

Abstract
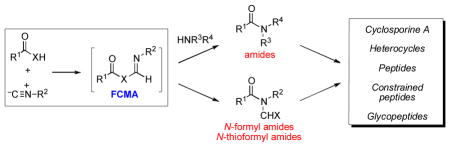
We describe herein our recent explorations in the field of isonitrile chemistry. An array of broadly useful coupling methodologies has been developed for the formation of peptidyl and glycopeptidyl amide bonds. We further describe the application of these methods to the syntheses of complex systems, including the cyclic peptide cyclosporine A, constrained peptide systems, and heterocycles.
Introduction
In recent years, our laboratory has been concerned with the development of improved methods for the synthesis of polypeptides and glycopeptides of biological importance. As a result of methodological advances from many quarters,i including our own,ii it is now possible to gain access to complex glycopeptides and even modestly-sized glycoproteins through purely synthetic means. Drawing inspiration and insights from established principles of mechanistic chemistry, we continue to pursue fresh ideas and strategies for the efficient formation of amide bonds housed in complex “biologic” settings. Along these lines, we recently developed a novel mechanistic platform, with potentially far-reaching applications, which exploits the rather unique reactivity of the isonitrile functional group.iii This Account is offered to summarize the body of advances that have been realized by revisiting the chemistry of isonitriles.
We begin with a few introductory observations. The impressive growth in the capacity of chemists to synthesize complex structures has arisen less from exceptional new insights into abstract issues of strategy than from explosive developments in enabling synthetic methodology. It can hardly be disputed that the major driving force in the growth of increasingly powerful new reactions in synthesis, has been the recognition of the potential of mechanistically inspired organometallic chemistry to allow for bond constructions that were previously difficult to achieve. This review will deal with what we regard as a significant advance in enabling methodology, pursued from a dramatically different type of perspective. What we describe below was initially undertaken to feed our collective curiosities about a class of compounds, namely isonitriles, which date from the early history of systematized organic chemistry.iv
As will be seen, the subject of isonitriles is a very old one in organic and organometallic chemistry. Moreover, isonitriles are of considerable contemporary interest with respect to their tendencies in cycloaddition and insertion reactions, as well as in reactions with various “standalone” and Lewis acid – activated electrophiles. It is not our intention here to provide a review of the chemistry of isonitriles.iii Rather, what we offer the reader is a perspective of how our particular research laboratory became interested in isonitriles, and a travelogue describing our own personal journey. Moreover, our story focuses strictly on recently reported uses of isonitrile chemistry with respect to amide bond formation. These, now logical, reactions might well have been discovered a century ago but, in fact, weren’t (at least as judged by published information which survived scrutiny). That these advances were still ripe for discovery in 2006, arose from a failure of earlier work to address some simple core questions. Curiously, earlier chemistry in this subfield had been posed in a more complex form. In retrospect, it seems that the now-obvious chemistry we describe below was obscured by the higher complexity-level at which the questions had been posed (as well as by several serious, if well intentioned, errors at the level of structure). The reader is urged to peruse our primary publications, provided below, for the epistemology which governed these isonitrile-amide bond related discoveries.
Before launching into our exploration of isonitriles, it is perhaps appropriate to begin by noting that the nitrogen counterpart of an acylium ion is called a nitrilium ion. In principle, two obvious routes for the generation of nitrilium ions present themselves. One begins with N-protonation (or N-alkylation) of a nitrile. The most celebrated reaction of this type, which well reflects the highly exploitable electrophilicity of nitrilium ions, is the Ritter Reaction.v Another obvious possible route to a nitrilium ion, though less well appreciated, would be initiated by the carbon of an isonitrile attacking an electrophile. Two notable examples of this mode of reactivity were devised by Passerinivi and Ugi (Scheme 1).vii Each of these transformations provides access to the mechanistic equivalent of a nitrilium ion through electrophilic functionalization of the carbon atom of an isonitrile. For the sake of completeness, we also note the emergence of nitrilium character in the Beckmann rearrangement of activated oximes.viii However, this route was not pursued in our study.
Scheme 1.

Synthesis of nitrilium ion; Passerini reaction and Ugi reaction; Type Ia coupling of isonitriles and carboxylic acids.
In examining the literature pertinent to the feasibility of isonitriles as precursors of nitrilium ions, we were surprised to find a paucity of indications concerning an obvious possibility, namely, a simple two-component reaction of isonitriles with carboxylic acids. With the exception of a few isolated and seemingly curious entries (vide infra), this obvious question had not been properly addressed. It seemed to us (a priori) that a carboxylic acid (5) might initially combine with an isonitrile (6), even in the absence of a catalyst or a basic additive, to generate a high-energy formimidate carboxylate mixed anhydride (7), which we came to describe as a FCMA. The formation of FCMA 7 may occur either via a step-wise process, commencing with protonation of the isonitrile by the carboxylic acid (see Scheme 1) or, alternatively, through direct insertion of the isonitrile into the carboxylic O–H bond through an Alderene like mechanism.ix In any event, we envisioned that this FCMA intermediate might subsequently undergo spontaneous rearrangement to generate an N-formyl amide adduct of the type 8.x Clearly, we were drawing precedent from the [1,4]-O→O transfer step in the Passerini process (1 → 2) and from the [1,4]-O→N acyl transfer step in the Ugi cascade (3 → 4). However, what we were contemplating was something rather more venturesome, i.e. a [1,3]-O→N acyl transfer step (7 → 8). Furthermore, given the absence of external acidic or basic agents in our projected reaction, epimerization issues that often compromise the value of amide bond-forming reactions could well be attenuated.
As will be shown, we have successfully reduced these concepts to practice. Through extension of the original premises, an array of broadly useful coupling methodologies has been developed for the formation of peptidyl and glycopeptidyl amide bonds. We review herein these recent advances and provide applications of emerging methodologies to help tackle complex synthetic challenges.
Type Ia Coupling of Isonitriles and Carboxylic Acids
We first explored the concept of 1:1 isonitrile –carboxylic acid coupling in the context of benzoic acid (9) and benzyl isonitrile (10). Although no product was observed at room temperature, the desired coupling did occur under thermally induced microwave irradiation conditions (ca 130 °C), to provide N-formyl amide 11 in 83% yield (Scheme 2). In order to classify the variants of isonitrile-based coupling reactions that have subsequently been developed in our laboratory, we assign the generic term “Type Ia”xi isonitrile-based coupling to the original carboxylic acid/isonitrile form of the transformation. We next demonstrated the ability of the Type Ia coupling to enable the construction of asparagine-linked glycosylamino acids. As shown in Scheme 2, a range of glycosyl isonitriles (12–14) were successfully merged with aspartate 15 to provide N-formyl glycosylamino acids (16–18).
Scheme 2.

