

Abstract
To identify new aminothiol radioprotectors that are active when applied topically and have fewer side effects when administered systemically, a new family of aminothiol radioprotectors was designed and synthesized. Three key elements in the aminothiol design were, (1) small size for efficient transmembrane diffusion, (2) positive charged amines in alkyl backbone for strong ionic interaction with DNA backbone, and (3) a perpendicular, alkyl side-chain with a terminal thiol that is projected away from the DNA backbone to enable reactive oxygen species scavenging around DNA. Several in vitro assays were used to characterize the prototype aminothiol, PrC-210, for efficacy: protection against reactive oxygen species-induced plasmid DNA nicking, mass spectrometry to detect aminothiol-reactive oxygen species by-products, S. typhimurium mutagenesis, human cell growth inhibition, Western blot for p21 expression, and FACS analysis. Additionally, two in vivo assays were used to assess radioprotective efficacy; a Sprague-Dawley rat dorsal skin radiodermatitis assay was developed to screen for aminothiol efficacy when topically applied, and ICR mouse survival was scored after systemic PrC-210 administration and whole-body radiation. PrC-210 efficiently scavenged reactive oxygen species and completely protected supercoiled plasmid DNA against reactive oxygen species-induced damage. Neither PrC-210 nor its analog PrC-211 were bacterial mutagens. In cell culture, PrC-210 application to diploid human fibroblasts showed: (1) inhibition of cell growth with an IC70 of 4.1 mM, (2) induced levels of p21 expression, and (3) a G1/S-cell cycle block that was reversed after washout of PrC-210-containing medium. In rodents, PrC-210 was an effective radioprotector showing: (1) complete prevention of Grade 2–3 radiodermatitis when applied topically (370 mM in ethanol:propylene glycol:water solution) prior to skin irradiation, (2) complete prevention of Grade 2–3 radioder-matitis when administered by i.p. injection (200 μg/g of body weight) before skin irradiation, (3) 100% survival of mice from an otherwise 100% lethal dose of whole-body radiation (8.75 Gy) when administered by i.p. injection (252 μg/g of body weight = 0.5 × maximum tolerated dose) before irradiation, and (4) a dose reduction factor of 1.6, the same as amifostine. These data suggest that the PrC-210 aminothiol is a plausible candidate for drug development as a human pre-exposure radioprotector.
INTRODUCTION
Worldwide, nearly 5 million patients receive cancer therapy each year, of which 70% will receive fractionated radiotherapy. An estimated 3.5 million courses of cancer radiotherapy are administered every year, and many of these patients will develop radiation dermatitis (1, 2). Radiation dermatitis can lead to severe irritation and pain, skin infections, and cessation or gaps in radiotherapy treatment. Hendry et al. (3) and Bese et al. (4) reported a 1.4–1.6% increase in local tumor recurrence for each missed day or gap in treatment within a standard fractionated radiotherapy protocol.
The use of a new topically applied radioprotector such as PrC-210 could have significant impact on the treatment of breast cancer patients. For example, to prevent radiation dermatitis in post-surgical breast cancer patients, clinical staff would apply PrC-210 shortly before radiotherapy each day. Prevention of radiation dermatitis would allow breast cancer patients to continue with a radiotherapy course uninterrupted. Additionally, because of its protective efficacy, a topical radioprotector such as PrC-210 may allow breast cancer patients to receive a higher dose of radiation. Studies have shown significantly improved five-year-survival rates in breast cancer patients who receive 20–32% increases in radiation dose (5).
Amifostine, an aminothiophosphonate radioprotector developed by the U.S. Antiradiation Drug Development Program from 1959 to 1988, has been the focus of a large number of important preclinical and clinical studies, including prevention of some cancer therapy side effects (6, 7). Basic concepts governing thiol usage and positively charged amine(s) to foster DNA interaction emerged from this and later work (8). Previous efforts to use amifostine or other agents for prevention of radiation dermatitis in breast cancer patients have not been successful (9, 10).
To identify new radioprotectors, which could be applied topically before irradiation of cancer patients, we designed and synthesized a new family of thiolated polyamines (11). This was followed by an extended effort to evolve the larger polyamines into a small family of low-molecular-weight aminothiols that we expected would be more efficiently delivered into skin and mucosal surfaces to protect skin stem cell populations prior to irradiation (12). The prototype aminothiol from this group is PrC-210 shown in Fig. 1.
FIG. 1.

PrC-210 and PrC-211 aminothiol structures and design parameters.
MATERIALS AND METHODS
Chemicals
Synthesis of the PrC-210 aminothiol molecule is described separately (12, 13). Purities of the PrC-210 and PrC-211 tested here were each greater than 98% as assayed by mass spectrometry and magnetic resonance (NMR) spectroscopy. In addition to the PrC-210 and PrC-211 structures shown in Fig. 1, 15 additional aminothiol and thiolated polyamine analogs have been synthesized. Mass spectrometry of aminothiols and their oxidative metabolites was performed at the Mass Spectrometry facility in the University of Wisconsin Biotechnology Center.
Animals
Sprague-Dawley female rats (35–40 g) and ICR female mice (20–25 g) were purchased from Harlan Laboratories (Indianapolis, IN). Both rats and mice were maintained on a 12 h light/dark cycle and provided water ad libitum and Harlan 8604 (rats) or 5305 (mice) lab chow. Before irradiation, rats were anesthetized with isoflurane and the fur on their backs was shaved using Oster clipper. Before irradiation, to firmly position rats on the lead plate within the irradiator, rats received i.p. injections of pentobarbital, 30 μg/g of body weight (μg/g BW). Rats received one dose of 17.3 Gy to their backs and 13 days later they were anesthetized using isoflurane and their backs were photographed. Animal procedures were approved by the University of Wisconsin (Protocol no. M0476).
Doses of the aminothiol drug administered to mice are provided in units of μg/g of body weight (e.g., 422 μg/g), because the mice are small, generally 20–30 g in body weight rather than ferrets or small primates that will exceed 1 kg in weight. Conversion of μg/g to mg/kg is straightforward, the number is unchanged just the milligram/kilogram descriptor is interchanged.
