERK (PD98059), JNK (SP600125) or DMSO for 45 minutes at 37°C prior to adding to polymerized matrigel. CCL21 (10ng/ml) was then added to the wells and the plate was incubated for 16h at 37°C (in triplicate). Photomicrographs taken of representative wells treated with PBS (E), FGF (20 ng/ml) (F), CCL21 (10ng/ml) plus DMSO (G), CCL21 (10ng/ml) plus LY294002 (5 μM) (H), CCL21 (10ng/ml) plus PD98059 (5 μM) (I) and CCL21 (10ng/ml) plus SP600125 (5 μM) (J). K. demonstrates mean number of tubes/well determined in E-J. Values are the mean ± SE, n=3. * represents p <0.05.
CCL21 and CCR7 are involved in RA synovial fluid-mediated HMVEC chemotaxis
Next we asked whether the CCL21 identified in human RA synovial fluid is chemotactic for HMVECs. We found that RA synovial fluid immunodepleted with anti-CCL21 significantly reduced HMVEC chemotaxis (p<0.05), compared with control IgG-treated fluids (Fig. 5A). Neutralization of CCR7 on HMVECs was effective in suppressing RA synovial fluid-mediated HMVEC migration (Fig. 5B) and tube formation (Figs. 5C-G) by 30%. These results suggest that CCL21 and CCR7 may play an important role in RA angiogenesis.
Figure 5. RA synovial fluid-induced HMVEC chemotaxis and/or tube formation is mediated by CCL21 ligation to CCR7.
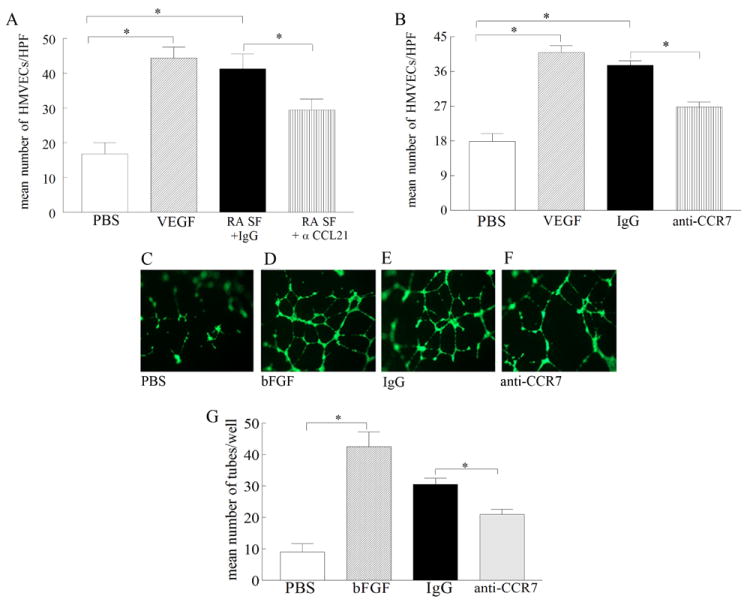
A. anti-CCL21 (10 μg/ml) or control IgG was added to RA synovial fluids from 8 patients (1:20) (1h at 37°C) prior to performing HMVEC chemotaxis in response to RA synovial fluids. B. HMVECs were incubated with antibodies to CCR7 (10 μg/ml), as well as isotype control for 1h prior to performing HMVEC chemotaxis in response to 8 RA synovial fluids (1:20). HMVECs were incubated with anti-CCR7 or IgG for 45 minutes at 37°C prior to adding to polymerized matrigel. RA synovial fluid (1:20) was then added to the wells and was incubated for 16h at 37°C (in triplicate). Photomicrographs taken of representative wells treated with PBS (C), FGF (20 ng/ml) (D), IgG (10μg/ml) plus RA synovial fluid (E), anti-CCR7 (10μg/ml) plus RA synovial fluid (F). G. demonstrates mean number of tubes/well determined in C-F. Values are the mean ± SE, n=9 synovial fluid examined in 3 independent tube formation studies. * represents p <0.05.