Assembly of glycosylamino acids through Type Ia coupling.
As seen above, the Type Ia coupling reaction delivers an N-formyl moiety to the nitrogen atoms of the resultant amide bond. We anticipated that we would be able to achieve selective deformylation of the N-formyl bond, which was expected to be particularly vulnerable to nucleophilic cleavage due to its mixed imide-like character. Indeed, in a simple model series, we have demonstrated the selective conversion of the N-formyl group (11) to a variety of potentially useful functionalities, including N-H (19), N-homoallyl (20), and N-Me (21). In a separate system, the N-formyl amide adduct arising from a Type Ia coupling was readily cyclized to deliver the cyclic vinylogous imide, 22 (Scheme 3).
Scheme 3.

Methods for the selective derivatization of the N-formyl functionality arising from Type Ia coupling.
Type Ia Coupling: Mechanistic Studies
Having demonstrated the gross feasibility of our Type Ia coupling sequence, we next sought to undertake mechanistic studies into the nature of the transformation. As described above, we had postulated that the reaction would proceed through the intermediacy of a highly reactive FCMA species, which would spontaneously undergo [1,3]-O→N acyl transfer, generating N-formyl amide products (5 + 6 → 7 → 8, Scheme 4). We had also observed that in the presence of external nucleophilic trapping agents (RXH), there also arise products of type 23, in which the putative FCMA intermediate has presumably been intercepted by the nucleophile prior to rearrangement. In a generic sense, we consider the formation of these interrupted coupling products to arise through what we term a “Type IIa” coupling reaction.
Scheme 4.

Type Ia and Type IIa coupling: Mechanistic considerations.
In light of our experiences with the high-energy FCMA species, we were skeptical of a number of reports from other groups claiming to have accomplished the isolation of such intermediates as crystalline products under standard conditions.xii We were particularly curious about a disclosure in which alleged FCMA species were reported to have been isolated from various benzoic acids and isonitriles in the presence of methanol at room temperature.xiia Based on our experiences, we predicted that even if the FCMA species were to be formed, it would have rapidly undergone nucleophilically mediated solvolysis under these conditions. We thus set out to explore these claims, and found that in each case, the observed product had been misassigned as a FCMA species.xiii Thus, to our knowledge, no FCMA has yet been isolated under usual reaction conditions.xiv
The formation of the observed products is consistent with the intermediacy of a highly reactive FCMA species (7), which rapidly undergoes either [1,3]-O→N acyl transfer to provide an N-formyl amide, 8 (Type Ia), or in the presence of an external nucleophile, acylation to generate 23 and 24 (Type IIa).xv Of interest to us from a mechanistic standpoint was the product distribution observed when the FCMA is formed in the presence of an exogenous nucleophile (RXH). Under these conditions, a mixture of Type Ia (rearrangement) and Type IIa (acylation) products is obtained. It may be that a single FCMA intermediate is formed, which can either undergo rearrangement (8) or nucleophilic trapping (23). Alternatively, the observed product ratio might be a consequence of concurrent formation of two FCMA isomers –the anti (7a) and syn (7s) isomers. The former would be well positioned to undergo rapid [1,3]-O→N acyl transfer, and might be already “committed” to forming 8. In contrast, the non-competent syn isomer would be susceptible to nucleophilic attack in the presence of an appropriate nucleophile. Presumably, in the absence of a suitable acyl acceptor, the non-rearranging 7s isomer would revert to the rearranging 7a form. We have obtained some evidence in support of this view. Under our standard coupling conditions, we observed the formation of rearranged and intercepted products in the presence of either 1 or 5 equivalents of external nucleophile (MeOH). Interestingly, a large increase in methanol did not lead to any significant increase in formation of the Type IIa methyl ester product (~40%). We best interpret these results as suggesting that there are produced two geometrically isomeric FCMA intermediates: 7s, which is highly susceptible to methanolysis, and 7a, which is committed to undergoing intramolecular rearrangement, even in the presence of excess nucleophile.
In attempting to elucidate the mechanistic intricacies of our Type Ia coupling reaction, we also took note of the possibility of an alternative FCMA – based pathway, which would not involve a [1,3]-O→N acyl rearrangement to produce N-formyl amide products. As outlined in Scheme 5, FCMA intermediate 7 would be attacked by carboxylic acid 1 to produce symmetrical anhydride 25 and formamide 24. N-acylation of 24 with 25 would furnish the observed product (8). This potential pathway was examined in the context of the N-formyl amide adduct 28, which is readily formed under our coupling conditions. Thus, if the – symmetrical anhydride pathway were viable, the intermediates formed from addition of acid to FCMA, 26 and 27, would be expected to combine to produce amide 28 under standard coupling conditions. In the event, however, only a 16% yield of 28 was obtained from coupling of 26 and 27 under microwave irradiation. These results imply that the symmetrical anhydride pathway is certainly not dominant under our reaction conditions.
Scheme 5.

Evaluation of a potential formamide symmetrical anhydride pathway.
Finally, we sought to evaluate the reactivity of the intermediate FCMA species. In this context, we wondered whether a weakly nucleophilic formamide would be capable of nucleophilic attack on a FCMA intermediate. As described above (Scheme 5), we had previously demonstrated that an intermediate formamide would not be sufficiently nucleophilic to efficiently trap a symmetrical anhydride (26 + 27 → 28). As shown in Scheme 6, carboxylic acid 29, isonitrile 30, and formamide 31 were subjected to standard reaction conditions. After 30 minutes, the N-formyl amide 33, arising from [1,3]-O→N acyl transfer of the presumed FCMA (32), was isolated in 50% yield. However, we also obtained a substantial amount (20%) of cross-over product, 34, presumably arising from interception of the intermediate species by formamide 31. These data lend support to the supposition that a species which is a more powerful acylating agent than anhydride (presumably FCMA 32) is produced in the course of the reaction.
Scheme 6.

