

Abstract
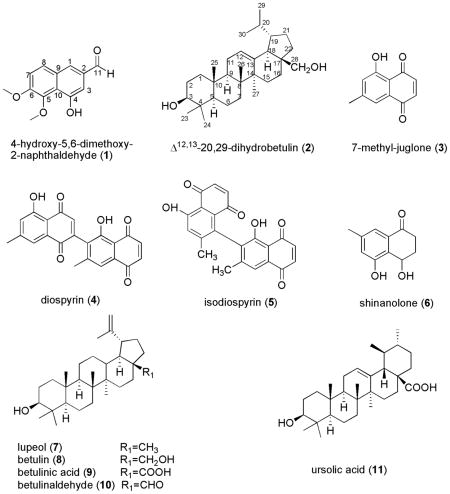
A preparative overpressure layer chromatography (OPLC) method was successfully used for the separation of two new natural compounds, 4-hydroxy-5,6-dimethoxy-2-naphthaldehyde (1) and Δ12,13-20,29-dihydrobetulin (2) together with nine known compounds including 7-methyl-juglone (3), diospyrin (4), isodiospyrin (5), shinanolone (6), lupeol (7), betulin (8), betulinic acid (9), betulinaldehyde (10), and ursolic acid (11) from the acetone extract of the roots of Diospyros virginiana. Their identification was performed with mono and bidimensional NMR spectroscopy and HR-ESI-MS methods. All the isolated compounds were evaluated for their antifungal activity against Colletotrichum fragariae, C. gloeosporioides, C. acutatum, Botrytis cinerea, Fusarium oxysporum, Phomopsis obscurans, and P. viticola using in vitro micro-dilution broth assay. The results indicated that compounds 3 and 5 showed high antifungal activity against P. obscurans at 30 μM with 97.0 % and 81.4 % growth inhibition and moderate activity against P. viticola (54.3 % and 36.6 %). It appears that an optimized OPLC system offers a rapid and efficient method of exploiting bioactive natural products.
Introduction
Discovery of new crop protections from natural products has received considerable interest as alternatives to synthetic agrochemicals for use as pest and disease control agents. This stems from these generally being safer for human health and the environment. Biologically active natural products can be used in modern crop protections or can serve as lead structures for the development of new semi-synthetic analogs. Our current research efforts are to identify natural product-based fungicides. In this research program, we evaluated 37 plant extracts using a direct bioautography bioassay to detect antifungal activity against Colletotrichum. Of these, Diospyros virginiana root extract showed the most promising activity. Based on preliminary screening results, a bioassay-guided fractionation of D. virginiana root extract was conducted in order to isolate and identify the pure metabolites possessing antifungal activity. Diospyros belongs to the family Ebenaceae that contains approximately 500 species of trees and shrubs [1], of which more than 350 species are distributed in tropical and subtropical regions worldwide [2–3]. The most well-known species is Diospyros kaki, which originated in East China and has been cultivated in Japan for centuries [4]. Some of the Diospyros species, such as D. peregrina and D. melanoxylon, have been used in folk medicine for the treatment of inflammation, urinary discharges and enrichment of blood [5]. Diospyros virginiana, the American persimmon, is native to North America and the fruits were reported to show cholesterol lowering activity, and usefulness to treat bloody stools, thrush and sore throats [6]. The literature shows reports of nine compounds being isolated and identified from either the leaves or the wood of D. virginiana [7–9]. However, there are no reports in the literature on chemical fractions from the roots of D. virginiana.
OPLC was developed by Tyihák and Mincsovics in the late seventies [10]. In this process, a TLC plate is covered by a sheet of flexible material and subjected to a high external pressure. The high external pressure, which is generated by a programmable pump, pushes the mobile phase through the analytical or preparative adsorbent layer. This allows a faster separation and more compact spots than conventional TLC. Generally, the Rf of the migrated compounds is two to five times greater than in conventional TLC. OPLC also offers an advantage over traditional TLC as it is a sealed system, which results in less solvent loss through evaporation [11–12]. As such, OPLC has been widely used in analytical and preparative applications [13–15]. Compared to conventional methods, OPLC offers a more rapid and reliable method for the isolation of naturally occurring compounds. In the present paper, we report the use of a preparative OPLC technique to isolate co-migrating bioactive metabolites from the roots of Diospyros virginiana.