CCL21 induces angiogenesis in vivo in matrigel plugs
The role of CCL21 on angiogenesis in vivo was assessed by determining its effect on blood vessel formation in matrigel plugs by quantifying hemoglobin levels or histological staining. The hemoglobin content of the CCL21 treated group was 14 times greater (p<0.05) than the PBS control (Fig. 6A). Matrigel blood vessel formation was also examined histologically employing H&E (Figs. 6C, 6F and 6I) and Masson’s trichrome staining (Figs. 6D, 6G and 6J). The histological analysis demonstrated that CCL21 markedly enhances (p<0.05) blood vessel growth compared to the control group (Fig. 6B). Concentration of CCL21 quantified from day 10 matrigel plugs was 405 pg/ml ± 89 pg/ml which is within the range of CCL21 levels detected in RA synovial fluid (519 ± 38 pg/ml) and tissue (824 ± 104 pg/mg). We also asked whether matrigel plugs containing CCL21 were capable of recruiting EPCs. We document that CCL21 induced EPC extravasation was significantly greater than the PBS group suggesting that both endothelial cells and EPCs contribute to CCL21 in vivo blood vessel formation (Figs. 6E, 6H and 6K). These results support the role of CCL21 in angiogenesis in vivo at concentrations available in RA synovial fluid and tissue.
Figure 6. CCL21 enhances blood vessel growth in matrigel plugs in vivo.

A. CCL21 (4 μg) induced angiogenesis in the matrigel plugs to a significantly greater degree compared to PBS control. Matrigel containing bFGF (100 ng) served as positive control. The values represent the concentration of hemoglobin (g/dl) / plug weight (g) ± SE, with n=10. B. The histology was quantified on a score of 0-4 scale in matrigel plugs that contained PBS, CCL21 or bFGF employing H&E and Masson’s trichrome staining. A representative assay shows H&E (C, F and I) and Masson’s trichrome (D, G and J) staining of blood vessels in paraffin sections of matrigel plugs containing PBS (C and D), CCL21 (F and G) and bFGF (I and J) that was histologically scored in B (original magnification × 200). Matrigel plugs containing PBS (E), CCL21 (H) or bFGF (K) were stained with VEGFR2 to distinguish EPCs recruitment in this study. Values demonstrate mean ± SE, with n=5. * represents p<0.05.
DISCUSSION
We show that CCR7 and CCL21 are coexpressed on synovial tissue endothelial cells and that CCR7 ligands CCL19 and CCL21 are capable of inducing macrophages and RA fibroblasts to produce potent proangiogenic factors (6). Therefore, studies were performed to determine whether CCL19 or CCL21 ligation to CCR7 may directly contribute to angiogenesis in RA. Our data demonstrate that while CCL21 induces HMVEC chemotaxis, CCL19 was not effective in this process. We further demonstrate that CCL21-mediated HMVEC chemotaxis and/or tube formation are mediated through ligation to CCR7 on HMVECs and activation of the PI3K pathway. Lastly, we document that CCL21 can form blood vessels at concentrations detected in the RA joint. Hence, these results support a novel role for CCL21 in RA angiogenesis.
CCL21 and CCR7 colocolize on RA endothelium where their expression is correlated. Conversely, results from previous studies show that CCL21 was only weakly expressed on RA blood vessels while demonstrating that 100% of CCL21 producing vessels were LYVE-1+ in the RA synovium (24). Our data demonstrate that close to 50% of LYVE-1+ fields positively stained for CCL21 while CCR7 immunostaining was present on all LYVE-1+ fields. The variability of our results may be due in part to heterogeneity of RA disease or patient treatment. Hence RA synovium utilized by Manzo et al. (24) may have been obtained from patients with less disease activity since only 12 of 27 patients were on substantial treatment and as a result inflammatory factors such as IL-17 that elevate expression of CCL21 may not have been present or may have been expressed in lower concentrations. Thus, CCL21 immunostaining on blood vessels demonstrated a weak intensity. Unlike Manzo et al. (24), our lower percentage of CCL21 staining in LYVE-1+ cells may be due to selection of RA synovial tissues. While these investigators selected 9 RA tissues that were rich in lymphoid aggregates in our study CCL21 and LYVE-1 immunostaining were performed in random RA synovium.
