

Abstract
The caudal homeobox factor 1 (CDX1) is an essential transcription factor for intestinal differentiation. Its aberrant expression in intestinal metaplasia of the upper gastrointestinal tract is a hallmark within the gastritis-metaplasia-carcinoma sequence. CDX1 expression is influenced by certain pathways, such as Wnt, Ras, or NF-κB signaling; however, these pathways alone cannot explain the transient expression of CDX1 in intestinal metaplasia or the molecular inactivation mechanism of its loss in cases of advanced gastric cancer. In this study, we investigated the epigenetic inactivation of CDX1 by promoter methylation, as well as the functional link of CDX1 promoter methylation to the inflammatory NF-κB signaling pathway. We identified methylation-dependent NF-κB binding to the CDX1 promoter and quantified it using competitive electrophoretic mobility shift assays and chromatin immunoprecipitation. A methylated CDX1 promoter was associated with closed chromatin structure, reduced NF-κB binding, and transcriptional silencing. Along the gastritis-metaplasia-carcinoma sequence, we observed a biphasic pattern of tumor necrosis factor-α (TNF-α) protein expression and an inverse biphasic pattern of CDX1 promoter methylation; both are highly consistent with CDX1 protein expression. The stages of hyper-, hypo-, and hyper-methylation patterns of the CDX1 promoter were inversely correlated with the NF-κB signaling activity along this sequence. In conclusion, these functionally interacting events drive CDX1 expression and contribute to intestinal metaplasia, epithelial dedifferentiation, and carcinogenesis in the human stomach.
Gastric carcinogenesis evolves in a stepwise manner1 in which the development of intestinal metaplasia, within the gastritis-metaplasia-carcinoma sequence is a pivotal step that remains poorly understood. Chronic infection with Helicobacter pylori (H.p.), the main cause of gastric inflammation, may lead to mucosal atrophy and intestinal metaplasia.2 Further accumulation of mutagenic events finally leads to dysplasia and invasive gastric cancer. This 20-year-old model initially proposed by Correa has been proven in several long-term follow-up studies. Transitions from intestinal metaplasias to low-grade dysplasias have been found, as well as a slight reversibility of intestinal metaplasia, especially of the complete type after H.p. eradication. The unpredictable transitions from low- to high-grade lesions, however, remains, and many attempts were undertaken to stratify the group of low-grade lesions for reasons of clinical surveillance.3–7 In this model of gastric carcinogenesis, milieu factors such as acidic pH or pro-inflammatory cytokines are able to influence this process.8,9 The underlying cell signaling cascades, however, are yet to be discovered. Detailed analyses further distinguish between complete and incomplete types of intestinal metaplasia or present oxyntic atrophy of the mucosa as a different kind of metaplasia, a so-called spasmolytic polypeptide-expressing metaplasia, which seems to be a separate mechanism in the development of gastric cancer.5,10–12
Beyond genetic studies, which revealed, for example, the background of E-cadherin mutations in gastric cancer,13,14 an increasing number of epigenetic studies on DNA methylation have been performed in gastroenterological research in the last years. A PubMed search (using keywords for gastric cancer and methylation, which was performed on November 20, 2011) revealed over 800 articles concerning gastric cancer and methylation. These studies showed loss of gene expression, particularly of tumor suppressor genes, bearing prognostic values.15,16 Such genes include p16, RUNX3, PCDH10 (Protocadherin 10), and DKK-3, which have biological relevance in the context of gastric cancer.16–19 Nevertheless, only a minority of these studies focused on pre-malignant stages in gastric cancer, such as H.p. gastritis, intestinal metaplasia, and dysplasia.20–22
The aberrant expression of evolutionarily conserved intestinal transcription factors, such as the homeobox caudal-type homeobox factor 1 (CDX1) and caudal-type homeobox factor 2 (CDX2), seem to be crucial for inflammation-associated intestinal transdifferentiation of stem cells in the human stomach.23 It has been previously reported that bile acids activate the CDX2 promoter via NF-κB and that CDX1 seems to play an important role in the development of Barrett's esophagus.24 It has yet to be investigated, if epigenetic alterations play a functional role in the dysregulation of these intestine-specific tumor suppressor genes.
With this study, we describe the promoter methylation level of CDX1 along the gastritis-metaplasia-carcinoma sequence. We analyzed DNA-protein interactions and verified an interactive functional link between CDX1 promoter methylation and NF-κB signaling. These interesting findings for CDX1 could not be transferred to CDX2, which seems to be transcriptionally unaffected by promoter methylation.
Materials and Methods
Sequences, CDX1 Promoter Characterization, CpG Island Detection (European Molecular Biology Open Software Suite), rVISTA-Analysis
Analyzed sequences refer to GenBank entries: CDX1-mRNA (NM_001804.2), TNFAIP3-mRNA (NM_006290), CDX2-mRNA (NM_001265), CDX1 gene and promoter (NC_000005.9), CDX2 gene and promoter (NC_000013.10), NFKBIA gene and promoter (NG_007571), organic cation transporter 1 (OCT1) gene and promoter (NM_003057.2). Promoter characterization was performed using open source software, such as rVISTA 2.0 and European Molecular Biology Open Software Suite (European Molecular Biology Open Software Suite and rVISTA Anaylsis).25,26
Cell Culture
The human gastric cancer cell lines AGS, 23132/87, KATO3, and HGT1 were maintained in Dulbecco's modified Eagle's medium or Dulbecco's minimal essential medium containing 10% fetal calf serum. As indicated, 24 hours before treatments, cells were plated at optimal density. Chinese hamster ovary cells were grown in RPMI 1640 medium with 10% fetal calf serum, 1% pyruvate, 1% penicillin/streptomycin, and 1% glutamine (all from Invitrogen, Darmstadt, Germany) at 5% CO2 humidified atmosphere.
5-Aza Treatment and TNF-α Stimulation
Cell lines were seeded to reach confluence in 96 hours and treated unstarved. After 24 hours of initial adherence, cells were cultured for 72 hours with 5-aza-2′-deoxycytidine (5-aza) (Sigma-Aldrich, Taufkirchen, Germany) at 0.5, 1.0, 2.0, 3.0, 4.0, and 5.0 μmol/L. Untreated cells served as controls. Cytotoxicity was measured by cell counting. For all further experiments, a concentration of 3 μmol/L 5-aza was chosen. In addition, AGS cells were either stimulated 24 hours before harvesting with a solution of 10 ng/mL of TNF-α (R&D Systems, Wiesbaden, Germany), or were left unstimulated. Cells were lysed in RLT buffer, and processed for RNA isolation.
RT-PCR
RNA was isolated with the RNeasy Micro Kit (Qiagen, Hilden, Germany). The primers used for RT-PCR were: CDX1: forward, 5′-ACAATCCGGCGGAAATCAG-3′, reverse, 5′-TTCACTTTGCGCTCCTTTGC-3′; CDX2: forward, 5′-CTGGAGCTGGAGAGGAGTTTC-3′, reverse, 5′-ATTTTAACCTGCCTCTCAGAGAGC-3′; TNFAIP3: forward, 5′-CTGCCCAGGAATGCTACAGATAC-3′, reverse, 5′-ACAAGTGGAACAGCTCGGATTT-3′; GAPDH: forward, 5′-GAAGATGGTGATGGGATTTC-3′, reverse, 5′-GAAGGTGAAGGTCGGAGTC-3′ (Eurofins MWG, Ebersberg, Germany). All RT-PCR was performed as previously described.9 In quantitative PCR experiments, the targets were compared to glyceraldehydes-3-phosphate dehydrogenase. qPCR was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA). Results were calculated using the ΔΔCT-method.
Electrophoretic Mobility Shift Assay
Nuclear proteins of inflammatory activated, NF-κB enriched Chinese hamster ovary cells were used in electrophoretic mobility shift assay (EMSA), as previously described.27 NF-κB binding activity was investigated with IRDye 700-labeled oligos.
As control probes for specific NF-κB binding, we used the consensus NF-κB binding site of the NFKBIA promoter, and as a loading control, the promoter of the organic cation transporter 1 (OCT1), as published previously.28 For the CDX1 promoter, we generated the following probe: sense 5′-CCCCCGACGGGTTTCCCCCTTTGATTCGCG-3′. This was obtained in a native unmethylated form and in a methylated variant with methyl-cytosine bases in sense and anti-sense oligonucleotides at position X: sense 5′-CCCCXGAXGGGTTTCCCCCTTTGATTXGXG-3′ (Eurofins MWG, Ebersberg, Germany). The 20 μL reaction contained 2 μL binding buffer (100 mMTris, 500 mMKCl, 10 mmol/L DTT), 1 μL of double-stranded poly (dI-dC; 2 μg/μL), 1 μL of 2.5% Tween 20, 1 μL of 1% NP-40, 2 μg bovine serum albumin (10 mg/mL), and 0.5 μL of 700-infared labeled-double-stranded NF-κB consensus oligonucleotide (50 nmol/L) supplemented with 10 μg nuclear protein in H2O. The mixture was incubated at room temperature for 30 minutes, and then applied on a 4% Lipage Gel (4°C for 2 hours at 150 V), analyzed, and quantified by the Odyssey system (Licor; Biosciences GmbH, Bad Homburg, Germany). For specification, co-incubation experiments with unlabeled consensus and mutant NF-κB oligonucleotides (Santa Cruz Biotechnology, Santa Cruz, CA) were performed.