Results and Discussion
The roots of D. virginiana were extracted using acetone in a Soxhlet extractor. The constituents of this extract were separated by flash chromatography, OPLC and preparative TLC. A new naphthyl derivative, 4-hydroxy-5,6-dimethoxy-2-naphthaldehyde (1), and a triterpene, Δ12,13-20,29-dihydrobetulin (2) were isolated from a natural source for the first time, along with nine known compounds: 7-methyl-juglone (3) [16], diospyrin (4) [17], isodiospyrin (5) [16–18], shinanolone (6) [19], lupeol (7) [20], betulin (8) [21], betulinic acid (9) [22], betulinaldehyde (10) [23], and ursolic acid (11) [24]. Although they are common in most of the Ebenaceae family [5], compounds 10 and 11 were isolated for the first time from D. virginiana. The structures of the known compounds were confirmed by comparison of their spectroscopic data (MS, 1H and 13C NMR) with literature values.
The HR-ESI-MS of compound 1 indicated a molecular formula C13H12O4 and its IR spectrum exhibited characteristic absorption bands for hydroxyl and conjugated carbonyl groups. The 1H NMR spectrum showed signals for an aldehyde group at δ 10.03 ppm, one aromatic AB system at δ 7.37 (d, J = 9.0 Hz) and 7.78 (d, J = 9.0 Hz). The above data suggested a 2-naphthaldehyde structure with the presence of two methoxyl groups at δ 4.06, and 4.13 ppm, and a hydroxyl substituent at δ 9.75 ppm. The 13C-NMR spectrum showed thirteen signals with ten typical aromatic carbons. Assignment of structure was accomplished by HMBC experiment. The NOESY experiment showed a correlation between the methoxyl group at δ 4.06 and H-7 (δH 7.37, d) and methoxyl group at δ 4.13 and the hydroxyl signal at δ 9.75 ppm, corroborating the location of the methoxyl groups at C(5) and C(6). Thus, 1 was characterized as 4-hydroxy-5,6-dimethoxy-2-naphthaldehyde.
Compound 2 was obtained as a white solid. The molecular formula was determined as C30H51O2 by HR-ESI-MS ([M + H]+ at m/z = 443.3874). The 13C NMR data of 2 indicated the presence of 30 carbons comprising seven methyl groups, ten methylenes, seven methines and six quaternary carbons. The seven methyl groups resonated at δH 0.79 (s, 6H), 0.93 (d, 6H, J = 6.8), 0.98 (s, 3H), 0.99 (s, 3H), and 1.10 (s, 3H) in the 1H NMR spectrum indicating the lupane triterpene skeleton for 2 [25]. The 13C NMR spectrum also revealed the presence of one olifenic methine (δ(C) 125.0) correlated in the HMQC spectrum with the proton at δ(H) 5.20 (H-C(12)). The last proton showed HMBC correlation with C(9), C(14), and C(18), this correlation led to the placement of the double bond between C(12) and C(13). This was confirmed by the COSY correlation between H-C(12) (δ(H) 5.20) and H2-C(11) (δ(H) 1.90) and further supported by the HMBC correlation of H3-C(27) (δ(H) 1.10) and C(13) (δ(C) 136.7) (Fig. 1). From these data, the compound 2 was deduced to be Δ12,13-20,29-dihydrobetulin. This is the first report for the full spectral data and isolation of 2 from a natural source, although it has been prepared synthetically and only the 1H NMR data was reported [26].
Fig. 1.
