

Polyarylated methanes are attracting considerable attention due to their growing importance in developing medicinal agents for cancer[1] and vascular disease,[2, 3] as well as in leuco dye precursors,[4, 5] photochromic agents, and applications in materials science.[6, 7] Most syntheses of polyarylmethanes involve Friedel-Crafts-type (F-C) electrophilic aromatic substitution reactions,[5,6, 8–13] although there are some limited exceptions.[4, 14, 15] Recently, modified F-C processes,[9, 16] including a novel copper-catalyzed aza-Friedel-Crafts variant,[17] have been used in the preparation of differentially substituted triarylmethanes. The F-C approach, however, is limited by both reactivity and selectivity: the nucleophile must be electron-rich and unhindered for adequate reactivity and the selectivity is controlled by the relative directing abilities of the substituents. Thus, triarylmethanes with certain electron withdrawing groups and those with meta substitution, are largely inaccessible. A complementary and general route to polyarylated methanes that enables the synthesis of currently inaccessible members of this important structural class is needed.
A strategy that would circumvent the limitations of the F-C reaction is based on benzylic anion synthons. Traditionally, reagents for this synthon are based on metals such as boron, tin or zinc to temper reactivity.[18–20] An alternative approach to attenuate the reactivity of benzylic nucleophiles is η6-coordination to a metal fragment such as Cr(CO)3.[21–24] Along these lines, Kalinin and co-workers generated (η6-C6H5CH2Li)Cr(CO)3, but were unable to effect palladium catalyzed cross-coupling reactions. After transmetallation to zinc the resulting (η6-C6H5CH2ZnCl)Cr(CO)3 underwent palladium catalyzed single coupling with aryl halides to furnish diarylmethanes in low yield (average 37%).[25] The intermediate transmetallation to zinc prevents the realization of the tricarbonylchromium group’s full potential: to activate more than one benzylic C–H bonds and open the door to multiple functionalizations via sequential deprotonation/coupling events.[26]
Herein we disclose a general, high-yielding cross-coupling method between Cr(CO)3-activated toluene derivatives and aryl bromides to afford a broad range of di- and triarylmethanes that are difficult or impossible to access by known methods.
Our initial studies used tricarbonylchromium-complexed diphenylmethane (1, Scheme 1) and 4-bromotoluene as coupling partners. Use of alkoxide bases at room temperature resulted in no product (Table 1, entries 1 and 2). In contrast, LiN(SiMe3)2 combined with PdCl2(PPh3)2 (3 mol %) afforded 70% isolated yield of the coupled product 2a after 20 h at RT (entry 3) and 91% yield in 45 min at 60 °C (entry 4). On the other hand, stronger amide bases such as LDA, NaN(SiMe3)2, or KN(SiMe3)2 (entries 5–7), or related catalysts, including Cl2Pd(dppf)(entries 8–10) were less effective. The optimized conditions in entry 4 were used to determine the substrate scope (Table 2).
Scheme 1.

Coupling of diphenylmethane tricarbonylchromium 1 with aryl bromide
Table 1.
Optimization of reaction conditions (Scheme 1)
| Catalyst | mol % | Base | time (h)[a] | yield[b] | SM[c] |
|---|---|---|---|---|---|
| 1 PdBr2(PPh3)2 | 10 | LiOt-Bu | 24 | - [d] | - |
| 2 PdBr2(PPh3)2 | 5 | NaOt-Bu | 24 | - [d] | 67 |
| 3 PdCl2(PPh3)2 | 3 | LiN(SiMe3)2 | 20 | 70 [d] | trace |
| 4 PdCl2(PPh3)2 | 3 | LiN(SiMe3)2 | 0.75 | 91 | 0 |
| 5 PdCl2(PPh3)2 | 3 | LDA | 12 | 50 | trace |
| 6 PdCl2(PPh3)2 | 5 | NaN(SiMe3)2 | 18 | 26 | 38 |
| 7 PdCl2(PPh3)2 | 5 | KN(SiMe3)2 | 20 | 9 | 48 |
| 8 NiCl2(PPh3)2 | 5 | LiN(SiMe3)2 | 20 | 0 | 29 |
| 9 Pd(PPh3)4 | 5 | LiN(SiMe3)2 | 17 | 67 | 12 |
| 10 PdCl2(dppf) | 5 | LiN(SiMe3)2 | 19 | 78 | 14 |
Conducted at 55–60 °C in THF solvent, except where otherwise noted
% isolated yield
% recovered SM
Reaction conducted at RT.