EMSA Competition Assay
The CDX1-specific double-stranded oligonucleotide sense 5′-AGTTGAGGGGACTTTCCCAGG-3′ was used in a non-dye-labeled variant (Eurofins MWG, Ebersberg, Germany). This unlabeled, unmethylated probe was incubated in descending 10% steps with either the 700-dye-labeled unmethylated CDX1-specific ds-oligo or the 700-dye-labeled methylated CDX1-specific ds-oligo. Vice versa, the amount of dye-labeled oligos was raised in 10% steps. Thus, the affinity of the nuclear extracts to the dye-labeled and non-dye-labeled probe was semi-quantitatively calculated by measuring the decrease of fluorescence intensity in comparison to the total input probe intensity.
Chromatin Immunoprecipitation-Assay for Histone Modification and NF-κB Binding
Chromatin in confluent AGS cells was cross-linked and harvested, and the protein-chromatin lysate was fragmented to a length between 200 and 1000 bp by sonication. Histone modifications in the CDX1 promoter region were checked in AGS cells after 5-aza treatment, as previously described. Chromatin was immunoprecipitated with the following antibodies overnight at 4°C with rotation: dimethylated histone H3K4 (1:50, H3K4me2; Cell Signaling, Danvers, MA), dimethylated histone H3K9 (1:25, H3K9me2; Cell Signaling, Danvers, MA). A chromatin immunoprecipitation (ChIP) DNA Clean & Concentrator kit (ZYMO Research, Orange, CA) was used to clean and concentrate the chromatin. We designed primers to amplify the CDX1 promoter area that flanks the NF-κB binding site: forward 5′-CGGCAATTTGTCTCCTTTTGAACC-3′, reverse 5′-CCCACCCAGGCCTTTTATAGCTC-3′ (Eurofins MWG, Ebersberg, Germany).
Then AGS cells were treated with 5-aza and TNF-α, as previously described, and were then transferred to NF-κB ChIP experiments using the ChIP-IT Express Kit (Active Motif, Rixensart, Belgium). The chromatin was incubated with 3 μg/mL p65-NF-κB-antibody. The same specific PCRs flanking the NF-κB binding sites were used in this setting. The NFKBIA promoter served as positive control for ChIP, as it is a well-known target in NF-κB-signaling: forward, 5′-GCAGAGGACGAAGCCAGTTCT-3′, reverse, 5′-GACTGCTGTGGGCTCTGCA-3′ (Eurofins MWG, Ebersberg, Germany).
Each ChIP experiment was performed three times. DNA was eluted in 50 μL final volume, and 2 μL of each ChIP-DNA was subjected to qPCR. Fold enrichment in the binding of histones and in the binding of NF-κB to the CDX1 promoter were determined. The PCRs after ChIP compared the precipitated genomic fragment covering the NF-κB binding site in the CDX1 promoter to the input DNAs before precipitation. qPCR was performed on a 7500 Fast Real-time PCR System (Applied Biosystems, Carlsbad, CA). Results were calculated using the ΔΔCT-method.
Tissue Samples
Human formalin-fixed, paraffin-embedded (FFPE) tissue specimens from the diagnostic files of the Institute of Pathology of the University Hospital Erlangen were analyzed.
For DNA preparation, a collection of 66 gastric mucosa specimens from 50 patients were investigated (age range, 24–92; mean, 65; sex distribution M:F, 35:15). From each sample 1 μg DNA was microdissected. The specimens included 10 cases of normal gastric mucosa, 12 cases of H.p. gastritis, 17 cases of intestinal metaplasia, 6 cases of low grade, and 5 cases of high-grade intraepithelial neoplasias, as well as 16 advanced gastric adenocarcinomas.
The immunohistochemical study included 121 gastric mucosa specimens from 51 patients, which underwent gastric resection (age range, 28–85; mean, 67; sex distribution m:w, 31:20). The specimens included 33 cases of normal gastric mucosa, 14 cases of H.p. gastritis, 32 cases of intestinal metaplasia, 12 cases of low-grade, and 6 cases of high-grade intraepithelial neoplasias, as well as 24 advanced gastric adenocarcinomas. The study was stated to be in accordance with local and national ethical principles as of January 25, 2005.
Bisulfite Sequencing and Pyrosequencing
Bisulfite treatment was performed using the Epitect Bisulfite Kit (Qiagen, Hilden, Germany). Nonselective methylation specific PCRs were designed: forward 5′-TTGTGTGAAGTTGGTTTAGAATTT-3′, reverse 5′-ACACATAACCCACATACATAATAAC-3′ (Eurofins MWG, Ebersberg, Germany). The amplicon was 505 bp long and covered 49 CG sites. Amplicons from AGS cells were cloned with the TOPO-TA-Cloning Kit (Invitrogen, Darmstadt, Germany), 10 clones were picked and analyzed by Qiagen Sequencing Services (Qiagen, Hilden, Germany).
A retrograde pyrosequencing assay was designed for FFPE material. The primer sequences were as follows: forward 5′-GGTAATTTGTTTTTTTTTGAATTTTT-3′, reverse 5′-CCCCACCCAAACCTTTTATAAC-3′ (Eurofins MWG). The pyrosequencing pipette scheme was designed by Varionostic (Ulm, Germany) and was as follows: CCRAACCRCCCRCRACCCCAACCRAAACRAATTTACATTTCAAAACRAAAAAAAACCTCCRAACCRCRAATCAAAAAAAAAAACCCRTCRAAAACRAAAAATT.
Immunohistochemistry
Pretreatment of tissue samples was performed as previously described.9 For CDX1 staining endogenous peroxidase was blocked by 3% H2O2 in Tris buffer for 15 minutes. The sections were incubated for 30 minutes at room temperature with a goat anti-CDX1 antibody (1:20, Santa Cruz Biotechnology, Santa Cruz, CA) or overnight with a mouse anti-TNF-α antibody (1:100; Abcam, Cambridge, UK) diluted in Antibody Diluent (Dako, Glostrup, Denmark).
CDX1 detection was established on duodenal and colonic mucosa and performed with an horseradish peroxidase-labeled anti-goat Polymer (B-Bridge, Mountain View, CA). Staining was visualized with diaminobenzidine chromogen (Dako, Glostrup, Denmark). For TNF-α, a macrophage-rich resorptive inflammation (seroma) served as positive control. Here, an AP labeled anti-mouse Polymer (Dako, Glostrup, Denmark) was used with Fast Red visualization afterward. All sections were counterstained with Mayer's hemalaun (Merck, Darmstadt, Germany). Negative controls without primary antibody were run for each sample.
Statistics
Two-way analysis of variance, two-sided t-tests, χ2-tests and Pearson correlations were used. Statistical significance was set at P values <0.05.
Results
Demethylation of Highly Methylated CDX1 Promoter Restores CDX1 Expression in Four Human Gastric Cancer Cell Lines
We treated AGS, HGT-1, 23132/87, and KATO-III cells with different concentrations of 5-aza-2′-deoxycytidine (5-aza). A concentration of 3 μmol/L 5-aza was used further on as it showed the lowest cytotoxicity (data not shown). Bisulfite sequencing covering the whole CpG island with 49 CG sites in 10 treated versus untreated clones from the cell culture experiments, which showed an average decrease in methylation from 95.1% to 62.6% (χ2 test, P < 0.001) according to data from the literature.29,30 After 5-aza treatment RT-PCR showed a general increase in CDX1-mRNA expression in all four cell lines (data not shown). The higher susceptibility of the demethylated CDX1 promoter is per se an unspecific process attributable to several possibly basic active pathways influencing CDX1, which we specified exemplarily further on.
In comparison, a basal CDX2-mRNA expression was generally visible in all four cell lines, even in those known to have a methylated CDX2 promoter like AGS cells. 5-aza treatment did not consistently increase the amount of CDX2 PCR product. This is consistent with literature, suggesting a lack of correlation between CDX2 methylation status and its transcriptional activity.31
The CDX1 Promoter Contains a Highly Conserved NF-κB Site in a CpG Island
Open source software tools like the EMBOSS and rVista 2.0 were used to further characterize the CDX2 and CDX1 promoter. For CDX2, EMBOSS confirmed a first putative CpG island −3800 bp to −3400 bp upstream, which might be located too far from the coding region to influence the transcriptional activity. In comparison, EMBOSS revealed a closer putative CpG island within the CDX1 promoter (Figure 1A) from −400 bp to + 100 bp covering 49 CG sites around the transcription start site (Figure 1C). The previously achieved bisulfite sequencing data verified highly methylated CGs in the conserved NF-κB site (Figure 1B). Previous studies claimed seven putative NF-κB sites in this CpG island.30 However, rVista 2.0 analyses verified only one highly conserved NF-κB site existing in humans, as well as rodents, at −138 bp (Figure 1C). AGS cells were selected as a model for the following studies.
