

Ectopic expression of the four transcription factors Oct4, Klf4, Sox2 and c-Myc reprograms adult somatic cells to induced pluripotent stem (iPS) cells.[1] Although iPS cells hold considerable promise as tools in research and drug discovery, the clinical application of iPS cells is hindered by the use of viruses that deliver the exogenous factors and modify the host genome. It is therefore of great interest to replace virally transduced factors with either proteins or small molecules. To date a number of compounds have been identified that facilitate reprogramming of somatic cells. Among these are kenpaullone[2], valproic acid[3] and inhibitors of TGFβ-signaling.[4] Here we have exploited a reporter based screen[2] to identify a new class of compounds that functionally replace Sox2: inhibitors of the Src family of kinases. These molecules provide novel tools to study the molecular mechanism of Sox2 in reprogramming.
To screen for small molecule replacements of Sox2, mouse embryonic fibroblasts (MEFs) harboring the firefly luciferase (Fluc) gene in the Nanog locus[2] (NL-MEFs) were transduced with Oct4, Klf4 and c-Myc (OKM), seeded into 1536-well plates in standard growth media and assayed against a large chemical library[5] (750,000 compounds; 2.2 μM). Compounds that reproducibly and dose-dependently activated the NL reporter >2.5-fold over vehicle-treated controls (Figure 1a) were then counter-screened in a cell based SV40-driven Fluc assay to rule out false positives that directly and non-specifically induce luciferase signal.[2, 6]
Figure 1. Chemical complementation of Sox2.
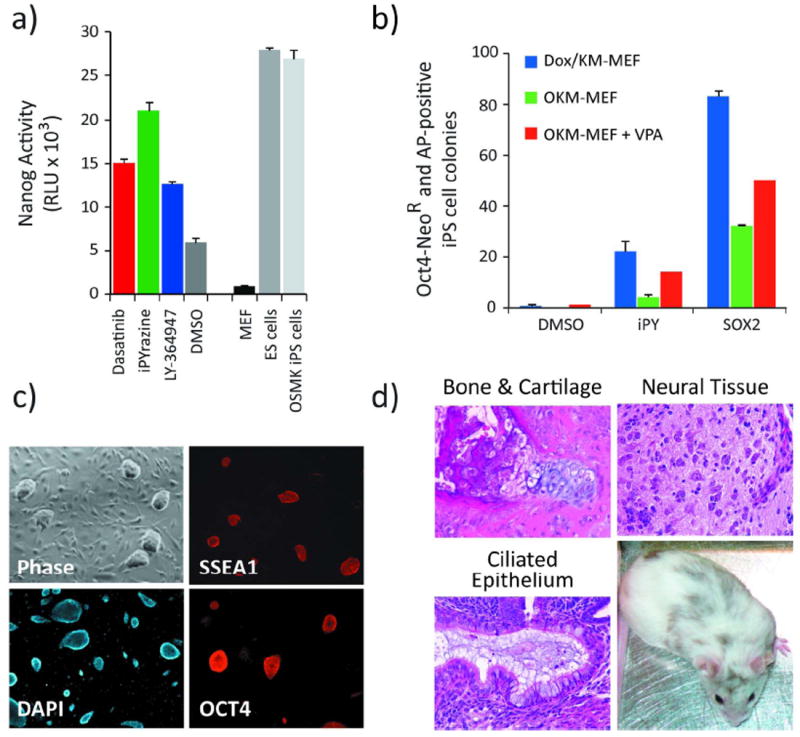
(a) OKM-transduced NL-MEFs were treated for 7 days with Dasatinib (0.5 μM), iPYrazine (10 μM), LY-364947 (a positive control; 1 μM) or vehicle (0.1% DMSO, v/v). The Nanog signal from treated cells is compared to that of non-transduced NL-MEFs, NL-ES cells and NL-iPS cells. Nanog activity is reported in relative light units (RLU). Error bars, standard deviation (n = 3). (b) O4NR-MEFs were transduced with Klf4 and c-Myc and grown in Dox (blue bars); transduced with OKM (no Dox; green bars); or transduced with OKM (no Dox) and grown in 1 mM VPA (red bars). Oct4-transduced O4NR-MEFs were used in order to take advantage of the stringent pluripotency marker, the Oct4-NeoR selection cassette. The OKM-expressing MEFs were treated with iPY (10 μM), DMSO (0.1%), or transduced with Sox2. At day 12, resultant colonies were selected upon supplementation of growth media with neomycin. Colonies that survived were stained for AP and counted 3 days later. Error bars, standard deviation (n = 3). (c) iPS cells derived from KM-transduced, Dox and iPY-treated O4NR-MEFs stain positive for the pluripotency-associated markers Oct4 and SSEA-1. (d) iPY-derived iPS cells form teratomas consisting of all three germ layers and contribute to live chimeras.