Irradiation and Aminothiol Treatment
Rats were irradiated in a J. L. Shepherd 137Cs irradiator. Two anesthetized rats were positioned on a lead plate that allowed the two 1.5 × 3.0 cm windows in the plate to irradiate the rats’ backs. The standard radiation exposure (5.15 min/17.3 Gy), or longer exposures (up to 12.5 min/41.7 Gy) to inflict more severe radiation dermatitis, were calibrated with TLDs and administered at a rate of 3.34 Gy/min. For topical aminothiol treatments, PrC-210 was dissolved in a delivery vehicle of ethanol:propylene glycol:water (50:30:20 or a variations of this) and in the standard administration, aliquots of 30 μl, 25 μl, 25 μl and 25 μl were applied and uniformly spread over the 1.5 × 3.0 cm area to be irradiated (marked with ink dots) at −2 h, −1 h,− 30 min and −10 min. In one experiment, subsets (1 to 4 applications) of the four topical doses were applied to determine their individual efficacy. Rats received i.p. pentobarbital for anesthesia at 10 min and were irradiated at 0 min. Some rats received i.p. injections of PrC-210 dissolved in water (pH adjusted to 5.5) 30 min before irradiation. In the standard radiodermatitis assay for screening drug efficacy, rats received 17.3 Gy radiation to the skin from Cs137 source at a rate of 3.34 Gy/min. Severity of radiation dermatitis in rats was scored using the same criteria used to score human radiation dermatitis (i.e., http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf). Digital images of the irradiated site, 13 days after irradiation, were scored visually as to the percentage of the irradiated field not covered by scab or wet desquamation.
For mouse aminothiol toxicity assays, mice received i.p. injections of PrC-210 dissolved in water (pH 5.5). Mice were observed continuously for 8 h to record initial, nonlethal aminothiol toxicities and were observed after an additional 16 h to determine their death or survival at 24 h after injection. There were 4–8 mice per dose level group.
For mouse radiation survival assays, unanesthetized mice received 8.63 or 8.75 Gy of whole-body radiation. Mice received either i.p. water or i.p. PrC-210 dissolved in water 30 min prior to irradiation. There were either 16 or 20 mice per individual treatment group. Mice were observed for 40 days after irradiation.
To determine whether topically applied PrC-210 had a tumor protective effect, xenograft tumors growing directly beneath the topical drug site were irradiated. Nude mice (female, nu/nu, 20–24 g) were obtained from Harlan Laboratory and maintained in sterilized bedding and cages on a 12 h light/dark cycle. Tumorigenic cell lines, B16 melanoma and A431 epidermoid carcinoma, were grown in DME with 10% fetal bovine serum and gentamycin, and 5 × 106 cells were injected subcutaneously on the dorsal backs of the mice. Experimental details are provided in the figure legends.
Distribution of [14C] PrC-210 Topically Applied to Rat Skin
[14C]-methyl-PrC-210 was synthesized by GE Healthcare (Amersham) as previously described (26).
[ 14C]-PrC210 distribution studies were carried out by Covance Laboratories (Madison, WI) on a fee-for-service basis under review of the Covance IACUC.
PrC-210 at concentrations of 370 mM or 1850 mM, in a delivery vehicle of 50:30:20 (ethanol:propylene glycol:water), each containing a radioactive dose of 100 μCi, was applied to shaved skin on the dorsal back of 300 g rats within a 12.4 cm2 rectangular enclosure. The dose was applied in a volume of 200 μl using a syringe with a ball-tipped gavage needle and spread with a glass rod. The dosing site was then protected with a nonocclusive cover, and Elizabethan collars were fitted to the rats. Blood samples were collected with an indwelling venous catheter at 0.5, 1, 2, 4, 6, 8, 24 and 48 h after application. Following the last blood collections, rats were sacrificed at 48 h. The occlusive covers were retained for radioanalysis and the entire skin dose site was washed with water and gauze, both of which were retained for radioanalysis. The entire skin dose site and an appropriate amount of skin surrounding the dose site were excised and retained for radioanalysis. Urine, urine wipes of the metabolic cage, and fecal material were also collected for radioanalysis.
Cell Culture, Flow Cytometry, p21 Analysis, and Bacterial Mutagenesis
Normal, diploid human fibroblasts were grown in DME containing 20% fetal bovine serum and penicillin/streptomycin (14). Cells were harvested by trypsinization, fixed in ethanol-containing phosphate-buffered saline and stained with propidium iodide as described (15). Flow cytometry analysis of the propidium iodide-stained cells was done using a FACSCaliber flow cytometer to quantify the percentages of cells in each cell cycle compartment.
To quantify p21 induction in aminothiol-treated human fibroblasts, p21 Western blot analysis was carried out using cells harvested and nuclear extracts were prepared as described (15). Proteins were separated on 0–12% SDS-PAGE gels and immunoblotted on PVDF membranes using a p21 antibody (no. 556431; BD-Pharmingen, Inc.).
To determine if the DNA-associating PrC-210 and/or PrC-211 molecules were mutagens, they were tested using S. typhimurium strains TA98 (frameshift mutations) and TA100 (point mutations). All materials for assays were provided by Moltoxt® Molecular Toxicology, Inc., Boone, NC.
RESULTS
New Aminothiol Radioprotector-ROS Scavengers
Existing aminothiols, like the amifostine prodrug and its thiol metabolite WR-1065, continue to be widely studied in research and clinical settings. However, side effects and lack of efficacy when amifostine is applied topically have limited their use. To identify new aminothiol radioprotector-chemoprotector molecules, we used a directed evolution strategy, beginning with a family of thiolated polyamines (11) and the design goals summarized in Fig. 1. Over three years, there were nine iterative cycles of molecule design, synthesis, and testing of candidates for radioprotective efficacy. Two final structures are shown in Fig. 1, and 15 other analogs have been synthesized and tested to date.
New Molecules are Efficient ROS Scavengers
Known protective aminothiols associate ionically with DNA and efficiently scavenge oxygen free radicals (8). Therefore, these two chemical characteristics were assessed first. Figure 2 shows that adding PrC-210 (0–70 mM) to supercoiled plasmid DNA in a standard hydroxyl radical generating system provided complete protection with no detectable nicking of the DNA that was seen in the “0 mM PrC-210” incubations (lanes 4 and 5). Consistent with its ROS scavenging role, mass spectrometry of aliquots taken from the aminothiol-ROS incubations (Fig. 3) showed only the free PrC-210 thiol before addition of the ROS generator, and the oxidized disulfide PrC-210 form and an MW = 164 ion (PrC-210-S-O) after ROS production.