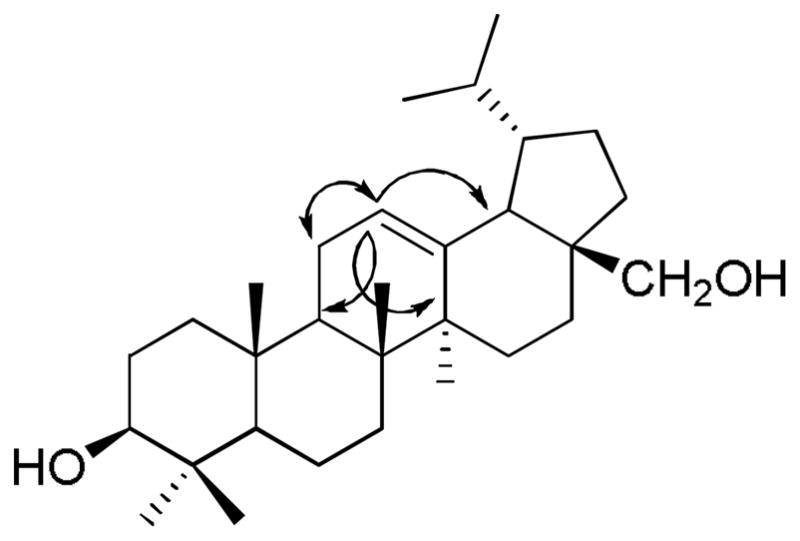
Relevant 1H-13C HMBC (→) and 1H-1H COSY (↔) correlations of 2
The antifungal activity of compounds 1–11 was examined using a 96-well micro-dilution broth assay against the plant pathogens: Botrytis cinerea, Fusarium oxysporum, Phomopsis obscurans, P. viticola and three Colletotrichum species. Phomopsis species were the most sensitive fungi to these compounds. 7-methyl-juglone (3) and isodiospyrin (5) showed the most antifungal activity against P. obscurans. Compound 3 showed 97.0 % growth inhibition of P. obscurans at 120 h at 30 μM, whereas compound 5 showed 81.4 % growth inhibition. The antifungal activity of 3 and 5 at 30 μM against P. viticola was 53.4 % and 57.7 %, respectively.
The newly reported compounds, 4-Hydroxy-5, 6-dimethoxy-2-naphthaldehyde (1) and Δ12,13-20,29-dihydrobetulin (2) at 30 μM showed weak antifungal activity with 26.9 % and 22.1 %, respectively (Fig. 2). Compound 3 and compound 5 at 30 μM caused 54.3 % and 36.6 % growth inhibition of P. viticola at 120 h. Any test compound possessing < 50 % growth inhibition at 30 μM is considered to have weak antifungal activity in this bioassay. Compound 1 was more active against P. viticola at 120 h than at 144 h. This response is often seen when an inducible enzyme system is turned on and a compound is detoxified by the fungus. However, the upward slope of the graph for compound 1 in Fig. 3 from the low to high concentration is indicative of precipitation in the aqueous microdilution broth assay. Because the microtiter plate reader measures changes in optical density it does not discriminate between fungal growth and precipitation. Compound 1 appears to have come out of solution at the higher concentration (30 μM) in the P. viticola testing (Fig. 3). Lipophilic compounds are problematic in in vitro aqueous bioassays and follow-up antifungal testing will take place using a detached leaf bioassay [27]. Interpretation of the graphical results indicated that compounds 2, 3, and 5 appear to remain soluble at 120 h and 144 h.
Fig. 2.

Mean fungal growth inhibition (%) of Phomopsis obscurans after exposure to 1, 2, 3 and 5 using a dose-response format at 120 and 144 h. Abreviations: Cap: Captan; Azo: Azoxystrobin; Ben: Benomyl
Fig. 3.