Figure 1.

A: The CDX1 promoter analyzed by a CpG-island detection tool offered by the European Molecular Biology Open Software Suite (EMBOSS). In the Promoter region starting 1000 bp upstream, the CAG unit represents a putative CpG island at 600 bp to 1100 bp corresponding to a range from −400 bp to 100 bp in direct relation to the transcription site. B: Bisulfite sequencing of the highly conserved and validated NF-κB site at −138 bp within the CDX1 promoter in native (left) and 5-aza treated AGS cells (right). Note the thymidine replaced cytosines in the unmethylated state (arrows). C: This CpG-island in detail shows 49 CGs (black diamonds) also covering an inter species conserved NF-κB binding site at −138 bp. Sequencing primers for cell line analysis overdrew the whole CpG island, whereas pyrosequencing primers due to the degraded shorter DNA lengths in FFPE material were designed to be representative and to detect the NF-κB site as the region of interest.
Stimulation with TNF-α Induces a Methylation-Dependent CDX1-mRNA Expression
We used TNF-α as a pro-inflammatory stimulus for activation of NF-κB signaling to investigate the epigenetic mechanisms of CDX1 inactivation. Experiments were performed three times in triplicates. The two-way analysis of variance showed that both kinds of treatment were followed by a significant increase of the basal CDX1-mRNA expression in qPCR (P < 0.001 for 5-aza and P = 0.011 for TNF-α). We observed an additive effect on analyzing the interaction of both treatments (P = 0.008).
Therefore, the basal CDX1-mRNA expression in unmethylated 5-aza-treated AGS cells could be significantly increased on TNF-α treatment (Figure 2A). In contrast, methylated 5-aza untreated cells were not susceptible to TNF-α.
Figure 2.

TNF-α significantly promotes CDX1-mRNA levels in unmethylated 5-aza treated AGS cells. A: CDX1 mRNA expression is blocked in methylated 5-aza untreated AGS cells and not inducible by TNF-α. B: CDX2 mRNA is lowered during TNFα treatment independent of its methylation status. C: TNF-α (as control) induces TNFAIP3 mRNA expression, independent of the methylation-status in 5-aza treated and untreated AGS cells. *P = 0.026; †P < 0.001; ‡P = 0.12; §P = 0.86; ¶P = 0.92; ∥P = 0.002.
In comparison CDX2-mRNA expression is known to be slightly decreased after TNF-α treatment due to the interaction with the PTEN pathway.32 The two-way analysis of variance confirmed that repression of CDX2-mRNA expression in qPCR in tendency, but showed no increase of the basal CDX2-mRNA expression after 5-aza treatment (P = 0.12 for TNF-α and P = 0.35 for 5-aza) (Figure 2B). AGS cells are known to have a methylated CDX2 promoter.31 Independent from the methylation status due to 5-aza treatment, the CDX2 expression is restored in the same way if TNF-α is absent. We, conclude, therefore that the transcriptional reactivity of the CDX2 promoter is independent from its methylation status, which is consistent with data from the literature.31 To study certain epigenetic events in gastric carcinogenesis, we further focused on CDX1, but not CDX2.
In addition, a methylation-independent induction of the specific NF-κB target gene TNFAIP3 excludes a generally diminished NF-κB transcriptional transactivation in highly methylated cells. A two-way analysis of variance on TNFAIP3 showed a significant increase of mRNA expression in qPCR, only on TNF-α treatment (P < 0.001), but no significant interaction on 5-aza treatment (P = 0.277). t-tests for groups of interest of these experiments are outlined in Figure 2.
Basal restoration of CDX1 after 5-aza treatment is not attributable to NF-κB signaling, but presumably other pathways might play a role, as we did not observe a response in TNFAIP3 gene (Figure 2C). Therefore, NF-κB-mediated differences in CDX1 expression seemed to be specifically dependent on the methylation status of the CDX1 promoter.
NF-κB Binds to Its CDX1 Promoter Site in a Methylation-Dependent Manner
We performed EMSAs to determine NF-κB binding to the CDX1 promoter. In establishing NF-κB specific EMSAs, the NFKBIA promoter, a well-known downstream target of NF-κB, served as a positive control. Then we generated a fluorescence 800-dye-labeled double-stranded oligonucleotide probe from the CDX1 promoter sequence containing the putative NF-κB binding site. We confirmed a sequence-specific DNA-binding activity of the NF-κB protein complex to the CDX1 promoter on incubating it with nuclear extracts from activated cells enriched with NF-κB protein. Three DNA-protein complexes appeared on the gel. The ability of NF-κB to form heteromeric DNA complexes has been previously published (Figure 3).33–35
Figure 3.
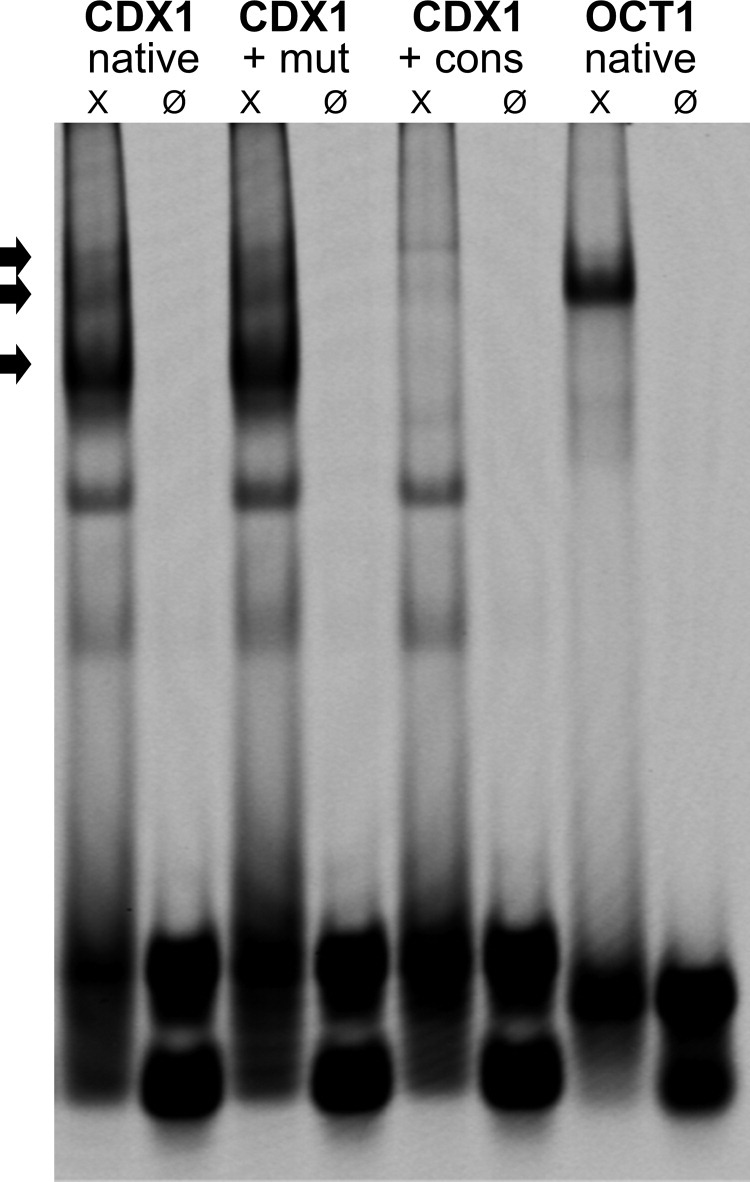
Electrophoretic mobility shift assay with DNA probes for the CDX1 promoter containing the putative NF-κB binding site and OCT1 probe as loading control. Each probe was added alone (Ø) or incubated with NF-κB enriched nuclear extracts (X). The specificity of the three bands showing NF-κB bindings (arrows) was confirmed by co-incubation either with a cold NF-κB consensus probe (+ cons) or with a cold NF-κB mutant probe (+mut).
The specificity of the EMSA experiment was outlined by co-incubation of the CDX1 probe with NF-κB specific cold consensus and mutant probes. Incubation with the consensus probe led to extinction of the specific three bands, whereas incubation with the mutant probe led to no change (Figure 3).
Next, we investigated whether methylation alters NF-κB binding to the CDX1 promoter, in vitro, using a modified CDX1 probe with methylated cytosines. Both complexes were again observed in EMSA, but with decreased signal intensity, possibly due to a direct effect of the CDX1 oligonucleotide methylation status (Figure 4, A and C).
Figure 4.