FIG. 2.

Agarose gel separation of supercoiled (S) and nicked (N) forms of pUC19 plasmid DNA after exposure of plasmid DNA to reactive oxygen species generator reaction. Supercoiled DNA was pre-incubated with water (lanes 2, 4, 5) or increasing concentrations of PrC-210 aminothiol (lanes 3, 6–12) and then exposed to ROS generator (lanes 4–12). Aliquots of each incubation were applied to gel, electrophoresed, stained with EtBr and photographed. Duplicate gels gave the same result.
FIG. 3.

Mass spectrograms showing that before the •OH generation reaction you see only the PrC-210 thiol (149.4 amu), but after the •OH generation reaction you no longer see the 149.4 amu thiol signal, but do see 3 peaks with amu signatures that represent either the oxidized PrC-210 disulfide (295.4 and 148.4) or a 163.2 amu peak that indicates direct modification of the PrC-210 sulfur with an oxygen atom.
Based upon these initial results showing DNA protection, we assumed that PrC-210 and PrC-211 were associating ionically with DNA and were scavenging ROS from the DNA environs. To initially determine whether there were negative consequences to this ionic association in living, replicative cells, the two aminothiols were tested for their mutagenicity in standard Salmonella test strains. The data of Table 1 show that they produce neither frameshift mutations (strain TA98) nor point mutations (strain TA100) at concentrations up to 7 mM.
TABLE 1. Lack of Mutagenicity of PrC-210 and PrC-211 Aminothiols in Frameshift (TA98) and Point Mutation (TA100) Salmonella typhimurium Reporter Strains.
| Molecule | Concentration (μM) |
Colonies/plate (mean ± SD) |
|||
|---|---|---|---|---|---|
| Strain TA98 |
Strain TA100 |
||||
| −S9 | +S9 | −S9 | +S9 | ||
| 0 | 52 ± 11* | 59 ± 9 | 133 ± 6 | 165 ± 20 | |
| PrC-210 | |||||
| 30 | 43 ± 2 | 6 ± 10 | 121 ± 8 | 119 | |
| 100 | 51 ± 6 | 64 ± 4 | 116 ± 22 | 127 ± 8 | |
| 400 | 58 ± 14 | 55 ± 3 | 128 | 144 ± 14 | |
| 1000 | 53 ± 3 | 54 ± 6 | 101 ± 19 | 148 ± 5 | |
| 4100 | 55 ± 5 | 55 | 134 ± 20 | 115 ± 33 | |
| 7000 | 51 ± 5 | 65 ± 9 | 128 ± 11 | 149 ± 18 | |
| PrC-211 | |||||
| 30 | 50 ± 2 | 61 ± 4 | 125 | 137 | |
| 100 | 47 ± 8 | 52 ± 5 | 139 | 114 | |
| 400 | 49 ± 6 | 52 ± 9 | 93 | 135 | |
| 1000 | 54 ± 12 | 53 ± 4 | 121 | 135 | |
| 4100 | 53 ± 3 | 61 ± 6 | 130 | 148 | |
| 7000 | 58 ± 9 | 60 | 133 | 126 | |
| Aminoanthracene | 87 | >1800 | 177 | 570 ± 90 | |
| Daunomycin | 804 | ||||
Mean and standard deviation of at least three plates.
A Reversible Cell-Cycle Block
To determine what effect(s) PrC-210-DNA interaction might have upon normal human cell cycle progression, we added PrC-210 to the medium of normal diploid human fibroblasts growing in cell culture. Figure 4A shows significant inhibition of cell growth when PrC-210 was added, with an IC70 dose of 4.1 mM. p21 induction after polyamine treatment has been shown to be the cause of a G1/S cell cycle block (15). To assess a possible p21 response to PrC-210 aminothiol exposure, nuclear extracts were prepared from both untreated normal human fibroblasts and the same cells treated with 4.1 mM PrC-210 for 30 h. p21 was found to be significantly elevated in PrC-210-treated cells (Fig. 4B). To determine whether a cell cycle block was associated with the induced levels of p21, control fibroblasts that were expanding exponentially (doubling time ~20 h) and fibroblasts exposed to PrC-210 for 30 h were harvested, fixed and stained with propidium iodide, and analyzed by flow cytometry to determine their cell-cycle distribution. Figure 4C (control) shows that exponentially expanding cells had a large S-phase fraction (53%) indicating their unrestrained growth. A 30 h presence of PrC-210 at 4.1 mM almost depleted (8%) the S-phase fraction of cells (Fig. 4C, +PrC-210). Significantly, washout of the PrC-210 medium (Fig. 4C, +PrC-210+washout) almost completely restored the S-phase compartment (40%) within 48 h. This is in contrast to the G2/M cell cycle block that has been reported in one setting for the active aminothiol metabolite of amifostine, WR-1065 (16).
FIG. 4.

Panel A: Growth inhibition of diploid human fibroblasts by the addition of increasing concentrations of PrC-210 aminothiol for the 30 h prior to cell counting. Mean ± SE of multiple wells at each point are plotted. Panel B: Western blot analysis of p21 protein in nuclear extracts from log-phase diploid human fibroblasts that received either control medium or medium containing 4.1 mM PrC-210 (IC70) for the 30 h before cell harvest and isolation of nuclear extracts. Panel C: Flow cytometry analysis of diploid human fibroblasts. Log-phase cells were exposed either to: (1) medium alone (control), (2) medium containing 4.1 mM PrC-210 for the last 30 h before harvest (+PrC-210), or (3) medium containing PrC-210 for 30 h followed by washing (3 medium rinses) and fresh medium for the last 48 h before harvest (+PrC-210 washout). Replicate treatment groups showed the same outcomes.