Mean fungal growth inhibition (%) of Phomopsis viticola after exposure to 1, 2, 3 and 5 using a dose-response format at 120 and 144 h. Abreviations: Cap: Captan; Azo: Azoxystrobin; Ben: Benomyl
Commercial fungicides standards captan and benomyl are significantly more active than any of the compounds tested. Azoxystrobin, which is commercially used to control Colletotrichum and Botrytis diseases, shows poor activity against P. viticola. Both captan and azoxystrobin show 100% growth inhibition at 3 μM against P. obscurans. Although once an excellent agent for controlling anthracnose and other diseases of strawberries and ornamentals, resistance developed by pathogens has resulted in benomyl being less useful [28]. While the test compounds appear inherently antifungal, we hypothesize that these compounds are probably present in the plant as constitutive defense compounds that act to deter infection or fungal growth. Since these compounds were found without elicitation they are probably constitutive in nature [29–30] and may have a potential role in preventing fungal infection in D. virginiana.
Conclusions
Using a bioassay-guided fractionation of the acetone extract D. virginiana were able to isolate the antifungal constituents of the roots of this plant. This research demonstrates that OPLC is a powerful technique that can be used to separate and isolate co-migrating natural products produced by plants. These constituents were tested for the first time against Colletotrichum spp, B. cinerea, F. oxysporum, P. obscurans, and P. viticola. The most promising agricultural lead compounds against the pathogen P. obscurans are 3 and 5. Phomopsis leaf blight and fruit rot is a serious disease with strawberries and causes serious economic loss of this fruit each year. Our results suggest that compounds 3 and 5 warrant further in vivo testing as plant protectants to control Phomopsis species [27].
Experimental Part
Chemistry
General
UV spectra were obtained in MeOH using a Varian Cary 50 spectrophotometer and IR spectra were recorded using Bruker Tensor 27 spectrophotometer. 1H- and 13C-NMR spectra were obtained on Bruker model AMX-500 and 400 NMR spectrometers with standard pulse sequences, operating at 500 and 400 MHz for 1H and 125 and 100 MHz for 13C. High resolution electrospray ionization mass spectroscopy (HR-ESI-MS) was recorded on a Micromass Q-Tof Micro mass spectrometer with a lock spray source. OPLC separations were performed with the Personal OPLC 50 instrument (OPLC-NIT, Budapest, Hungary) at 50 bars external pressure. OPLC silica gel layer, F254 20 × 20 cm on glass plate (LG 011, OPLC-NIT Ltd, Budapest Hungary) with 200 μm sorbent thickness, 11 μm particle size, 6 nm pore size, and 20 × 20 cm on aluminum sheet (BSLA 001, OPLC-NIT Ltd, Budapest Hungary) with 200 μm sorbent thickness, 5 μm particle size, 6 nm pore size. An AS-30 sample applicator (DESAGA, Wiesloch, Germany) was used for the sample application. Classical TLC analysis was performed on silica gel 60 F254 20 × 20 cm on aluminum sheet (Gibbstown, New Jersey, USA). Detection was carried out under UV light (254 nm, 366 nm) and visualization made with vanillin-H2SO4 (1 g vanillin in 100 ml of 20 % H2SO4 in EtOH) reagent followed by heating at 105 °C for 5 min. ACS-grade solvents, acetonitrile, acetone, chloroform, dichloromethane (DCM), diethyl ether, ethyl acetate (EtOAc), EtOH, n-hexane, isopropanol (IPA), methanol and toluene from Fisher Scientific (New Jersey, USA) were used for silica gel column chromatography and TLC separations. HPLC-grade solvents (acetonitrile, chloroform and DCM) from Sigma-Aldrich (St. Louis, USA) were used for OPLC chromatograms.
Plant materia
The roots of D. virginiana were collected from the Missouri Botanical Garden, U.S.A., in June 2009. The plant material was identified by Dr. Vaishali C. Joshi, and a voucher specimen CON310700-2-A was deposited at the National Center for Natural Products Research, School of Pharmacy, University of Mississippi.