We show that in macrophages, stimulation with RA synovial fluid modulates the expression of CCR7 and CCL21 (6). Further, activation with IL-17 and RA synovial fluid significantly increases the expression levels of CCR7 in RA fibroblasts and endothelium and concentrations of CCL21 in endothelial cells. While in HMVECs, TNF-α treatment had no effect on CCL21 expression (data not shown), levels of this chemokine were greatly elevated in human dermal lymphatic endothelial cells stimulated with TNF-α (25) suggesting that expression of CCL21 is differentially regulated in lymphatic compared to blood vessels. HIF-1α is a hypoxic transcription factor that has been shown to modulate CCR7 expression (8) and similar to its downstream target is expressed in RA synovial tissue macrophages and endothelial cells (26). Like CCR7, expression levels of HIF-1α are modulated by TLR4 ligation in RA synovial fluid macrophages (26) suggesting that TLR4 endogenous ligands in RA synovial fluid (27) may be important for regulation of HIF-1α and CCR7 pathway.
Consistent with our findings in RA synovial tissue, skin HMVECs express elevated levels of CCR7 compared to controls, therefore these cells were employed as surrogates for RA endothelial cells to examine the direct effect of CCL19 and CCL21 on angiogenesis. We demonstrate that CCL21 induces HMVEC chemotaxis at concentrations available in the human RA joint, which is due to its ligation to CCR7. Unlike CCL21, CCL19 had no effect on HMVEC migration. Although CCL19 and CCL21 have similar affinity to CCR7, ligation of these chemokines mediates different signaling effects. CCL19, but not CCL21, activates CCR7 phosphorylation and internalization, resulting in receptor desensitization (28, 29). This suggests that CCR7-induced cell responses to CCL19 may have a shorter time-span compared to those of CCL21. Consistently, previous studies have shown that while CCL19 was chemotactic for RA synovial tissue fibroblasts, CCL21 was unable to attract these cells (7). Although CCL19-activated RA synovial tissue fibroblasts produce VEGF, this effect was not noted with CCL21 stimulation (7). These results suggest that CCL19 or CCL21 ligation of CCR7 can differentially modulate angiogenesis through induction of different signaling pathways.
Next, experiments were performed to investigate signaling pathways that were associated with CCL21-induced HMVEC chemotaxis and tube formation. Inhibition of the CCL21-activated pathways in HMVECs demonstrated that only activation of PI3K significantly reduces CCL21-mediated chemotaxis and tube formation, and suppression of ERK and JNK pathways had no effect on this process. Consistently, B cell chemotaxis mediated by CCL21 was markedly reduced by inhibitors to PI3K while suppression of ERK and JNK pathways were ineffective (30). In contrast, monocyte derived dendritic cell migration to CCL21 was dependent on phospholipase C but not PI3K signaling pathway (31). Further, others have shown that PI3K signaling plays an important role in VEGF and FGF mediated endothelial migration (32-34), suggesting that PI3K is involved in the mediation of angiogenesis by various inflammatory factors.
Since CCL19 and CCL21 are both highly elevated in RA synovial fluid (6) the contribution of these two chemokines was examined in RA synovial fluid-mediated HMVEC chemotaxis. Neutralization of CCL21 and not CCL19 (data not shown) in RA synovial fluid partially reduced RA synovial fluid-mediated HMVEC chemotaxis. RA synovial fluid-mediated HMVEC chemotaxis was also mediated through CCR7, confirming the importance of this receptor in CCL21-mediated angiogenesis. Like CCL21 (6) other proangiogenic factors present in human RA synovial fluid are mostly produced by RA synovial tissue fibroblasts (VEGF, bFGF, VCAM1, IL-6 and ELR+ CXC chemokines) or macrophages (TNF-α, IL-8 and IL-1β) (35). Previous reports demonstrate that although both CCR7 ligands CCL19 and CCL21 play a key role in the migration of T cells (36), neutrophils (37) and dendritic cells (25, 38) expression of CCL21 in CCL19-/- mice was sufficient for dendritic cell trafficking, maturation and function suggesting that CCL21 may be the more critical CCR7 ligand in the inflammatory process (38).