Evidence for the existence of a highly reactive FCMA intermediate (32).
Development of a Modified Type Ia Coupling Protocol
We recognized that, in order for this method to find extensive application to complex biologic targets, we would need to develop a set of conditions that would allow for reaction under ambient temperatures. The high temperatures required for coupling (150 oC) could be problematic in the context of often sensitive, but biologically important products, such as many peptides and glycans. Along these lines, we took note of an earlier finding from our laboratory, wherein Type II products (of the type 23) could be obtained at room temperature. It seems that, although the FCMA intermediate is actually generated at ambient temperature, thermolysis is required to achieve [1,3]-O→N acyl rearrangement leading to the desired product. We had separately observed that a serine-derived isonitrile substrate, 35, presenting a free alcohol moiety, reacted with a carboxylic acid (36) at room temperature, to generate a rearrangement product, 37, in which the formyl group had transferred from the nitrogen to the hydroxyl group.xvi Interestingly, when the proximal alcohol was protected, no rearrangement product was observed. Seemingly, the resident free hydroxyl group had served to promote formation or activation of the FCMA species, perhaps via an activated cyclic intermediate, as shown.
In light of these observations, we sought to examine the consequences of adding an external nucleophile to the reaction, in the hopes of achieving a similar outcome in a generic substrate. Indeed, we were pleased to observe that, in the presence of thiophenol at 50 oC, carboxylic acid 38 underwent Type Ia coupling with cyclohexylisonitrile to afford a mixture of thioacetal (40) and N-formyl amide (41) products. The former may be converted to the latter in a single, high-yielding step (88%). The role of the thiophenol in facilitating the rearrangement has not yet been established. In principle, at least, the transformation may well proceed through a FCMA-derived tetrahedral intermediate, 39, which could well suffer more rapid intramolecular acyl transfer than is the case with the unmodified FCMA to generate the observed products.
Type Ib Coupling: Isonitriles and Thioacids
Further progress toward our goal of developing a mild and general route for amide bond formation was achieved with the discovery that thioacids, as opposed to carboxylic acids, readily undergo coupling with isonitrile substrates at room temperature (Scheme 8, eq. 1).xvii The merger of isonitriles and thioacids is termed a “Type Ib” coupling.xi A range of amino thioacids were found to react with amino acid isonitrile substrates to generate dipeptide products in good yields. Interestingly, product yields were generally observed to correlate positively with steric hindrance of the isonitrile. This trend may be attributed to suppression of thio-FCMA cleavage in hindered cases. Conceivably, these bulky thio-FCMA intermediates may also be more prone to undergo rapid syn→anti isomerization, thereby enhancing the rate of the [1,3]-S→N acyl transfer. Dethioformylation of the dipeptide products was achieved through a two-step protocol, involving treatment with mCPBA followed by sodium bicarbonate (44→ 45, Scheme 8).
Scheme 8.

Type Ib coupling of thioacids and isonitriles.
Thioacid substrates were also found to be competent coupling partners in reactions with glycan isonitriles.xviii As shown in Scheme 8 (eq. 2), thioacid 47 and isonitrile 46 readily underwent coupling at room temperature in the presence of 2,6-dimethylthiophenol activator, to afford the desired glycosylamino acid 48 in 88% yield.
Our attempts to extend Type Ia and Type 1b couplings to highly hindered substrates were unsuccessful. In a telling example, exposure of pivalic acid 49 and hindered isonitrile 50 to microwave irradiation under thermal conditions did not lead to formation of the rearrangement product expected on the basis of precedent (Scheme 9).xix It seems that either the neopentyl coupling partners could not form the FCMA species or, alternatively, a FCMA intermediate, so formed, would be unable to undergo the [1,3]-O→N migration step, presumably due to excessive steric hindrance. Interestingly, when an external nucleophile (54) was added to the reaction, amide 55, arising from interception of an intermediate FCMA species, was obtained. This finding would seem to establish that the neopentyl FCMA species is indeed generated under the reaction conditions, but is then unable to undergo the requisite [1,3]-O→N acyl transfer to deliver Type Ia adducts. An analogous finding was observed in Type Ib settings. The critical observation that highly hindered isonitrile substrates form FCMA intermediates that are impervious to rearrangement yet susceptible to external attack prompted us to pursue a new approach to isonitrile-based acylation (vide infra).
Scheme 9.

Coupling of Hindered Isonitriles.
Type IIb Coupling: The Use of thio-FCMA Intermediates as Particularly Strong Acyl Donors
As described above, in the presence of a nucleophile, FCMA and thio-FCMA intermediates can undergo reaction by either of two competing pathways: intramolecular acyl migration (Type Ia or Ib) or bimolecular reaction with an acyl acceptor (Type IIa or IIb).xx,xi Our initial efforts had focused mainly on promoting the acyl migration pathway, wherein the two substrates– i.e. thioacid 57 and isonitrile 6 –are both represented in the final product (61, Scheme 10). Although we have developed methods for the efficient syntheses of a variety of value – added isonitrile substrates,xxi we recognized that the need to pre-assemble complex isonitrile substrates could be a disincentive to the broad applicability of this approach, particularly in the context of large peptidic or glycan substrates.
Scheme 10.