Extraction and Isolation
Dried powdered roots (66 g) were extracted in a Soxhlet extractor with acetone (600 ml) for 8 h and the extract was subsequently evaporated under vacuum to yield the dry residue (2 g). The crude extract was fractionated on a silica gel column (100 g, 50 × 5 cm, ChemGlass) using n-hexane (500 ml), 5 % EtOAc in n-hexane (600 ml), chloroform (600 ml), 5 % IPA in chloroform (600 ml) and methanol (600 ml), respectively. Four fractions were obtained in total F-1 to F-4. Direct-bioautography guided assay showed that the activity was found to reside in F-1 (120 mg) and F-2 (240 mg), with some activity in F-3 (710 mg), while F-4 (800 mg) did not show activity against three notorious plant pathogenic fungi of the Colletotrichum species. A portion of F-1 (10 mg) showed one major product and was purified using preparative TLC (SiO2, n-hexane-chloroform 1:1) to furnish 7-methyl juglone (3, 1.5 mg). A portion of F-2 (20 mg) was purified using OPLC with DCM as the eluent. Elution conditions were as follows: flash volume, 300 μl; eluent volume, 30,000 μl; flow-rate, 500 μl min−1; development time, 3606 s; external pressure, 50 bars. Eighty-eight sub-fractions (1 ml/fraction) were obtained in total, of which sub-fractions 49–54 gave betulinaldehyde (10, 0.8 mg). Sub-fractions 72–88 afforded ursolic acid (11, 1.3 mg). Sub-fractions 28–37 were combined (7.9 mg) and further purification was performed by OPLC with n-hexane-ether (6/4, v/v, 20,000 μl; flash volume, 300 μl; flow-rate, 500 μl min−1; development time, 2406 s) as eluent and 66 sub-fractions (1 ml/fraction) were obtained, among which sub-fractions 10′–18′ furnished lupeol (7, 2 mg). Sub-fractions 19′–66′ (marked as fraction A) were combined to give a mixture of 3.6 mg containing four compounds with close Rf values in several TLC systems (SiO2, n-hexane-EtOAc 6:4, DCM-IPA 9:1, CHCl3-MeOH 19:1). In order to obtain enough material for further purification, OPLC procedures were repeated six times for F-2 using the same conditions, then fraction A (19.6 mg) was obtained and purified by OPLC with DCM (100 %, 20,000 μl; flash volume, 300 μl; flow-rate, 400 μl min−1; development time, 3007 s) as mobile phase. Sub-fraction 12″ yielded a new product 4-hydroxy-5, 6-dimethoxy-2-naphthaldehyde (1, 1 mg). Sub-fractions 9″–11″ gave diospyrin (4, 2 mg). Sub-fractions 14″–21″ afforded isodiospyrin (5, 2 mg). Fraction 3 (24 mg) was chromatographed using OPLC with CHCl3-EtOAc (95/5, v/v, 40,000 μl) with flash volume 300 μl, flow rate 500 μl min−1 and an elution time of 4806 s. One hundred and twenty-three sub-fractions (1 ml/fraction) were collected. Sub-fractions 54–66 yielded betulin (8, 3.4 mg). Sub-fractions 67–90 (7.4 mg), containing at least four compounds, were purified by OPLC with CHCl3-acetonitrile (98/2, v/v) and flash volume 300 μl, flow rate 300 μl min−1 and a total elution time of 4806 s. Sub-fractions 79′–87′ gave shinanolone (6, 1.5 mg). The other minor compounds were in such small quantity that they were impossible to isolate. In order to obtain enough material for further purification, flash chromatography of F-3 (400 mg) using a BIOTAGE (Isolera One) with CHCl3-EtOAc (0–5 %, 200 ml) and CHCl3 (100 %, 201 ml–640 ml), and flow rate, 5 ml min−1 (Si, SNAP 25 g column)was conducted. A total of 81 sub-fractions (1 ml/fraction) were collected. Subfractions 38–51 were combined to yielded a 30 mg mixture. This mixture was subjected to the OPLC system with CHCl3-acetonitrile (98/2, v/v, 40,000 μl) with flash volume 300 μl, flow rate 250 μl/min and total elution time 9624 s. One hundred and eighty-three subfractions (1 ml/fraction) were obtained. Subfractions 100–112 were combined and identified as betulin (8, 2 mg). Subfractions 136–144 yielded betulinic acid (9, 1 mg). Subfraction 114–126 gave a new triterpene, Δ12,13-20,29-dihydrobetulin (2, 1 mg) as a natural product.