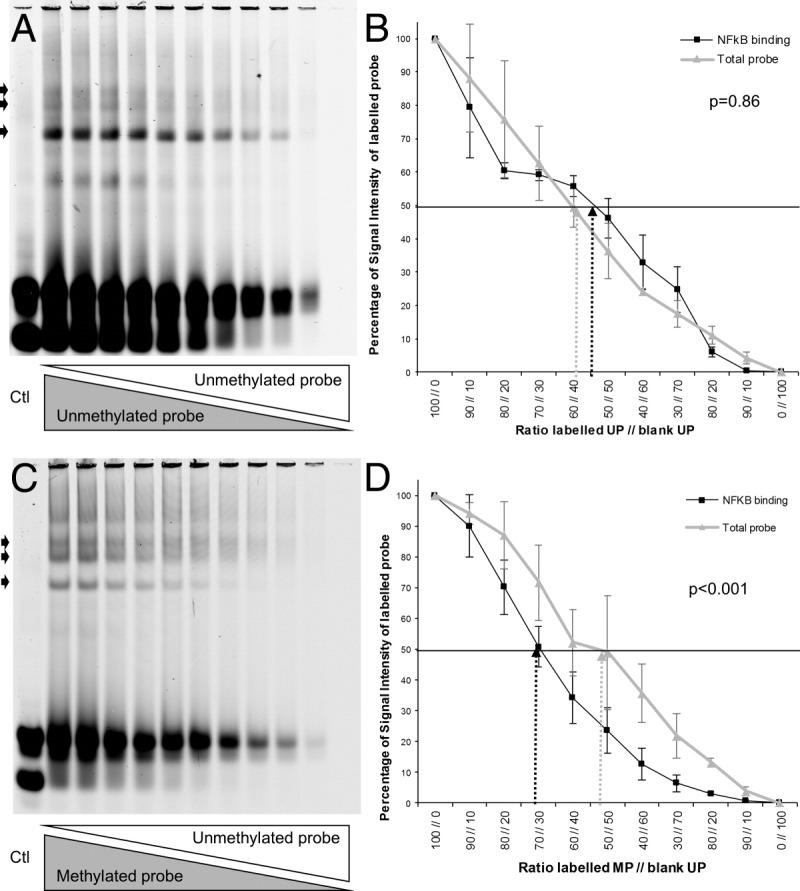
A: Competitive EMSA of a labeled unmethylated probe with an unlabeled unmethylated probe of the CDX1 promoter mixed in decreasing ratios of 10% steps. B: Quantitative analysis of the signal intensity of the unmethylated probe compared to the total inserted probe. The affinity of NF-κB to both probes was the same reaching half values at the 50:50 mixing ratio. C: Competitive EMSA of a labeled methylated probe with an unlabeled unmethylated probe of the CDX1 promoter in a descending mixing ratio in 10% steps. Note the fast extinction of the NF-κB binding site (arrows) of the methylated probe due to the lesser affinity of the protein compared to the unlabeled unmethylated probe. D: Quantitative analysis of the signal intensity of the methylated probe in comparison to the total inserted probe. The expected bisection of the intensity is not reached at the 50:50 mixing ratio but at a ratio of 70:30, indicating a twofold stronger binding of NF-κB to the unmethylated probe.
We further developed the EMSA to an innovative competitive binding assay, which allows us to quantify the NF-κB binding differences between the methylated and unmethylated CDX1 probes. To measure different NF-κB affinities, several CDX1 probe mixtures were investigated. In the first setting, unmethylated labeled and unlabeled probes were mixed. As expected, the fluorescence intensity was directly proportional to the mixing ratio. Addition of an excess of the unlabeled unmethylated probe resulted in extinction of all three bands, thereby again confirming the specificity of the EMSA (Figure 4A). In a second setting, the unmethylated labeled probe was replaced by a methylated labeled probe. With competition, the visible DNA-protein complex of the methylated CDX1 probe showed a significant faster extinction already at low mixing ratios (Figure 4B). We also quantified the signal intensities (Figure 4D). At a mixing ratio of 50:50, the ratio of specific signal intensity to input signal intensity was 46% for the unmethylated probe; and the ratio was significantly (P < 0.001) reduced to 23% for the methylated probe. Thus, the affinity of NF-κB to the unmethylated CDX1 probe was two-fold higher compared to the methylated CDX1 probe.
5-Aza Treatment Opens Chromatin at the CDX1 Promoter for NF-κB Binding
We used ChIP to validate the results of the EMSA experiments in vital cells. Demethylation by 5-aza treatment is known to switch between repressive and permissive chromatin status. Particular histone modifications lead to altered folding of the nucleosomal fiber that renders chromosomal domains more accessible.36 Histone methylation patterns should indicate chromatin activity status at the CDX1 promoter after 5-aza treatment. We used an antibody against dimethylated histone H3 lysine 4 (H3Me2Lys4) and against dimethylated histone H3 lysine 9 (H3Me2Lys9) to confirm an open or closed chromatin, respectively.37 Genomic qPCRs experiments were performed measuring the differences between Ct values of precipitated DNA versus input DNA. Indeed, the ratio of precipitated CDX1 promoter-to-input chromatin increased from 0.62 to 1.26 after 5-aza treatment for H3me2Lys4 IP, whereas the ratio of precipitated CDX1 promoter-to-input chromatin decreased from 1.62 to 0.80 for H3me2Lys9 IP. These experiments confirm permissive chromatin formation along the CDX1 promoter by 5-aza treatment.
5-Aza Treatment Induces CDX1 Up-Regulation by Increased NF-κB Binding to the CDX1 Promoter
To determine in vivo whether CDX1 up-regulation is due to direct transcriptional activation by NF-κB promoter binding, we used the p65-NF-κB antibody to immunoprecipitate the chromatin from AGS cells with or without 5-aza treatment and with or without TNF-α stimulation (Figure 5, A and B). The experiment was performed three times and measured in triplicates.
Figure 5.

A: ChIP assay using an NF-κB-p65 antibody and PCR directed to the CDX1promoter. B: The same PCR assay in the native nuclear chromatin before precipitation is shown (input control). C: The fold-change of the precipitated genomic fragment containing the NF-kB binding site of the CDX1 promoter was determined via quantitative qPCR, showing a significant 10-fold stronger protein-DNA interaction of NF-κB with the CDX1 promoter in the unmethylated TNF-α stimulated cells as an additive effect. *P = 0.018; †P < 0.001.
The two-way analysis of variance showed that both treatments were followed by a significant enrichment (P < 0.001) of the genomic fragment containing the NF-κB binding site. We observed an additive effect on analyzing the interaction of both treatments (demethylation and enhanced inflammation) (P = 0.004) (Figure 5, A–C).
We observed a maximal difference of 9.8-fold increase in NF-κB binding to the CDX1 promoter between unmethylated/TNF-α-treated versus methylated/TNFα-untreated AGS cells. This factor can be split into two categories. A 5.5-fold increase is attributable to the demethylated status due to 5-aza treatment alone, whereas a factor of 1.8 is due to additional TNF-α stimulation. Considering the methylated status, there was no significant difference between TNF-α treated or untreated AGS cells (P = 0.41) (Figure 5C). These ChIP experiments confirmed experimentally the methylation-dependent genomic binding of NF-κB to the CDX1 promoter at the proposed position. Furthermore these epigenetically modified protein-DNA-interactions are highly consistent with the previously shown mRNA-expression experiments (Figure 1).
A Biphasic Pattern of CDX1 Promoter-Methylation Is Observed in Human Tissues Along the Gastritis-Metaplasia-Carcinoma Sequence
We analyzed the CDX1 promoter methylation status in human FFPE samples by pyrosequencing along the gastritis-metaplasia-carcinoma sequence. Due to DNA fragmentation in FFPE material, the assay had to be shortened and covers 13 CG sites within the CpG island, including the conserved NF-κB site of the CDX1 promoter. The bisulfite sequencing data in this area regarding the 10 treated versus untreated clones from the cell culture experiments were chosen to judge the representativeness of this shortened assay. Here a decrease in methylation from 92.3% to 68.0% (χ2-test, P < 0.001) within the analyzed amplicon was found and thus indicated values that match in comparison to the methylation status of the whole CDX1 promoter as previously described.
No statistic correlations to sex or age were detectable. Indeed, the highest degree of methylation was observed in normal gastric mucosa, whereas the lowest degree was noticed in specimens with low-grade intraepithelial neoplasia. The level of hypermethylation increases again in gastric cancer (Figure 6). Here, higher degrees of methylation at the CDX1 promoter were associated with loss of differentiation and higher tumor grade.
Figure 6.