A Topical Radioprotector
Because prevention of radiation dermatitis in humans was the focus of this work, we developed a straightforward radiation dermatitis assay using 4–6-week-old rats. The assay tested drug uptake/efficacy and radiation response in the epidermis of the rat’s dorsal back, whose structure is very similar to human chest, axilla and neck epidermis where human radiation dermatitis commonly occurs. The Fig. 5A sketch shows how rat backs were irradiated through a 1.5 × 3.0 cm window in a 1-inch-thick lead plate that was positioned as close as possible to the Cs137 source in the irradiator. Figure 5B shows how increasing radiation doses produced the same increases in rat radiation dermatitis severity as seen in human radiotherapy patients. We empirically chose 17.3 Gy as a standard dose because it routinely induced Grade 2–3 dermatitis in irradiated rats, and prevention of this provided a stringent test of a candidate radioprotector’s efficacy. In Fig. 5C and E, a Grade 3 dermatitis is seen in irradiated control rats, but at or above 370 mM topical PrC-210, dermatitis is completely prevented. Because topical PrC-210 completely prevented radiodermatitis that sometimes included wet desquamation in the irradiated field we speculate that PrC-210 might also protect radiotherapy patients who receive boost doses above the standard 50–60 Gy fractionated course.
FIG. 5.

Complete prevention of radiation dermatitis conferred by topical or systemic administration of PrC-210 prior to irradiation. Panel A: Schematic showing placement of Sprague-Dawley rats on lead plate with 2 windows. Panel B: Increasing radiation dose gave radiation dermatitis of increasing severity (Grade 1–>4) scored 13 days after irradiation. Panel C: Topical application of 370 mM PrC-210 in 50:30:20 (ethanol:propylene glycol:water) delivery vehicle to skin before irradiation completely prevented the Grade 2–3 radiation dermatitis. Panel D: A single i.p. injection of PrC-210 at −30 min before the irradiation completely prevented the Grade 2–3 radiation dermatitis. At least 3 rats were present in each of the C–F treatment groups. Panel E: Dose-dependent prevention of radiation dermatitis by topical application of PrC-210 before irradiation. With topical application of PrC-210 in an early delivery vehicle of 0:95:5 (ethanol:propylene glycol:water), dose-dependent prevention of Grade 2–3 radiation dermatitis can be seen, but much higher concentrations of PrC-210 are required. Panel F; PrC-210 dissolved at either 370 mM or 740 mM in a 50:30:20 delivery vehicle was topically-applied at the indicated times before irradiation. Severity of radiation dermatitis was scored 13 days later.
Four topical applications of PrC-210 over 2 h were required to achieve 98% prevention of radiation dermatitis (Fig. 5F), and 1 to 2 applications of the ROS scavenger conferred much less protection. As a possible explanation for this, in a separate study (17) we show that the PrC-210 thiol is capable of reacting in vitro with both cysteine-thiol and glutathione-thiol to form mixed disulfides. When applied to skin, competing reactions with these physiologic thiols would require higher applied doses of PrC-210 to maintain a labile pool of PrC-210 thiol available for ROS-scavenging.
Studies to optimize PrC-210 delivery were also performed with radiodermatitis prevention as the scored endpoint (not shown). To summarize: (1) PrC-210 (e.g., 370 mM) dissolved in vehicles with a higher alcohol percentage (e.g., 50:30:20) achieved the greatest potency, (2) PrC-210 (e.g., 1500 mM) dissolved in vehicles containing 75% water still conferred protection, and (3) PrC-210 provided complete prevention of radiation dermatitis when the drug was administered i.p. (200 μg/g BW) 30 min before a 17.3 Gy irradiation, thus providing initial proof of concept for PrC-210 as a systemic radioprotector. (Fig. 5D).
Dissolution of the PrC-210-HCl salt in water results in a pH of ~3.0. Aliquots of this aqueous solution were adjusted to pH 5.5 and 8.0, and we found that after 1 week at room temperature in the dark, there was, <1%, and >98%, respectively, of the PrC-210 disulfide in the two solutions when assayed by mass spectrometry. Therefore, aqueous PrC-210 solutions for i.p. injection were adjusted to pH 5.5 prior to use to avoid an injected bolus of pH 3.0 liquid.
Although PrC-210 was known to be an aminothiol, as a new molecule we wanted to corroborate its mechanism of action. In a brief PrC-210 structure-activity study (Fig. 6), individual structural elements of the PrC-210 molecule were each tested (at 370 mM) to determine their respective contribution to its radioprotective phenotype. Only when combining both the ROS-scavenging thiol function and the positive charged alkyl backbone was 100% radiodermatitis prevention achieved, thus confirming the value of pairing a thiol with a flexible, alkyl-amine backbone.
FIG. 6.

Functional contribution of each PrC-210 design element to its radioprotective phenotype. Rats received either topical vehicle or topical vehicle containing 370 mM of the indicated compounds. Following irradiation, radiodermatitis severity was scored 13 days later. Each treatment group contained the pictured rats.
Distribution of Topically Applied [14C] PrC-210
To ascertain the pharmacokinetic fate of PrC-210 that was applied to rat skin [14C]-PrC-210 was synthesized, purified, added to unlabeled PrC-210 and applied to a defined area of dorsal skin on large rats. The rats had in-dwelling venous catheters installed to enable blood sampling at set times. Three test groups, with 3 rats per group were explored. The pharmacokinetics and recovery data are summarized in Table 2. Either a PrC-210 dose representing a single application (Group 1, 370 mM) or a single dose representing all four standard applications of PrC-210 (Groups 2 and 3, 1850 mM PrC-210) were applied. In Group 3, 300 mM (−) norepinephrine-bitartrate was also added to the vehicle formulation to test whether co-application of the vasoconstrictor would increase retention of PrC-210 at the application site.
TABLE 2
| Group | n | Dose and route |
mg PrC-210 applied |
NEp | Cmax (ng-eq/g) | Tmax (h) | Recoverya (excreta) |
Recovery (carcass) |
Recoveryb (systemic) |
Recoveryc (skin) | Recoveryd (total) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 370 mM 100 μCi Dermal |
16.28 | - | 257 | 4 | 6.18% | 0.52% | 6.70% | 41.54% | 49.4% |
| 2 | 3 | 1850 mM 100 μCi Dermal |
81.40 | 1,630 | 8 | 13.28% | 1.84% | 15.12% | 47.16% | 66.0% | |
| 3 | 3 | 1850 mM 100 μCi Dermal |
81.40 | 726 | 48 | 6.88% | 1.53% | 8.41% | 60.46% | 70.0% | |
| 4 | 3 | 370 mM 100 μCi Dermal |
16.28 | 0.22% of topically administered dose was recoverable in expired air over 48 h | |||||||
Recovery (excreta) = urine + urine wipes + feces.