4-hydroxy-5,6-dimethoxy-2-naphthaldehyde (1)
Yellow amorphous solid; UV (MeOH): λmax nm (log ε): 375 (4.1), 320 (4.2), 270 (3.8); IR (neat): νmax 3332, 2923, 2851, 2360, 2341, 1687, 1372, 1274, 1055 cm−1; 1H- NMR (500 MHz, CDCl3): δ 4.06 (3H, s, OMe), 4.13 (3H, s, OMe), 7.29 (1H, s br, H-C(3)), 7.37 (1H, d, J= 9.0 Hz, H-C(7)), 7.78 (1H, d, J=9.0 Hz, H-C(8)), 7.81 (1H, s br, H-C(1)), 9.75 (1H, OH-C(4)), 10.03 (1H, s, H-C(11)); 13C-NMR (125 MHz, CDCl3): δ 56.67 (OMe), 62.20 (OMe), 106.14 (C(3)), 115.46 (C(7)), 121.49 (C(10)), 126.02 (C(1)), 127.19 (C(8)), 130.120 (C(9)), 134.09 (C(2)), 143.46 (C(5)), 149.95 (C(6)), 154.28 (C(4)), 191.79 (C(11)); HR-ESI-MS: m/z = 233.0812 ([M + H]+, C13H13O4; calc. 233.0814).
Δ12,13-20,29-dihydrobetulin (2)
White amorphous solid; [α]20D = +79.2 (c 0.05, MeOH ); IR (KBr): νmax 3340, 2940, 2867, 1446, 1372, 1027 cm−1; 1H-NMR (400 MHz, CDCl3): δ 0.73 (1H, d, J=7.2 H-C(5)), 0.79 (6H, s, Me-24, Me-26), 0.89 (1H, m, H-C(20)), 0.93 (6H, d, J= 6.8 Hz, Me-29, Me-30), 0.98 (3H, s, J=9.0 Hz, Me-25), 0.99 (3H, s, Me-23), 1.10 (3H, s, Me-27), 1.20-1.81 (8CH2), 1.90 (2H, m, H2-C(11)), 3.19 (1H, d, J=10.4, H-C(28a)), 3.22 (1H, m, H-C(3)), 3.53 (1H, d, J=10.4, H-C(28b)), 5.13 (1H, bs, H-C(12)); 13C NMR (100 MHz, CDCl3): δ 15.6 (C(29)), 15.7 (C(24)), 16.7 (C(25)), 17.3 (C(26)), 18.3 (C(6)), 21.3 (C(30)), 23.2 (C(21)), 23.3 (C(27)), 23.4 (C(11)), 25.9 (C(2)), 27.2 (C(15)), 28.1 (C(23)), 30.6 (C(16)), 32.8 (C(7)), 35.2 (C(1)), 36.9 (C(14)), 38.7 (C(22)), 38.0 (C(4)), 39.3 (C(20)), 39.4 (C(19)), 40.0 (C(10)), 42.0 (C(8)), 42.3 (C(17)), 47.6 (C(18)), 54.0 (C(9)), 55.1 (C(5)), 69.9 (C(28)), 79.0 (C(3)), 125.0 (C(12)), 136.7 (C(13)). HR-ESI-MS m/z = 443.3874 ([M + H]+, C30H51O2; calc. 443.3889).