Type IIb coupling: Thio-FCMA intermediates as strong acyl donors.
Armed with the knowledge that hindered isonitriles do not undergo [1,3]-acyl transfer, a promising variation presented itself. We considered the possibility of employing such hindered isonitriles to effectively suppress the Type Ib [1,3]-S→N acyl migration pathway from thio-FCMA 58, thus favoring the bimolecular acylation (Type IIb) pathway from thio-FCMA 58. If the failure of hindered isonitriles to undergo intramolecular acyl transfer was not contingent on the thioacid coupling partner also being hindered, it should be possible to accomplish the direct coupling of substrates such as 57 with external nucleophiles (NuH) to produce adducts of the type 59. Under this plan, the hindered isonitrile (6) would serve as a simple “throwaway” (but recyclable!) reagent, and following interception by the amine nucleophile, an isonitrile-derived recyclable debris product 60 would be extruded along with the product. Needless to say, the benefit of such a plan would be that the amine substrate could be employed directly in the coupling, obviating the need to pre-assemble a high value – added isonitrile function.
We were gratified to find that, as expected from our earlier results, treatment of thioacid 62 with the bulky tert-butylisonitrile (50) and aniline readily resulted in coupling at room temperature to provide amide adduct 63 in 77% yield.
This powerful new acylation method nicely accommodates a range of substrates, including bulky thioacids and amines (eq. 2–3), secondary amines (eq. 4–5) and even alcohol nucleophiles (eq. 6). Of particular note is the smooth formation of products in which both sides of the resultant amide bond are highly hindered. Needless to say, the formation of isopropyl ester 74, under these conditions, is particularly convincing testimony to the high reactivity of the intermediate acyl donor (presumably the corresponding thio-FCMA).
Application: Cyclosporine
To further evaluate the complexity-building potential of our isonitrile-based findings under “battlefield realities”, we sought to apply them to a total synthesis of cyclosporine A.xxii Isolated from the fungus Tolypocladium inflatum gams,xxiii cyclosporine A exhibits potent immunosuppressive properties and, as such, is used to facilitate organ transplantation as well as in the treatment of immune system-related disorders including psoriasis and rheumatoid arthritis.xxiv Cyclosporine A is a cyclic peptide containing seven sites of N-methylation, which are apparently essential to the observed biological activity. Our isonitrile-based methods, which can deliver N-methyl functionality at the site of ligation, were seen to be well suited to the synthetic challenges posed by cyclosporine A.
Our plan for accomplishing the total synthesis of cyclosporine A envisioned the assembly of two peptidic fragments, 81 and 91, through recourse to the recently discovered isonitrile-based methods. In the concluding stages of the synthesis, fragments 81 and 91 were to be joined and ultimately subjected to a late-stage macrolactamization. Indeed, we also hoped to accomplish cycloamidation through application of our Type IIa acylation method.
In the event, the synthesis of peptide fragment 81 was achieved through a sequence featuring two Type Ib coupling reactions between thioacid and isonitrile amino acid substrates (Scheme 12). Similarly, fragment 91, representing the bulk of the peptidic backbone, was assembled via Type IIb acylation between intermediates 85 and 89, themselves prepared via Type Ia and Type IIb methods (Scheme 13).
Scheme 12.

Synthesis of tetrapeptide 81 en route to cyclosporine A.
Scheme 13.

Synthesis of intermediate 91 en route to cyclosporine A.
We were now prepared to merge fragments 81 and 91 in anticipation of the key macrolactamization step. In the event, following removal of the benzyl ester of 81, the two peptide fragments combined to afford polypeptide 92. As shown in Scheme 14, we were able to successfully extend the logic of the FCMA acylation to the macrolactamization reaction. Thus, when treated with HOBt and cyclohexylisonitrile, the deprotected carboxylic acid derived from 92 underwent macrolactamization to deliver the natural product, cyclosporine A, in 54% yield from 81. The success of the macrolactamization in the context of this highly complex system is presumably attributable, at least in part, to the pre-organization of the substrate through intra-strand hydrogen bonding.
Scheme 14.

Completion of the total synthesis of cyclosporine A.
In a further demonstration of the broad utility of the emerging isonitrile-based strategy toward amide bond formation, we have recently completed a second-generation route to cyclosporine A.xxv In designing our second-generation synthesis, we took recourse to a bond-disconnection strategy not unlike that employed in our first route. As shown in Scheme 15, dipeptide 76 (see Scheme 12) was readily advanced to tetrapeptide 81 through iterative Type IIb couplings. Hydrogenolysis of 81 delivered intermediate 94 in excellent overall yield.
Scheme 15.

Second generation route to cyclosporine A: Synthesis of tetrapeptide 94.
The synthesis of the heptapeptide substrate, 91, is outlined in Scheme 16. Thus, a series of high-yielding iterative Type IIb coupling and deprotection reactions gave rise to hexapeptide 101b. In a key transformation, peptide 101b underwent Type IIa coupling with unnatural amino acid 102, bearing a free alcohol functionality, to deliver heptapeptide 91 in good yield without any apparent epimerization (Scheme 16).
Scheme 16.

Second generation route to cyclosporine A: Synthesis of hexapeptide 91.
With the key fragments in hand, we next investigated the proposed isonitrile-based Type II coupling between 94 and 91. Interestingly, under isonitrile-activated Type II conditions, we obtained a 71% yield of epi-92, wherein the α-carbon of MeVal (see asterisk) had suffered complete inversion (Scheme 17). In light of this finding, the requisite fragment condensation of 94 and 91 was instead accomplished, as precedented,xxii through the use of PyBOP and NMM. With the desired undecapeptide 92 in hand, we next sought to explore the possibility of enhancing the efficiency of the Type II macrolactamization reaction by incorporating a C-terminal thioacid in lieu of the carboxylic acid at the site of cyclization. Our first-generation route had employed a Type IIa carboxylic acid-based cyclization, and the elevated temperatures required for cyclization had led to an erosion of yield, presumably due to starting material decomposition and formation of by-products. We postulated that a Type IIb thioacid-based macrolactamization would proceed under ambient temperatures, minimizing the opportunity for competing side reactions. Thus, 92 was converted to 103 in 68% overall yield. Removal of the Boc protecting group, followed by exposure to cyclohexylisonitrile in the presence of HOBt at room temperature resulted in nearly complete conversion to cyclosporine A within 10h, with minimal formation of byproducts.
Scheme 17.