Table 2.
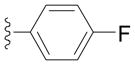
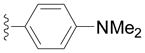
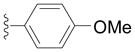
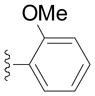
| Entry | Ar = | yield[b] |
|---|---|---|
| 2a |
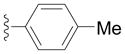
|
91 |
| 2b |
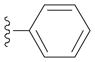
|
92 |
| 2c |
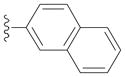
|
90 |
| 2d |
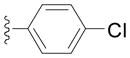
|
83 |
| 2e |
|
88 |
| 2f |
|
91 |
| 2g |
|
94 |
| 2h |
|
91 |
| 2j |
|
81 |
| 2k |
|
89 |
Conducted at 55–60 °C using 1.5 equiv. LiN(SiMe3)2, 1.5 equiv. ArBr, 3–5 mol % PdCl2(PPh3)2, and THF as solvent
Isolated yields.
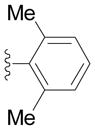
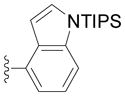
The diphenylmethane complex 1 readily undergoes cross-coupling reactions with a variety of aryl bromides in 81–94% isolated yield of triarylmethane complexes 2a–2k (Table 2). Electron-withdrawing (2d, 2e), donating (2f–2h), and ortho substituted aryl bromides (2h–2k) were all successful coupling partners. Of medicinal relevance, our method enabled installation of a 4-substituted indole (2k).[27] In contrast, F-C chemistry strongly favours reaction of indole at the 3-position (or 2-position if the 3 is blocked).[5, 11–13, 17] Polyarylmethane derivatives bearing privileged indoles have attracted interest due to their widespread occurrence in biologically active compounds.
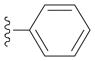
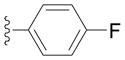
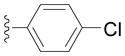
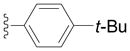
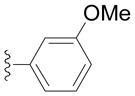
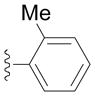
Next we examined reactions of (η6-toluene)Cr(CO)3 (3, Scheme 2). Although these reactions did not proceed well at room temperature, conversion to triarylmethane products 5a–f (Table 3) was observed upon heating at 60 °C. Note that reaction with diarylmethane intermediate 4 is faster than the first coupling with 3, probably due to the greater acidity of the benzlic C–H’s of 4 over 3.[28] In contrast, bulky 2,6-disubstitued aryl bromides afforded mono-coupled products 4g and 4h, likely due to steric hindrance.
Scheme 2.

Cross-Coupling Reactions with (η6-toluene)Cr(CO)3 (3).
Table 3.
Double and Single Couplings of 3 with ArBr[a]
| Entry | Ar = | yield[b] |
|---|---|---|
| 5a |
|
90 |
| 5b |
|
83 |
| 5c |
|
78 |
| 5d |
|
82 |
| 5e |
|
86 |
| 5f |
|
88 |
| 4g |
|
86 |
| 4h |
|
81 |
Conducted at 55–60 °C using 2.8 equiv. LiN(SiMe3)2, 3 equiv. ArBr, 4–8 mol % PdCl2(PPh3)2, and THF as solvent.
Isolated yields.