We next demonstrate that CCL21 can directly contribute to blood vessel formation at concentrations detected in RA synovial fluid and tissue through extravasation of endothelial cells as well as EPCs as the matrigel plugs are acellular and other proangiogenic factors can not be produced.
In conclusion, endothelial migration and tube formation induced by CCL21 were mediated through activation of the PI3K pathway and ligation to CCR7. Neutralization of CCL21 or CCR7 significantly downregulated RA synovial fluid mediated endothelial migration, suggesting that CCL21 plays an important role in RA angiogenesis.
Supplementary Material
Acknowledgments
This work was supported in part by awards from the National Institutes of Health (AR056099, AR055240), Arthritis National Research Foundation, grant from Within Our Reach from The American College of Rheumatology and funding provided by Department of Defense PR093477.
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Shahrara had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Shahrara, Pickens.
Acquisition of data. Pickens, Chamberlain, Shahrara, Talarico, Volin, Mandelin.
Analysis and interpretation of data. Shahrara, Pickens, Chamberlain, Pope, Volin
References
- 1.Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A. 1998;95(1):258–63. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189(3):451–60. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crola Da Silva C, Lamerant-Fayel N, Paprocka M, Mitterrand M, Gosset D, Dus D, et al. Selective human endothelial cell activation by chemokines as a guide to cell homing. Immunology. 2009;126(3):394–404. doi: 10.1111/j.1365-2567.2008.02906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weninger W, Carlsen HS, Goodarzi M, Moazed F, Crowley MA, Baekkevold ES, et al. Naive T cell recruitment to nonlymphoid tissues: a role for endothelium-expressed CC chemokine ligand 21 in autoimmune disease and lymphoid neogenesis. J Immunol. 2003;170(9):4638–48. doi: 10.4049/jimmunol.170.9.4638. [DOI] [PubMed] [Google Scholar]
- 5.Wengner AM, Hopken UE, Petrow PK, Hartmann S, Schurigt U, Brauer R, et al. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007;56(10):3271–83. doi: 10.1002/art.22939. [DOI] [PubMed] [Google Scholar]
- 6.Pickens SR, Chamberlain ND, Volin MV, Pope RM, Mandelin AM, 2nd, Shahrara S. Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheum. 2011;63(4):914–22. doi: 10.1002/art.30232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruhl H, Mack M, Niedermeier M, Lochbaum D, Scholmerich J, Straub RH. Functional expression of the chemokine receptor CCR7 on fibroblast-like synoviocytes. Rheumatology (Oxford) 2008;47(12):1771–4. doi: 10.1093/rheumatology/ken383. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Qiu X, Zhang S, Zhang Q, Wang E. Hypoxia induced CCR7 expression via HIF-1alpha and HIF-2alpha correlates with migration and invasion in lung cancer cells. Cancer Biol Ther. 2009;8(4):322–30. doi: 10.4161/cbt.8.4.7332. [DOI] [PubMed] [Google Scholar]
- 9.Ruth JH, Volin MV, Haines GK, III, Woodruff DC, Katschke KJ, Jr, Woods JM, et al. Fractalkine, a novel chemokine in rheumatoid arthritis and in rat adjuvant-induced arthritis. Arthritis and Rheumatism. 2001;44:1568–1581. doi: 10.1002/1529-0131(200107)44:7<1568::AID-ART280>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 10.Koch AE, Nickoloff BJ, Holgersson J, Seed B, Haines GK, Burrows JC, et al. 4A11, a monoclonal antibody recognizing a novel antigen expressed on aberrant vascular endothelium. Upregulation in an in vivo model of contact dermatitis. American Journal of Pathology. 1994;144(2):244–259. [PMC free article] [PubMed] [Google Scholar]