Biological Assay
Direct-bioautography assay
Bioautography procedures were described in our previous studies [31–32]. The acetone extract of D. virginiana roots was applied at 80 and 160 μg/spot in chloroform onto a silica plate. Technical fungicide grade standards benomyl, cyprodinil, azoxystrobin, and captan (Chem Service Inc., West Chester, PA) were used as positive controls at 2 mM in 2 μl of 95% ethanol. TLC profiles of F-1 to F-4 in chloroform were tested against Colletotrichum spp and mild polar compounds appear to be responsible for antifungal activity.
Micro-dilution broth assay
A standardized 96-well micro-dilution broth assay developed by Wedge and Kuhajek [33] was used to evaluate the antifungal activity of pure compounds from D. virginiana that were identified as active by bioautography.
Isolates of Colletotrichum acutatum Simmonds, Colletotrichum fragariae Brooks, Colletotrichum gloeosporioides (Penz.) Penz & Sacc. In Penz, Botrytis cinerea Pers.:Fr, Fusarium oxysporum Schlechtend:Fr, Phomopsis obscurans (Ellis and Everh.) B. sutton, and P. viticola Sacc., were used to evaluate the antifungal activity of the test compounds using in vitro micro-dilution broth assay. Each fungus was challenged in a dose-response format using test compounds where the final treatment concentrations were 0.3, 3.0 and 30.0 μM. Technical grade commercial fungicides captan, azoxystrobin, and benomyl, which represent three different modes of actions, were used as positive fungicide standards. Each compound was evaluated in duplicate and the experiment was performed three times in time. Mean absorbance and standard errors were used to evaluate fungal growth after 48 and 72 h, except for P. obscurans and P. viticola (120 and 144 h).
Acknowledgments
The project described was supported by Grant Number 5P20RR021929 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. This investigation was conducted in a facility constructed with support from research facilities improvement program C06 RR-14503-01 from the NIH National Center for Research Resources. Francisco León was supported by JAE-Postdoctoral Program from the Ministerio de Ciencia e Innovación, Spain. The authors thank J. Linda Robertson and Ramona Pace for assistance in performing various bioassays and J’Lynn Howell for the photographic expertise information.
Contributor Information
David E. Wedge, Email: dwedge@olemiss.edu.
Stephen J. Cutler, Email: cutler@olemiss.edu.
References
- 1.Brummitt RK. Vascular Plant Families and Genera. Kew Publishing; UK: 1992. p. 804. [Google Scholar]
- 2.Bailey LH. Manual of Cultivated Plants. Mcmillan Publishing Company; New York: 1949. p. 791. [Google Scholar]
- 3.Chopra RN, Badhwar RL, Ghosh S. Poisonous Plants of India. Indian Council of Agricultural Research; New Delhi: 1965. [Google Scholar]
- 4.Kawase M, Motohashi N, Satoh K, Sakagami H, Nakashima H, Tani S, Shirataki Y, Kurihara T, Spengler G, Wolfard K, Molnar J. Phytother Res. 2003;17:495. doi: 10.1002/ptr.1183. [DOI] [PubMed] [Google Scholar]
- 5.Mallavadhani UV, Panda AK, Rao YR. Phytochemistry. 1998;49:901. doi: 10.1016/s0031-9422(97)01020-0. [DOI] [PubMed] [Google Scholar]