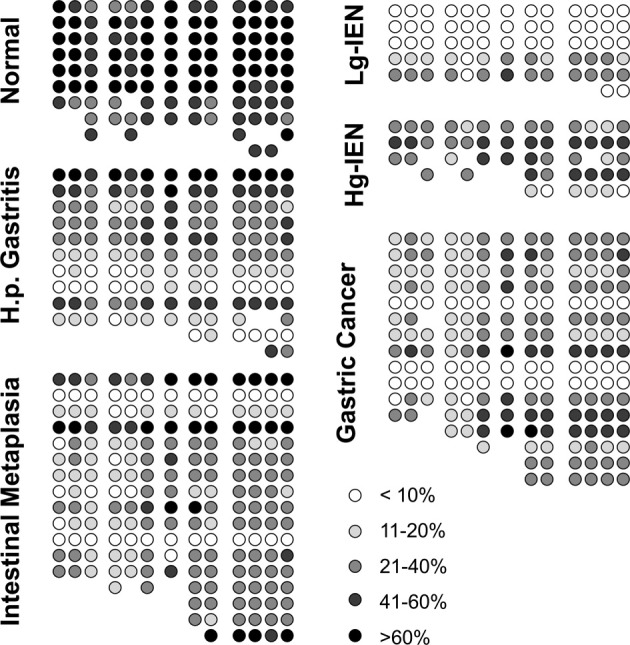
Semi-quantitative depiction of the methylation pattern along the inflammation-metaplasia-carcinoma sequence including the NF-κB binding site of the CDX1 promoter. High degrees of methylation were detectable in the normal human gastric mucosa. The initial hypo-methylation in Helicobacter gastritis and intestinal metaplasia reaches the lowest levels in low-grade dysplasia. Hyper-methylation gains importance in high-grade dysplasia and gastric carcinoma. Pyrosequencing data that was not interpretable due to background signals or shortened fragments were omitted (missing circles). IEN, intraepithelial neoplasia.
In a subgroup of nine patients, material was available from carcinomas and corresponding intestinal metaplasias. In these cases, a significant lower methylation level in intestinal metaplasia (22,6%) than in gastric cancer (30,4%) was detectable (paired t-test, P = 0.02), which indicated specific methylation patterns in certain lesions along the gastritis-metaplasia-carcinoma-sequence rather than general methylation effects of the whole mucosa.
The Biphasic CDX1 Protein Expression Along the Gastritis-Metaplasia-Carcinoma Sequence Reflects the Inflammatory Activity and the CDX1 Promoter-Methylation Pattern
We performed TNF-α and CDX1 immunohistochemical staining to correlate the observed epigenetic alterations with the protein expression in human tissues. The observed expression levels of normal, gastritis metaplasia, dysplasia, and carcinoma samples are shown in Figure 7. In brief, cytoplasmatic TNF-α expression was absent in normal gastric tissue, reaching highest levels in intestinal metaplasia and decreased again in carcinoma (Figure 8A). No statistic correlations to sex or age were detectable. As previously reported,23 normal gastric mucosa did not show any CDX1 protein expression. The strongest nuclear staining of the CDX1 protein was observed in intestinal metaplasia, as well as in low-grade dysplasia. In gastric carcinoma, the CDX1 expression was remarkably decreased. The inflammatory activity measured by TNF-α expression showed a highly significant direct correlation to CDX1 expression at Pearson's value of 0.419 (P < 0.001).
Figure 7.

CDX1- and TNF-α immunohistochemical staining in normal (A, E), gastritis-metaplasia (B, F), dysplasia (C, G) and carcinoma (D, H) samples, respectively. Note the strong CDX1 nuclear staining in intestinal metaplasia (B, right side) and dysplasia (C) in contrast to normal gastric or gastritis mucosa (A, B, left side). Loss of nuclear staining is observed in invasive gastric cancer (D). Epithelial cytoplasmatic TNF-α staining is enhanced in gastritis-metaplasia and dysplasia (F, G). Reduced levels of TNF-α are found in invasive gastric cancer (H). Magnification ×100.
Figure 8.
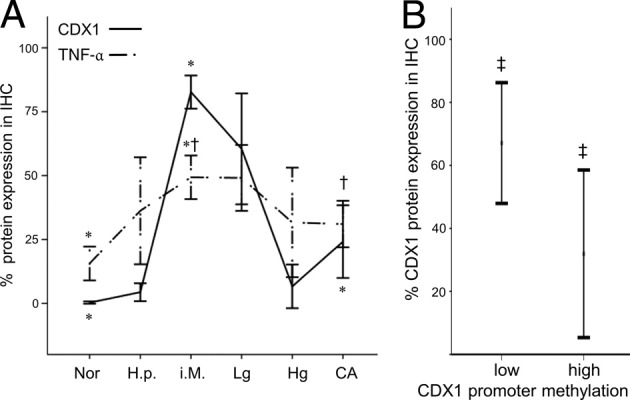
A: Immunohistochemical protein expression of nuclear CDX1 and cytoplasmatic TNF-α along the gastritis-metaplasia-carcinoma sequence from the left to the right: 33 cases of normal gastric mucosa (Nor), 14 cases of Helicobacter pylori (H.p.) gastritis (H.p.), 32 cases of intestinal metaplasia (i.M.), 12 cases of low-grade (Lg) intraepithelial neoplasia, 6 cases of high grade (Hg) intraepithelial neoplasia, and 24 gastric adenocarcinomas (CA). Note the parallel dome shape of increase and decrease of both proteins along the sequence. B: At a cut-off of 26%, the CDX1 promoter methylation clearly distinguishes between samples with high and low CDX1 protein expression levels, independent of the point along the gastritis-metaplasia-carcinoma sequence (data taken from an overlap of n = 25 samples, in which the molecular and immunohistochemical data were available). Error bars depict a confidence interval of 95%. *P < 0.001; †P < 0.01; ‡P = 0.03.
In addition, the two groups for pyrosequencing and immunohistochemistry shared samples from the same patients in n = 25 cases, mainly in the category of metaplasia and carcinoma. These overlapping cases showed a significantly negative correlation between the methylation level and protein expression at Pearson value −0.427 (P = 0.033). At a cut-off level of 26% for the CDX1 promoter methylation, a significant (P = 0.03) decrease in CDX1 protein expression could be observed (Figure 8B). The protein expression profile of CDX1 seems to follow the CDX1 epigenetic profile along the gastritis-metaplasia-carcinoma sequence.
Discussion
CDX1 and CDX2 are intestine-specific transcription factors of the caudal homeobox transcription factor family and essential for embryonal gut development.23 In close interaction, CDX2 transcriptionally activates CDX1 by direct promoter binding.38 Inversely, Cdx1 overexpression in transgenic mice reduces Cdx2 expression on mRNA and protein levels.39 Functionally, CDX1 and CDX2 are able to substitute each other in the maintenance of the crypt-villus-axis and are frequently co-expressed.40 We, as well as others, found that CDX1 and CDX2 are indistinguishably present in different types of intestinal metaplasia.41 In our study, therefore, complete and incomplete types of intestinal metaplasia were grouped together. But also differences between CDX1 and CDX2 exist. So Cdx2 transgenic mice showed intestinal metaplasia, as well as oxyntic atrophy of the gastric mucosa, whereas in Cdx1 transgenic mice intestinal metaplasia was seen alone.42 Inflammatory signals like TNF-α can promote CDX1 expression, but diminish CDX2 expression through the PTEN pathway.32 Whereas the responsiveness of the CDX1 promoter is dependent from its methylation state, this does not account for CDX2. In our study, CDX2 maintained a basal transcriptional activity and responsiveness of its promoter independent from its methylation state, which is consistent with literature.31 To study functionally epigenetic events in gastric carcinogenesis, we therefore focused on CDX1.
The loss of CDX1 causes dedifferentiation of gastrointestinal cancers associated with a higher tumor grade,43 and therefore stresses its tumor suppressive role. Beyond this tumor suppressive role an oncogenic role has been found for CDX1 as well. CDX1 can promote intestinal cell proliferation mediated through the induction of PAP I up-regulation of the proliferating cell nuclear antigen and down-regulation of the cdk inhibitor p21WAF.31–32,44–46 CDX1, furthermore, is a target of oncogenic pathways of early colon cancerogenesis, such as the APC/β-catenin and the Ras pathways.47–49 However, these effects did not alter chemically induced colon carcinogenesis in Cdx1 knockout or Cdx1-villin transgenic mice.39,40 These data question the role of CDX1 as an oncogene.
A tumor suppressor function of CDX1 is seen, as it can counteract proliferation in colon cancer cell lines by reducing cyclin D1 gene expression through altered Rb-pathway signaling.50 Noteworthy, CDX1 can induce a panel of intestinal differentiation genes, such as Muc2, CK20, aminopeptidase N, villin, and peroxisome proliferator-activated receptor-γ.44,51–53 In addition, CDX1 increases E-cadherin-mediated cell adhesion.54
In summary, CDX1 plays a key role in regulating intestinal epithelial differentiation, permeation of cell adhesion, and control of proliferation.39
Focusing on the upper gastrointestinal tract, CDX1 is aberrantly expressed in intestinal metaplasias of the stomach, as well as the gastroesophageal junction,23,30,55 which is consistent with our immunohistochemical findings. Intestinal metaplasia in the stomach is induced by chronic inflammation caused by Helicobacter pylori or by reflux disease in Barrett's mucosa, respectively. Bile salts and inflammatory signals caused by Helicobacter pylori promote this metaplastic CDX1 and CDX2 expression.8,30 In transgenic mouse models, Cdx1 and Cdx2 independently induce intestinal metaplasia of the stomach.42,55 Hence, the metaplastic expression of both caudal homeobox genes is strongly linked to inflammatory stimuli.