Second-generation total synthesis of cyclosporine A.
Further Application: Synthesis of Constrained Peptides
Peptides often exhibit high affinity and selectivity for key biotargets, and thus represent, at least in principle, an important class of lead compound for drug development. However, a serious impediment to the broad success of peptides as clinical agents is their high susceptibility to protease-induced degradation.xxvi In the context of various efforts to address this kind of problem, it has been found that the introduction of conformational constraint elements to these peptidic scaffolds can serve to enhance both pharmacostability and biological activity.xxvii In this context, we took note that our Type I coupling protocols, which deliver an N-formyl amide at the site of ligation, could potentially be exploited to allow access to biologically active peptidic fragments incorporating conformational constraints. In addition to exploiting partial conformational immobilization to influence the affinities of ligands for their receptors, we envisioned that a temporary molecular constraint might serve to mitigate the aggregation tendencies often exhibited by unfolded polypeptides. Of course, we also sought to ensure that the constraint could be readily removed to restore the native peptide sequence.xxviii In this way, a temporary molecular constraint might be used in the context of a total synthesis to stabilize otherwise aggregation-prone peptides en route to the eventual more stable target. More specifically, as outlined in Scheme 18 below, we envisioned iterative Type Ia coupling reactions to generate a polypeptide of the type 108, bearing N-formyl groups on adjacent “imide – like” nitrogens. We further anticipated conversion, by two-fold reduction, of these proximal formyl groups to generate a constrained tripeptide, 109. Elongation of the peptide in the N-and C-terminal directions would provide a target polypeptide, 110, bearing a site-specific cyclic constraint. Finally, removal of the CH2–O–CH2 bridge would reveal the native peptide system, 111, at a strategically propitious stage.
Scheme 18.

Synthesis of constrained peptides through isonitrile coupling.
This plan has indeed been reduced to practice. Thus, in a representative example, carboxylic acid 82 and isonitrile 83 underwent Type Ia coupling to afford, following cleavage of the t-butyl ester, dipeptide 112 in 82% yield (Scheme 19). A second Type Ia reaction with isonitrile 43 generated tripeptide 113, containing proximal N-formyl functionalities. Interestingly, the resident N-formyl group of 112 had played a key role in ensuring the success of this second coupling reaction, by stabilizing the intermediate FCMA against non-productive oxazolone formation. Next, treatment of 113 with LiBH4 followed by TFA provided constrained tripeptide 114. The N-terminal amine, which had until now been housed as an azide functionality, was exposed by reduction, and the N- and C- terminii were extended to generate 115. Finally, treatment with 0.1M HCl and 1,3-propanedithiol delivered the target linear heptapeptide 116 in 95% yield. We note that, in this study, constrained peptides (cf. 110) were generally found to be much more solubilizable and less prone to aggregation than were their linear counterparts (cf. 111).
Scheme 19.

Application of Type Ia coupling to the synthesis of a temporary peptide constraint.
Beyond isonitriles: Development of a general method for the ligation of peptides
Despite the broad utility of the isonitrile-based Type I and Type II coupling reactions described above, we were not unaware of the difficulties inherent in extending our peptide-based coupling to encompass more complex systems. In fact, the Type II coupling of even dipeptidyl thioacids is often low-yielding, apparently due to the rapid conversion of the intermediate dipeptide thio-FCMA to the non-productive oxazolone intermediate (Scheme 20). In exploring potential solutions to this conundrum, we considered the possibility of achieving in situ conversion of the activated thio-FCMA intermediate to a still more productive acyl donor. Along these lines, we took note of the fact that peptide bonds formed via HOBt ester intermediates tend to exhibit significantly lower levels of C-terminal, oxazolone- induced epimerization.xxix We thus envisioned a reaction setting wherein a thio-FCMA, derived from a peptidyl thioester and a “throwaway” isonitrile (i.e. cyclohexylisonitrile), would progress to become an HOBt ester. The latter would hopefully undergo ligation with the N-terminal coupling partner.
Scheme 20.

Thio-FCMA ligation: The challenge.
As shown in Scheme 21, this proposed solution was indeed successful.xxx Thus, in the presence of both HOBt and cyclohexylisonitrile, thioacid peptide 117 and peptide 118 underwent ligation to deliver adduct 119, within 20h in 60% yield (eq. 1), presumably via the intermediacy of both the thio-FCMA and HOBt ester species (Scheme 20). To our surprise, however, a control experiment revealed that even in the absence of isonitrile, the coupling of 117 and 118 in the presence of HOBt proceeded in 82% yield over only 12h (eq. 2). Moreover, these HOBt -based conditions were demonstrated to effectively promote ligation of a range of peptidyl substrates, including those bearing epimerizable C-terminal amino acid residues. In several direct comparisons, ligations performed in the presence of only HOBt actually proceeded with higher yields and faster reaction times than those executed under isonitrile/HOBt conditions. Moreover, the HOBt-promoted reaction could be extended to the ligation of glycopeptide substrates, to provide access to di-glycosylated peptide adducts (Scheme 22, 120 + 121→ 122).
Scheme 21.

Type Ib (1) and oxidative (2) ligations of peptides.
Scheme 22.

Oxidative ligation of glycopeptides.
From a mechanistic perspective, these results are quite interesting, albeit not yet explicated. Of course, the finding that the isonitrile is not required for ligation certainly does not preclude the possibility that the mechanism proposed above (Scheme 20) is, in fact, operational when the isonitrile is present. However, the efficient formation of product in the absence of isonitrile does establish the existence of a pathway to ligation from thioacid and amine starting materials, which is not dependent on the intermediacy of a FCMA species. It is, in principle, possible that the thioacid peptide is inherently capable of participating in direct ligation with an amine coupling partner; in other words, that SH is a sufficiently powerful leaving group to allow for acylation. However, consistent with the findings of Sheehan and Johnsonxxxi and, more recently, those of Orgel and Liu,xxxii it is perhaps more likely that these HOBT-mediated ligations are proceeding through some, as yet unspecifiable, oxidative mechanism. A series of mechanistic studies have provided support for this theory. Thus, when attempts were made to minimize opportunities for fortuitous oxidation (i.e., by performing the reaction under an argon atmosphere) the yield of amide was notably reduced. Conversely, the ligations proceeded more rapidly in the presence of iodine, an established oxidizing agent, than in its absence. Although the exact nature of the transformation has not yet been defined, one can envision a reaction pathway, initiated by low levels of thioacid oxidation, with turnover to restore the C-terminal acyl donor. Any number of plausible pathways could well serve to sustain a high-turnover oxidation – initiated route. One such potential model, involving trace formation of a diacyl disulfide intermediate (124), is outlined in Scheme 23. However, we have certainly not ruled out that the oxidative component of the pathway in fact operates via “stoichiometric” rather than catalytic levels of oxidizing agent with high turnover.
Scheme 23.