Recovery (systemic) = Recovery (excreta) + Recovery (carcass).
Recovery (skin) = skin (dose site) + skin wash.
Recovery (total) = Recovery (systemic) + Recovery (skin) + cage wash + cage wipe + enclosure + nonocclusive cover.
Forty-eight hours after topical application, 40–60% of the administered radioactivity was recovered from the application site, either in the skin wash or in the skin. This skin recovery was highest in Group 3 with vasoconstrictor added to the topical vehicle formulation. The main route of elimination of absorbed radioactivity was past through the urine.
For the low dose, the mean Cmax of radioactivity in plasma was 257 ng equivalents of [14C] PrC-210/g at 4 h (Group 1). For the high dose without norepinephrine, the mean Cmax of radioactivity was 1630 ng equivalents [14C] PrC-210/g at 8 h (Group 2). For the high dose rats with norepinephrine (Group 3), the mean Cmax of radioactivity in plasma was delayed to 48 h and was lower; the mean Cmax was 726 ng equivalents [14C] PrC-210/g.
Based on the observation that the total recoveries of radiolabeled PrC-210 were lower than expected for a mass balance study, an additional study group was added to test the hypothesis that the low recoveries might have been caused by PrC-210 metabolism and the excretion of radiolabel in expired air. The data from Group 4 indicated that volatile radiolabeled compounds were present in expired air after dermal administration, but the percentage was minimal (0.22%) and did not account for the missing radioactivity. Additional investigations were then performed to determine the amount of radioactivity binding to glass and the enclosures. The results showed that high levels of radioactivity were associated with the enclosures and with the attached skin, indicating that drug-related radioactivity was bound to both and was not extractable for counting.
An estimate of the amount (mass) of PrC-210 that crossed the skin barrier over 48 h and entered the systemic circulation is 1.1 mg for Group 1 rats (i.e., 16.28 mg × 6.7%), 9.8 mg for Group 2 rats and 6.8 mg for Group 3 rats. For 300 g rats, this gives systemic doses ranging from 1.1 mg/300 g BW (or 3.7 μg/g BW) to 9.8 mg/g BW (or 32.7 μg/g BW). These systemic doses, administered over a 48 h after topical application period are in comparison to the i.p. 0.5 maximum tolerated dose (MTD) doses of PrC-210 of 422–504 μg/g BW given as a single i.p. bolus.
Absence of Tumor-Protective Effect
When applying a radioprotector topically to skin above a to-be-irradiated tumor mass a logical concern is whether it will also protect the underlying tumor mass during a radiotherapy course. To address this question, tumor protection studies with two tumor cell lines (B16 melanoma and A431 epidermoid carcinoma) were done. These tumor cell lines grow aggressively as xenograft tumors so several replicate nude mice carrying tumor cell plaques (3 days post-injection) of subcutaneous B16 or A431 tumor cells received increasing doses of Cs137 radiation to the tumor plaques. Initially, post-irradiation growth (Fig. 7A and B) of tumors provided a rough estimation of I70 radiation doses for both tumor types. In a subsequent experiment, multiple, replicate mice carrying either B16 or A431 xenograft plaques (3 days post tumor cell injection) either had four applications of topical vehicle or four applications of 1850 mM PrC-210 in 50:30:20 vehicle (ethanol:propylene glycol:water) applied to skin above the subcutaneous tumor over a 2 h period. Mice then received either 9.2 Gy to the B16 tumor plaques or 12.1 Gy to the A431 tumor plaques. Growth of the irradiated tumors and unirradiated control tumors was monitored for 14 days. The averages ± SEM for areas of the xenografts are graphed in Fig. 7C and D. There was no significant difference in tumor growth when PrC-210 was applied above either B16 or A431 xenografts, even when PrC-210 was applied at 5× its normal effective concentration of 370 mM (Fig. 7C and D).
FIG. 7.

Lack of tumor-protective effect of a topically applied, 5× dose of PrC-210 to skin directly over the subcutaneous xenograft tumors in nude mice. Panel A: B16 melanoma cell and panel B: A431 epidermoid carcinoma cell xenograft tumors in nude mice were allowed to grow 3 days post-injection to small, subcutaneous plaques, and they were then exposed to increasing doses of external radiation from a Cs137 source. Tumors were positioned within a 1.5 × 3.0 cm hole in a one-inch-thick lead plate for tumor irradiation. The radiation doses indicated by the respective arrows in panels A and B were chosen because they gave ~70% reduction in tumor growth rate, compared to unirradiated control tumors. In a subsequent experiment, either topical vehicle (50:30:20, ethanol:propylene glycol:water) or topical vehicle containing 1850 mM PrC-210 was applied 4 times in 2 h before irradiation. In panels C and D, no significant difference was seen (P > 0.05) between radiation ± in mice treated with topical vehicle or topical, 5X PrC-210 before tumor irradiation. There were 4 nude mice in each treatment group. Mean ± SE values are plotted.
A Systemic Pre-Exposure Radioprotector
An i.p. injection of PrC-210 was found to prevent radiation dermatitis (Fig. 5D), thus we decided to test the broader utility of PrC-210 as a systemic radioprotector in mice exposed to whole-body radiation. Assays to first determine the i.p. PrC-210 MTD were done with ICR mice. A high dose (700 μg/g BW) was 100% lethal; the mice rapidly (min) developed severe muscle spasm followed by death (min). A lower i.p. dose (500 μg/g BW) causes either a transient response in which the mice are sleepy but then rebound over several hours and survive, or they die within 1–2 h. The i.p. PrC-210 MTD, which equals the i.p. LD10 dose of this aminothiol (18), was 422 μg/g BW when extrapolated from the best fit curve (Fig. 8A). The LD10 dose based upon a subsequent probit analysis (19) of the percentage survival data provided a higher LD10 dose of 504 μg/g BW (not shown). It should be noted that these i.p. MTD of 422 and 504 μg/g BW are similar to a published i.p. MTD of 800 μg/g BW for amifostine, and that the muscle spasm seen with near MTD of PrC-210 are similar but not the same as the muscle spasms observed in mice that received a near MTD of amifostine.
FIG. 8.
