

Abstract
In this article we review the evolution of cancer research involving PPARgamma, including mechanisms, target genes, and clinical applications. For the last thirteen years, the effects of PPARgamma activity on tumor biology have been studied intensely. Most of this research has focused upon the potential for employing agonists of this nuclear receptor in cancer treatment. As a monotherapy such agonists have shown little success in clinical trials, while they have shown promise as components of combination treatments both in culture and in animal models. Other investigations have explored a possible role for PPARgamma as a tumor suppressor, and as an inducer of differentiation of cancer stem cells. Whereas early studies have yielded variable conclusions regarding the prevalence of PPARgamma mutations in cancer, the protein level of this receptor has been more recently identified as a significant prognostic marker. We predict that indicators of PPARgamma activity may also serve as predictive markers for tailoring treatments.
Keywords: PPARgamma, cancer, thiazolidinediones, lipoxygenases, COX
2. INTRODUCTION
PPARgamma is a member of the peroxisome proliferator activated receptor (PPAR) family, a subfamily of the nuclear receptor superfamily. The name derives from the fact that the first identified member, mPPAR, was observed to respond to a group of hepatocarcinogens that up-regulate the proliferation of peroxisomes (1). The gamma variant was first cloned from a Xenopus cDNA library, along with the alpha and beta variants (2). In the same study, all three receptors were observed to increase the activity of the acyl coenzyme A oxidase promoter, evidencing a role in the regulation of beta-oxidation. The same year, a sequence identified on the acyl coenzyme A oxidase promoter that bound to one of the PPARs was found to be indispensible for this induction and was, thus, termed a “peroxisome proliferator response element” (PPRE) (3). A number of additional genes containing active PPREs in their promoters have since been characterized, and most of these are similarly involved in lipid metabolism (4). Not long after the discovery of the three PPAR variants, a murine version of the gamma variant (mPPARgamma1) was cloned (5). This was followed by a second murine form (mPPARgamma2) which, through a screening of several genes, was discovered to be part of an mRXR heterodimer known to be necessary for the transcription of aP2, a lipoprotein expressed only in adipocytes (6). Accordingly, the same study found mPPARgamma2 mRNA expression to be at least 20 fold higher in murine adipose tissue than in several other organs. mPPARgamma2 was, thus, believed to be involved in adipocyte differentiation. Indeed, it was later shown that murine fibroblasts could be differentiated into adipocytes in vitro through retroviral expression of mPPARgamma2 (7).
PPARgamma1 and PPARgamma2, which arise from separate promoters on the same gene, are also present in humans (8). The PPARgamma2 form contains 28 extra N-terminal amino acids that increase the ligand independence of its activity (9). At least two dominant-negative isoforms of PPARgamma have also been identified, emerging through the inclusion of introns with stop codons. Among these are gammaORF4, which ends in the fourth intron (10), and PPARgamma1tr, which ends in the third intron (11). Both of these lose their activity through exclusion of the C-terminal ligand-binding domain, a common alternative splicing strategy for deactivating members of the nuclear receptor superfamily (12). Other transcripts also arise from distinct promoters, but it is currently understood that the proteins they encode are not unique. A third promoter, for example, results in mRNA transcript PPARgamma3, which yields PPARgamma1 after translation (13). Two additional promoters are known to produce mRNA transcripts PPARgamma4 and PPARgamma6 (14, 15), which both also presumably yield PPARgamma1 after translation (15), although any data verifying this have not been published.
Tissue localization experiments on the active forms of PPARgamma have found that the PPARgamma1 form is expressed ubiquitously at the mRNA level, whereas PPARgamma2 is expressed primarily in adipocytes like its murine homologue (16). The same experiments have also found that the mRNA level of PPARgamma2 in adipose tissue is positively associated with obesity and can be down-regulated through caloric restriction. Shortly after PPARgamma’s role in adipocyte differentiation was characterized, the thiazolidinedione (TZD) drug troglitazone was discovered to be a potent ligand (17). Currently, PPARgamma is understood to be the primary mediator of TZDs’ ability to reverse insulin resistance in patients with type II diabetes (18). PPARgamma is also believed to play a role in suppressing the development of atherosclerosis (19). More recently, PPARgamma has been characterized in vitro as an antagonist of the differentiation of mesenchymal stem cells into osteoblasts, while PPARgamma antagonists have been proposed for use as therapeutic enhancers of osteogenesis in patients with osteoporosis (20).
Shortly after PPARgamma’s role in adipocyte differentiation was characterized, differentiation and growth inhibition effects were demonstrated in cell lines from several types of cancer, both in vitro and in xenografts (21–24). For the last decade much research has, therefore, focused upon characterizing the role of this receptor in cancer biology, as well as the potential for exploiting its agonists as clinical treatments. Early studies, however, collectively failed to establish either mutations of PPARgamma or changes in PPARgamma mRNA level as prevalent events in tumorigenesis (25–27). Additionally, speculation emerged that the anticarcinogenic effects elicited by synthetic agonists are independent of this receptor’s activity (28, 29). Clinical trials also failed to demonstrate the effectiveness of such agonists as a monotherapy for cancer treatment, a fact which fueled a search for combination treatments which to enhance their effects (30). Other researchers directed their focus toward developing optimized compounds derived from thiazolidinediones, with or without PPARgamma activation activity (31). Within the last 2–3 years, a significant shift in the understanding of PPARgamma’s role in tumor biology has occurred; studies employing immunohistochemistry staining have shown that the alteration of PPARgamma protein level is, in fact, significantly correlated with both tumor progression and patient prognosis in several types of carcinomas. An understanding of PPARgamma’s activity, as well as the post-genomic regulation of this activity, therefore, holds much promise for both the development of future treatments and the classification of tumors.
3. HISTORY OF CANCER RESEARCH INVOLVING PPARgamma
3.1. Characterization of the Anti-Carcinogenic Effects of PPARgamma Agonists
3.1.1. Early Experiments with Troglitazone
PPARgamma’s potential tumor suppressive role was first explored when the same research group that characterized its role in adipocyte differentiation demonstrated that a human liposarcoma cell line could be re-differentiated through treatment with the PPARgamma agonist troglitazone (22). The following year, the same research group confirmed similar effects in carcinoma cell lines from colon cancer and breast cancer (23, 24), while another group repeated them in prostate cancer (21). Differentiation was observed in each of these studies; however, the type of differentiation and the methods of measurement varied. The colon cancer group observed re-differentiation into normal colon mucosa through both an increase in carcinoembryonic antigen staining (immunohistochemistry) and changes in the expression profile of several mRNAs associated with malignancy (northern blot). The breast cancer study observed suppression of both K19 and Muc-1, while changes in gene expression usually observed during adipocyte differentiation were absent. The prostate cancer group observed differentiation through several markers including a decrease in PSA. This group then carried out a morphological analysis of TEM images from the treated cells. Interestingly, the cytoplasmic volume increased, along with the number and volume of vacuoles. Many of these cells contained vacuoles large enough to displace the nuclei as in adipocytes; however, unlike the storage vacuoles of such cells, they did not stain positively for lipids. Growth inhibition was also observed in each of these studies, and the timescale for this effect was much longer than for a cytotoxic agent. The colon cancer study measured growth after 7 days of continuous culturing, while the prostate cancer study examined colony formation on soft agar. In xenografts of highly malignant PC-3 (prostate cancer) cells in immuno-compromised mice, continuous treatment with troglitazone similarly halted the growth of tumors for about a week before they resumed the growth curve characteristic of the control group (21). It is worth noting that troglitazone, the PPARgamma agonist used in all of these experiments, is no longer used clinically in the United States on account of liver toxicity (32).
3.1.2. A Growing Repertoire of Qualitative Effects on Tumors
As more studies have been performed with other agonists, such as rosiglitazone, ciglitazone, pioglitazone, and edogenous ligants, an increasing number of qualitative effects on tumor biology have since been documented. Among these is the promotion of apoptosis, although this does not occur with all agonists or in all conditions (29). The reasons for this will be discussed in the Mechanisms of PPARgamma Activity section. PPARgamma agonists may also influence tumors in vivo by inhibiting angiogenesis through their activity in endothelial cells (33). In glioblastomas, PPARgamma agonists also reportedly block the promotion of the cancer stem cell subpopulation by growth factors in vitro, an important effect since this subpopulation is responsible for both metastasis and resistance to chemotherapy (34).
3.2. Characterizing PPARgamma Activity in Tumors
3.2.1. Early Attempts to Characterize PPARgamma Activity in Tumors
The effects of PPARgamma activation in early experiments with troglitazone led researchers to search for a loss of PPARgamma expression in both cell lines and clinical samples. In one of the early studies, 4 out of 55 clinical samples of sporadic colon cancer were found to have either missense or nonsense mutations, all of which resulted in a loss of PPARgamma activity (25). Another study found non-intragenic hemizygous deletions of PPARgamma in 8 of 38 prostate tumor samples and noted a reduction of PPARgamma protein levels in a few of these samples relative to the normal prostate tissue (26). A following study, however, failed to find either mutations or losses of expression at the mRNA level in both cell lines and clinical samples from several types of cancer including carcinomas of the colon, breast, lung and prostate; gliomas; osteosarcomas; myelodysplastic syndrome; and several leukemias (27). This was mirrored at the protein level through a western blot analysis of numerous cell lines, but not clinical samples. When compared with patient prognosis, however, a clear correlation emerged as PPARgamma mRNA transcript levels were found to be a favorable indicator in lung carcinomas (35). Only within the last 2–3 years have reliable correlations linking markers of PPARgamma activity to patient prognosis been constructed for other cancers.
3.2.2. Recent Immunohistochemistry Correlations
While most clinicopathological correlations involving PPARgamma appear to be inconclusive at the DNA/RNA level, those involving immunohistochemistry (IHC) data have been much more informative. In breast cancer, for example, the level of PPARgamma staining in clinical tumor samples is strongly positively correlated with the patients’ survival (p<0.001), with a higher independent predictive value than tumor size, axillary lymph node status, TNM stage, Histologic grade, ER, or Ki-67 in a combined model (36). Likewise, in colon cancer overall survival is significantly higher in patients whose tumors demonstrate higher PPARgamma staining (37). Similar findings have also been demonstrated in mobile tongue squamous cell carninoma (38). As for prostate cancer, IHC staining is detectable in the nuclei of tumor cells in clinical samples, whereas it is absent in the adjacent, non-malignant epithelial tissue; however, the expression level is highly inversely correlated with PSA level (39). This would suggest that in prostate cancer PPARgamma functions as an inducible tumor suppressor triggered by malignancy, while its expression is down-regulated during tumor progression. A direct survival correlation, however, was not constructed in this study. In pancreatic cancer the expression of PPARgamma is inversely associated with patients’ survival time (40). Most likely, PPARgamma exerts similar tumor-suppressive activity in these tumors, while this activity is down-regulated through mechanisms independent of changes in its protein level.
3.2.3. Expression of Dominant Negative Splice Variants
Studies examining the alternative splicing of PPARgamma have yielded results consistent with a tumor suppressive role in all tumor types studied. In one such study, mRNA transcripts encoding the dominant negative splice variant gammaORF4 were expressed in 16 of 25 clinical specimens of sporadic colorectal cancer at high levels relative to the adjacent nonmalignant tissue (10). It is unknown, however, whether the protein expression profile mirrored that of the mRNA. Shortly after this study, another group identified PPARgamma1tr (another dominant-negative splice variant) and noted its expression in the cell lines K562 (myelogenous leukemia), THP-1 (monocytic leukemia), A549 (alveolar adenocarcinoma), and HeLa (cervical cancer) (11). Expression of this splice variant was later detected in 8 of 9 clinical lung tumor samples at the protein level, whereas it was absent in the adjacent nonmalignant tissue (41). Importantly, there is no conclusive evidence that the expression level of either dominant negative isoform in clinical samples was sufficient to significantly reduce wild-type PPARgamma activity. Several hypothesized mechanisms for the repression of nuclear receptors by their respective dominant-negative splice variants have been reviewed by others and may lead to a method for conclusively confirming their suppression of PPARgamma activity in vivo (12).