- 11.Shahrara S, Proudfoot AE, Woods JM, Ruth JH, Amin MA, Park CC, et al. Amelioration of rat adjuvant-induced arthritis by Met-RANTES. Arthritis Rheum. 2005;52(6):1907–19. doi: 10.1002/art.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shahrara S, Proudfoot AE, Park CC, Volin MV, Haines GK, Woods JM, et al. Inhibition of monocyte chemoattractant protein-1 ameliorates rat adjuvant-induced arthritis. J Immunol. 2008;180(5):3447–56. doi: 10.4049/jimmunol.180.5.3447. [DOI] [PubMed] [Google Scholar]
- 13.Pickens SR, Chamberlain ND, Volin MV, Pope RM, Talarico NE, Mandelin AM, 2nd, et al. Characterization of interleukin-7 and interleukin-7 receptor in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2011;63(10):2884–93. doi: 10.1002/art.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pickens SR, Chamberlain ND, Volin MV, Gonzalez M, Pope RM, Mandelin AM, 2nd, et al. Anti-CXCL5 therapy ameliorates IL-17-induced arthritis by decreasing joint vascularization. Angiogenesis. 2011;14(4):443–55. doi: 10.1007/s10456-011-9227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shahrara S, Pickens SR, Mandelin AM, 2nd, Karpus WJ, Huang Q, Kolls JK, et al. IL-17-mediated monocyte migration occurs partially through CC chemokine ligand 2/monocyte chemoattractant protein-1 induction. J Immunol. 2010;184(8):4479–87. doi: 10.4049/jimmunol.0901942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shahrara S, Park CC, Temkin V, Jarvis JW, Volin MV, Pope RM. RANTES modulates TLR4-induced cytokine secretion in human peripheral blood monocytes. J Immunol. 2006;177(8):5077–87. doi: 10.4049/jimmunol.177.8.5077. [DOI] [PubMed] [Google Scholar]
- 17.Shahrara S, Huang Q, Mandelin AM, 2nd, Pope RM. TH-17 cells in rheumatoid arthritis. Arthritis Res Ther. 2008;10(4):R93. doi: 10.1186/ar2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shahrara S, Pickens SR, Dorfleutner A, Pope RM. IL-17 induces monocyte migration in rheumatoid arthritis. J Immunol. 2009;182(6):3884–91. doi: 10.4049/jimmunol.0802246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koch AE, Halloran MM, Haskell CJ, Shah MR, Polverini PJ. Angiogenesis mediated by soluble forms of E-selectin and vascular cell adhesion molecule-1. Nature. 1995;376(6540):517–519. doi: 10.1038/376517a0. [DOI] [PubMed] [Google Scholar]
- 20.Park CC, Morel JC, Amin MA, Connors MA, Harlow LA, Koch AE. Evidence of IL-18 as a novel angiogenic mediator. Journal of Immunology. 2001;167(3):1644–1653. doi: 10.4049/jimmunol.167.3.1644. [DOI] [PubMed] [Google Scholar]
- 21.Pickens SR, Volin MV, Mandelin AM, 2nd, Kolls JK, Pope RM, Shahrara S. IL-17 contributes to angiogenesis in rheumatoid arthritis. J Immunol. 2010;184(6):3233–41. doi: 10.4049/jimmunol.0903271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shahrara S, Castro-Rueda HP, Haines GK, Koch AE. Differential expression of the FAK family kinases in rheumatoid arthritis and osteoarthritis synovial tissues. Arthritis Res Ther. 2007;9(5):R112. doi: 10.1186/ar2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haas CS, Amin MA, Ruth JH, Allen BL, Ahmed S, Pakozdi A, et al. In vivo inhibition of angiogenesis by interleukin-13 gene therapy in a rat model of rheumatoid arthritis. Arthritis Rheum. 2007;56(8):2535–48. doi: 10.1002/art.22823. [DOI] [PubMed] [Google Scholar]