To confirm that filtered hit compounds which activate Nanog gene expression also replace Sox2, iPS cell colony formation was used as a secondary assay. Specifically, Klf4 and c-Myc were delivered retrovirally to O4NR-MEFs[1b] (cells harboring a Doxycycline (Dox)-inducible Oct4 cDNA in the collagen locus and the neomycin-resistance gene in the Oct4 locus), and Oct4 expression was induced by addition of Dox to the culture media (day 0). Two days later, positive screen hits (1-10 μM) were added to OKM-expressing MEFs in place of Sox2. After 10 days of compound treatment, growth media was supplemented with neomycin to select for colonies that reactivated the endogenous Oct4 locus. The reactivation of epigenetically silenced pluripotency-associated genes is required for somatic cells to transition to the iPS cell state.[7]
Dox-independent, neomycin resistant colonies were not observed in DMSO-treated (0.1%, v/v) controls, indicating that vehicle-treated cells had not removed the epigenetic silencing marks from the Oct4 promoter (which drives NeoR) and were thus not pluripotent. Among the compounds tested, one compound, iPYrazine (iPY; 10 μM), promoted the formation of neomycin-resistant iPS cell colonies (Figure 1b, blue bars) that survived and could be cultured in the absence of Dox. Transgenic Oct4 independent (minus Dox) growth of the iPY-treated iPS cells demonstrated that they had reactivated and relied on endogenous Oct4 to maintain the pluripotent state. In addition, OKM transduction combined with iPY treatment of MEFs carrying a GFP reporter under control of the endogenous Oct4 locus[8] also gave rise to stable, GFP-positive iPS cell lines (Figure S1, Supporting Information).
iPS cells derived from O4NR-MEFs with iPY, Dox and KM-transduction grew as pluripotent stem cell colonies in the absence of Dox and iPY. Moreover, these cells were indistinguishable from ES cells by morphological criteria and expressed the pluripotency-associated markers Oct4 and SSEA1 (Figure 1c). We next tested the differentiation potential of the iPY-derived iPS cells in a teratoma assay by injecting 106 cells subcutaneously into NOD-SCID mice. Tumors were isolated 3 weeks later and histological analyses demonstrated that cell types of all three germ layers were present; these included neural tissues, bone, cartilage and ciliated epithelium (Figure 1d). Furthermore, iPY-derived iPS cells contributed to live chimeras, as shown in Figure 1d. The results from this series of analyses indicate that the iPY-derived, Sox2-free iPS cells are pluripotent.
In order to identify the biological target of iPY, we profiled the compound against a biochemical panel of tyrosine kinases (51 kinases; Table S1). From this analysis, we found that iPY potently inhibited a number of tyrosine kinases at 5 μM. Commercially available inhibitors (Figures 2a-b and Table S2) of these candidate kinase targets were then assayed for their ability to replace Sox2 in the iPS cell colony formation assay. As shown in Figure 2b, the pan-Src family kinase (SFK) inhibitors Dasatinib[9] and PP1[10] (Figure 2b) were able to recapitulate the activity of iPY. Interestingly, both Dasatinib and PP1 were >2-fold more active than iPY and efficiently replaced Sox2 (Figure 2b). Moreover, the pan-SFK inhibitors gave rise to colonies with a similar efficiency to TGFβ inhibitors (SB-431542 and LY-364947). The latter have been reported to replace Sox2 and served as a positive control in this study.[4] In addition to TGFβ inhibitors, Ichida et al. have also reported that the SFK inhibitor PP1 is able to replace Sox2.[4a] Together with our work, these results indicate that iPY is likely playing a role in reprogramming by inhibiting Src kinases, although additional mechanisms cannot be excluded.
Figure 2. Src family kinase and TGFβ-inhibitors recapitulate the Sox2 replacement activity of iPY.