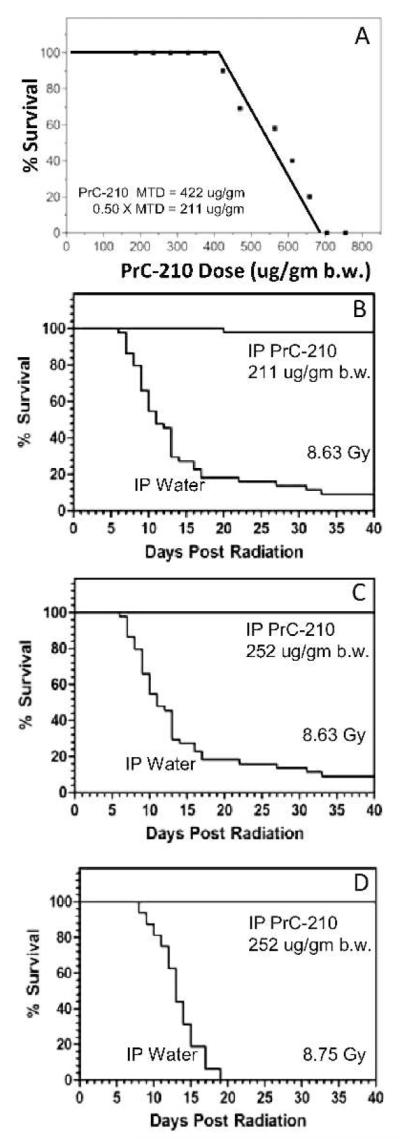
Radioprotective effect of PrC-210 in pre-exposure mouse models. Panel A: To establish PrC-210 toxicity and MTD dose, mice (4–8 per data point) received a single i.p. PrC-210 injection at the indicated doses. Panel B: Mouse survival either after i.p. PrC-210 (211 μg/g BW) or water 30 min before 8.63 Gy whole-body radiation. Panel C, Mouse survival after either i.p. PrC-210 (252 μg/g BW) or water 30 min before 8.63 Gy whole-body radiation. Panel D: Mouse survival after either i.p. PrC-210 (252 μg/g BW) or water 30 min before 8.75 Gy whole-body radiation. Each treatment group contained 20 mice.
To determine the radioprotective efficacy of PrC-210 when it was administered systemically before whole-body radiation, groups of 16 mice first received an i.p. injection of PrC-210, then were exposed to whole-body radiation, and were monitored for 35–40 days to record percentage survival. Whole-body radiation of 8.63 Gy gave a 7% survival rate for mice (Fig. 8B), which enabled χ2 statistical analysis of survival groups compared to vehicle controls. With survival parameters defined, the radioprotective efficacy of PrC-210 was measured. Treatment of mice with the 0.5× MTD of PrC-210 defined by best fit analysis (211 μg/g BW) 30 min before irradiation (8.63 Gy) resulted in 98% (49/50) survival (Fig. 8B). Treatment of mice with half of the PrC-210 MTD defined by probit analysis (252 μg/g BW) gave 100% survival for mice receiving the 8.63 Gy dose (Fig. 8C), but it also gave 100% survival for mice that received an otherwise 100% lethal dose of radiation, i.e., 8.75 Gy (Fig. 8D).
To better define PrC-210 radioprotective efficacy and enable its comparison to other known radioprotectors, 13 groups with16 mice each were either first treated with i.p. vehicle or PrC-210 (211 μg/g BW) and were then exposed to increasing doses of radiation to enable calculation of the PrC-210 dose reduction factor (DRF) (Fig. 9). The DRF for PrC-210 was found to equal 1.6, a number that is currently shared by amifostine and the CBLB502 apoptosis inhibitor, thus, PrC-210 is a strong pre-exposure radioprotector. The data also suggest that had we used the probit analysis 0.5 MTD of 252 μg/g BW, the DRF would be even greater.
FIG. 9.

Sixteen mice per group received either i.p. water or i.p. water containing the 0.5× MTD of PrC-210 (211 μg/bm body weight) 30 min before irradiation by Cs137 source. Survival was scored through day 35 post-irradiation. Dose reduction factor value of 1.6 for PrC-210 is indicated.
DISCUSSION
The prototype structure of a new family of aminothiols, PrC-210, was found to be an efficient in vitro ROS scavenger. When added to exponentially expanding bacterial or human cells, it was not a bacterial mutagen, but at millimolar concentrations it inhibited normal human cell growth. The PrC-210-associated G1/S cell cycle block presumably resulted from the induced levels of p21 that we saw, and when PrC-210 was washed out, the cells resumed normal growth. These data support the idea that PrC-210 could be used as a transient, systemic radioprotector in healthy humans and then washed out. Topically applied PrC-210 completely prevented the Grade 2–3 radiation dermatitis seen in control irradiated rats, and topical application of a 5× dose of PrC-210 to skin above growing tumor xenografts had no impact upon radiation killing of the tumor xenograft cells. This lack of systemic effect from topically applied PrC-210 is consistent with the pharmacokinetic data that showed only 6–15% transfer of topically applied PrC-210 into systemic circulation over 48 h period following topical administration. These data support the use of PrC-210 as a topical radioprotector to prevent radiation dermatitis in human cancer radiotherapy patients. An important part of a Phase I clinical study of PrC-210 would be to determine whether there was any skin toxicity at the application site over the course of a multi-week application schedule analogous to a standard fractionated radiotherapy course.
The first demonstration of a chemical radioprotector in animals was in 1949 when Patt and colleagues (20) showed that intravenous administration of cysteine significantly improved rat and mouse survival from a subsequent 80% lethal dose of whole-body radiation. This observation was greatly expanded in subsequent U.S. Army and Soviet programs to identify chemical radioprotectors (21). Many useful thiol radioprotectors emerged from these studies, and the most well studied is the aminothiophosphonate prodrug, amifostine, and its thiol metabolite, WR-1065 (22). Amifostine (Ethyol) is used clinically today to help reduce radiotherapy and chemotherapy side effects, and several clinical studies are ongoing in an effort to further expand its effectiveness in humans (23).