3.3. Animal Models
3.3.1. Early Controversy over PPARgamma’s Influence in Colorectal Cancer
Given the complexity of clinicopathological data regarding PPARgamma, observations drawn from controlled experiments using animal models should be more informative. Such studies examining PPARgamma’s role in colorectal cancer, however, have yielded mixed results, although more recent evidence appears to support a tumor suppressive role. In two nearly identical early studies, long-term treatment of APC (+/−) mice with PPARgamma agonists increased the frequency of spontaneous polyps in the colon (42, 43). Saez and colleagues used troglitazone exclusively, while the others used both troglitazone and rosiglitazone. In a subsequent model, however, treatment of mice with the PPARgamma agonist pioglitazone was able to reduce the number of spontaneous tumors induced by dimethylhydrazine (44). Taken together without further evidence, these studies might have suggested that PPARgamma activation generally suppresses tumorigenesis in tumors driven by random mutations, whereas it contributes to malignancy when coupled to an APC mutation. Subsequent studies, however, have rejected this conclusion. Another group of researchers using APC(+/−) mice replaced PPARgamma agonists with intestine-specific PPARgamma haploinsufficiency and found that this increased the rate of tumor formation (45), although a previous theoretically redundant experiment had failed to observe this difference (46). In yet another study employing the agonist MCC-555 in APC(+/−) mice, PPARgamma activation reduced the rate of tumor-formation (32). Other studies employing animal models have established the efficacy of PPARgamma-specific agonists in suppressing colitis, an established risk factor for colorectal cancer (47–49). Rosiglitazone has also been approved as a clinical treatment for colitis, although it has not become a mainstream treatment because of adverse effects on the cardiovascular system (50). Even more recently, a diet rich in conjugated linoleic acid (another PPARgamma ligand) has been shown to suppress tumor formation (in addition to colitis) in mice during long-term azoxymethane treatment, an effect which is abrogated by PPARgamma deficiency (50).
3.3.2. Animal Models for Carcinomas, Aside from Colon Cancer
Studies employing animal models for other types of cancer have similarly characterized PPARgamma as a tumor suppressor. The agonists rosiglitazone and fenretinide, for example, have been shown to exert a protective effect in a rat model for mammary carcinogenesis (51). Similarly, in a gastric carcinoma chemoprevention model, PPARgamma (+/−) mice displayed an enhanced generation of tumors during treatment with N-methyl-N-nitrosourea (52). In lung cancer, lung-specific overexpression of PPARgamma in transgenic mice reduced the formation of tumors induced by ethyl carbamate treatment (53).
3.4. Epidemiological Correlations and Clinical Trials
Clinical data regarding the effects of PPARgamma activation clearly indicate the potential for modulation of PPARgamma as a chemopreventive strategy, although an effective treatment using PPARgamma agonists against established tumors has not been developed. One study employing data on 87,678 male diabetic patients correlated the use of TZDs (PPARgamma ligands) with the risk of developing cancer, finding a risk factor-adjusted 29% reduction in lung cancer for all patients, a 45% reduction in colorectal cancer for African Americans, an insignificant reduction in colorectal cancer for whites, and an increase in prostate cancer at 15% (P=0.03) for whites and 18% (not statistically significant) for African Americans (54). Unfortunately, the data for this study included patients treated with troglitazone, which can no longer be used clinically and may not accurately reflect the effects of other PPARgamma agonists. In an early clinical trial with prostate cancer, long-term treatment with troglitazone prevented the rise of PSA levels, especially for androgen-dependent tumors (26). For more advanced, late stage tumors that are refractory to either chemotherapy or hormone therapy, however, clinical trials using PPARgamma agonists alone have shown little success. In one such study, troglitazone treatment of patients with metastatic colorectal cancer resulted in no partial responses (55). In a separate trial, breast cancer patients with either stage IV or stage IIIB tumors also showed no partial responses to troglitazone treatment, although this study was discontinued prematurely because the drug was withdrawn from the market (56). In yet another trial using the PPARgamma agonist rosiglitazone to treat recurrent prostate cancer, a small, statistically insignificant increase in survival time was observed relative to a placebo group (57).
3.5. Combination Therapies
Subsequent attempts have been made to develop combination therapies in which PPARgamma activation synergizes or potentiates other drugs. Since much of the genomic activity of PPARgamma is achieved through a heterodimer with RXRalpha, treatment strategies employing agonists for both receptors have been proposed (58). A phase I trial using this approach, however, failed to demonstrate any partial responses in a group of patients with refractory solid tumors (59). The most widely accepted explanation for this failure will be discussed in the PPARgamma-RXR part of the Mechanisms of PPARgamma Activity section.
Another promising combination treatment strategy is co-administration with conventional chemotherapy drugs (30). Treatments combining rosiglitazone with the platinum-based drug carboplatin, for example, result in a marked improvement over either individual compound both in cell lines and in vivo in a murine model with KRAS-induced tumors (60, 61). Cisplatin also shows synergistic combination effects with rosiglitazone in DMBA-induced murine mammary tumors (62), and with troglitazone in a mesothelioma cell line (63). A similar combination effect occurs between rosiglitazone and 5-fluorouracil in colon cancer and liver cancer cell lines (64, 65). 15d-PGJ2, an endogenous PPARgamma ligand, also reportedly acts synergistically with docetaxel in lung cancer cell lines (66). Antagonism, however, likely occurs for compounds removed from the cell by the ATP cassette transporter ABCG2, since PPARgamma-RXR directly transactivates this gene (67). Such drugs include irinotecan (68), daunorubicin, doxorubicin, and mitoxantrone (69). Among these, doxorubicin is antagonized by rosiglitazone in HT-29 cells (70), while, to our knowledge, no report exists regarding combination effects between specific PPARgamma agonists and the remaining compounds from this group. The agonists ciglitazone and rosiglitazone have also been reported to antagonize gamma radiation-induced cell death in Caco2 cells in vitro by up-regulating Hsp70 (71). Additional proposals for combination treatments have been reviewed by others (30). Many more of these approaches will likely emerge as the mechanisms of PPARgamma activity are further elucidated.
4. REGULATION OF PPARgamma
4.1. Ligands
4.1.1. Lipoxygenase Products
PPARgamma is activated by several known endogenous ligands emerging from the metabolism of arachadonic acid and linoleic acid. A map of these pathways is shown in Figure 1. Among the ligands represented are the lipoxygenase products 13(S)HODE (produced from linoleic acid by 15-LOX-1) and 15(S)HETE (produced from arachidonic acid by both 15-LOX-1 and 15-LOX-2, although 15-LOX-2 catalyzes this reaction much more efficiently) (72). Induction of 15-LOX-1 activity has actually been shown to mediate the promotion of apoptosis by non-steroidal anti-inflammatory drugs (NSAIDs) in certain tumor cell lines and to occur independently of COX-2 inhibition (73, 74). Although this finding was originally demonstrated only in colon cancer cell lines, it has been repeated in cell lines from both esophageal cancer and gastric cancer (75, 76). Supposedly, apoptosis is achieved through the activity of 13(S)HODE because, unlike 15(S)HETE, its level is induced by these treatments, while inhibition of 15-LOX-1 reverses this effect (73, 74). Additional compounds produced either in parallel to these products by 15-LOX-1 or downstream of them, however, may account for this effect. 13(S)HODE is, in fact, degraded by 13-HODE dehydrogenase to form 13-oxooctadecadienoic acid (13-OXO) (77), another PPARgamma ligand (78). Seeing as the expression of 13-HODE dehydrogenase is reportedly reduced in colon cancer samples relative to nonmalignant tissue (79), 13-OXO likely influences additional mechanisms. In contrast to its role as a ligand, 13(S)HODE can also suppress PPARgamma activity through a poorly understood, context-dependent MAPK activation (80). Because a pro-tumorigenic effect has been demonstrated for 15-LOX-1 in PC-3 cells, it has been hypothesized that this activity reverses its role in prostate cancer, causing it to enhance, rather than suppress, tumorigenesis (81). Importantly, the role of 15-LOX-1 has not been firmly established in prostate cancer.
Figure 1. Bioactive lipids and PPARgamma ligands.
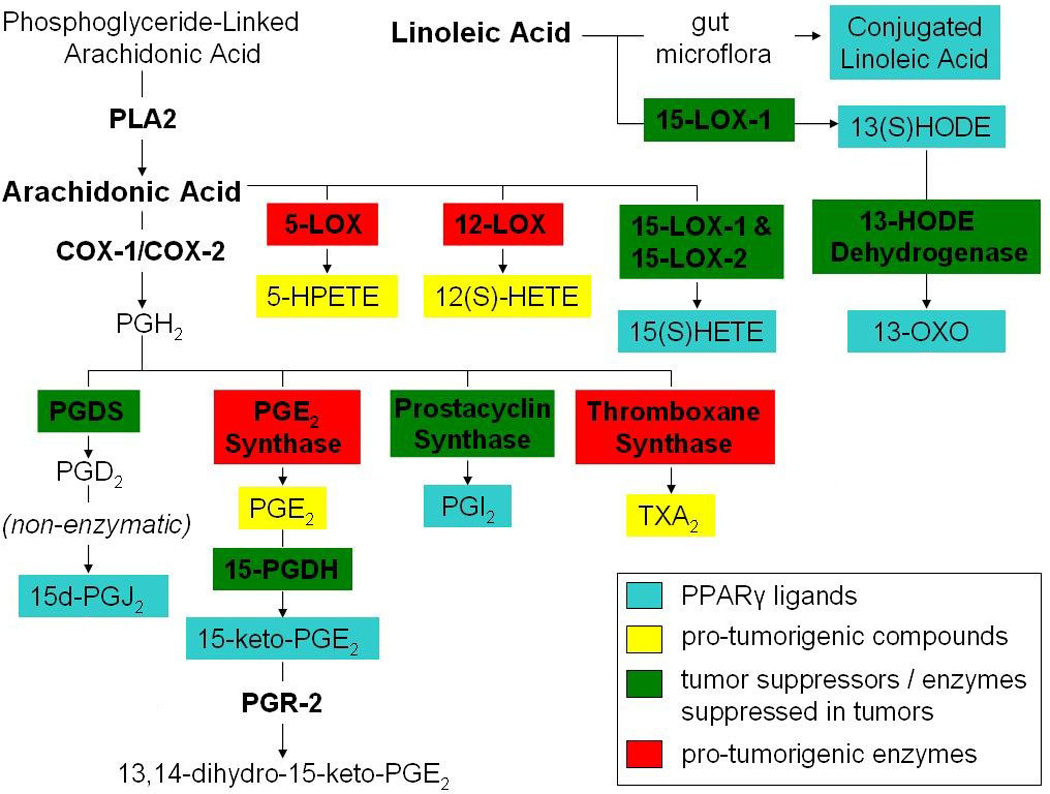
Naturally occurring PPARgamma ligands arising from the metabolism of arachidonic acid and linoleic acid are shown. Several of the enzymes in these pathways are associated with tumorigenesis, and some of them have well described tumor-suppressive functions.
15(S)HETE is believed to play a less significant role in activating PPARgamma, at least in colorectal cancer (73, 74). While the ability of 13(S)HODE and 15(S)HETE to activate PPARgamma at concentrations of 5µM appears to be roughly comparable (82), the endogenous concentration of 13(S)HODE has been determined to be about 4–5 times that of 15(S)HETE in clinical samples from both colorectal tumors and paired non-malignant epithelium (83). Roughly the same ratio has been reported for mice lungs, both for non-malignant tissue and for tumors generated through treatment with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (84). This difference has been attributed to a much higher availability of linoleic acid than arachidonic acid in the diet (83). A decrease of 13(S)HODE production has also been observed in clinical colorectal tumor samples relative to paired nonmalignant epithelium samples, whereas no discernable difference has been observed with respect to the concentration of 15(S)HETE, suggesting that 15(S)HETE plays a less significant role in suppressing these tumors (83). In a lung carcinogenesis model, however, a statistically significant decrease was detected in the levels of both 13(S)HODE and 15(S)HETE (84). While the concentration of 15(S)HETE appears to be low in tumors, it is possible that the downstream metabolites are more prevalent. Among these, both 15-oxo-ETE and 5-oxo-15(S)-OH-ETE have been characterized as PPARgamma ligands, although extremely high concentrations are required for their activity (85). It is unlikely that the local concentrations of these compounds are ever sufficient to activate PPARgamma.
Interestingly, in assays measuring the endogenous production of 15(S)HETE, about 95% of this product is detected in the medium, although the efficiency of its secretion appears to vary between cell lines (72). Such variation may indicate that this process is regulated, in part, by an active transport mechanism. Recently, it has also been demonstrated that 15(S)HETE preferentially activates PPARbeta/delta over PPARgamma (86). While the expression and activation of PPARbeta/delta are down-regulated by 13(S)HODE during the induction of 15-LOX-1 in colon cancer cells (where PPARbeta/delta activity favors malignant characteristics) (87), activation may occur within neighboring cells that lack 13(S)HODE. Interestingly, PPARbeta/delta activation specifically within stromal cells has been shown to alter cytokine production so as to reduce mitogenic paracrine signals when cultured with multiple myeloma cells (88). Although 15(S)HETE may not play a significant role in the activation of PPARgamma in most tumors, it could conceivably exert a similar tumor-suppressive effect through such paracrine signaling.