- 6.Briand CH. Huntia. 2005;12:71. [Google Scholar]
- 7.Fallas AL, Thomson RH. J Chem Soc. 1968;18:2279. [Google Scholar]
- 8.Carter FL, Garlo AM, Stanley JB. J Agric Food Chem. 1978;26:869. [Google Scholar]
- 9.Shukla YN, Kapadia GJ. Indian J Pharm Sci. 1989;51:73. [Google Scholar]
- 10.Tyihák E, Mincsovics E, Kalász H. J Chromatogr. 1979;174:75. [Google Scholar]
- 11.Nyiredy-Mikita S, Erdelmeier CAJ, Dallenbachtoelke K, Nyiredymikita K, Sticher O. J Nat Prod. 1986;49:885. [Google Scholar]
- 12.Dallenbach-toelke K, Nyiredy S, Meier B, Sticher O. J Chromatogr. 1986;365:63. [Google Scholar]
- 13.Mincsovics E. J Planar Chromatogr. 2004;17:411. [Google Scholar]
- 14.Galand N, Ernouf D, Montigny F, Dollet J, Pothier J. J Chromatogr Sci. 2004;42:130. doi: 10.1093/chromsci/42.3.130. [DOI] [PubMed] [Google Scholar]
- 15.Gombosuren N, Novak Z, Kotschy A, Mincsovics E, Dibo G. J Biochem Biophys Methods. 2007;69:239. doi: 10.1016/j.jbbm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Khan RM, Rwekika E. Phytochemistry. 1998;49:2501. [Google Scholar]
- 17.Yoshida M, Mori K. Eur J Org Chem. 2000;7:1313. [Google Scholar]
- 18.Mothilal KK, Inbaraj JJ, Chignell CF, Gandhidasan R, Murugesan R. J Photochem Photobiol A, Chem. 2004;163:141. [Google Scholar]
- 19.Hussein AA, Barberena I, Capson TL, Kursar TA, Coley PD, Solis PN, Gupta MP. J Nat Prod. 2004;67:451. doi: 10.1021/np030223d. [DOI] [PubMed] [Google Scholar]
- 20.Pinheiro MLB, Fernandes AF, Monte FJQ, Villar JDF, Cruz ER. Quim Nova. 2004;27:188. [Google Scholar]
- 21.Sholichin M, Yamasaki K, Kasai R, Tanaka O. Chem Pharm Bull. 1980;28:1006. [Google Scholar]
- 22.Peng C, Bodenhausen G, Qiu S-X, Fong HHS, Farnsworth NR, Yuan S-G, Zheng C-Z. Magn Reson Chem. 1998;36:267. [Google Scholar]
- 23.Monaco P, Previtera L. J Nat Prod. 1984;47:673. [Google Scholar]
- 24.Guvenalp Z, Ozbek H, Kuruuzum-Uz A, Kazaz C, Demirezer LO. Turk J Chem. 2009;33:667. [Google Scholar]
- 25.Du Y-C, Lin A-S, Wu C-C, Hsieh PW, Chen Y-H, Chen I-H, Chen S-L, Yen H-F, Lubken T, Chang F-R, Wu Y-C. Planta Med. 2009;75:848. doi: 10.1055/s-0029-1185438. [DOI] [PubMed] [Google Scholar]
- 26.Bohlmann F, Knoll KH, Zdero C, Mahanta PK, Grenz M, Suwita A, Ehlers D, Van NL, Abraham WR, Natu AA. Phytochemistry. 1977;16:965. [Google Scholar]
- 27.Wang X-N, Wedge DE, Tabanca N, Johnson RD, Cutler SJ, Pace PF, Smith BJ, Zhou L-G. Natural Products Communications. 2008;3:1079. [Google Scholar]
- 28.Wedge DE, Smith BJ, Quebedeaux JP, Constantin RJ. Crop Protection. 2007;26:1449. [Google Scholar]
- 29.Cutler HG, Cutler SJ. Biologically active natural products: Agrochemicals. CRC Press; Boca Raton: 2000. p. 1. [Google Scholar]
- 30.Wedge DE, Duke SO. Natural Products for Pest Management. American Chemical Society Symposium Book Series. 2006;927:152. [Google Scholar]
- 31.Tabanca N, Bedir E, Ferreira D, Slade D, Wedge DE, Jacob MR, Khan SI, Kirimer N, Baser KHC, Khan IA. Chem Biodivers. 2005;2:221. doi: 10.1002/cbdv.200590005. [DOI] [PubMed] [Google Scholar]
- 32.Meazza G, Dayan FE, Wedge DE. J Agric Food Chem. 2003;51:3824. doi: 10.1021/jf0343229. [DOI] [PubMed] [Google Scholar]
- 33.Wedge DE, Kuhajek JM. SAAS Bull Biochem Biotech. 1998;11:1. [Google Scholar]