(a) Chemical structures of selected kinase inhibitors used in this study. (b) Inhibition of Src-kinase signaling by dasatinib (0.5 μM) or PP1 (10 μM) replaces Sox2 during reprogramming. O4NR-MEFs were transduced with Klf4 and c-Myc and grown in Dox (blue bars); transduced with OKM (no Dox; green bars); or transduced with OKM (no Dox) and grown in 1 mM VPA (red bars). OKM-expressing MEFs were transduced with Sox2 or treated with kinase inhibitors or vehicle (0.1% DMSO, v/v) for 10 days. At day 12, resultant colonies were selected upon supplementation of growth media with neomycin. Colonies that survived were stained for AP and counted 3 days later. Error bars, standard deviation (n = 3). Complete names, descriptions and concentrations of the kinase inhibitors used in this assay are provided in Table S2. (c) Activation of c-Src signaling by JK239 (10 μM) or TGFβ signaling with TGFβ1 ligand (10 ng/mL) inhibits 4-factor reprogramming. O4NR-MEFs were transduced with Sox2, c-Myc, Klf4 and treated with Dox to initiate Oct4 expression. 12 days later, resultant colonies were selected upon supplementation of growth media with neomycin. Colonies that survived were AP stained and counted 3 days later. Error bars, standard deviation (n = 3).
SFKs are a class of proto-oncogene tyrosine kinases that include nine mammalian members (i.e., c-Src, Yes, Fyn, Fgr, Lck, Hck, Blk, Lyn and Frk).[11] Several members of the SFK family have been reported to influence embryonic stem (ES) cell self-renewal and differentiation.[12] For example, activation of c-Src signaling promotes ES cell differentiation.[13] Consistent with this observation we find that the activation of Src signaling in MEFs with JK239[14] potently inhibits 4-factor reprogramming (Figure 2c). Together, our results suggest that SFK signaling is an important mediator of somatic cell reprogramming, where activation of the SFK pathway prevents reprogramming and inhibition allows for reprogramming in the absence of exogenous Sox2.
Previously, Ichida et al. demonstrated that small molecule mediated inhibition of TGFβ-signaling with LY-364947 or E-616452 can replace Sox2 through the activation of Nanog expression.[4a] The results from our screen, which rely on Nanog activation as a surrogate for the replacement of Sox2, suggest that the inhibition of SFK- and TGFβ-signaling may converge on a similar mechanism; that is, the function of Sox2 can be replaced during direct reprogramming by activating Nanog expression. Another potential scenario comes from the observation that both Nanog[15] and SFK inhibition[13] are capable of maintaining the self-renewing pluripotent state in ES cells. Thus, TGFβ inhibitor-mediated Nanog activation and pan-SFK inhibition may instead converge on a common mechanism in which the differentiation of newly formed iPS cells is prevented, thereby assisting in the transition to an undifferentiated state. In either case, it is interesting to note that inhibition of distinct signaling responses converge on a common end point.
In summary, we applied a cell-based, high-throughput chemical screen to identify small molecules that replace Sox2 during somatic cell reprogramming. The identification of novel SFK inhibitors provides new chemical tools to study the mechanisms underlying direct reprogramming and may ultimately help to bring iPS cell technology one step closer to clinical application.
Supplementary Material
Footnotes
We thank Dr. Xu Wu for assistance facilitating chemical analyses; Raaji Alaggapan and Ping Xu for support with tissue culture; and Jessie Dausman, Ruth Flannery and Dongdong Fu for help with animal husbandry and teratoma processing. We thank members of the Jaenisch lab for critical reading of the manuscript. This work was supported by a long-term HFSP postdoctoral fellowship (to J.S.), a National Science Foundation Predoctoral Fellowship (to C.A.L.), a Canadian Institutes for Health Research Postdoctoral Fellowship (to L.L.L.), NIH grants HD 045022 and 5 R37CA084198 (to R. J.) and grants from the Skaggs Institute for Chemical Biology of The Scripps Research Institute (to P.G.S.).
Supporting information for this article is available on the WWW under http://www.angewandte.org
Contributor Information
Dr. Judith Staerk, Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, 9 Cambridge Center, Cambridge, MA 02142 (USA)
Dr. Costas A. Lyssiotis, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA, 92037 (USA)
Lea A. Medeiro, Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, 9 Cambridge Center, Cambridge, MA 02142 (USA)
Michael Bollong, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA, 92037 (USA).
Dr. Ruth K. Foreman, Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, 9 Cambridge Center, Cambridge, MA 02142 (USA)
Dr. Shoutian Zhu, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA, 92037 (USA)
Michael Garcia, The Genomics Institute of the Novartis Research Foundation, 10675 John Jay Hopkins Drive, San Diego, CA 92121 (USA).