4.1.2. Prostaglandins
Several arachidonic acid metabolites generated downstream of the COX enzymes can also act as PPARgamma ligands. Among these, 15d-PGJ2 appears to be the most prevalent in the literature, probably because it was the first endogenous ligand discovered for PPARgamma (89). Prostacyclin (PGI2), another downstream product of the COXs, is also believed to activate PPARgamma as its stable analog iloprost has been shown to activate a PPARgamma reporter in both a bronchial epithelial cell line and two lung adenocarcinoma cell lines (53). Furthermore, it has been suggested that this activity mediates prostacyclin’s well-established tumor suppressive activity, since knock-down of the IP receptor (a G-protein-coupled receptor for prostacyclin) fails to reduce the protective effect of prostacyclin synthase over-expression (53). The evidence for this, however, is still inconclusive because the effect of PPARgamma knockdown on prostacyclin synthase-mediated suppression of tumorigenesis has never been determined. Increased activity of a PPARgamma reporter has also been observed upon treatment of 3T3-L1 cells with 15-keto-PGE2, an arachidonic acid product generated through conversion of PGE2 by 15-PGDH (90). This particular ligand is believed to be indispensible for at least a subset of endogenous processes that require PPARgamma activation, seeing as exogenous over-expression of the downstream enzyme PGR-2 (which metabolizes 15-keto-PGE2 to form 13,14-dihydro-15-keto-PGE2) blocks hormone-induced differentiation of 3T3-L1 cells into adipocytes (90). On the other hand, the activity of 13,14-dihydro-15-keto-PGE2 may be responsible for this effect, rather than the absence of 15-keto-PGE2.
4.1.3. Importance of Arachidonic Acid Products
Given PPARgamma’s role in adipocyte differentiation, it is interesting that siRNA-mediated knockdown of phospholipase A2, the initiator of arachidonic acid signaling, is able to block hormone-induced differentiation of 3T3-L1 preadipocytes (91). It is also worth noting that this differentiation is rescued through treatment with the PPARgamma agonist troglitazone. These observations provide highly suggestive, circumstantial evidence that at least one of the known PPARgamma ligands generated through arachidonic acid metabolism is necessary for endogenous induction of PPARgamma activity. This conclusion is also consistent with in vivo observations in animal models, although these can be readily explained by at least one other mechanism. In the aforementioned adipocyte-specific PPARgamma double knockout study in mice, for example, increases in body weight due to a high fat diet were attenuated relative to a control group, as was the total body fat weight (92). Similarly, adipocyte-specific phospholipase A2 knockout mice had a normal body weight when fed a standard diet, but they failed to develop obesity in response to a high fat diet (93). Although the results of the latter study were consistent with the effects of decreased PPARgamma activity, the researchers explained them through an elevation of lipolysis due to decreased PGE2 production. Nonetheless, current evidence appears to support the hypothesis that ligands originating from arachidonic acid metabolism are the principal means of inducing PPARgamma activity for most organs in vivo, whereas other mechanisms only modulate this activation. In the colon, however, an exception emerges in that natural PPARgamma ligands from the diet complement those from the arachidonic acid pathway. As mentioned previously, dietary supplementation with conjugated linoleic acid, has been shown to reduce colorectal tumorigenesis induced by azoxymethane in mice, presumably through an observed suppression of colitis (a known risk factor for colorectal cancer), whereas this effect is absent in PPARgamma-deficient mice (50).
4.2. Phosphorylation
Phosphorylation of PPARgamma also plays a significant role in its regulation. Not long after PPARgamma’s discovery, treatment of NIH3T3 and 293T cells with either EGF or PDGF was shown to down-regulate the activity of a PPARgamma luciferase reporter, an event which was attributed to MAPK-mediated phosphorylation of PPARgamma1 at Ser82 (94). In breast cancer, it has actually been hypothesized that a loss of PPARgamma activity is achieved entirely through MAPK activation, as evidenced by the fact that the growth inhibition effect of troglitazone treatment in an otherwise resistant cell line can be rescued through the addition of the MAPK inhibitor PD98059 (24). More recent studies have found that this effect can be achieved through phosphorylation of RXR (part of an important heterodimer with PPARgamma) under the same conditions, while it can also be reduced through transfection with an RXR mutant protected against phosphorylation (95, 96). The two mechanisms are, in fact, complementary; several lines of evidence confirming an independent role for the phosphorylation of PPARgamma in modulating its activity have been reviewed (97).
4.3. Corepressors
The activity of PPARgamma is also modulated through interaction with corepressors, either through a repression of PPARgamma activity or through PPARgamma’s enhancement of the ability of these corepressors to transrepress other genes (98). PPARgamma’s affinity for these proteins can be modulated by numerous factors. For example, most of these corepressors lose their affinity for nuclear receptors when these receptors are activated by their respective ligands (98), while some of these interactions are enhanced by ligands. The binding of the corepressor RIP140 to ternary complexes of PPARgamma and RXR, for example, is enhanced by the addition of RXR ligands (99). Additionally, at least one protein is known to block the interaction between PPARgamma and its corepressors. Direct interaction with phosphorylated extracellular signal-related kinase 5 (ERK5) has been observed to increase the activity of PPARgamma1 in reporter assays, presumably by blocking its interaction with the corepressor SMRT (100). Seeing as an elevation of this mitogenic kinase’s expression has been observed in prostate tumor samples relative to the corresponding nonmalignant tissue, this type of mechanism could be prevalent in cancer (101). This fact may also explain why a suppression of PPARgamma protein level would be necessary for highly malignant prostate tumors, seeing as ERK5 would reduce the ability of these corepressors to suppress PPARgamma activity.
4.4. Repression of PPREs by Other Proteins
The genomic activity of PPARgamma can also be blocked through binding of other proteins to PPREs. This behavior has been reported for the estrogen receptor (102). A similar repression of PPREs has been reported for a thyroid receptor beta mutant observed clinically, in which either a homodimer or heterodimers with either PPARgamma or RXR could bind and repress PPREs (103). Whether such a direct repression of PPREs is common clinically remains to be seen.
4.5. Proteasomal Degradation
PPARgamma activity is also regulated through proteasomal degradation. Targeting of PPARgamma to the proteasome occurs through ubiquitylation of the AF-2 segment of its ligand binding domain (104). This process is counterintuitively enhanced by the addition of TZDs, an effect which contrasts with their effect on the activation of PPREs (105). In cancer cell lines, however, the influence of this process often appears to be overcome by a competing mechanism. PC-3 cells, for example, show no change in the protein level of PPARgamma upon treatment with troglitazone (21), while a similar treatment elicits an increase in PPARgamma protein in 21MT cells (24).
4.6. Transcriptional Regulation
Little is known regarding control of PPARgamma transcription in epithelial cells. The C2H2 zinc finger transcription factor Zac, which is known to play a role in apoptosis, has been reported to activate the transcription of PPARgamma and is expressed in some colon cancer cell lines (106). Additionally, during the well studied process in which NIH-3T3 cells are differentiated into adipocytes through activation of PPARgamma, a positive feedback loop emerges in which PPARgamma and its target gene C/EBPalpha up-regulate the transcription of each other (107). In light of the effect ligands have in promoting PPARgamma degradation (105), this process likely proceeds after a transient induction by endogenous ligands. A similar loop may be active during the treatment of carcinomas with synthetic PPARgamma agonists. Two transcription factors modulated by TGFbeta, Egr-1 and AP1, are also considered to be important regulators of PPARgamma transcription in smooth muscles; however, their importance in epithelial cells has not been established (108). Likewise, the circadian clock transcription factors DBP and E4BP4 have been shown to regulate the PPARgamma6 mRNA transcript level (15), while their role in PPARgamma regulation in epithelial cells and tumors has not been characterized.
5. MECHANISMS OF PPARgamma ACTIVITY
5.1. Is PPARgamma Responsible for the Antitumor Effects of its Agonists?
Many researchers have speculated that the anticancer effects elicited by PPARgamma agonists are independent of this receptor’s activity. This conclusion was reached in an early study after growth inhibition was elicited by both troglitazone and ciglitazone in PPARgamma (−/−) mouse ES cells (28). Another group of researchers extended this finding to other agonists upon finding that the anticancer effects elicited by troglitazone, ciglitazone, and 15-d-PGJ2 (an endogenous ligand) on several prostate and bladder carcinoma cell lines varied qualitatively (growth inhibition vs. apoptosis), while rosiglitazone and pioglitazone elicited no effects whatsoever (29). An interpretation of these discrepancies, however, should account for the fact that different agonists can influence independent mechanisms of PPARgamma activity. In one study, for example, treatment of a myeloid leukemia cell line with either 15-d-PGJ2 or troglitazone resulted in a repression of STAT3 reporter activity, an effect which was reversed by the PPARgamma antagonist GW9662 (109). 15-d-PGJ2, however, caused direct binding of PPARgamma to STAT3, whereas troglitazone increased the binding of NCoR to STAT3, presumably through the observed release of this corepressor from PPARgamma. Considering that all of the known mechanisms of PPARgamma’s activity are also context-dependent, much of the variability in the effects of PPARgamma ligands is likely caused by an interaction between off-target mechanisms and the activity of PPARgamma.
5.2. PPARgamma-RXR Heterodimer
Among the mechanisms of PPARgamma activity, the most prevalent in the literature is that of the PPARgamma-RXR heterodimer, which is known to promote the transcription of genes whose promoters contain peroxisome proliferator response elements (PPREs) (98). In adipocytes several genes, such as aPC, are constitutively bound to PPARgamma-coactivator complexes, among which PPARgamma-RXR is highly prevalent (110). The significance of the activity of this heterodimer in clinical tumors, however, has not been established. This uncertainty stems from the fact that treatments combining ligands for PPARgamma and RXR result in an additive or synergistic induction of the dimer’s activity (6). In early studies on the effects of PPARgamma activation in cancer, this behavior was observed in a liposarcoma cell line (22). Recently, however, the activity of this heterodimer has been shown to be absent in several cancer cell lines due to growth factor-triggered phosphorylation of RXRalpha, while the synergy between ligands for the two receptors can be restored through either the withdrawal of growth factors or the addition of MAPK inhibitors (95, 96). An elevation of RXRalpha phosphorylation has actually been observed in clinical colorectal tumor samples relative to nonmalignant epithelial colon tissue (95). Much research has, therefore, focused upon the prospect of developing combination therapies that reduce the phosphorylation of RXR to potentiate the effect of PPARgamma agonists (58). Importantly, the activity resulting from PPARgamma’s interaction with RXR is not restricted to the induction of PPREs. The oncogenic protein beta-Catenin, for example, has been shown to interact with both RXR and PPARgamma (111–113). Moreover, treatment of cells with PPARgamma ligands during exogenous expression of PPARgamma and RXR decreases the ability of beta-Catenin to transactivate its target genes (114). Much of PPARgamma’s anticancer activity, however, is achieved through mechanisms independent of RXR. This is evident in the antitumor effects elicited by troglitazone in MDA-MB-231 breast cancer cells (115), which lack a detectable level of RXRalpha expression (116).
5.3. Coactivators
The transcription activity of PPARgamma’s target genes can also be achieved through complexes containing PPARgamma and various coactivators, although the specificity of the target genes may vary. A complex with PGC-1alpha, for example, activates the transcription of UCP-1, which is inaccessible to PPARgamma-RXR (117). Different coactivators also have different modes of activity. Many of the classical coactivators, such as CBP/p300 and SRC-1, achieve this activity through their histone acetyl transferase (HAT) activity, while others like PGC-1alpha simply recruit other coactivators to complexes with PPARgamma (118). The influence of PPARgamma ligands on the activity of these coactivators also varies. In most cases, interaction between PPARgamma and its coactivators is enhanced by ligand activation (98). Some coactivators, however, activate PPARgamma constitutively. Among these are ARA70 and SHP (119, 120). Additionally, interaction with certain coactivators may be enhanced by only a subset of the PPARgamma ligands. For example, binding to PGC-1alpha is reportedly enhanced by the addition of 15d-PGJ2 and troglitazone (121), but not rosiglitazone (117). This contrast, however, may not be valid because the effects of 15d-PGJ2 and troglitazone were determined through an endogenous immunoprecipitation of PGC-1alpha, whereas the absence of this activity for rosiglitazone was determined through an exogenous immunoprecipitation of GST-tagged PGC-1alpha. Importantly, many of PPARgamma’s co-activators are promiscuous, a fact exemplified by PGC-1alpha’s binding to NF-kappaB (121), nuclear respiratory factor-1 (NRF-1) (118), estrogen receptor alpha (ERalpha), the retinoic acid (RA) receptor, and thyroid receptor beta (117). Clearly the availability of these coactivators is an important determinant of PPARgamma’s activity in tumors. Madjalaweih and Ro provide a much more extensive review of PPARgamma’s coactivators, as well as its corepressors (122).
5.4. Corepressors
Any PPARgamma that is not part of an active complex normally exists in an inactive form, bound to a corepressor, such as SMRT or NCoR (98). Under most circumstances, interaction with a ligand causes PPARgamma to dissociate from the corepressor and bind to a coactivator (98). The corepressor, in turn, may be freed to bind to other transcription factors, thereby transrepressing their target genes, an effect which, as previously mentioned, was observed during treatment of a multiple myeloma cell line with troglitazone (109). Ligand activation also enhances SUMOylation of PPARgamma, a change which initiates the recruitment of PPARgamma to NCoR-HDAC3 complexes associated with specific genes, thereby repressing their transcription by blocking the ubiquitylation-mediated destruction of the complexes (123). An extensive list of PPARgamma corepressers is included in a review by Madjalaweih and Ro (122).