After analyzing the extensive amifostine and amifostine analog literature, we undertook this project to develop a small family of multifunctional aminothiol analogs that would be different from amifostine in several ways, including: (1) an alkyl backbone with one or more amines that enabled efficient binding and disruption of normal B DNA helical structure, (2) with binding and helical DNA disruption, induced expression of the p21 cell cycle regulator and its disappearance when aminothiol was washed out, (3) a thiol group displaced away from the DNA backbone on an alkyl side chain to scavenge ROS from the DNA environs, (4) a topically applied radioprotector (to skin) that did not require prodrug conversion to achieve radioprotective efficacy, (5) a molecule that could be administered orally to mammals and show systemic radioprotection, and (6) a molecule that elicited neither the nausea/emesis or hypotension/fainting side effects that are associated with systemic administration of amifostine.
Amifostine has not been effective when applied topically to skin or mucosa in humans, and significant systemic toxicities (nausea/emesis, hypotension/fainting) are seen with human amifostine use (24, 25). Because the mechanism(s) underlying amifostine-induced nausea and hypotension are unknown, predicting which candidates in our new aminothiol family would be free of these side effects was purely speculative. Though it’s beyond the scope of this paper, in a separate report (26), we show that PrC-210 elicits neither the retch/vomit response nor hypotension response in appropriate ferret and rat preclinical models, whereas amifostine elicits both responses in the same animal models at the same functional drug dose (i.e., 0.5× mouse MTD) as PrC-210. The structural and mechanistic basis for this difference is presently unknown.
Radiation dermatitis has been an observed side effect of radiation exposure for more than 100 years (27). In 1901 Pierre Curie produced a burn on his arm by intentionally holding a tube of radium close to the skin. Marie Curie was described in 1934 as having severely damaged fingers and hands as a result of radiation dermatitis. In this historical context, it would be very useful if we had a radioprotector formulation that could be applied daily before the 30+ radiation exposures in current, fractionated radiotherapy regimens that would diminish or eliminate radiation dermatitis. There are currently no effective clinical measures to prevent or diminish its severity. However, topical application of increasing doses of either PrC-210 or PrC-211 to rat skin before irradiation completely prevented the Grade 2–3 radiation dermatitis seen in vehicle controls (Fig. 5). Reducing radiation dermatitis severity also depended upon both delivery vehicle composition and application schedule. For example, the IC100 dose of PrC-210 for radiation dermatitis went from 1500 mM in 0:95:5 to 370 mM in 50:30:20 (ethanol:propylene glycol:water).
Following a topically applied dose of PrC-210 to skin, 41–60% of unabsorbed radioactivity was recovered from the dose site, in the skin wash and in the excised skin. Of the radioactivity absorbed, the main route of elimination was by the urine (90% or greater). Smaller amounts of radioactivity were excreted in the feces and in the expired air as CO2. Relatively little radioactivity remained in the carcass 48 h after a dermal dose. Dermal absorption was clearly reduced and slowed when the vasoconstrictor norepinephrine was co-administered with PrC-210. Preliminary analysis (not shown) of rat blood samples showed that the large majority of the 14C cpm in blood was precipitable by TCA and that only a small percentage, less than 10%, remained soluble. The implication is that the 14C-labeled PrC-210 was covalently attached to precipitable blood proteins, most likely through disulfide formation with cysteine residues in blood proteins. This low blood presence of soluble PrC-210, combined with its low crossover rate for skin makes it very unlikely that topically administered PrC-210, a radio-protectant with a mass-action mechanism of action, can reach distant tumor sites in a biologically active form (free thiol) and protect such tumor sites from radiation-induced killing.
When administered systemically before whole-body irradiation, PrC-210 was as effective a radioprotector as amifostine; both have DRF of 1.6. However, the observations that PrC-210 is active when taken orally (26) and does not induce the retch/vomit or hypotension side effects observed in preclinical models with amifostine (26), opens new possibilities for the use of aminothiol radioprotectors in healthy humans. Some examples of healthy subjects who could benefit from this radioprotection include: (1) healthy patients who receive extensive medical radiation from extended or recurrent CT scans (28, 29) and (2) space travelers and military personnel in settings where fainting or impaired judgment are not acceptable side effects (30).
The utility of PrC-210 as a topical and systemic radioprotector makes it a plausible candidate for development as a human pre-exposure radioprotector.
ACKNOWLEDGMENTS
This work was supported by grants from NIH (R44-CA099307), the Wisconsin Alumni Research Foundation (WARF, University of Wisconsin-Madison) and ProCertus BioPharm, Inc. (Madison, WI).
REFERENCES
- 1.Kumar S, Juresic E, Barton M, Shafiq J. Management of skin toxicity during radiation therapy: a review of the evidence. J Med Imag Radiat Oncol. 2010;54:264–79. doi: 10.1111/j.1754-9485.2010.02170.x. [DOI] [PubMed] [Google Scholar]
- 2.Krasin MJ, Hoth KA, Hua C, Gray JM, Wu S, Xiong X. Incidence and correlates of radiation dermatitis in children and adolescents receiving radiation therapy for the treatment of paediatric sarcomas. Clin Oncol (R Coll Radiol) 2009;21:781–5. doi: 10.1016/j.clon.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 3.Hendry JH, Bentzen SM, Dale RG. A modeled comparison of the effects of using different ways to compensate for missed treatment days in radiotherapy. Clin Oncol. 1996;8:297–307. doi: 10.1016/s0936-6555(05)80715-0. [DOI] [PubMed] [Google Scholar]
- 4.Bese NS, Hendry JH, Jeremic B. Effects of prolongation of overall treatment time due to unplanned interruptions during radiotherapy of different tumor sites and practical methods for compensation. Int J Radiat Oncol Biol Phys. 2007;68:654–61. doi: 10.1016/j.ijrobp.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Poortmans PM, Collette L, Bartelink H, Struikmans H, Van den Bogaert WF, Fourquet A, et al. EORTC Radiation Oncology and Breast Cancer Groups The addition of a boost dose on the primary tumour bed after lumpectomy in breast conserving treatment for breast cancer. A summary of the results of EORTC 22881-10882 “boost versus no boost” trial. Cancer Radiother. 2008;12:565–70. doi: 10.1016/j.canrad.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Weiss JF, Landauer MR. History and development of radiation-protective agents. Int J Radiat Biol. 2009;85:539–73. doi: 10.1080/09553000902985144. [DOI] [PubMed] [Google Scholar]
- 7.Winczura P, Jassem J. Combined treatment with cytoprotective agents and radiotherapy. Cancer Treat Rev. 2010;36:268–75. doi: 10.1016/j.ctrv.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Zheng S, Newton GL, Gonick G, Fahey RC, Ward JF. Radioprotection of DNA by thiols: relationship between the net charge on a thiol and its ability to protect DNA. Radiat Res. 1988;114:11–27. [PubMed] [Google Scholar]
- 9.Montana GS, Anscher MS, Mansbach CM, Daly N, Delannes M, Carke-Pearson D, et al. Topical application of WR-2721 to prevent radiation-induced proctosigmoiditis. A Phase I/II trial. Cancer. 1992;69:2826–30. doi: 10.1002/1097-0142(19920601)69:11<2826::aid-cncr2820691131>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 10.Salvo N, Barnes E, van Draanen J, Stacey E, Mitera G, et al. Prophylaxis and management of acute radiation-induced skin reactions: a systematic review of the literature. Curr Oncol. 2010;17:94–112. doi: 10.3747/co.v17i4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fahl WE, Copp RR, Ochsner CE, Peebles DD, Fahl KL. Polyamine compounds and compositions for use in conjunction with cancer therapy. US patent US 7,414,154. http://patft.uspto.gov.