A relevant regulatory mechanism mainly attributed to CDX1 expression is methylation. In colon cancer cell lines, CDX1 gene expression is down-regulated by methylation and restorable via 5-aza treatment.29,56,57 Demethylation alone, for instance, through deprivation of folat donors, however, does not successfully restore CDX1 expression.58 Beyond these descriptive data, only a few studies combine the promoter methylation status of CDX1 with functional cell signaling cascades. In colon cancer cell lines, CDX1-mRNA levels increased dose-response-related after inducing nuclear NF-κB-p65 signaling.29 In Barrett's mucosa bile salts and acid provoke an inflammatory response. Here, methylation of the CDX1 promoter was correlated with an altered susceptibility to NF-κB signaling.30 Methylation of the CDX1 promoter further influences the synergistic interaction of Cdx2 on the Cdx1 promoter in gastric mouse models.38
We investigated the level and significance by which NF-κB signaling on the CDX1 promoter is impaired by its methylation. We showed that the CDX1 promoter is highly methylated in gastric cancer cell lines and CDX1 expression is restorable after 5-aza treatment, which may be attributable to a basal activity of other already mentioned CDX1 inducing pathways. We observed a significant increase of CDX1 expression after an inflammatory stimulus in case of an unmethylated promoter. EMSAs confirmed experimentally the existence of a highly conserved NF-κB binding site within a CpG island of the CDX1 promoter. Competitive EMSAs were designed as a new quantitative tool for the investigation of methylation-modified protein-DNA interactions. In detail, methylation of the NF-κB binding site significantly impaired direct NF-κB-p65 binding by two-fold. In ChIP experiments the binding ability of NF-κB-p65 to the methylated CDX1 promoter was even stronger reduced (ie, up to 10-fold). Here the shown methylation-dependent heterochromatin formation enhances the methylation-specific transcriptional silencing.36,37
In summary, NF-κB signaling is able to transcriptionally activate CDX1 expression by direct binding on its implicitly unmethylated promoter. In normal gastric mucosa, CDX1 is not expressed due to high levels of CDX1 promoter methylation and absence of inflammation. As shown here, the methylation level of the CDX1 promoter decreases during Helicobacter pylori gastritis and intestinal metaplasia, and reaches the lowest level in low-grade intraepithelial neoplasia of the stomach. This effect is attributable to the phenomenon of global hypomethylation, as seen in the early stages of cancer.59 Indeed, a decrease to a methylation level of 26% still allows detectable CDX1 protein expression. This threshold is reached between gastritis and intestinal metaplasia. It could be presumed that there several additional factors that might influence the transdifferentiation process, such as the endurance of the inflammatory process, other signaling cascades, or the intensity of inflammation. For example, TNF-α expression, which is known to mediate NF-κB signaling in Helicobacter pylori infection,60 might contribute to this phenomenon, as we noticed an inversely dome-shaped expression profile along the gastritis-metaplasia-carcinoma sequence (Figure 8A) in comparison to the CDX1 methylation pattern. We suggest that in intestinal metaplasia the synergistic effect of low methylation and increased inflammation now activates the CDX1 promoter and initiates intestinal transdifferentiation. During further tumor progression in gastric carcinogenesis, the level of CDX1 promoter methylation raises again, whereas TNF-α expression decreases. Therefore, the inflammatory NF-κB signaling again loses its impact on the CDX1 promoter and CDX1 expression diminishes. This loss of an intestine-specific transcription factor has already been described for colorectal cancer,29 indicating dedifferentiation of carcinoma, and could therefore be transferred to gastric cancer. In our study, CDX1 methylation was significantly lower in metaplasia than in associated carcinoma. In our opinion, this is a strong argument for lesion associated methylation patterns rather than generally altered methylation levels of the whole organism, which is discussed as highly controversely.31,61 Long-term follow-up studies showed that the progression from low-grade lesions to high-grade lesions still could not be predicted, which is what leads to problems in healthcare and surveillance recommendations.3,5,62 Here, the raising CDX1 methylation pattern from low- to high-grade lesions might serve as a suitable predictive biomarker.
In conclusion, the methylation dependent NF-κB binding on the CDX1 promoter points out that epigenetic DNA-modification fits into the context of cell signaling cascades. This helps to explain certain events in carcinogenesis, as in our case, inflammation-associated intestinal metaplasia. For the first time, we described a biphasic CDX1 promoter methylation pattern along the gastritis-metaplasia-carcinoma sequence, in which stages with hypermethylation are interrupted with intervals of hypomethylation. Further studies on DNA-methylation should therefore be kept in mind for the possible co-existence of hyper- and hypo-methylation events, and rely on functional regulatory aspects between descriptive methylation data and cell signaling.
Acknowledgments
We thank Dr. Uwe Gerstenmaier (Varionostic Gmbh, Ulm, Germany) for excellent technical support and design of the methylation-specific pyrosequencing assays, and Angela Neumann and Rudolf Jung for their excellent technical assistance.
Footnotes
Supported by the Interdisciplinary Center for Clinical Research and the ELAN-Fonds at the University Hospital of the University Erlangen-Nuremberg (T.T.R.) and National Cancer Institute (grant no. cR01CA93999 to W.E.R.).
R.E.V. and R.S.S. contributed equally to this work.
T.T.R., A.R., and M.F. conceived and performed experiments. A.J., A.D., and G.F. initiated the project and designed technical approach. R.E.V. and B.S. contributed excellent experience in NF-κB signaling, electrophoretic mobility shift assay (EMSA), and chromatin immunoprecipitation (ChIP) techniques. T.T.R., R.S.S., R.E.V., and A.H. conceived experiments and analyzed data. M.F. performed experiments. P.C.K. contributed clinical data. W.E.R. contributed ChIP experiments and expertise in methylation analyses. All authors were involved in writing the paper and gave the final approval of the submitted and published versions.
References
- 1.Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988;48:3554–3560. [PubMed] [Google Scholar]
- 2.Konturek P.C., Konturek S.J., Brzozowski T. Helicobacter pylori infection in gastric cancerogenesis. J Physiol Pharmacol. 2009;60:3–21. [PubMed] [Google Scholar]
- 3.Adamu M.A., Weck M.N., Gao L., Brenner H. Incidence of chronic atrophic gastritis: systematic review and meta-analysis of follow-up studies. Eur J Epidemiol. 2010;25:439–448. doi: 10.1007/s10654-010-9482-0. [DOI] [PubMed] [Google Scholar]
- 4.Cho S.J., Choi I.J., Kim C.G., Lee J.Y., Kook M.C., Park S., Ryu K.W., Lee J.H., Kim Y.W. Risk of high-grade dysplasia or carcinoma in gastric biopsy-proven low-grade dysplasia: an analysis using the Vienna classification. Endoscopy. 2011;43:465–471. doi: 10.1055/s-0030-1256236. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez C.A., Pardo M.L., Liso J.M., Alonso P., Bonet C., Garcia R.M., Sala N., Capella G., Sanz-Anquela J.M. Gastric cancer occurrence in preneoplastic lesions: a long-term follow-up in a high-risk area in Spain. Int J Cancer. 2010;127:2654–2660. doi: 10.1002/ijc.25273. [DOI] [PubMed] [Google Scholar]
- 6.Kodama M., Murakami K., Okimoto T., Sato R., Uchida M., Abe T., Shiota S., Nakagawa Y., Mizukami K., Fujioka T. Ten-year prospective follow-up of histological changes at five points on the gastric mucosa as recommended by the updated Sydney system after Helicobacter pylori eradication. J Gastroenterol. 2012;47:394–403. doi: 10.1007/s00535-011-0504-9. [DOI] [PubMed] [Google Scholar]