- 24.Manzo A, Bugatti S, Caporali R, Prevo R, Jackson DG, Uguccioni M, et al. CCL21 expression pattern of human secondary lymphoid organ stroma is conserved in inflammatory lesions with lymphoid neogenesis. Am J Pathol. 2007;171(5):1549–62. doi: 10.2353/ajpath.2007.061275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson LA, Jackson DG. Inflammation-induced secretion of CCL21 in lymphatic endothelium is a key regulator of integrin-mediated dendritic cell transmigration. Int Immunol. 2010;22(10):839–49. doi: 10.1093/intimm/dxq435. [DOI] [PubMed] [Google Scholar]
- 26.Westra J, Brouwer E, van Roosmalen IA, Doornbos-van der Meer B, van Leeuwen MA, Posthumus MD, et al. Expression and regulation of HIF-1alpha in macrophages under inflammatory conditions; significant reduction of VEGF by CaMKII inhibitor. BMC Musculoskelet Disord. 2010;11:61. doi: 10.1186/1471-2474-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang QQ, Pope RM. The role of toll-like receptors in rheumatoid arthritis. Curr Rheumatol Rep. 2009;11(5):357–64. doi: 10.1007/s11926-009-0051-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bardi G, Lipp M, Baggiolini M, Loetscher P. The T cell chemokine receptor CCR7 is internalized on stimulation with ELC, but not with SLC. Eur J Immunol. 2001;31(11):3291–7. doi: 10.1002/1521-4141(200111)31:11<3291::aid-immu3291>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 29.Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279(22):23214–22. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- 30.Cuesta-Mateos C, Lopez-Giral S, Alfonso-Perez M, de Soria VG, Loscertales J, Guasch-Vidal S, et al. Analysis of migratory and prosurvival pathways induced by the homeostatic chemokines CCL19 and CCL21 in B-cell chronic lymphocytic leukemia. Exp Hematol. 38(9):756–64. 764 e1–4. doi: 10.1016/j.exphem.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 31.Scandella E, Men Y, Legler DF, Gillessen S, Prikler L, Ludewig B, et al. CCL19/CCL21-triggered signal transduction and migration of dendritic cells requires prostaglandin E2. Blood. 2004;103(5):1595–601. doi: 10.1182/blood-2003-05-1643. [DOI] [PubMed] [Google Scholar]
- 32.Jiang BH, Liu LZ. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets. 2008;8(1):19–26. doi: 10.2174/156800908783497122. [DOI] [PubMed] [Google Scholar]
- 33.Hayashi H, Nakagami H, Takami Y, Koriyama H, Mori M, Tamai K, et al. FHL-2 suppresses VEGF-induced phosphatidylinositol 3-kinase/Akt activation via interaction with sphingosine kinase-1. Arterioscler Thromb Vasc Biol. 2009;29(6):909–14. doi: 10.1161/ATVBAHA.108.178541. [DOI] [PubMed] [Google Scholar]
- 34.Rieck PW, Cholidis S, Hartmann C. Intracellular signaling pathway of FGF-2-modulated corneal endothelial cell migration during wound healing in vitro. Exp Eye Res. 2001;73(5):639–50. doi: 10.1006/exer.2001.1067. [DOI] [PubMed] [Google Scholar]
- 35.Szekanecz Z, Koch AE. Angiogenesis and its targeting in rheumatoid arthritis. Vascul Pharmacol. 2009;51(1):1–7. doi: 10.1016/j.vph.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwan J, Killeen N. CCR7 directs the migration of thymocytes into the thymic medulla. J Immunol. 2004;172(7):3999–4007. doi: 10.4049/jimmunol.172.7.3999. [DOI] [PubMed] [Google Scholar]
- 37.Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood. 2011;117(4):1196–204. doi: 10.1182/blood-2009-11-254490. [DOI] [PubMed] [Google Scholar]
- 38.Britschgi MR, Favre S, Luther SA. CCL21 is sufficient to mediate DC migration, maturation and function in the absence of CCL19. Eur J Immunol. 2010;40(5):1266–71. doi: 10.1002/eji.200939921. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.