Proposed mechanistic pathway for oxidative peptide ligation.
A powerful new route for the formation of N-glycosyl asparagine glycopeptides
We have been pursuing the development of improved methods for the merger of oligosaccharide and peptide domains. Currently, the standard approach to glycopeptide assembly involves appendage of an anomeric β-glycosylamine via Lansbury aspartylation to a properly protected aspartate acyl donor, which is housed within the target peptide fragment.xxxiii Several complicating factors tend to erode the efficiency of the Lansbury aspartylation, starting with the weak nucleophilicity of the glycan amine at the reducing end of the glycan. Further complicating aspartylation is a competing base-induced decomposition pathway, wherein the putative aspartyl donor undergoes cyclization with the NH of the adjacent amino acid to generate a non-productive aspartimide side product (Scheme 24). In certain peptide substrates, particularly those bearing glycine, serine, or alanine residues at the C-terminus of the aspartate, the tendency toward aspartimide formation can become prohibitive.xxxiv
Scheme 24.

Merger of glycan and peptide domains through aspartylation; undesired base-induced aspartimide formation.
We recognized that the mild, base-free acylation methods recently discovered in our laboratory, as described above, could well be suited to the formidable challenge of glycan aspartylation. As shown in Scheme 25, we were indeed able to accomplish convergent aspartylation under Type IIb coupling conditions. Thus, in the presence of the “sacrificial” cyclohexylisonitrile, peptide thioacid 128 and glycosylamine 129 were joined to yield glycopeptide 130 in 70% yield, accompanied by low levels of peptide carboxylic acid, aspartimide, and products presumably arising from Type Ib intramolecular S→N rearrangement of the thio-FCMA. In light of the discovery, described above, that HOBt can promote thioacid coupling, presumably via the oxidative mechanism described above, we next examined the reaction of 128 and 129 in the presence of HOBt and in the absence of isonitrile mediation. We were indeed pleased to observe that aspartylation occurred rapidly, to yield glycopeptide 130, in 85% yield, along with quite low levels of carboxylic acid and aspartimide side products. It is possible that this reaction also proceeds through an oxidative mechanism, analogous to those proposed above (see Scheme 23, R = glycan).xxxv
Scheme 25.

Isonitrile-based and oxidative aspartylation methods.
Further Application: Generation of chiral heterocycles
The biological and pharmaceutical importance of heterocyclic compounds in medicinal chemistry needs little elaboration. We have recently begun the pursuit of methods for the generation of pharma type heterocycles with pendant enantiodefined chirality, from the N-formyl amide products of our Type Ia coupling reactions. This work arose from a fundamental interest in exploring the reactivity of the previously little known N-formyl amide functional group, which is now available through the Type I reaction. We have found that the azide moiety in these products offers an excellent handle for the selective functionalization of the formyl carbonyl group (Scheme 26). In considering the particularly reactive character of the imide-like N-formyl group, the possibility of a Staudinger reaction of a resident azide group followed by aza-Wittig reaction presented itself. In the event, we found that treatment of 131 (obtained via coupling of the azido acid with glycine isonitriles) with trimethylphosphine in benzene resulted in formation of imidazolinones such as 132 in good yield. We were anticipating an alternative method for activation of the azide for reaction, which would be initiated via a presumed radical reduction of the azido group. Indeed, reaction of 133 with n-SnBu3H gave rise to hydantoin 134 in good yield, seemingly as a single stereoisomer. A similar conversion of 135 to 136 demonstrated applicability of the reaction to a methionine type side chain. We are currently studying the scope of this reaction, not to mention its non-obvious mechanism. These results suggest that a broad study of peptidyl N-formylamides could well be productive.
Scheme 26.

Application of Type Ia coupling to the synthesis of heterocycles.
Conclusion
In summary, we have described herein recent advances from our laboratory in the field of acylation chemistry, arising from revisiting a classical functional group in organic chemistry with the benefit of contemporary mechanistic insights. We have thus come upon new isonitrile-based coupling methods and applied them to the syntheses of complex “biologic” targets of potential therapeutic import. Finally, these investigations have led to the somewhat fortuitous discovery of a widely general oxidative coupling pathway between thioacid and amine substrates, which even encompasses aspartylation of highly complex “reducing end” glycosylamines. We anticipate that continuing research following these new directions could well lead to further discoveries of value to chemical synthesis.
Scheme 7.

Mild conditions for the Type Ia coupling reaction.
Scheme 11.