- 7.Vannella L., Lahner E., Osborn J., Bordi C., Miglione M., Delle Fave G., Annibale B. Risk factors for progression to gastric neoplastic lesions in patients with atrophic gastritis. Aliment Pharmacol Ther. 2010;31:1042–1050. doi: 10.1111/j.1365-2036.2010.04268.x. [DOI] [PubMed] [Google Scholar]
- 8.Burnat G., Rau T., Elshimi E., Hahn E.G., Konturek P.C. Bile acids induce overexpression of homeobox gene CDX-2 and vascular endothelial growth factor (VEGF) in human Barrett's esophageal mucosa and adenocarcinoma cell line. Scand J Gastroenterol. 2007;42:1460–1465. doi: 10.1080/00365520701452209. [DOI] [PubMed] [Google Scholar]
- 9.Faller G., Dimmler A., Rau T., Spaderna S., Hlubek F., Jung A., Kirchner T., Brabletz T. Evidence for acid-induced loss of Cdx2 expression in duodenal gastric metaplasia. J Pathol. 2004;203:904–908. doi: 10.1002/path.1590. [DOI] [PubMed] [Google Scholar]
- 10.Dimmler A., Brabletz T., Hlubek F., Hafner M., Rau T., Kirchner T., Faller G. Transcription of sonic hedgehog, a potential factor for gastric morphogenesis and gastric mucosa maintenance, is up-regulated in acidic conditions. Lab Invest. 2003;83:1829–1837. doi: 10.1097/01.lab.0000101729.25140.0c. [DOI] [PubMed] [Google Scholar]
- 11.Goldenring J.R., Nomura S. Differentiation of the gastric mucosa III: Animal models of oxyntic atrophy and metaplasia. Am J Physiol Gastrointest Liver Physiol. 2006;291:G999–G1004. doi: 10.1152/ajpgi.00187.2006. [DOI] [PubMed] [Google Scholar]
- 12.Suh Y.S., Lee H.J., Jung E.J., Kim M.A., Nam K.T., Goldenring J.R., Yang H.K., Kim W.H. The combined expression of metaplasia biomarkers predicts the prognosis of gastric cancer. Ann Surg Oncol. 2012;19:1240–1249. doi: 10.1245/s10434-011-2125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humar B., Blair V., Charlton A., More H., Martin I., Guilford P. E-cadherin deficiency initiates gastric signet-ring cell carcinoma in mice and man. Cancer Res. 2009;69:2050–2056. doi: 10.1158/0008-5472.CAN-08-2457. [DOI] [PubMed] [Google Scholar]
- 14.Jiang L., Gonda T.A., Gamble M.V., Salas M., Seshan V., Tu S., Twaddell W.S., Hegyi P., Lazar G., Steele I., Varro A., Wang T.C., Tycko B. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 2008;68:9900–9908. doi: 10.1158/0008-5472.CAN-08-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jee C.D., Kim M.A., Jung E.J., Kim J., Kim W.H. Identification of genes epigenetically silenced by CpG methylation in human gastric carcinoma. Eur J Cancer. 2009;45:1282–1293. doi: 10.1016/j.ejca.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 16.Yu J., Tao Q., Cheng Y.Y., Lee K.Y., Ng S.S., Cheung K.F., Tian L., Rha S.Y., Neumann U., Rocken C., Ebert M.P., Chan F.K., Sung J.J. Promoter methylation of the Wnt/beta-catenin signaling antagonist Dkk-3 is associated with poor survival in gastric cancer. Cancer. 2009;115:49–60. doi: 10.1002/cncr.23989. [DOI] [PubMed] [Google Scholar]
- 17.Yu J., Cheng Y.Y., Tao Q., Cheung K.F., Lam C.N., Geng H., Tian L.W., Wong Y.P., Tong J.H., Ying J.M., Jin H., To K.F., Chan F.K., Sung J.J. Methylation of protocadherin 10, a novel tumor suppressor, is associated with poor prognosis in patients with gastric cancer. Gastroenterology. 2009;136:640–651. doi: 10.1053/j.gastro.2008.10.050. [DOI] [PubMed] [Google Scholar]
- 18.Hou P., Ji M.J., Shen J.Y., He N.Y., Lu Z.H. Detection of p16 hypermethylation in gastric carcinomas using a seminested methylation-specific PCR. Biochem Genet. 2005;43:1–9. doi: 10.1007/s10528-005-1062-8. [DOI] [PubMed] [Google Scholar]
- 19.Kim T.Y., Lee H.J., Hwang K.S., Lee M., Kim J.W., Bang Y.J., Kang G.H. Methylation of RUNX3 in various types of human cancers and premalignant stages of gastric carcinoma. Lab Invest. 2004;84:479–484. doi: 10.1038/labinvest.3700060. [DOI] [PubMed] [Google Scholar]
- 20.Perri F., Cotugno R., Piepoli A., Merla A., Quitadamo M., Gentile A., Pilotto A., Annese V., Andriulli A. Aberrant DNA methylation in non-neoplastic gastric mucosa of H. Pylori infected patients and effect of eradication. Am J Gastroenterol. 2007;102:1361–1371. doi: 10.1111/j.1572-0241.2007.01284.x. [DOI] [PubMed] [Google Scholar]
- 21.Kim H.C., Kim J.C., Roh S.A., Yu C.S., Yook J.H., Oh S.T., Kim B.S., Park K.C., Chang R. Aberrant CpG island methylation in early-onset sporadic gastric carcinoma. J Cancer Res Clin Oncol. 2005;131:733–740. doi: 10.1007/s00432-005-0017-0. [DOI] [PubMed] [Google Scholar]
- 22.Schneider B.G., Peng D.F., Camargo M.C., Piazuelo M.B., Sicinschi L.A., Mera R., Romero-Gallo J., Delgado A.G., Bravo L.E., Wilson K.T., Peek R.M., Jr., Correa P., El-Rifai W. Promoter DNA hypermethylation in gastric biopsies from subjects at high and low risk for gastric cancer. Int J Cancer. 2010;127:2588–2597. doi: 10.1002/ijc.25274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silberg D.G., Furth E.E., Taylor J.K., Schuck T., Chiou T., Traber P.G. CDX1 protein expression in normal, metaplastic, and neoplastic human alimentary tract epithelium. Gastroenterology. 1997;113:478–486. doi: 10.1053/gast.1997.v113.pm9247467. [DOI] [PubMed] [Google Scholar]
- 24.Kazumori H., Ishihara S., Kinoshita Y. Roles of caudal-related homeobox gene Cdx1 in oesophageal epithelial cells in Barrett's epithelium development. Gut. 2009;58:620–628. doi: 10.1136/gut.2008.152975. [DOI] [PubMed] [Google Scholar]
- 25.Loots G.G., Ovcharenko I. rVISTA 2.0: evolutionary analysis of transcription factor binding sites. Nucleic Acids Res. 2004;32:W217–W221. doi: 10.1093/nar/gkh383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice P., Longden I., Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh S., Baltimore D. Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature. 1990;344:678–682. doi: 10.1038/344678a0. [DOI] [PubMed] [Google Scholar]
- 28.Yang C.H., Murti A., Pfeffer L.M. STAT3 complements defects in an interferon-resistant cell line: evidence for an essential role for STAT3 in interferon signaling and biological activities. Proc Natl Acad Sci USA. 1998;95:5568–5572. doi: 10.1073/pnas.95.10.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong N.A., Britton M.P., Choi G.S., Stanton T.K., Bicknell D.C., Wilding J.L., Bodmer W.F. Loss of CDX1 expression in colorectal carcinoma: promoter methylation, mutation, and loss of heterozygosity analyses of 37 cell lines. Proc Natl Acad Sci USA. 2004;101:574–579. doi: 10.1073/pnas.0307190101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong N.A., Wilding J., Bartlett S., Liu Y., Warren B.F., Piris J., Maynard N., Marshall R., Bodmer W.F. CDX1 is an important molecular mediator of Barrett's metaplasia. Proc Natl Acad Sci USA. 2005;102:7565–7570. doi: 10.1073/pnas.0502031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pereira B., Oliveira C., David L., Almeida R. CDX2 promoter methylation is not associated with mRNA expression. Int J Cancer. 2009;125:1739–1742. doi: 10.1002/ijc.24544. [DOI] [PubMed] [Google Scholar]
- 32.Kim S., Domon-Dell C., Wang Q., Chung D.H., Di Cristofano A., Pandolfi P.P., Freund J.N., Evers B.M. PTEN and TNF-alpha regulation of the intestinal-specific Cdx-2 homeobox gene through a PI3K: PKB/Akt, and NF-kappaB-dependent pathway. Gastroenterology. 2002;123:1163–1178. doi: 10.1053/gast.2002.36043. [DOI] [PubMed] [Google Scholar]
- 33.Kashima L., Toyota M., Mita H., Suzuki H., Idogawa M., Ogi K., Sasaki Y., Tokino T. CHFR, a potential tumor suppressor, downregulates interleukin-8 through the inhibition of NF-kappaB. Oncogene. 2009;28:2643–2653. doi: 10.1038/onc.2009.123. [DOI] [PubMed] [Google Scholar]
- 34.Panikashvili D., Mechoulam R., Beni S.M., Alexandrovich A., Shohami E. CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab. 2005;25:477–484. doi: 10.1038/sj.jcbfm.9600047. [DOI] [PubMed] [Google Scholar]