6. PPARgamma TARGET PATHWAYS
6.1. Overview
Microarray studies have hinted at numerous pathways that may be modulated by PPARgamma ligands. The first of these papers characterized the effect on known markers of differentiation, such as RegIA and keratin20, in Moser and COS7 colon cancer cells, finding that these differentiation markers were mostly up-regulated (124). A more recent paper profiled the influence of three different PPARgamma agonists on several pathways in HCT116 colon cancer cells, based on changes in the levels of mRNA transcripts of representative genes (125). Some of the processes characterized and the accompanying genes used as markers are as follows: the cell cycle (MDM2 decreased, CHEK2 and RBX1 somehow altered), cytokine-cytokine receptor interaction (IL18 and VEGFB somehow altered), ubiquitin-mediated proteolysis (UBE2E1 and RBX1 somehow altered), phosphotydlinositol signaling (INPP5D decreased), Wnt signaling (RBX1 somehow altered), the “colorectal cancer pathway” (APPL decreased), and calcium signaling (SLC25A4 somehow altered). Several genes involved in apoptosis, such as BCL-X(L), MCL-1, BAX (126), XIAP (127), and Survivin (128), are also reportedly modulated by PPARgamma so as to favor this process. In contrast, PPARgamma has been shown to up-regulate Hsp70 in Caco2 colon cancer cells and MCF7 breast cancer cells (71, 129), an affect which prevents radiation-induced apoptosis (71). Although these observations provide promising directions for future research, their reliability is unknown. Numerous mechanisms of PPARgamma activity are already known to be context-dependent and may, therefore, vary widely between individual tumors. Accordingly, the development of clinical treatments exploiting this receptor will depend upon understanding the precise mechanisms by which these pathways are influenced. The pathways that follow are more thoroughly researched and include discussions of their mechanisms.
6.2. Well Studied Non-Genomic Targets
6.2.1. beta-Catenin
PPARgamma is believed to inhibit signaling by the beta-Catenin pathway, whose contribution to tumor biology is well established. The activity of beta-Catenin is important in promoting epithelial-mesenchymal transition (130), an important process in tumor progression. The protumorigenic activity of Wnt/beta-Catenin signaling arises from the nuclear localization of beta-Catenin, which activates the TCF/LEF families of transcription factors by displacing them from the corepressor Groucho/TLE (131). This results in an up-regulation of cyclin D and c-myc, both of which exert pro-tumorigenic effects (132). In non-malignant cells this event is prevented by a destruction complex containing Axin, APC, and the kinase GSK-3beta, which phosphorylates beta-Catenin, thereby targeting it for proteasomal degradation (133). Cannonical Wnt signaling, however, leads to the formation of a membrane-bound complex containing Fz, LRP6, Dvl, and Axin (134), effectively removing Axin from the destruction complex and deactivating it (135).
Speculation regarding PPARgamma’s influence on the Wnt/beta-Catenin pathway began when early APC(+/−) mice models failed to demonstrate a chemopreventive effect for PPARgamma ligands against colorectal carcinogenesis, whereas a protective effect was observed during treatment of wild-type mice with mutagenic agents (46). Importantly, APC is an established tumor suppressor that is necessary for the proper function of the beta-catenin destruction complex (136); a loss-of-function mutation of this gene increases the nuclear localization of beta-Catenin within a subset of cells in the tumor (137). In one study seeking to resolve this issue, tumors were induced in both wild-type and PPARgamma(+/−) mice through treatment with azoxymethane (46). Tumor formation was enhanced in the PPARgamma(+/−) mice, relative to the wild-type mice, while beta-Catenin was up-regulated in their colon epithelial tissue prior to the onset of these tumors. The next year, PPARgamma activation by both troglitazone and GW7845 was found to increase the proteasomal degradation of beta-Catenin in NIH 3T3-L1 cells (138). Interaction between beta-catenin and PPARgamma was also soon demonstrated through immunoprecipitation in two other laboratories (112, 113). Another group of researchers failed to repeat the PPARgamma-mediated degradation of beta-Catenin; however, they found that the PPARgamma ligands troglitazone, rosiglitazone, and GW1929 all repressed the activity of a beta-Catenin transcription complex during co-expression of PPARgamma and RXR (114). Repression of the Wnt signaling pathway, therefore, would appear to require sufficient expression of both PPARgamma and RXR. Importantly, the nuclear localization of beta-Catenin is strongly modulated by both intrinsic and paracrine signals, even in the presence of an APC mutation (139); therefore, PPARgamma’s influence on such signals may partially mediate PPARgamma’s impact on beta-Catenin activity. On the other hand, such signals may completely abrogate PPARgamma’s influence on this pathway. It should be mentioned that in clinical colorectal cancer samples, nuclear-localized beta-Catenin immunohistochemistry staining reportedly shows no significant correlation with overall PPARgamma staining intensity (37).
The process of sorting through the mechanisms of PPARgamma activity on this pathway is further complicated by its effect on E-Cadherin. E-Cadherin is a membrane protein whose expression is inversely correlated with malignancy, while restored expression of this receptor reduces malignant characteristics (140). Importantly, E-Cadherin sequesters beta-Catenin at the cell membrane, thereby preventing its localization to the nucleus (140). A recent paper has found that ciglitazone and linoleic acid treatments in several prostate cancer cell lines lead to an up-regulation of E-Cadherin, along with an even more reliable down-regulation of N-Cadherin (141). The effects of these ligands, moreover, are PPARgamma dependent, since they are reversed by the PPARgamma antagonist GW9662 (141). They also lead to a repression of both beta-Catenin and c-myc expression, indicating that the full pathway is, in fact, influenced by these treatments. The clinical relevance of this mechanism is supported by the fact that the overall PPARgamma immunoreactivity in clinical breast tumor samples is positively correlated with the localization of beta-Catenin to the membrane as well as the cytoplasm (36). One group has attributed this effect to a repression of Snail observed in non-small cell lung carcinoma cell lines (142). Snail otherwise directly represses the transcription of E-Cadherin (142). This repression is believed to have been somehow mediated by an inhibition of ERK through an unknown phosphatase, although the mechanism connecting these events is unknown.
6.2.2. STAT3/NF-kappaB
PPARgamma’s ability to inhibit IL-6/STAT3/NF-kappaB signaling has been reported by numerous laboratories in a wide variety of cell types. The prospect of targeting this pathway is particularly exciting because it is part of an important epigenetic positive feedback loop (IL6→STAT3→NF-kappaB→Lin28→Let-7→IL6) that has been found to be active in half of all cancer cell lines screened (143). Among PPARgamma’s effects on this pathway is an inhibition of STAT3’s promotion of its target genes (109). These treatments, however, do not appear to impact STAT3’s ability to activate NF-kappaB (109). STAT3-mediated processing of NF-kappaB requires the acetylation of STAT3 (144), an event which PPARgamma has not been shown to disrupt. Importantly, PPARgamma ligands appear to influence the binding of proteins to STAT3, rather than STAT3 phosphorylation (109).
Treatments with PPARgamma agonists have also been observed to reduce the ability of NF-kappaB to promote its target genes, an effect which has been established both in vitro in cell lines from multiple tissue sources (47, 121, 145, 146), and in vivo in an experimentally induced colitis model in mice (147). Among these agonists, 15d-PGJ2 is unique in that it has been observed to block NF-kappaB-mediated transcription in the absence of PPARgamma (although repression is enhanced through exogenous expression of PPARgamma), supposedly through direct inhibition of IKK (145). Importantly, this mechanism reduces the nuclear localization of NF-kappaB in the cell (145). In contrast, activation of PPARgamma by all ligands tested appears to block the interaction between NF-kappaB and its target genes (145). One mechanism that may explain this activity is a ligand-induced SUMOylation of PPARgamma that causes it to bind corepressor complexes associated with NF-kappaB target genes, an event which inhibits the transcription of these genes by blocking the degradation of the complexes (123). Alternatively, ligand activation of PPARgamma has been reported to promote its association with PGC-1, thus, reducing the availability of this coactivator for binding with NF-kappaB (121). While it has been reported that PPARgamma ligands can block the induction of IL-6 by stromal cells in a multiple myeloma cell line (supposedly through inhibition of NF-kappaB) (121), such treatments have not been reported to disrupt autocrine loops involving IL-6 in carcinoma cell lines. This effect would lead to a much more pronounced inhibition of their proliferation. PPARgamma may repress only a subset of NF-kappaB target genes that does not include Lin28. Importantly, the inhibition effects of PPARgamma agonists on both STAT3 and NF-kappaB are context-dependent, since repression of STAT3 through troglitazone depends upon the level of SMRT (109), while the trans-repression of NF-kappaB target genes is attenuated by knockdown of NCoR (123). Interference with STAT3/NF-kappaB signaling through these mechanisms, therefore, may be highly variable and unreliable in a clinical setting if not somehow guided by molecular profiling.
Alternatively, PPARgamma agonists can also block Tyk2-mediated phosphorylation of STAT3 through an unknown mechanism (34, 148). This mechanism is believed to mediate the growth inhibition effects that PPARgamma agonists have on the CD133+ subpopulation of glioblastoma cell lines during treatment with specific growth factors in vitro (34). Some other studies have established that Tyk2 expression contributes to metastasis (149, 150). Tyk2 is also reportedly expressed in an appreciable fraction of clinical prostate tumors, although thorough clinicopathological correlations have not been constructed (149). More recently, a head and neck cancer cell line has been reported to dependent upon the combination of pSrc and Tyk2 for STAT3 activation; therefore, it is likely that manipulation of this mechanism by PPARgamma agonists would depend upon combinations with other treatments for its effectiveness (151). Importantly, the inhibition of Tyk2 activity by PPARgamma agonists has not been reported in carcinoma cell lines.
6.2.3. Androgen Receptor
For prostate cancer, the most attractive effect of PPARgamma ligands is their interference with androgen receptor signaling. Upon stimulation by androgen, this membrane-bound nuclear receptor internalizes and acts as a transcription factor for genes whose promoters contain androgen responsive elements (152). Among these is PSA (152), an important marker for detecting prostate cancer (153). Suppression of this activity was first noted when treatment of the androgen-dependent LNCaP cells with troglitazone caused a marked reduction of their PSA level (21). Likewise, an early clinical trial found that patients with androgen-dependent prostate cancer, as well as a subset of patients with androgen-refractory prostate cancer, encountered a drop in their PSA levels during troglitazone treatment (26). In yet another study, treatment of LNCaP cells with the PPARgamma agonists troglitazone, pioglitazone, and 15-d-PGJ2 reduced both the PSA level and the activity of an ARE reporter without altering the androgen receptor protein level (154). It has also been demonstrated through chromatin immunoprecipitation experiments that both troglitazone and ciglitazone block the recruitment of the androgen receptor to androgen responsive elements on the promoters of its target genes (155). The authors who performed the latter study have argued that this activity is independent of PPARgamma because the same effect was achieved by structurally similar analogues that failed to “activate” PPARgamma. In a previously mentioned study examining immunohistochemistry staining of PPARgamma in clinical tumor samples, however, it was argued that the tight inverse correlation observed between PPARgamma immunoreactivity and PSA level suggests that the effects of PPARgamma ligands on PSA are dependent upon binding of these ligands to PPARgamma (39).
6.3. Verified Genomic Targets
6.3.1. P53
Rosiglitazone reportedly promotes p53 expression at the transcriptional level in MCF7 breast cancer cells through recruitment of PPARgamma to an NF-kappaB site on its promoter (156). A well described tumor suppressor gene, p53 is important for the induction of apoptosis, cell cycle arrest, and autophagy during various types of stress (157). This activity is likely mediated by its transrepression activity on Bcl-2, survivin, IGFR, Mcl-1, and PIK3CA and its transactivation of such targets such as p21, PTFG, Gadd45, and 14-3-3σ (157). This mechanism, however, is unlikely to account for many of the effects previously observed, seeing as loss-of-function p53 mutations are ubiquitous in most types of cancer. Certain types of tumors usually lack this mutation, and may, therefore, be more susceptible to this mechanism.
6.3.2. PTEN
Among the most attractive antitumor mechanisms arising from PPARgamma’s genomic activity is a direct activation of the PTEN promoter by the PPARgamma-RXR heterodimer, an event which should exert pleotropic effects through a suppression of Akt (158). As with p53, mutations of PTEN are highly frequent in most types of cancer. For pancreatic cancer, however, it has been reported that activation of PPARgamma in the cell line PANC-1 through treatment with rosiglitazone increases the protein level of PTEN, while the reverse is true for the addition of PPARgamma antagonist GW9662 (159). The growth suppression effects elicited by rosiglitazone in this cell line are especially pronounced, with almost no measureable growth in xenografts after the initiation of treatment (160). Although the immunoreactivity of PTEN has been found to be significantly reduced in clinical samples of pancreatic cancer (161), the cancer genome project has found that loss-of-function mutations of this gene are a rare event, occurring in only 1 out of 66 clinical samples in the COSMIC_pancreas data set of the Cancer Genome Workbench. It is, therefore, conceivable that the repression of PTEN may be overcome through activation of PPARgamma in these tumors. If population mutation rates prove to be a reliable predictor of PTEN induction through PPARgamma activity, then data from the Cancer Genome Workbench Datadump indicate that carcinomas of the oesophagus, head and neck, ovary, and lung may also be potential candidates for this mechanism, with PTEN mutation rates ≤ 2%. The directions for accessing this site are detailed in the Acknowledgements section.