- 12.Fahl WE, Peebles DD, Copp RR. Amino thiol compounds and compositions for use in conjunction with cancer therapy. US patent US 7,314,959. http://patft.uspto.gov.
- 13.Copp RR, Peebles DD, Fahl WE. Synthesis and growth regulatory activity of a prototype member of a new family of aminothiol radioprotectors. Bioorg Medic Chem Letters. 2011;21:7426–30. doi: 10.1016/j.bmcl.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens CW, Brondyk WH, Burgess JA, Manoharan TH, Haene BG, Fahl WE. Partially transformed, anchorage-independent human diploid fibroblasts result from overexpression of the c-sis oncogene: mitogenic activity of an apparent monomeric platelet-derived growth factor 2 species. Mol Cell Biol. 1988;8:2089–96. doi: 10.1128/mcb.8.5.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kramer DL, Fogel-Petrovic M, Diegelman P, Cooley JM, Bernacki RJ, Porter CW. Effects of novel spermine analogues on cell cycle progression and apoptosis in MALME-3M human melanoma cells. Cancer Res. 1997;57:5521–7. [PubMed] [Google Scholar]
- 16.Grdina DJ, Constantinou A, Shigematsu N, Murley JS. Inhibition of topoisomerase II alpha activity in CHO K1 cells by 2-[(aminopropyl)amino]ethanethiol (WR-1065) Radiation Res. 1994;38:44–52. [PubMed] [Google Scholar]
- 17.Copp RR, Peebles DD, Soref CM, Fahl WE. Radioprotection conferred by PrC-210 aminothiol and its family of analogs. submitted. [Google Scholar]
- 18.Weiss JF. Pharmacologic approaches to protection against radiation-induced lethality and other damage. Envir Hlth Perspect. 1997;105:1473–8. doi: 10.1289/ehp.97105s61473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finney DJ. Statistical aspects of monitoring for dangers in drug therapy. Methods Inf Med. 1971;10:1–8. [PubMed] [Google Scholar]
- 20.Patt HM, Tyree EB, Straube RL, Smith DE. Cysteine protection against X irradiation. Science. 1949;110:213–4. doi: 10.1126/science.110.2852.213. [DOI] [PubMed] [Google Scholar]
- 21.Sweeney TR. A survey of compounds from the antiradiation drug development program of the U.S. Army Medical Research Development Command. Walter Reed Army Institute of Research; Washington: 1979. [Google Scholar]
- 22.Grdina DJ, Shigematsu N, Dale P, Newton GL, Aguilera JA, Fahey RC. Thiol and disulfide metabolites of the radiation protector and potential chemopreventive agent WR-2721 are linked to both its anti-cytotoxic and anti-mutagenic mechanisms of action. Carcinogenesis. 1995;16:767–74. doi: 10.1093/carcin/16.4.767. [DOI] [PubMed] [Google Scholar]
- 23.A service of the US. National Institutes of Health ClinicalTrials.gov. ( http://www.clinicaltrials.gov/ct2/results?term=ethyol)
- 24.Rose PG. Amifostine cytoprotection with chemotherapy for advanced ovarian carcinoma. Semin Oncol. 1996;23:83–9. [PubMed] [Google Scholar]
- 25.Kligerman MM, Turrisi AT, 3rd, Urtasun RC, Norfleet AL, Phillips TL, Barkley T, et al. Final report on phase I trial of WR-2721 before protracted fractionated radiation therapy. Int J Radiat Oncol Biol Phys. 1988;14:1119–22. doi: 10.1016/0360-3016(88)90387-2. [DOI] [PubMed] [Google Scholar]
- 26.Soref CM, Hacker TA, Fahl WE. A New Orally Active, Aminothiol Radioprotector-Free of Nausea and Hypotension Side Effects at its Highest Radioprotective Doses. Int J Radiat Oncol Biol Phys. 2012;82:701–7. doi: 10.1016/j.ijrobp.2011.11.038. [DOI] [PubMed] [Google Scholar]
- 27.Ellis H. Marie Curie: discoverer of radium. doi: 10.1177/175045890901900105. http://findarticles.com/p/articles/mi_m0748/is_1_19/ai_n31944651/ [DOI] [PubMed] [Google Scholar]
- 28.Einstein AJ, Moser KW, Thompson RC, et al. Radiation dose to patients from cardiac diagnostic imaging. Circulation. 2007;116:1290–305. doi: 10.1161/CIRCULATIONAHA.107.688101. [DOI] [PubMed] [Google Scholar]
- 29.Bogdanich W. Radiation offers new cures, and ways to do harm. The New York Times; Jan 23, 2010. Available from: http://www.nytimes.com/2010/01/24/health/24radiation.html. [Google Scholar]
- 30.Langell J, Jennings R, Clark J, et al. Pharmacological agents for the prevention and treatment of toxic radiation exposure in spaceflight. Aviat Space Environ Med. 2008;79:651–60. doi: 10.3357/asem.2113.2008. [DOI] [PubMed] [Google Scholar]