Having demonstrated that two couplings could occur on a single methyl group, we wondered if one tricarbonylchromium center could activate multiple methyl groups in complexes of xylene and mesitylene. We were concerned that after the first methyl had undergone two arylations, the newly-formed triarylmethane moiety would be deprotonated and the resulting anion would inhibit further deprotonation and coupling at the remaining methyl groups. As shown in eq 2 and Table 4, this was not a serious issue. Subjecting (η6-p-xylene)Cr(CO)3 (6) to cross-coupling conditions with the bulky 1-bromo-2,4,6-triisopropylbenzene furnished di-coupled 7 in 73% yield. Employing 4-bromofluorobenzene gave tetracoupled 8 in 70% yield. Furthermore, the mesitylene complex 9 was coupled with 4-bromoanisole (Scheme 3) resulting in formation of the hexacoupled product 10 in 43% yield (over 86% for each C–C bond-forming event). The structure of the hexacoupled product is illustrated in Figure 1.[29] In the case of (η6-o-xylene)Cr(CO)3 (11), bulky 1-bromo-2,6-dimethylbenzene as coupling partner resulted in single arylation of each benzylic methyl to give the dicoupled product (12), in 85% yield. Use of the less sterocally demanding aryl bromide 4-bromo-N,N-dimethylaniline, however, led to either unsymmetrical di-coupled product (13, in which both aryl groups couple to a single methyl), or unsymmetrical tri-coupled product (14) by varying the amount of base and reaction time. Steric barriers to both deprotonation and transmetallation likely account for the absence of tetraarylated (symmetrical) coupling product. These polyarylation reactions highlight a significant advantage of this approach over others: the possibility of multiple coupling reactions.[16]
Table 4.
Coupling reaction at multiple arylation sites[a]
|
Conducted at 50–60 °C using 4–11 equiv. LiN(SiMe3)2, 3–10 equiv. ArBr, 10–20 mol % PdCl2(PPh3)2, and THF as solvent.
Isolated yields.
Scheme 3.

Cross-Coupling at multiple benzylic reaction sites.
Figure 1.

ORTEP of hexa-arylated product (10).
We also examined the possibility of coupling in the presence of α-heteroatoms and β–hydrogens using tricarbonylchromium-coordinated benzyl ether (15), N,N-dimethylbenzylamine (16), and ethylbenzene (17). Benzyl ether complex 15 underwent coupling employing conditions similar to those in Scheme 2 to afford monocoupled products 18a–e in 58–80% yield (Scheme 4). Surprisingly, further reaction, whether in the presence of the original aryl bromide or with a different aryl bromide after isolation, was found to give triarylmethyl ether complexes 19a–e. N,N-Dimethylbenzyl amine complex 16 underwent single coupling to form diarylamines 20a–d in 68–82% yield. No doubly coupled products were observed, possibly due to larger size of the dimethylamino group over the n-propyl ether in 15. It is noteworthy that diarylmethylamines are important pharmacophores and are present in numerous medications.[30] Ethylbenzene complex 17 underwent single-coupling to generate the 1,1-diarylethane derivative 21 in 70% yield, despite the presence of β-hydrogens in the intermediate palladium complex. Interestingly, this reaction also produced 6% of the triarylmethane quaternary product 22.
Scheme 4.

Coupling in the presence of heteroatoms and β–H‘s. [a] Product obtained directly from 15 [b] From isolated 18b. [c] From isolated 18a. [d] 6% doubly coupled product was also isolated.
To exploit of the ability of Cr(CO)3 to shield one face of the coordinated arene,[21] we also examined cross-coupling reactions with complexes of indane (23) and tetrahydronaphthalene (24) (Scheme 5). By adjusting the conditions, either mono- or diarylated product could be obtained, though yields of 25 and 26 were diminished due to partial conversion to diarylated product. In the case of the diarylation reactions to form 27 and 28, cis-diaryl products were obtained in all cases (determined by 1H NMR and X-ray diffraction of 27b,[29] see Supporting Information).