7. PERSPECTIVE
Aside from its role as a specific target for anticancer treatments, PPARgamma is now emerging as a potential biomarker for predicting patient prognosis. PPARgamma staining may also become a useful tool for selecting treatments. Activation of PPARgamma, for example, should lead to the up-regulation of ABCG2, which antagonizes several prominent chemotherapy drugs (67). PPARgamma might also regulate additional multidrug resistance (MDR) proteins. A role for the PPARs in regulating MDR proteins has been reviewed by others (161). Considering the fact that other drugs are synergized by PPARgamma activity (60, 62, 64, 66), if additional multidrug resistance proteins are not influenced by PPARgamma, then PPARgamma staining is most likely an independent predictor of drug resistance when combined with these other markers. No correlation, however, has been constructed at present. PPARgamma may also be useful for predicting resistance to radiotherapy, since it reportedly activates Hsp70 (71). Additionally, as the drive to develop inhibitors for targets downstream of COX-1/COX-2 progresses, an understanding of both the effect of such inhibitors on the expression of the lipoxygenases and the clinical significance of the various prostaglandins as PPARgamma ligands will be important for optimizing such treatments. Biomarkers should also be established to predict which PPARgamma target pathways are responsive to PPARgamma activation within a given tumor, since nearly all of these pathways are context-dependent. Establishing the exact mechanisms of PPARgamma activity in each target pathway, therefore, will likely be necessary.
ACKNOWLEDGEMENT
We gratefully acknowledge efforts of those who are compiling data for the Cancer Genome Project. The data used in this article can be found in the Cancer Genome Workbench at https://cgwb.nci.nih.gov/. We are also grateful to SIU-School of Medicine for their support. We also thank Dr. Gayle E Woodson and Dr. Kevin T Robbins for offering their comments on our manuscript.
Abbreviations
- PPREs
peroxisome proliferator responsive elements
- TZDs
thiazolidinediones
- TEM
transmission electron microscopy
- IHC
immunohistochemistry
- ER
estrogen receptor
- COX
cyclooxygenase
- AR
androgen receptor
REFERENCES
- 1.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. PMid:2129546. [DOI] [PubMed] [Google Scholar]
- 2.Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- 3.Tugwood JD, Issemann I, Anderson RG, Bundell KR, McPheat WL, Green S. The mouse peroxisome proliferator activated receptor recognizes a response element in the 5' flanking sequence of the rat acyl CoA oxidase gene. Embo J. 1992;11:433–439. doi: 10.1002/j.1460-2075.1992.tb05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. PMid:8240342. [DOI] [PubMed] [Google Scholar]
- 5.Chen F, Law SW, O'Malley BW. Identification of two mPPAR related receptors and evidence for the existence of five subfamily members. Biochem Biophys Res Commun. 1993;196:671–677. doi: 10.1006/bbrc.1993.2302. [DOI] [PubMed] [Google Scholar]
- 6.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2:tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 7.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 8.Stanton LA, Li JR, Beier F. PPARgamma2 expression in growth plate chondrocytes is regulated by p38 and GSK-3. J Cell Mol Med. 2010;14:242–256. doi: 10.1111/j.1582-4934.2008.00396.x. PMid:15857827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deeb SS, Fajas L, Nemoto M, Pihlajamaki J, Mykkanen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet. 1998;20:284–287. doi: 10.1038/3099. PMid:16842753. [DOI] [PubMed] [Google Scholar]
- 10.Sabatino L, Casamassimi A, Peluso G, Barone MV, Capaccio D, Migliore C, Bonelli P, Pedicini A, Febbraro A, Ciccodicola A, Colantuoni V. A novel peroxisome proliferator-activated receptor gamma isoform with dominant negative activity generated by alternative splicing. J Biol Chem. 2005;280:26517–26525. doi: 10.1074/jbc.M502716200. [DOI] [PubMed] [Google Scholar]
- 11.Kim HJ, Woo IS, Kang ES, Eun SY, Lee JH, Chang KC, Kim JH, Seo HG. Identification of a truncated alternative splicing variant of human PPARgamma1 that exhibits dominant negative activity. Biochem Biophys Res Commun. 2006;347:698–706. doi: 10.1016/j.bbrc.2006.06.147. PMid:11554739. [DOI] [PubMed] [Google Scholar]
- 12.van der Vaart M, Schaaf MJ. Naturally occurring C-terminal splice variants of nuclear receptors. Nucl Recept Signal. 2009;7:e007. doi: 10.1621/nrs.07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fajas L, Fruchart JC, Auwerx J. PPARgamma3 mRNA, a distinct PPARgamma mRNA subtype transcribed from an independent promoter. FEBS Lett. 1998;438:55–60. doi: 10.1016/s0014-5793(98)01273-3. PMid:20093779. [DOI] [PubMed] [Google Scholar]
- 14.Sundvold H, Lien S. Identification of a novel peroxisome proliferator-activated receptor (PPAR) gamma promoter in man and transactivation by the nuclear receptor RORalpha1. Biochem Biophys Res Commun. 2001;287:383–390. doi: 10.1006/bbrc.2001.5602. PMid:9153284 PMCid:508081. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi S, Inoue I, Nakajima Y, Seo M, Nakano T, Yang F, Kumagai M, Komoda T, Awata T, Ikeda M, Katayama S. A promoter in the novel exon of hPPARgamma directs the circadian expression of PPARgamma. J Atheroscler Thromb. 2010;17:73–83. doi: 10.5551/jat.2410. [DOI] [PubMed] [Google Scholar]
- 16.Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A, Pories WJ, Caro JF, Flier JS. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest. 1997;99:2416–2422. doi: 10.1172/JCI119424. PMid:8922349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. PMid:9665460 PMCid:1852950. [DOI] [PubMed] [Google Scholar]
- 18.Saltiel AR, Olefsky JM. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes. 1996;45:1661–1669. doi: 10.2337/diab.45.12.1661. PMid:20150512. [DOI] [PubMed] [Google Scholar]
- 19.Marx N, Sukhova G, Murphy C, Libby P, Plutzky J. Macrophages in human atheroma contain PPARgamma: differentiation-dependent peroxisomal proliferator-activated receptor gamma(PPARgamma) expression and reduction of MMP-9 activity through PPARgamma activation in mononuclear phagocytes in vitro. Am J Pathol. 1998;153:17–23. doi: 10.1016/s0002-9440(10)65540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krause U, Harris S, Green A, Ylostalo J, Zeitouni S, Lee N, Gregory CA. Pharmaceutical modulation of canonical Wnt signaling in multipotent stromal cells for improved osteoinductive therapy. Proc Natl Acad Sci U S A. 2010;107:4147–4152. doi: 10.1073/pnas.0914360107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubota T, Koshizuka K, Williamson EA, Asou H, Said JW, Holden S, Miyoshi I, Koeffler HP. Ligand for peroxisome proliferator-activated receptor gamma (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res. 1998;58:3344–3352. [PubMed] [Google Scholar]
- 22.Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD, Brun RP, Mueller E, Altiok S, Oppenheim H, Evans RM, Spiegelman BM. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc Natl Acad Sci U S A. 1997;94:237–241. doi: 10.1073/pnas.94.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, Holden SA, Chen LB, Singer S, Fletcher C, Spiegelman BM. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med. 1998;4:1046–1052. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- 24.Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C, Singer S, Spiegelman BM. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell. 1998;1:465–470. doi: 10.1016/s1097-2765(00)80047-7. PMid:10984506. [DOI] [PubMed] [Google Scholar]
- 25.Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, de la Chapelle A, Spiegelman BM, Eng C. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell. 1999;3:799–804. doi: 10.1016/s1097-2765(01)80012-5. PMid:11431375. [DOI] [PubMed] [Google Scholar]
- 26.Mueller E, Smith M, Sarraf P, Kroll T, Aiyer A, Kaufman DS, Oh W, Demetri G, Figg WD, Zhou XP, Eng C, Spiegelman BM, Kantoff PW. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc Natl Acad Sci U S A. 2000;97:10990–10995. doi: 10.1073/pnas.180329197. PMid:11507074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikezoe T, Miller CW, Kawano S, Heaney A, Williamson EA, Hisatake J, Green E, Hofmann W, Taguchi H, Koeffler HP. Mutational analysis of the peroxisome proliferator-activated receptor gamma gene in human malignancies. Cancer Res. 2001;61:5307–5310. [PubMed] [Google Scholar]
- 28.Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001;61:6213–6218. [PubMed] [Google Scholar]
- 29.Chaffer CL, Thomas DM, Thompson EW, Williams ED. PPARgamma-independent induction of growth arrest and apoptosis in prostate and bladder carcinoma. BMC Cancer. 2006;6:53. doi: 10.1186/1471-2407-6-53. PMid:18790559 PMCid:2712818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veliceasa D, Schulze-Hoepfner FT, Volpert OV. PPARgamma and Agonists against Cancer: Rational Design of Complementation Treatments. PPAR Res. 2008;2008:945275. doi: 10.1155/2008/945275. PMid:18790758 PMCid:2597004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei S, Yang J, Lee SL, Kulp SK, Chen CS. PPARgamma-independent antitumor effects of thiazolidinediones. Cancer Lett. 2009;276:119–124. doi: 10.1016/j.canlet.2008.08.008. PMid:18464916 PMCid:2366048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi K, Cekanova M, McEntee MF, Yoon JH, Fischer SM, Renes IB, Van Seuningen I, Baek SJ. Peroxisome proliferator-activated receptor ligand MCC-555 suppresses intestinal polyps in ApcMin/+ mice via extracellular signal-regulated kinase and peroxisome proliferator-activated receptor-dependent pathways. Mol Cancer Ther. 2008;7:2779–2787. doi: 10.1158/1535-7163.MCT-08-0173. PMid:19018263 PMCid:2607234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giaginis C, Tsantili-Kakoulidou A, Theocharis S. Peroxisome Proliferator-Activated Receptor-gamma Ligands: Potential Pharmacological Agents for Targeting the Angiogenesis Signaling Cascade in Cancer. PPAR Res. 2008;2008:431763. doi: 10.1155/2008/431763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chearwae W, Bright JJ. PPARgamma agonists inhibit growth and expansion of CD133+ brain tumour stem cells. Br J Cancer. 2008;99:2044–2053. doi: 10.1038/sj.bjc.6604786. PMid:19495794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki H, Tanahashi M, Yukiue H, Moiriyama S, Kobayashi Y, Nakashima Y, Kaji M, Kiriyama M, Fukai I, Yamakawa Y, Fujii Y. Decreased perioxisome proliferator-activated receptor gamma gene expression was correlated with poor prognosis in patients with lung cancer. Lung Cancer. 2002;36:71–76. doi: 10.1016/s0169-5002(01)00449-4. PMid:19186181 PMCid:2663601. [DOI] [PubMed] [Google Scholar]
- 36.Jiang Y, Zou L, Zhang C, He S, Cheng C, Xu J, Lu W, Zhang Y, Zhang H, Wang D, Shen A. PPARgamma and Wnt/beta-Catenin pathway in human breast cancer: expression pattern, molecular interaction and clinical/prognostic correlations. J Cancer Res Clin Oncol. 2009;135:1551–1559. doi: 10.1007/s00432-009-0602-8. PMid:19432669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogino S, Shima K, Baba Y, Nosho K, Irahara N, Kure S, Chen L, Toyoda S, Kirkner GJ, Wang YL, Giovannucci EL, Fuchs CS. Colorectal cancer expression of peroxisome proliferator-activated receptor gamma (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology. 2009;136:1242–1250. doi: 10.1053/j.gastro.2008.12.048. PMid:19396032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Theocharis S, Klijanienko J, Giaginis C, Rodriguez J, Jouffroy T, Girod A, Point D, Tsourouflis G, Satre-Garau X. Peroxisome proliferator-activated receptor-gamma in mobile tongue squamous cell carcinoma: associations with clinicopathological parameters and patients survival. J Cancer Res Clin Oncol. 2010 doi: 10.1007/s00432-010-0882-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura Y, Suzuki T, Sugawara A, Arai Y, Sasano H. Peroxisome proliferator-activated receptor gamma in human prostate carcinoma. Pathol Int. 2009;59:288–293. doi: 10.1111/j.1440-1827.2009.02367.x. PMid:17473186. [DOI] [PubMed] [Google Scholar]