Scope of Type IIb coupling reaction.
Acknowledgments
Support was provided by the National Institutes of Health (CA103823 and CA28824 to SJD). P.K.P. was supported by a postdoctoral fellowship (PF-11-014- 01-CDD) from the American Cancer Society.
Biographies
Rebecca M. Wilson. Rebecca M. Wilson received her undergraduate training in biochemistry at Tufts University. She went on to the University of California at Berkeley and then to Caltech, where she conducted research with Professor David MacMillan, with a focus on the development of organocatalytic enantioselective intramolecular Diels-Alder reactions. After receiving her masters with Prof. MacMillan, she worked in the small-molecule project group at Amgen Pharmaceuticals, where she dealt with analytical and regulatory matters. In 2004, she joined the program of Professor Samuel Danishefsky at Memorial Sloan-Kettering Cancer Center, where she is involved in various consultative and editorial undertakings in chemical synthesis and its emerging role in vaccines and small-molecule drugs.
Jennifer L. Stockdill. Jennifer L. Stockdill was born in Mankato, Minnesota, USA in 1981. She obtained a BS in Chemistry at Virginia Polytechnic Institute and State University in 2003. In 2009, she obtained a PhD under the direction of Professor Brian M. Stoltz at Caltech as a Novartis Women and Minorities Research Fellow. She is currently a K99 NIH postdoctoral fellow in the laboratories of Prof. Samuel J. Danishefsky. Her research interests include the design of new reaction methods for the total synthesis of medically important natural products and proteins.
Paul A. Vadola. Paul A. Vadola was born in Staten Island, New York, USA in 1982. Following the completion of his undergraduate studies at Fordham University in 2004 he began his graduate work at Columbia University. In 2010 he received his PhD in chemistry under the direction of Prof. Dalibor Sames. He is currently pursuing his postdoctoral research in the laboratories of Prof. Samuel J. Danishefsky.
Xiangyang Wu. Xiangyang Wu received his MS in organic chemistry at NanKai University in China. He went on to Konstanz University in Germany, where he conducted research with Professor Richard R. Schmidt, with a focus on the synthesis of complex carbohydrate on polymer support. After receiving his PhD with Prof. Schmidt in 2003, he moved to Canada, and worked as a post-doctoral research associate with Professor David R. Bundle at the University of Alberta. He is currently pursuing his postdoctoral research in the laboratories of Prof. Samuel J. Danishefsky. His research interests include the synthesis of biologically important molecules based on isonitrile chemistry.
Xuechen Li. Xuechen Li studied Chemistry for a BSc in Nankai University, China. After the completion of his MSc at University of Alberta (2000–2003) and Ph.D at Harvard University (2003–2006), he worked as a postdoctoral fellow in the laboratory of Prof. Samuel Danishefsky at Memorial Sloan-Kettering Cancer Center. Since 2009, he joined the Department of Chemistry at The University of Hong Kong as an assistant professor. His current research interests include chemical biology and drug discovery.
Peter K. Park. Peter K. Park was born in Taegu, South Korea in 1979. He received his A.B. degree in chemistry in 2001 from Harvard College, where he worked in the laboratory of Professor Stuart L. Schreiber. He pursued graduate studies at Columbia University under the auspices of the NIH Medical Scientist Training Program, and received a combined M.D., Ph.D. degree in 2009. His doctoral work, in which he completed the first total synthesis of the marine macrolide dolabelide D, was conducted under the guidance of Professor James L. Leighton. He is currently an American Cancer Society postdoctoral fellow with Professor Samuel J. Danishefsky at Memorial Sloan-Kettering Cancer Center.
Samuel J. Danishefsky. Samuel J. Danishefsky received his B.S. degree at Yeshiva University. He did his graduate research under the late Professor Peter Yates. He then joined the laboratory of Professor Gilbert Stork at Columbia University as an NIH Postdoctoral Associate. His first academic position was at the University of Pittsburgh, where he joined as Assistant Professor in 1963. He was promoted to Associate Professor, Professor, and University Professor. In January 1980, he moved to Yale University and was named Eugene Higgins Professor in 1981. Appointed by President A. Bartlett Giamatti as Chairman of the Department of Chemistry, he served until 1987. He became Sterling Professor at Yale in 1990. In 1993, Professor Danishefsky moved back to New York, where he is currently Centennary Professor of Chemistry at Columbia University and the Eugene Kettering Chair and Head of the Laboratory of Bioorganic Chemistry at Memorial Sloan-Kettering Cancer Center. In 1996, he shared the Wolf Prize in Chemistry with Professor Gilbert Stork. In 2006, he was the recipient of the Franklin Medal in Chemistry, the Bristol Myers Squibb Lifetime Achievement Award in Chemistry, and the National Academy of Sciences Award in the Chemical Sciences.
References
- i.For a recent review, see: Gamblin DP, Scanlan EM, Davis BG. Chem Rev. 2009;109:131. doi: 10.1021/cr078291i.
- ii.a) Kan C, Danishefsky SJ. Tetrahedron. 2009;65:9047. doi: 10.1016/j.tet.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yuan Y, Chen J, Wan Q, Wilson RM, Danishefsky SJ. Biopolymers: Peptide Science. 2010;94:373. doi: 10.1002/bip.21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iii.For recent literature reviews on isonitrile chemistry, see: Tobisu M, Chatani N. Chem Lett. 2011;40:330.Lygin AV, de Meijere A. Angew Chem. 2010;122:9280. doi: 10.1002/anie.201000723.Angew Chem Int Ed. 2010;49:9094.Gulevich AV, Zhdanko AG, Orru RVA, Nenajdenko VG. Chem Rev. 2010;110:5235–5331. doi: 10.1021/cr900411f.
- iv.a) Hofmann AW. Justus Liebigs Ann Chem. 1867;144:114. [Google Scholar]; b) Gautier A. Justus Liebigs Ann Chem. 1868;146:119. [Google Scholar]