Scheme 5.

Access to 1-Aryl-indanes and 1,3-cis-diaryl indanes. [a]40:60 Toluene:THF was used instead of pure THF to improve selectivity for monoarylation. [b] From isolated 25b.
To demonstrate the practical advantages to our method, double coupling of indane tricarbonylchromium (23) with PhBr, followed by exposure to light and air provided 79% isolated yield of cis-1,3-diphenylindane (29, Scheme 6). Existing syntheses of 1,3-diaryl indanes provide cis/trans mixtures.[31] The cis-1,3-diarylindane moiety is present in potent nonpeptide endothelin receptor antagonists.[2, 3] Performing the coupling of (η6-toluene)Cr(CO)3 (3) with 3-bromoanisole on a 5 mmol scale, followed by exposure to air and light afforded 1.4 g PhCH(3-C6H4-OMe)2 (30) in 93% yield, a product inaccessible by standard F-C procedures with anisole.[32]
Scheme 6.

One-pot coupling and direct decomplexation of (η6-Arene)Cr(CO)3 complexes. [a] Reaction is exposed to light prior to purification. [b] Single (cis-) isomer observed. [c] Reaction was conducted on a 5 mmol scale.
In summary, we have introduced an efficient method for benzylic coupling reactions leading to polyarylated methanes. This method is complementary to F-C approaches and enables the synthesis of polyarylmethanes that are not accessible through electrophilic aromatic substitution reactions alone. Activation of the benzylic protons of (η6-C6H5CH2R)Cr(CO)3 allows a base of moderate strength, LiN(SiMe3)2, to be used for the in-situ generation of the benzyllithiums,[33] which directly participates in a palladium-catalyzed coupling reaction. Coordinated complexes can undergo multiple arylations, furnishing polyarylated products including unsymmetrically substituted triarylmethanes with quaternary carbon centers. Currently, efforts are underway to broaden the application of our method to the generation of quaternary polyarylmethanes and their enantioselective syntheses.
Supplementary Material
Footnotes
This research was supported by the National Institutes of Health, General Medical Sciences (GM058101) and National Science Foundation (CHE-0848467). JT is a Vagelos Scholar.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Palchaudhuri R, Nesterenko V, Hergenrother PJ. J Am Chem Soc. 2008;130:10274. doi: 10.1021/ja8020999. [DOI] [PubMed] [Google Scholar]
- 2.Elliott JD, Lago MA, Cousins RD, Gao A, Leber JD, Erhard KF, Nambi P, Elshourbagy NA, Kumar C. J Med Chem. 1994;37:1553. doi: 10.1021/jm00037a003. [DOI] [PubMed] [Google Scholar]
- 3.Ohlstein EH, Nambi P, Douglas SA, Edwards RM, Gellai M, Lago A, Leber JD, Cousins RD, Gao AM, Frazee JS, Peishoff CE, Bean JW, Eggleston DS, Elshourbagy NA, Kumar C, Lee JA, Yue TL, Louden C, Brooks DP, Weinstock J, Feuerstein G, Poste G, Ruffolo RR, Gleason JG, Elliott JD. Proc Natl Acad Sci U S A. 1994;91:8052. doi: 10.1073/pnas.91.17.8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muthyala R, Katritzky AR, Lan X. Dyes and Pigments. 1994;25:303. [Google Scholar]
- 5.Katritzky AR, Gupta V, Garot C, Stevens CV, Gordeev MF. Heterocycles. 1994;38:345. [Google Scholar]
- 6.Duxbury DF. Chem Rev. 2002;93:381. [Google Scholar]
- 7.Shchepinov MS, Korshun VA. Chem Soc Rev. 2003;32:170. doi: 10.1039/b008900l. [DOI] [PubMed] [Google Scholar]
- 8.Nair V, Thomas S, Mathew SC, Abhilash KG. Tetrahedron. 2006;62:6731. [Google Scholar]
- 9.Lin S, Lu X. J Org Chem. 2007;72:9757. doi: 10.1021/jo071232k. [DOI] [PubMed] [Google Scholar]
- 10.Das SK, Shagufta, Panda G. Tetrahedron Lett. 2005;46:3097. [Google Scholar]
- 11.Yadav JS, Reddy BVS, Sunitha S. Adv Syn & Catal. 2003;345:349. [Google Scholar]
- 12.Ramesh C, Barerjee J, Pal R, Das B. Adv Syn & Catal. 2003;345:557. [Google Scholar]
- 13.Nair V, Abhilash KG, Vidya N. Org Lett. 2005;7:5857. doi: 10.1021/ol052423h. [DOI] [PubMed] [Google Scholar]
- 14.For a complementary procedure involving vicarious nucleophilic substitution of nitroarenes see: Katritzky AR, Toader D. J Org Chem. 1997;62:4137.