- 40.Giaginis C, Katsamangou E, Tsourouflis G, Zizi-Serbetzoglou D, Kouraklis G, Theocharis S. Peroxisome proliferator-activated receptor-gamma and retinoid X receptor-alpha expression in pancreatic ductal adenocarcinoma: association with clinicopathological parameters, tumor proliferative capacity, and patients' survival. Med Sci Monit. 2009;15:BR148–BR156. [PubMed] [Google Scholar]
- 41.Kim HJ, Hwang JY, Choi WS, Lee JH, Chang KC, Nishinaka T, Yabe-Nishimura C, Seo HG. Expression of a peroxisome proliferator-activated receptor gamma 1 splice variant that was identified in human lung cancers suppresses cell death induced by cisplatin and oxidative stress. Clin Cancer Res. 2007;13:2577–2583. doi: 10.1158/1078-0432.CCR-06-2062. PMid:9734400. [DOI] [PubMed] [Google Scholar]
- 42.Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, Briggs M, Heyman R, Auwerx J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–1057. doi: 10.1038/2036. PMid:14601316. [DOI] [PubMed] [Google Scholar]
- 43.Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, Baird SM, Thomazy VA, Evans RM. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med. 1998;4:1058–1061. doi: 10.1038/2042. PMid:16858678. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Lv Y, Dong X, Jin Z. [Pioglitazone, a peroxisome proliferators-activated receptor gamma ligand, inhibits dimethylhydrazine (DMH) induced aberrant crypt foci in rats] Beijing Da Xue Xue Bao. 2003;35:537–539. [PubMed] [Google Scholar]
- 45.McAlpine CA, Barak Y, Matise I, Cormier RT. Intestinal-specific PPARgamma deficiency enhances tumorigenesis in ApcMin/+ mice. Int J Cancer. 2006;119:2339–2346. doi: 10.1002/ijc.22115. PMid:10449430 PMCid:408529. [DOI] [PubMed] [Google Scholar]
- 46.Girnun GD, Smith WM, Drori S, Sarraf P, Mueller E, Eng C, Nambiar P, Rosenberg DW, Bronson RT, Edelmann W, Kucherlapati R, Gonzalez FJ, Spiegelman BM. APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc Natl Acad Sci U S A. 2002;99:13771–13776. doi: 10.1073/pnas.162480299. PMid:12479648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest. 1999;104:383–389. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saubermann LJ, Nakajima A, Wada K, Zhao S, Terauchi Y, Kadowaki T, Aburatani H, Matsuhashi N, Nagai R, Blumberg RS. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflamm Bowel Dis. 2002;8:330–339. doi: 10.1097/00054725-200209000-00004. PMid:20089779. [DOI] [PubMed] [Google Scholar]
- 49.Katayama K, Wada K, Nakajima A, Mizuguchi H, Hayakawa T, Nakagawa S, Kadowaki T, Nagai R, Kamisaki Y, Blumberg RS, Mayumi T. A novel PPAR gamma gene therapy to control inflammation associated with inflammatory bowel disease in a murine model. Gastroenterology. 2003;124:1315–1324. doi: 10.1016/s0016-5085(03)00262-2. PMid:19380302. [DOI] [PubMed] [Google Scholar]
- 50.Evans NP, Misyak SA, Schmelz EM, Guri AJ, Hontecillas R, Bassaganya-Riera J. Conjugated linoleic acid ameliorates inflammation-induced colorectal cancer in mice through activation of PPARgamma. J Nutr. 2010;140:515–521. doi: 10.3945/jn.109.115642. PMid:15930296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kocdor H, Kocdor MA, Canda T, Gurel D, Cehreli R, Yilmaz O, Alakavuklar M, Guner G. Chemopreventive efficacies of rosiglitazone, fenretinide and their combination against rat mammary carcinogenesis. Clin Transl Oncol. 2009;11:243–249. doi: 10.1007/s12094-009-0347-5. PMid:19138979 PMCid:2680197. [DOI] [PubMed] [Google Scholar]
- 52.Lu J, Imamura K, Nomura S, Mafune K, Nakajima A, Kadowaki T, Kubota N, Terauchi Y, Ishii G, Ochiai A, Esumi H, Kaminishi M. Chemopreventive effect of peroxisome proliferator-activated receptor gamma on gastric carcinogenesis in mice. Cancer Res. 2005;65:4769–4774. doi: 10.1158/0008-5472.CAN-04-2293. PMid:17442990. [DOI] [PubMed] [Google Scholar]
- 53.Nemenoff R, Meyer AM, Hudish TM, Mozer AB, Snee A, Narumiya S, Stearman RS, Winn RA, Weiser-Evans M, Geraci MW, Keith RL. Prostacyclin prevents murine lung cancer independent of the membrane receptor by activation of peroxisomal proliferator--activated receptor gamma. Cancer Prev Res (Phila Pa) 2008;1:349–356. doi: 10.1158/1940-6207.CAPR-08-0145. PMid:12416897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada MV, Kim L, Kim PJ, Owens RJ, Lang NP. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J Clin Oncol. 2007;25:1476–1481. doi: 10.1200/JCO.2006.07.2777. PMid:12846423. [DOI] [PubMed] [Google Scholar]
- 55.Kulke MH, Demetri GD, Sharpless NE, Ryan DP, Shivdasani R, Clark JS, Spiegelman BM, Kim H, Mayer RJ, Fuchs CS. A phase II study of troglitazone, an activator of the PPARgamma receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer J. 2002;8:395–399. doi: 10.1097/00130404-200209000-00010. PMid:15468186. [DOI] [PubMed] [Google Scholar]
- 56.Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat. 2003;79:391–397. doi: 10.1023/a:1024038127156. PMid:1312391. [DOI] [PubMed] [Google Scholar]
- 57.Smith MR, Manola J, Kaufman DS, George D, Oh WK, Mueller E, Slovin S, Spiegelman BM, Small E, Kantoff PW. Rosiglitazone versus placebo for men with prostate carcinoma and a rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy. Cancer. 2004;101:1569–1574. doi: 10.1002/cncr.20493. PMid:8001151. [DOI] [PubMed] [Google Scholar]
- 58.Shimizu M, Moriwaki H. Synergistic Effects of PPARgamma Ligands and Retinoids in Cancer Treatment. PPAR Res. 2008;2008:181047. doi: 10.1155/2008/181047. PMid:18528526 PMCid:2408709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Read WL, Baggstrom MQ, Fracasso PM, Govindan R. A phase I study of bexarotene and rosiglitazone in patients with refractory cancers. Chemotherapy. 2008;54:236–241. doi: 10.1159/000140468. PMid:18560232. [DOI] [PubMed] [Google Scholar]
- 60.Girnun GD, Naseri E, Vafai SB, Qu L, Szwaya JD, Bronson R, Alberta JA, Spiegelman BM. Synergy between PPARgamma ligands and platinum-based drugs in cancer. Cancer Cell. 2007;11:395–406. doi: 10.1016/j.ccr.2007.02.025. PMid:17482130 PMCid:2564847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Girnun GD, Chen L, Silvaggi J, Drapkin R, Chirieac LR, Padera RF, Upadhyay R, Vafai SB, Weissleder R, Mahmood U, Naseri E, Buckley S, Li D, Force J, McNamara K, Demetri G, Spiegelman BM, Wong KK. Regression of drug-resistant lung cancer by the combination of rosiglitazone and carboplatin. Clin Cancer Res. 2008;14:6478–6486. doi: 10.1158/1078-0432.CCR-08-1128. PMid:18927287 PMCid:2696122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tikoo K, Kumar P, Gupta J. Rosiglitazone synergizes anticancer activity of cisplatin and reduces its nephrotoxicity in 7, 12-dimethyl benz{a}anthracene (DMBA) induced breast cancer rats. BMC Cancer. 2009;9:107. doi: 10.1186/1471-2407-9-107. PMid:19356226 PMCid:2676298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamaguchi N, Hamada H, Miyoshi S, Irifune K, Ito R, Miyazaki T, Higaki J. In vitro and in vivo therapeutic efficacy of the PPAR-gamma agonist troglitazone in combination with cisplatin against malignant pleural mesothelioma cell growth. Cancer Sci. 2010 doi: 10.1111/j.1349-7006.2010.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang YQ, Tang XQ, Sun L, Dong L, Qin Y, Liu HQ, Xia H, Cao JG. Rosiglitazone enhances fluorouracil-induced apoptosis of HT-29 cells by activating peroxisome proliferator-activated receptor gamma. World J Gastroenterol. 2007;13:1534–1540. doi: 10.3748/wjg.v13.i10.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cao LQ, Wang XL, Wang Q, Xue P, Jiao XY, Peng HP, Lu HW, Zheng Q, Chen XL, Huang XH, Fu XH, Chen JS. Rosiglitazone sensitizes hepatocellular carcinoma cell lines to 5-fluorouracil antitumor activity through activation of the PPARgamma signaling pathway. Acta Pharmacol Sin. 2009;30:1316–1322. doi: 10.1038/aps.2009.119. PMid:19684609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fulzele SV, Chatterjee A, Shaik MS, Jackson T, Ichite N, Singh M. 15-Deoxy-Delta12,14-prostaglandin J2 enhances docetaxel anti-tumor activity against A549 and H460 non-small-cell lung cancer cell lines and xenograft tumors. Anticancer Drugs. 2007;18:65–78. doi: 10.1097/CAD.0b013e3280101006. [DOI] [PubMed] [Google Scholar]
- 67.Szatmari I, Vamosi G, Brazda P, Balint BL, Benko S, Szeles L, Jeney V, Ozvegy-Laczka C, Szanto A, Barta E, Balla J, Sarkadi B, Nagy L. Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J Biol Chem. 2006;281:23812–23823. doi: 10.1074/jbc.M604890200. PMid:16785230. [DOI] [PubMed] [Google Scholar]
- 68.Kawabata S, Oka M, Shiozawa K, Tsukamoto K, Nakatomi K, Soda H, Fukuda M, Ikegami Y, Sugahara K, Yamada Y, Kamihira S, Doyle LA, Ross DD, Kohno S. Breast cancer resistance protein directly confers SN-38 resistance of lung cancer cells. Biochem Biophys Res Commun. 2001;280:1216–1223. doi: 10.1006/bbrc.2001.4267. PMid:11162657. [DOI] [PubMed] [Google Scholar]
- 69.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–15670. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tencer L, Burgermeister E, Ebert MP, Liscovitch M. Rosiglitazone induces caveolin-1 by PPARgamma-dependent and PPRE-independent mechanisms: the role of EGF receptor signaling and its effect on cancer cell drug resistance. Anticancer Res. 2008;28:895–906. [PubMed] [Google Scholar]
- 71.Pierzchalski P, Krawiec A, Gawelko J, Pawlik WW, Konturek SJ, Gonciarz M. Molecular mechanism of protection against chemically and gamma-radiation induced apoptosis in human colon cancer cells. J Physiol Pharmacol. 2008;59(Suppl 2):191–202. [PubMed] [Google Scholar]
- 72.Subbarayan V, Xu XC, Kim J, Yang P, Hoque A, Sabichi AL, Llansa N, Mendoza G, Logothetis CJ, Newman RA, Lippman SM, Menter DG. Inverse relationship between 15-lipoxygenase-2 and PPAR-gamma gene expression in normal epithelia compared with tumor epithelia. Neoplasia. 2005;7:280–293. doi: 10.1593/neo.04457. PMid:15799828 PMCid:1501140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shureiqi I, Chen D, Lotan R, Yang P, Newman RA, Fischer SM, Lippman SM. 15-Lipoxygenase-1 mediates nonsteroidal anti-inflammatory drug-induced apoptosis independently of cyclooxygenase-2 in colon cancer cells. Cancer Res. 2000;60:6846–6850. [PubMed] [Google Scholar]
- 74.Shureiqi I, Chen D, Lee JJ, Yang P, Newman RA, Brenner DE, Lotan R, Fischer SM, Lippman SM. 15-LOX-1: a novel molecular target of nonsteroidal anti-inflammatory drug-induced apoptosis in colorectal cancer cells. J Natl Cancer Inst. 2000;92:1136–1142. doi: 10.1093/jnci/92.14.1136. PMid:10904086. [DOI] [PubMed] [Google Scholar]
- 75.Shureiqi I, Xu X, Chen D, Lotan R, Morris JS, Fischer SM, Lippman SM. Nonsteroidal anti-inflammatory drugs induce apoptosis in esophageal cancer cells by restoring 15-lipoxygenase-1 expression. Cancer Res. 2001;61:4879–4884. [PubMed] [Google Scholar]
- 76.Wu J, Xia HH, Tu SP, Fan DM, Lin MC, Kung HF, Lam SK, Wong BC. 15-Lipoxygenase-1 mediates cyclooxygenase-2 inhibitor-induced apoptosis in gastric cancer. Carcinogenesis. 2003;24:243–247. doi: 10.1093/carcin/24.2.243. PMid:12584173. [DOI] [PubMed] [Google Scholar]