- 35.Pogliaghi G., Tacchini L., Anzon E., Radice L., Bernelli-Zazzera A. Heat shock activation of NFkB in rat liver is mediated by interleukin-1. FEBS Lett. 1995;372:181–184. doi: 10.1016/0014-5793(95)00973-d. [DOI] [PubMed] [Google Scholar]
- 36.Eberharter A., Becker P.B. Histone acetylation: a switch between repressive and permissive chromatin: Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinskaya M., Gourvennec S., Morillon A. H3 lysine 4 di- and tri-methylation deposited by cryptic transcription attenuates promoter activation. EMBO J. 2009;28:1697–1707. doi: 10.1038/emboj.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mutoh H., Hayakawa H., Sakamoto H., Sashikawa M., Sugano K. Transgenic Cdx2 induces endogenous Cdx1 in intestinal metaplasia of Cdx2-transgenic mouse stomach. FEBS J. 2009;276:5821–5831. doi: 10.1111/j.1742-4658.2009.07263.x. [DOI] [PubMed] [Google Scholar]
- 39.Crissey M.A., Guo R.J., Fogt F., Li H., Katz J.P., Silberg D.G., Suh E.R., Lynch J.P. The homeodomain transcription factor Cdx1 does not behave as an oncogene in normal mouse intestine. Neoplasia. 2008;10:8–19. doi: 10.1593/neo.07703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonhomme C., Calon A., Martin E., Robine S., Neuville A., Kedinger M., Domon-Dell C., Duluc I., Freund J.N. Cdx1, a dispensable homeobox gene for gut development with limited effect in intestinal cancer. Oncogene. 2008;27:4497–4502. doi: 10.1038/onc.2008.78. [DOI] [PubMed] [Google Scholar]
- 41.Barros R., Camilo V., Pereira B., Freund J.N., David L., Almeida R. Pathophysiology of intestinal metaplasia of the stomach: emphasis on CDX2 regulation. Biochem Soc Trans. 2010;38:358–363. doi: 10.1042/BST0380358. [DOI] [PubMed] [Google Scholar]
- 42.Mutoh H., Sakurai S., Satoh K., Osawa H., Hakamata Y., Takeuchi T., Sugano K. Cdx1 induced intestinal metaplasia in the transgenic mouse stomach: comparative study with Cdx2 transgenic mice. Gut. 2004;53:1416–1423. doi: 10.1136/gut.2003.032482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonhomme C., Duluc I., Martin E., Chawengsaksophak K., Chenard M.P., Kedinger M., Beck F., Freund J.N., Domon-Dell C. The Cdx2 homeobox gene has a tumour suppressor function in the distal colon in addition to a homeotic role during gut development. Gut. 2003;52:1465–1471. doi: 10.1136/gut.52.10.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moucadel V., Soubeyran P., Vasseur S., Dusetti N.J., Dagorn J.C., Iovanna J.L. Cdx1 promotes cellular growth of epithelial intestinal cells through induction of the secretory protein PAP I. Eur J Cell Biol. 2001;80:156–163. doi: 10.1078/0171-9335-00148. [DOI] [PubMed] [Google Scholar]
- 45.Oh E.J., Park J.H., Cho M., Lee W.J., Choi Y.H., Yoo M.A. The caudal-related homeodomain protein CDX1 activates proliferating cell nuclear antigen expression in hepatocellular and colorectal carcinoma cells. Int J Oncol. 2002;20:23–29. [PubMed] [Google Scholar]
- 46.Moucadel V., Totaro M.S., Dell C.D., Soubeyran P., Dagorn J.C., Freund J.N., Iovanna J.L. The homeobox gene Cdx1 belongs to the p53-p21(WAF)-Bcl-2 network in intestinal epithelial cells. Biochem Biophys Res Commun. 2002;297:607–615. doi: 10.1016/s0006-291x(02)02250-7. [DOI] [PubMed] [Google Scholar]
- 47.Lorentz O., Cadoret A., Duluc I., Capeau J., Gespach C., Cherqui G., Freund J.N. Downregulation of the colon tumour-suppressor homeobox gene Cdx-2 by oncogenic ras. Oncogene. 1999;18:87–92. doi: 10.1038/sj.onc.1202280. [DOI] [PubMed] [Google Scholar]
- 48.Domon-Dell C., Freund J.N. Stimulation of Cdx1 by oncogenic beta-catenin/Tcf4 in colon cancer cells; opposite effect of the CDX2 homeoprotein. FEBS Lett. 2002;518:83–87. doi: 10.1016/s0014-5793(02)02650-9. [DOI] [PubMed] [Google Scholar]
- 49.Lickert H., Domon C., Huls G., Wehrle C., Duluc I., Clevers H., Meyer B.I., Freund J.N., Kemler R. Wnt/(beta)-catenin signaling regulates the expression of the homeobox gene Cdx1 in embryonic intestine. Development. 2000;127:3805–3813. doi: 10.1242/dev.127.17.3805. [DOI] [PubMed] [Google Scholar]
- 50.Lynch J., Keller M., Guo R.J., Yang D., Traber P. Cdx1 inhibits the proliferation of human colon cancer cells by reducing cyclin D1 gene expression. Oncogene. 2003;22:6395–6407. doi: 10.1038/sj.onc.1206770. [DOI] [PubMed] [Google Scholar]
- 51.Chan C.W., Wong N.A., Liu Y., Bicknell D., Turley H., Hollins L., Miller C.J., Wilding J.L., Bodmer W.F. Gastrointestinal differentiation marker Cytokeratin 20 is regulated by homeobox gene CDX1. Proc Natl Acad Sci USA. 2009;106:1936–1941. doi: 10.1073/pnas.0812904106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mesquita P., Jonckheere N., Almeida R., Ducourouble M.P., Serpa J., Silva E., Pigny P., Silva F.S., Reis C., Silberg D., Van Seuningen I., David L. Human MUC2 mucin gene is transcriptionally regulated by Cdx homeodomain proteins in gastrointestinal carcinoma cell lines. J Biol Chem. 2003;278:51549–51556. doi: 10.1074/jbc.M309019200. [DOI] [PubMed] [Google Scholar]
- 53.Soubeyran P., Andre F., Lissitzky J.C., Mallo G.V., Moucadel V., Roccabianca M., Rechreche H., Marvaldi J., Dikic I., Dagorn J.C., Iovanna J.L. Cdx1 promotes differentiation in a rat intestinal epithelial cell line. Gastroenterology. 1999;117:1326–1338. doi: 10.1016/s0016-5085(99)70283-0. [DOI] [PubMed] [Google Scholar]
- 54.Keller M.S., Ezaki T., Guo R.J., Lynch J.P. Cdx1 or Cdx2 expression activates E-cadherin-mediated cell-cell adhesion and compaction in human COLO 205 cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G104–G114. doi: 10.1152/ajpgi.00484.2003. [DOI] [PubMed] [Google Scholar]
- 55.Almeida R., Silva E., Santos-Silva F., Silberg D.G., Wang J., De Bolos C., David L. Expression of intestine-specific transcription factors: CDX1 and CDX2, in intestinal metaplasia and gastric carcinomas. J Pathol. 2003;199:36–40. doi: 10.1002/path.1246. [DOI] [PubMed] [Google Scholar]
- 56.Suh E.R., Ha C.S., Rankin E.B., Toyota M., Traber P.G. DNA methylation down-regulates CDX1 gene expression in colorectal cancer cell lines. J Biol Chem. 2002;277:35795–35800. doi: 10.1074/jbc.M205567200. [DOI] [PubMed] [Google Scholar]
- 57.Pilozzi E., Onelli M.R., Ziparo V., Mercantini P., Ruco L. CDX1 expression is reduced in colorectal carcinoma and is associated with promoter hypermethylation. J Pathol. 2004;204:289–295. doi: 10.1002/path.1641. [DOI] [PubMed] [Google Scholar]
- 58.Lu X., Freund J.N., Muller M., Ravey J., Nicolas J.P., Gueant J.L., Namour F. Differential regulation of CDX1 and CDX2 gene expression by deficiency in methyl group donors. Biochimie. 2008;90:697–704. doi: 10.1016/j.biochi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 59.Cravo M., Pinto R., Fidalgo P., Chaves P., Gloria L., Nobre-Leitao C., Costa Mira F. Global DNA hypomethylation occurs in the early stages of intestinal type gastric carcinoma. Gut. 1996;39:434–438. doi: 10.1136/gut.39.3.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Senthilkumar C., Niranjali S., Jayanthi V., Ramesh T., Devaraj H. Molecular and histological evaluation of tumor necrosis factor-alpha expression in Helicobacter pylori-mediated gastric carcinogenesis. J Cancer Res Clin Oncol. 2011;137:577–583. doi: 10.1007/s00432-010-0921-9. [DOI] [PubMed] [Google Scholar]
- 61.Yuasa Y., Nagasaki H., Akiyama Y., Hashimoto Y., Takizawa T., Kojima K., Kawano T., Sugihara K., Imai K., Nakachi K. DNA methylation status is inversely correlated with green tea intake and physical activity in gastric cancer patients. Int J Cancer. 2009;124:2677–2682. doi: 10.1002/ijc.24231. [DOI] [PubMed] [Google Scholar]
- 62.Filomena A., Saieva C., Lucchetti V., Santacroce F., Falorni P., Francini V., Carrieri P., Zini E., Ridolfi B., Belli P., Orsini B., Mandi P., Palli D., Scheggi S. Gastric cancer surveillance in a high-risk population in tuscany (Central Italy): preliminary results. Digestion. 2011;84:70–77. doi: 10.1159/000322689. [DOI] [PubMed] [Google Scholar]