- 15.Katritzky AR, Lan X, Lam JN. Chem Ber. 1991;124:1809. [Google Scholar]
- 16.Li YZ, Li BJ, Lu XY, Lin S, Shi ZJ. Angew Chem, Int Ed. 2009;48:3817. doi: 10.1002/anie.200900341. [DOI] [PubMed] [Google Scholar]
- 17.Esquivias J, Arrayás RG, Carretero JC. Angew Chem, Int Ed. 2006;45:629. doi: 10.1002/anie.200503305. [DOI] [PubMed] [Google Scholar]
- 18.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 19.Liégault B, Renaud JL, Bruneau C. Chem Soc Rev. 2008;37:290. doi: 10.1039/b704255h. [DOI] [PubMed] [Google Scholar]
- 20.Imao D, Glasspoole BW, Laberge VS, Crudden CM. J Am Chem Soc. 2009;131:5024. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]
- 21.Uemura M. In: Org React. Overman LE, editor. Vol. 67. John Wiley; NJ: 2006. p. 217. [Google Scholar]
- 22.Merlic CA, Walsh JC, Tantillo DJ, Houk KN. J Am Chem Soc. 1999;121:3596. [Google Scholar]
- 23.Pfletschinger A, Dargel TK, Bats JW, Schmalz HG, Koch W. Chem Eur J. 1999;5:537. [Google Scholar]
- 24.Kündig EP. Topics in Organometallic Chemistry. Vol. 7. Berline; New York: 2004. [Google Scholar]
- 25.Kalinin VN, Cherepanov IA, Moiseev SK. J Organomet Chem. 1997;536–537:437. [Google Scholar]
- 26.Brewer ARE, Drake AF, Gibson SE, Rendell JT. Org Lett. 2007;9:3487. doi: 10.1021/ol070934t. [DOI] [PubMed] [Google Scholar]
- 27.Sundberg RJ. Indoles. Academic; San Diego: 1996. [Google Scholar]
- 28.When 1H NMR deprotonation studies were conducted in THF with one equivalent of LiN(SiMe3)2, 1 exhibited approximately 50% deprotonation, whereas 3 was only 10% deprotonated.
- 29.CCDC 775771 (10) and 765716 (27b) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 30.Yamada K-i, Tomioka K. Chem Rev. 2008;108:2874. doi: 10.1021/cr078370u. [DOI] [PubMed] [Google Scholar]
- 31.Lantaño B, Aguirre J, Finkielsztein ML, Alesso E, Brunet E, Moltrasio GY. Syn Commun. 2004;34:625. [Google Scholar]
- 32.Alternately, the coordinated arene can be purified first and then simply decomplexed by exposure to light and air. A solution of coordinated arene 2a exposed to light and air provided triphenylmethane in 98% isolated yield.
- 33.For cross-coupling reactions with organolithium reagents see: Murahashi SI. J Organomet Chem. 2002;653:27.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.