- 77.Earles SM, Bronstein JC, Winner DL, Bull AW. Metabolism of oxidized linoleic acid: characterization of 13-hydroxyoctadecadienoic acid dehydrogenase activity from rat colonic tissue. Biochim Biophys Acta. 1991;1081:174–180. doi: 10.1016/0005-2760(91)90023-b. [DOI] [PubMed] [Google Scholar]
- 78.Altmann R, Hausmann M, Spottl T, Gruber M, Bull AW, Menzel K, Vogl D, Herfarth H, Scholmerich J, Falk W, Rogler G. 13-Oxo-ODE is an endogenous ligand for PPARgamma in human colonic epithelial cells. Biochem Pharmacol. 2007;74:612–622. doi: 10.1016/j.bcp.2007.05.027. PMid:17604003. [DOI] [PubMed] [Google Scholar]
- 79.Silverman AL, Bronstein JC, Krymgold S, Kahlon D, Bull AW. Decreased levels of 13-hydroxyoctadecadienoic acid (13-HODE) dehydrogenase in neoplastic tissue of human colon biopsies. Cancer Epidemiol Biomarkers Prev. 1996;5:53–56. [PubMed] [Google Scholar]
- 80.Hsi LC, Wilson L, Nixon J, Eling TE. 15-lipoxygenase-1 metabolites down-regulate peroxisome proliferator-activated receptor gamma via the MAPK signaling pathway. J Biol Chem. 2001;276:34545–34552. doi: 10.1074/jbc.M100280200. PMid:11447213. [DOI] [PubMed] [Google Scholar]
- 81.Kelavkar UP, Nixon JB, Cohen C, Dillehay D, Eling TE, Badr KF. Overexpression of 15-lipoxygenase-1 in PC-3 human prostate cancer cells increases tumorigenesis. Carcinogenesis. 2001;22:1765–1773. doi: 10.1093/carcin/22.11.1765. PMid:11698337. [DOI] [PubMed] [Google Scholar]
- 82.Nixon JB, Kamitani H, Baek SJ, Eling TE. Evaluation of eicosanoids and NSAIDs as PPARgamma ligands in colorectal carcinoma cells. Prostaglandins Leukot Essent Fatty Acids. 2003;68:323–330. doi: 10.1016/s0952-3278(03)00023-1. [DOI] [PubMed] [Google Scholar]
- 83.Shureiqi I, Chen D, Day RS, Zuo X, Hochman FL, Ross WA, Cole RA, Moy O, Morris JS, Xiao L, Newman RA, Yang P, Lippman SM. Profiling lipoxygenase metabolism in specific steps of colorectal tumorigenesis. Cancer Prev Res (Phila Pa) 2010;3:829–838. doi: 10.1158/1940-6207.CAPR-09-0110. PMid:20570882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yuan H, Li MY, Ma LT, Hsin MK, Mok TS, Underwood MJ, Chen GG. 15-Lipoxygenases and its metabolites 15(S)-HETE and 13(S)-HODE in the development of non-small cell lung cancer. Thorax. 2010;65:321–326. doi: 10.1136/thx.2009.122747. PMid:20388757. [DOI] [PubMed] [Google Scholar]
- 85.O'Flaherty JT, Rogers LC, Paumi CM, Hantgan RR, Thomas LR, Clay CE, High K, Chen YQ, Willingham MC, Smitherman PK, Kute TE, Rao A, Cramer SD, Morrow CS. 5-Oxo-ETE analogs and the proliferation of cancer cells. Biochim Biophys Acta. 2005;1736:228–236. doi: 10.1016/j.bbalip.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 86.Naruhn S, Meissner W, Adhikary T, Kaddatz K, Klein T, Watzer B, Muller-Brusselbach S, Muller R. 15-hydroxyeicosatetraenoic acid is a preferential peroxisome proliferator-activated receptor beta/delta agonist. Mol Pharmacol. 2010;77:171–184. doi: 10.1124/mol.109.060541. PMid:19903832. [DOI] [PubMed] [Google Scholar]
- 87.Shureiqi I, Jiang W, Zuo X, Wu Y, Stimmel JB, Leesnitzer LM, Morris JS, Fan HZ, Fischer SM, Lippman SM. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-delta to induce apoptosis in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100:9968–9973. doi: 10.1073/pnas.1631086100. PMid:12909723 PMCid:187904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chong HC, Tan MJ, Philippe V, Tan SH, Tan CK, Ku CW, Goh YY, Wahli W, Michalik L, Tan NS. Regulation of epithelial-mesenchymal IL-1 signaling by PPARbeta/delta is essential for skin homeostasis and wound healing. J Cell Biol. 2009;184:817–831. doi: 10.1083/jcb.200809028. PMid:19307598 PMCid:2699156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 90.Chou WL, Chuang LM, Chou CC, Wang AH, Lawson JA, FitzGerald GA, Chang ZF. Identification of a novel prostaglandin reductase reveals the involvement of prostaglandin E2 catabolism in regulation of peroxisome proliferator-activated receptor gamma activation. J Biol Chem. 2007;282:18162–18172. doi: 10.1074/jbc.M702289200. PMid:15024020. [DOI] [PubMed] [Google Scholar]
- 91.Su X, Mancuso DJ, Bickel PE, Jenkins CM, Gross RW. Small interfering RNA knockdown of calcium-independent phospholipases A2 beta or gamma inhibits the hormone-induced differentiation of 3T3-L1 preadipocytes. J Biol Chem. 2004;279:21740–21748. doi: 10.1074/jbc.M314166200. PMid:14660788 PMCid:307633. [DOI] [PubMed] [Google Scholar]
- 92.He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky JM, Evans RM. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100:15712–15717. doi: 10.1073/pnas.2536828100. PMid:19136964 PMCid:2863116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jaworski K, Ahmadian M, Duncan RE, Sarkadi-Nagy E, Varady KA, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, Kim KH, de Val S, Kang C, Sul HS. AdPLA ablation increases lipolysis and prevents obesity induced by high-fat feeding or leptin deficiency. Nat Med. 2009;15:159–168. doi: 10.1038/nm.1904. PMid:9099735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Camp HS, Tafuri SR. Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J Biol Chem. 1997;272:10811–10816. doi: 10.1074/jbc.272.16.10811. PMid:19732722. [DOI] [PubMed] [Google Scholar]
- 95.Yamazaki K, Shimizu M, Okuno M, Matsushima-Nishiwaki R, Kanemura N, Araki H, Tsurumi H, Kojima S, Weinstein IB, Moriwaki H. Synergistic effects of RXR alpha and PPAR gamma ligands to inhibit growth in human colon cancer cells--phosphorylated RXR alpha is a critical target for colon cancer management. Gut. 2007;56:1557–1563. doi: 10.1136/gut.2007.129858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shankaranarayanan P, Rossin A, Khanwalkar H, Alvarez S, Alvarez R, Jacobson A, Nebbioso A, de Lera AR, Altucci L, Gronemeyer H. Growth factor-antagonized rexinoid apoptosis involves permissive PPARgamma/RXR heterodimers to activate the intrinsic death pathway by NO. Cancer Cell. 2009;16:220–231. doi: 10.1016/j.ccr.2009.07.029. PMid:15860251. [DOI] [PubMed] [Google Scholar]
- 97.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771:952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tan NS, Michalik L, Desvergne B, Wahli W. Multiple expression control mechanisms of peroxisome proliferator-activated receptors and their target genes. J Steroid Biochem Mol Biol. 2005;93:99–105. doi: 10.1016/j.jsbmb.2004.12.025. PMid:15367687 PMCid:516745. [DOI] [PubMed] [Google Scholar]
- 99.Treuter E, Albrektsen T, Johansson L, Leers J, Gustafsson JA. A regulatory role for RIP140 in nuclear receptor activation. Mol Endocrinol. 1998;12:864–881. doi: 10.1210/mend.12.6.0123. PMid:19855844 PMCid:2763222. [DOI] [PubMed] [Google Scholar]
- 100.Akaike M, Che W, Marmarosh NL, Ohta S, Osawa M, Ding B, Berk BC, Yan C, Abe J. The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells. Mol Cell Biol. 2004;24:8691–8704. doi: 10.1128/MCB.24.19.8691-8704.2004. PMid:16144913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Clape C, Fritz V, Henriquet C, Apparailly F, Fernandez PL, Iborra F, Avances C, Villalba M, Culine S, Fajas L. miR-143 interferes with ERK5 signaling, and abrogates prostate cancer progression in mice. PloS one. 2009;4:e7542. doi: 10.1371/journal.pone.0007542. PMid:16260719 PMCid:1283481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bonofiglio D, Gabriele S, Aquila S, Catalano S, Gentile M, Middea E, Giordano F, Ando S. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor gamma signaling in breast cancer cells. Clin Cancer Res. 2005;11:6139–6147. doi: 10.1158/1078-0432.CCR-04-2453. PMid:19148122 PMCid:2750041. [DOI] [PubMed] [Google Scholar]
- 103.Araki O, Ying H, Furuya F, Zhu X, Cheng SY. Thyroid hormone receptor beta mutants: Dominant negative regulators of peroxisome proliferator-activated receptor gamma action. Proc Natl Acad Sci U S A. 2005;102:16251–16256. doi: 10.1073/pnas.0508556102. PMid:10748014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kilroy GE, Zhang X, Floyd ZE. PPAR-gamma AF-2 domain functions as a component of a ubiquitin-dependent degradation signal. Obesity (Silver Spring) 2009;17:665–673. doi: 10.1038/oby.2008.616. PMid:17178896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, Spiegelman BM. Degradation of the peroxisome proliferator-activated receptor gamma is linked to ligand-dependent activation. J Biol Chem. 2000;275:18527–18533. doi: 10.1074/jbc.M001297200. [DOI] [PubMed] [Google Scholar]
- 106.Barz T, Hoffmann A, Panhuysen M, Spengler D. Peroxisome proliferator-activated receptor gamma is a Zac target gene mediating Zac antiproliferation. Cancer Res. 2006;66:11975–11982. doi: 10.1158/0008-5472.CAN-06-1529. PMid:12457461 PMCid:1223218. [DOI] [PubMed] [Google Scholar]
- 107.Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ, Spiegelman BM. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev. 2002;16:22–26. doi: 10.1101/gad.948702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fu M, Zhang J, Lin Y, Zhu X, Zhao L, Ahmad M, Ehrengruber MU, Chen YE. Early stimulation and late inhibition of peroxisome proliferator-activated receptor gamma (PPAR gamma) gene expression by transforming growth factor beta in human aortic smooth muscle cells: role of early growth-response factor-1 (Egr-1), activator protein 1 (AP1) and Smads. Biochem J. 2003;370:1019–1025. doi: 10.1042/BJ20021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang LH, Yang XY, Zhang X, Huang J, Hou J, Li J, Xiong H, Mihalic K, Zhu H, Xiao W, Farrar WL. Transcriptional inactivation of STAT3 by PPARgamma suppresses IL-6-responsive multiple myeloma cells. Immunity. 2004;20:205–218. doi: 10.1016/s1074-7613(04)00030-5. PMid:12771132. [DOI] [PubMed] [Google Scholar]
- 110.Guan HP, Ishizuka T, Chui PC, Lehrke M, Lazar MA. Corepressors selectively control the transcriptional activity of PPARgamma in adipocytes. Genes Dev. 2005;19:453–461. doi: 10.1101/gad.1263305. PMid:15665104 PMCid:547827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xiao JH, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N, Cordrey A, Zhao Y, Chandraratna RA. Adenomatous polyposis coli (APC)-independent regulation of beta-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem. 2003;278:29954–29962. doi: 10.1074/jbc.M304761200. PMid:16352713 PMCid:1317972. [DOI] [PubMed] [Google Scholar]
- 112.Jansson EA, Are A, Greicius G, Kuo IC, Kelly D, Arulampalam V, Pettersson S. The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc Natl Acad Sci U S A. 2005;102:1460–1465. doi: 10.1073/pnas.0405928102. PMid:8521498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu D, Cottam HB, Corr M, Carson DA. Repression of beta-catenin function in malignant cells by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A. 2005;102:18567–18571. doi: 10.1073/pnas.0509316102. PMid:17604322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lu D, Carson DA. Repression of beta-catenin signaling by PPAR gamma ligands. Eur J Pharmacol. 2010;636:198–202. doi: 10.1016/j.ejphar.2010.03.010. PMid:20303941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yu HN, Lee YR, Noh EM, Lee KS, Kim JS, Song EK, Han MK, Lee YC, Kwon KB, Lee SJ, Youn HJ, Jung SH. Induction of G1 phase arrest and apoptosis in MDA-MB-231 breast cancer cells by troglitazone, a synthetic peroxisome proliferator-activated receptor gamma (PPARgamma) ligand. Cell Biol Int. 2008;32:906–912. doi: 10.1016/j.cellbi.2008.04.011. PMid:18474441. [DOI] [PubMed] [Google Scholar]