Qing Gao, Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, 9 Cambridge Center, Cambridge, MA 02142 (USA).
Dr. Laure C. Bouchez, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA, 92037 (USA)
Dr. Luke L. Lairson, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA, 92037 (USA)
Bradley D. Charette, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA, 92037 (USA)
Dr. Lubica Supekova, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA, 92037 (USA)
Dr. Achim Brinker, The Genomics Institute of the Novartis Research Foundation, 10675 John Jay Hopkins Drive, San Diego, CA 92121 (USA)
Dr. Charles Y. Cho, The Genomics Institute of the Novartis Research Foundation, 10675 John Jay Hopkins Drive, San Diego, CA 92121 (USA)
Prof. Rudolf Jaenisch, Email: jaenisch@wi.mit.edu, Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, 9 Cambridge Center, Cambridge, MA 02142 (USA).
Prof. Peter G. Schultz, Email: schultz@scripps.edu, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA, 92037 (USA).
References
- 1.a) Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Cell. 2007;131:861. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]; b) Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. Nature. 2007;448:318. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 2.Lyssiotis CA, Foreman RK, Staerk J, Garcia M, Mathur D, Markoulaki S, Hanna J, Lairson LL, Charette BD, Bouchez LC, Bollong M, Kunick C, Brinker A, Cho CY, Schultz PG, Jaenisch R. Proc Natl Acad Sci U S A. 2009;106:8912. doi: 10.1073/pnas.0903860106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. Nat Biotechnol. 2008;26:795. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Ichida JK, Blanchard J, Lam K, Son EY, Chung JE, Egli D, Loh KM, Carter AC, Di Giorgio FP, Koszka K, Huangfu D, Akutsu H, Liu DR, Rubin LL, Eggan K. Cell Stem Cell. 2009;5:491. doi: 10.1016/j.stem.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Maherali N, Hochedlinger K. Curr Biol. 2009;19:1718. doi: 10.1016/j.cub.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, Nam TG, Gray NS, Chatterjee A, Janes J, Yan SF, Trager R, Caldwell JS, Schultz PG, Zhou Y, Winzeler EA. Proc Natl Acad Sci U S A. 2008;105:9059. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorne N, Auld DS, Inglese J. Curr Opin Chem Biol. 2010;14:315. doi: 10.1016/j.cbpa.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaenisch R, Young R. Cell. 2008;132:567. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lengner CJ, Camargo FD, Hochedlinger K, Welstead GG, Zaidi S, Gokhale S, Scholer HR, Tomilin A, Jaenisch R. Cell Stem Cell. 2007;1:403. doi: 10.1016/j.stem.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, Shen Z, Cook LS, Doweyko AM, Pitt S, Pang S, Shen DR, Fang Q, deFex HF, McIntyre KW, Shuster DJ, Gillooly KM, Behnia K, Schieven GL, Wityak J, Barrish JC. J Med Chem. 2006;49:6819. doi: 10.1021/jm060727j. [DOI] [PubMed] [Google Scholar]
- 10.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. J Biol Chem. 1996;271:695. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 11.Yeatman TJ. Nat Rev Cancer. 2004;4:470. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 12.a) Anneren C, Cowan CA, Melton DA. J Biol Chem. 2004;279:31590. doi: 10.1074/jbc.M403547200. [DOI] [PubMed] [Google Scholar]; b) Meyn MA, 3rd, Schreiner SJ, Dumitrescu TP, Nau GJ, Smithgall TE. Mol Pharmacol. 2005;68:1320. doi: 10.1124/mol.104.010231. [DOI] [PubMed] [Google Scholar]
- 13.Meyn MA, 3rd, Smithgall TE. Sci Signal. 2009;2:ra64. doi: 10.1126/scisignal.2000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Supekova L, Supek F, Lee J, Chen S, Gray N, Pezacki JP, Schlapbach A, Schultz PG. J Biol Chem. 2008;283:29. doi: 10.1074/jbc.M703988200. [DOI] [PubMed] [Google Scholar]
- 15.a) Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, Vrana J, Jones K, Grotewold L, Smith A. Nature. 2007;450:1230. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]; b) Vallier L, Mendjan S, Brown S, Chng Z, Teo A, Smithers LE, Trotter MW, Cho CH, Martinez A, Rugg-Gunn P, Brons G, Pedersen RA. Development. 2009;136:1339. doi: 10.1242/dev.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.