- v.a) Ritter JJ, Minieri PP. J Am Chem Soc. 1948;70:4045. doi: 10.1021/ja01192a022. [DOI] [PubMed] [Google Scholar]; b) Benson FR, Ritter JJ. J Am Chem Soc. 1949;71:4128. [Google Scholar]
- vi.a) Passerini M. Gazz Chim Ital. 1921;51:126. [Google Scholar]; b) Passerini M. Gazz Chim Ital. 1921;51:181. [Google Scholar]
- vii.a) Ugi I, Meyr R, Fitzer U, Steinbrücker C. Angew Chem. 1959;71:386. [Google Scholar]; b) Dömling A, Ugi I. Angew Chem. 2000;112:3300. doi: 10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2000;39:3168. [Google Scholar]
- viii.For a review of the Beckmann rearrangement, see: Gawley RE. Org React. 1988;35:1.
- ix.Jones GO, Li X, Hayden AE, Houk KN, Danishefsky SJ. Org Lett. 2008;10:4093. doi: 10.1021/ol8016287. [DOI] [PubMed] [Google Scholar]
- x.Li X, Danishefsky SJ. J Am Chem Soc. 2008;130:5446. doi: 10.1021/ja800612r.We were certainly not the first to conceive of the idea of an O-acylimidate to amide rearrangement. This type of rearrangement is inherent in the conversion of O-acylated to N-acylated ureas, and in the Mumm rearrangement. However, to the best of our knowledge, none of this chemistry had been connected to isonitriles via a FCMA. See Weygand F, Hoffmann D, Wunsch E. Z Naturforsch. 1966;B21:426.and Mumm O, Hesse H, Volquartz H. Ber. 1915;48:379.
- xi.In this nomenclature, the Roman numeral dictates the mode of reactivity and the letter indicates the nature of the carboxylic acid-derived component. Thus Type I reactions involve a direct coupling of an acid or thioacid and and an isonitrile, while Type II reactions involve the attack of a nucleophile on the FCMA intermediate. The designation “a” refers to a carboxylic acid, while “b” indicates a thioacid as the coupling partner.
- xii.Shaabani A, Soleimani E, Rezayan AH. Tetrahedron Lett. 2007;48:6137.Gloede J, Gross H. Zeitschrift fuer Chemie. 1968;8:219.Gloede J. J fuer Praktische Chemie. 1982;324:667.Shono T, Kimura M, Ito Y, Nishida K, Oda R. Bull Chem Soc Japan. 1964;37:635.The structure was presumed on the basis of its infrared spectrum; Isaka M, Boonkhao B, Rachtawee P, Auncharoen P. J Nat Prod. 2007;70:656. doi: 10.1021/np060509t.
- xiii.Li X, Yuan Y, Berkowitz WF, Todaro LJ, Danishefsky SJ. J Am Chem Soc. 2008;130:13222. doi: 10.1021/ja8047078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xiv.Spectroscopic suggestion of such an intermediate has been described. See: Hou JL, Ajami D, Rebek J., Jr J Am Chem Soc. 2008;130:7810. doi: 10.1021/ja802288k.Restorp P, Rebek J., Jr J Am Chem Soc. 2008;130:11850. doi: 10.1021/ja803854r.
- xv.Li X, Yuan Y, Kan C, Danishefsky SJ. J Am Chem Soc. 2008;130:13225. doi: 10.1021/ja804709s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvi.Wu X, Li X, Danishefsky SJ. Tetrahedron Lett. 2009;50:1523. doi: 10.1016/j.tetlet.2009.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvii.Yuan Y, Zhu J, Li X, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:2329. doi: 10.1016/j.tetlet.2009.02.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xviii.Wu X, Yuan Y, Li X, Danishefsky SJ. Tetrahedron Lett. 2009;50:4666. doi: 10.1016/j.tetlet.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xix.Stockdill JL, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:5152. doi: 10.1016/j.tetlet.2009.06.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xx.Rao Y, Li X, Danishefsky SJ. J Am Chem Soc. 2009;131:12924. doi: 10.1021/ja906005j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxi.Zhu J, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:577. doi: 10.1016/j.tetlet.2008.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxii.Wu X, Stockdill JL, Wang P, Danishefsky SJ. J Am Chem Soc. 2010;132:4098. doi: 10.1021/ja100517v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxiii.Rüegger A, Kuhn H, Lichti HR, Loosi R, Huguenin R, Quiquerez A, von Wartburg A. Helv Chim Acta. 1976;59:1075. doi: 10.1002/hlca.19760590412. [DOI] [PubMed] [Google Scholar]
- xxiv.a) Laupacis A, Keown PA, Ulan RA, McKenzie N, Stiller CR. Can Med Assoc J. 1982;126:1041. [PMC free article] [PubMed] [Google Scholar]; b) Dreyfuss M, Harri E, Hofmann H, Kobel H, Pache W, Tscherter H. Eur J Appl Microbiol. 1976;3:125. [Google Scholar]; c) Bueding E, Hawkins J, Cha Y-N. Agents and Actions. 1981:380. doi: 10.1007/BF01982474. [DOI] [PubMed] [Google Scholar]
- xxv.Wu X, Stockdill JL, Park PK, Danishefsky SJ. 2011 Submitted. [Google Scholar]
- xxvi.a) Marx V. Chem Eng News. 2005;83:17. [Google Scholar]; b) Nestor JJ., Jr Curr Med Chem. 2009;16:4399. doi: 10.2174/092986709789712907. [DOI] [PubMed] [Google Scholar]
- xxvii.a) Gentilucci L, De Marco R, Cerisoli L. Curr Pharm Des. 2010;16:3185. doi: 10.2174/138161210793292555. [DOI] [PubMed] [Google Scholar]; b) Hruby VJ. Nat Rev Drug Discov. 2002;1:847. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- xxviii.Wu X, Park PK, Danishefsky SJ. J Am Chem Soc. 2011;133:7700. doi: 10.1021/ja2023898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxix.König W, Geiger RA. Chem Ber. 1970;103:788. doi: 10.1002/cber.19701030319. [DOI] [PubMed] [Google Scholar]
- xxx.Wang P, Danishefsky SJ. J Am Chem Soc. 2010;132:17045. doi: 10.1021/ja1084628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxi.Sheehan JC, Johnson DA. J Am Chem Soc. 1952;74:4726. [Google Scholar]
- xxxii.Liu R, Orgel LE. Nature. 1997;389:52. doi: 10.1038/37944. [DOI] [PubMed] [Google Scholar]
- xxxiii.a) Cohen-Anisfeld ST, Lansbury PT. J Am Chem Soc. 1993;115:10531. [Google Scholar]; b) Anisfeld ST, Lansbury PT. J Org Chem. 1990;55:5560. [Google Scholar]
- xxxiv.a) Bodanszky M, Natarajan S. J Org Chem. 1975;40:2495. doi: 10.1021/jo00905a016. [DOI] [PubMed] [Google Scholar]; b) Bodanszky M, Kwei JZ. Int J Pept Protein Res. 1978;12:69. [PubMed] [Google Scholar]
- xxxv.Wang P, Li X, Zhu J, Chen J, Yuan Y, Wu X, Danishefsky SJ. J Am Chem Soc. 2011;133:1597. doi: 10.1021/ja110115a. [DOI] [PMC free article] [PubMed] [Google Scholar]