- 116.Crowe DL, Chandraratna RA. A retinoid X receptor (RXR)-selective retinoid reveals that RXR-alpha is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast Cancer Res. 2004;6:R546–R555. doi: 10.1186/bcr913. PMid:15318936 PMCid:549174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 118.Puigserver P, Adelmant G, Wu Z, Fan M, Xu J, O'Malley B, Spiegelman BM. Activation of PPARgamma coactivator-1 through transcription factor docking. Science. 1999;286:1368–1371. doi: 10.1126/science.286.5443.1368. PMid:10347167. [DOI] [PubMed] [Google Scholar]
- 119.Heinlein CA, Ting HJ, Yeh S, Chang C. Identification of ARA70 as a ligand-enhanced coactivator for the peroxisome proliferator-activated receptor gamma. J Biol Chem. 1999;274:16147–16152. doi: 10.1074/jbc.274.23.16147. PMid:11696534. [DOI] [PubMed] [Google Scholar]
- 120.Nishizawa H, Yamagata K, Shimomura I, Takahashi M, Kuriyama H, Kishida K, Hotta K, Nagaretani H, Maeda N, Matsuda M, Kihara S, Nakamura T, Nishigori H, Tomura H, Moore DD, Takeda J, Funahashi T, Matsuzawa Y. Small heterodimer partner, an orphan nuclear receptor, augments peroxisome proliferator-activated receptor gamma transactivation. J Biol Chem. 2002;277:1586–1592. doi: 10.1074/jbc.M104301200. PMid:17785586 PMCid:2234797. [DOI] [PubMed] [Google Scholar]
- 121.Wang LH, Yang XY, Zhang X, Farrar WL. Inhibition of adhesive interaction between multiple myeloma and bone marrow stromal cells by PPARgamma cross talk with NF-kappaB and C/EBP. Blood. 2007;110:4373–4384. doi: 10.1182/blood-2006-07-038026. PMid:16127449 PMCid:1464798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Majdalawieh A, Ro HS. PPARgamma1 and LXRalpha face a new regulator of macrophage cholesterol homeostasis and inflammatory responsiveness, AEBP1. Nucl Recept Signal. 2010;8:e004. doi: 10.1621/nrs.08004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. PMid:11397807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gupta RA, Brockman JA, Sarraf P, Willson TM, DuBois RN. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J Biol Chem. 2001;276:29681–29687. doi: 10.1074/jbc.M103779200. PMid:18360708 PMCid:2504864. [DOI] [PubMed] [Google Scholar]
- 125.Cekanova M, Yuan JS, Li X, Kim K, Baek SJ. Gene alterations by peroxisome proliferator-activated receptor gamma agonists in human colorectal cancer cells. Int J Oncol. 2008;32:809–819. [PMC free article] [PubMed] [Google Scholar]
- 126.Dionne S, Levy E, Levesque D, Seidman EG. PPARgamma ligand 15-deoxy-delta 12,14-prostaglandin J2 sensitizes human colon carcinoma cells to TWEAK-induced apoptosis. Anticancer Res. 2010;30:157–166. [PubMed] [Google Scholar]
- 127.Lee CJ, Han JS, Seo CY, Park TH, Kwon HC, Jeong JS, Kim IH, Yun J, Bae YS, Kwak JY, Park JI. Pioglitazone, a synthetic ligand for PPARgamma, induces apoptosis in RB-deficient human colorectal cancer cells. Apoptosis. 2006;11:401–411. doi: 10.1007/s10495-006-4003-z. PMid:15569667. [DOI] [PubMed] [Google Scholar]
- 28.Lu M, Kwan T, Yu C, Chen F, Freedman B, Schafer JM, Lee EJ, Jameson JL, Jordan VC, Cryns VL. Peroxisome proliferator-activated receptor gamma agonists promote TRAIL-induced apoptosis by reducing survivin levels via cyclin D3 repression and cell cycle arrest. J Biol Chem. 2005;280:6742–6751. doi: 10.1074/jbc.M411519200. PMid:18288403. [DOI] [PubMed] [Google Scholar]
- 129.Crabb SJ, Hague A, Johnson PW, Packham G. BAG-1 inhibits PPARgamma-induced cell death, but not PPARgamma-induced transcription, cell cycle arrest or differentiation in breast cancer cells. Oncol Rep. 2008;19:689–696. [PubMed] [Google Scholar]
- 130.Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26:463–476. doi: 10.1006/cbir.2002.0901. [DOI] [PubMed] [Google Scholar]
- 131.Daniels DL, Weis WI. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat Struct Mol Biol. 2005;12:364–371. doi: 10.1038/nsmb912. PMid:11839557 PMCid:1850660. [DOI] [PubMed] [Google Scholar]
- 132.Wong NA, Pignatelli M. Beta-catenin--a linchpin in colorectal carcinogenesis? Am J Pathol. 2002;160:389–401. doi: 10.1016/s0002-9440(10)64856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Reinacher-Schick A, Gumbiner BM. Apical membrane localization of the adenomatous polyposis coli tumor suppressor protein and subcellular distribution of the beta-catenin destruction complex in polarized epithelial cells. J Cell Biol. 2001;152:491–502. doi: 10.1083/jcb.152.3.491. PMid:17569865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science. 2007;316:1619–1622. doi: 10.1126/science.1137065. PMid:19633433. [DOI] [PubMed] [Google Scholar]
- 135.Ying Y, Tao Q. Epigenetic disruption of the WNT/beta-catenin signaling pathway in human cancers. Epigenetics. 2009;4:307–312. doi: 10.4161/epi.4.5.9371. PMid:10733430. [DOI] [PubMed] [Google Scholar]
- 136.Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus. Science. 2000;287:1606–1609. doi: 10.1126/science.287.5458.1606. PMid:9820866. [DOI] [PubMed] [Google Scholar]
- 137.Brabletz T, Jung A, Hermann K, Gunther K, Hohenberger W, Kirchner T. Nuclear overexpression of the oncoprotein beta-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract. 1998;194:701–704. doi: 10.1016/s0344-0338(98)80129-5. [DOI] [PubMed] [Google Scholar]
- 138.Moldes M, Zuo Y, Morrison RF, Silva D, Park BH, Liu J, Farmer SR. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem J. 2003;376:607–613. doi: 10.1042/BJ20030426. PMid:17306971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–158. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 140.Christofori G, Semb H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem Sci. 1999;24:73–76. doi: 10.1016/s0968-0004(98)01343-7. PMid:17466258. [DOI] [PubMed] [Google Scholar]
- 141.Laidler P, Dulinska J, Mrozicki S. Does the inhibition of c-myc expression mediate the anti-tumor activity of PPAR's ligands in prostate cancer cell lines? Arch Biochem Biophys. 2007;462:1–12. doi: 10.1016/j.abb.2007.03.013. PMid:20234816 PMCid:2838440. [DOI] [PubMed] [Google Scholar]
- 142.Choudhary R, Li H, Winn RA, Sorenson AL, Weiser-Evans MC, Nemenoff RA. Peroxisome proliferator-activated receptor-gamma inhibits transformed growth of non-small cell lung cancer cells through selective suppression of Snail. Neoplasia. 2010;12:224–234. doi: 10.1593/neo.91638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nadiminty N, Lou W, Lee SO, Lin X, Trump DL, Gao AC. Stat3 activation of NF-{kappa}B p100 processing involves CBP/p300-mediated acetylation. Proc Natl Acad Sci U S A. 2006;103:7264–7269. doi: 10.1073/pnas.0509808103. PMid:19409914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci U S A. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. PMid:12688511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jung WK, Park IS, Park SJ, Yea SS, Choi YH, Oh S, Park SG, Choi IW. The 15-deoxy-Delta12,14-prostaglandin J2 inhibits LPS-stimulated AKT and NF-kappaB activation and suppresses interleukin-6 in osteoblast-like cells MC3T3E-1. Life Sci. 2009;85:46–53. doi: 10.1016/j.lfs.2009.04.010. PMid:16737695. [DOI] [PubMed] [Google Scholar]
- 147.Takagi T, Naito Y, Tomatsuri N, Handa O, Ichikawa H, Yoshida N, Yoshikawa T. Pioglitazone, a PPAR-gamma ligand, provides protection from dextran sulfate sodium-induced colitis in mice in association with inhibition of the NF-kappaB-cytokine cascade. Redox Rep. 2002;7:283–289. doi: 10.1179/135100002125000802. PMid:17920038. [DOI] [PubMed] [Google Scholar]
- 148.Rajasingh J, Bright JJ. 15-Deoxy-delta12,14-prostaglandin J2 regulates leukemia inhibitory factor signaling through JAK-STAT pathway in mouse embryonic stem cells. Exp Cell Res. 2006;312:2538–2546. doi: 10.1016/j.yexcr.2006.04.010. PMid:18082134. [DOI] [PubMed] [Google Scholar]
- 149.Ide H, Nakagawa T, Terado Y, Kamiyama Y, Muto S, Horie S. Tyk2 expression and its signaling enhances the invasiveness of prostate cancer cells. Biochem Biophys Res Commun. 2008;369:292–296. doi: 10.1016/j.bbrc.2007.08.160. PMid:19223541. [DOI] [PubMed] [Google Scholar]
- 150.Schuster C, Muller M, Freissmuth M, Sexl V, Stoiber D. Commentary on H. Ide et al.,"Tyk2 expression and its signaling enhances the invasiveness of prostate cancer cells". Biochem Biophys Res Commun. 2008;366:869–870. doi: 10.1016/j.bbrc.2007.12.017. PMid:20388792. [DOI] [PubMed] [Google Scholar]
- 151.Sen B, Saigal B, Parikh N, Gallick G, Johnson FM. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009;69:1958–1965. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nadiminty N, Lou W, Sun M, Chen J, Yue J, Kung HJ, Evans CP, Zhou Q, Gao AC. Aberrant activation of the androgen receptor by NF-kappaB2/p52 in prostate cancer cells. Cancer Res. 2010;70:3309–3319. doi: 10.1158/0008-5472.CAN-09-3703. PMid:11034093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Paquette EL, Sun L, Paquette LR, Connelly R, McLeod DG, Moul JW. Improved prostate cancer-specific survival and other disease parameters: impact of prostate-specific antigen testing. Urology. 2002;60:756–759. doi: 10.1016/s0090-4295(02)01960-x. PMid:16452400. [DOI] [PubMed] [Google Scholar]
- 154.Hisatake JI, Ikezoe T, Carey M, Holden S, Tomoyasu S, Koeffler HP. Down-Regulation of prostate-specific antigen expression by ligands for peroxisome proliferator-activated receptor gamma in human prostate cancer. Cancer Res. 2000;60:5494–5498. [PubMed] [Google Scholar]
- 155.Yang CC, Ku CY, Wei S, Shiau CW, Chen CS, Pinzone JJ, Ringel MD. Peroxisome proliferator-activated receptor gamma-independent repression of prostate-specific antigen expression by thiazolidinediones in prostate cancer cells. Mol Pharmacol. 2006;69:1564–1570. doi: 10.1124/mol.105.018333. PMid:20165689 PMCid:2822448. [DOI] [PubMed] [Google Scholar]
- 156.Bonofiglio D, Aquila S, Catalano S, Gabriele S, Belmonte M, Middea E, Qi H, Morelli C, Gentile M, Maggiolini M, Ando S. Peroxisome proliferator-activated receptor-gamma activates p53 gene promoter binding to the nuclear factor-kappaB sequence in human MCF7 breast cancer cells. Mol Endocrinol. 2006;20:3083–3092. doi: 10.1210/me.2006-0192. PMid:16611854. [DOI] [PubMed] [Google Scholar]
- 157.Wang Z, Sun Y. Targeting p53 for Novel Anticancer Therapy. Transl Oncol. 2010;3:1–12. doi: 10.1593/tlo.09250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lee SJ, Yang EK, Kim SG. Peroxisome proliferator-activated receptor-gamma and retinoic acid X receptor alpha represses the TGFbeta1 gene via PTEN-mediated p70 ribosomal S6 kinase-1 inhibition: role for Zf9 dephosphorylation. Mol Pharmacol. 2006;70:415–425. doi: 10.1124/mol.106.022954. [DOI] [PubMed] [Google Scholar]
- 159.Bai MH, Ma HB, Wang XJ, Liu XX, Xue XH, Xue FJ. [Effects of activated PPARgamma on the expression of PTEN gene in pancreatic cancer cells] Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2008;24:717–720. [PubMed] [Google Scholar]
- 160.Dong YW, Wang XP, Wu K. Suppression of pancreatic carcinoma growth by activating peroxisome proliferator-activated receptor gamma involves angiogenesis inhibition. World J Gastroenterol. 2009;15:441–448. doi: 10.3748/wjg.15.441. PMid:19469651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tao J, Xiong J, Li T, Yang Z, Li X, Li K, Wu H, Wang C. Correlation between protein expression of PTEN in human pancreatic cancer and the proliferation, infiltration, metastasis and prognosis. J Huazhong Univ Sci Technolog Med Sci. 2006;26:444–447. doi: 10.1007/s11596-006-0417-7. PMid:9529258. [DOI] [PubMed] [Google Scholar]
- 162.Konieczna A, Lichnovka R, Erdosova B, Ehrmann J. The Role of PPARs in MDR - a lesson from embryonic development. Neoplasma. 2009;56:279–283. doi: 10.4149/neo_2009_04_279. PMid:19878981. [DOI] [PubMed] [Google Scholar]