

Abstract
The natural four-letter genetic alphabet, comprised of just two base pairs (dA-dT and dG-dC), is conserved throughout all life, and its expansion by the development of a third, unnatural base pair has emerged as a central goal of chemical and synthetic biology. We recently developed a class of candidate unnatural base pairs, exemplified by the pair formed between d5SICS and dNaM. Here, we examine the PCR amplification of DNA containing one or more d5SICS-dNaM pairs in a wide variety of sequence contexts. Under standard conditions, we show that this DNA may be amplified with high efficiency and greater than 99.9% fidelity. To more rigorously explore potential sequence effects, we used deep sequencing to characterize a library of templates containing the unnatural base pair as a function of amplification. We found that the unnatural base pair is efficiently replicated with high fidelity in virtually all sequence contexts. The results show that, for PCR and PCR-based applications, d5SICS-dNaM is functionally equivalent to a natural base pair, and when combined with dA-dT and dG-dC, it provides a fully functional six-letter genetic alphabet.
Keywords: expanded genetic alphabet, hydrophobic, artificial DNA, unnatural nucleotides, bioinformatics
Expansion of the genetic alphabet to include an unnatural base pair has emerged as a central goal of chemical and synthetic biology. Success would represent a remarkable integration of orthogonal synthetic components into a fundamental biological system and build the foundation for a semisynthetic organism with increased potential for information storage and retrieval (1). Moreover, the constituent unnatural nucleotides could be used to site-specifically label DNA or RNA with different functionalities of interest (2–4) and potentially revolutionize the already ubiquitous in vitro applications of nucleic acids, such as aptamer and DNA/RNAzyme selections (5, 6), PCR-based diagnostics (7, 8), and DNA-based nanomaterials and devices (9).
Although many candidate unnatural base pairs have been reported (10–21), only a few are actually replicable by DNA polymerases (10, 11, 13, 16). Moreover, it is clear that most applications will require that the unnatural base pair not only be replicated with high efficiency and fidelity but also, that replication be at least approximately independent of sequence context. Sequence dependencies would cause biased amplification and effectively preclude many uses of the unnatural base pair. No candidate unnatural base pair has been shown to be replicated without sequence bias, and thus, none can yet claim functional equivalence to a natural base pair.
In general, the most promising unnatural base pair candidates currently available have been developed by pursuing one of two different strategies. The first strategy, pioneered in the work by Benner and coworkers (22), relies on the use of nucleotide analogs bearing nucleobases that pair through complementary hydrogen bonding (H-bonding) patterns that are orthogonal to those patterns of the natural pairs. Early efforts along these lines were hindered by low-fidelity replication and chemical instability (23), but modifications have been found more recently that overcome these limitations, and DNA containing such unnatural pairs has been amplified by PCR (24). The amplification of DNA containing multiple, contiguous unnatural base pairs has also been shown (10), but the more general effects of sequence context have yet to be reported. The second strategy, originally pursued by our group (25), and in part inspired by the observation in the work by Kool and coworkers (26) that H-bonds are not absolutely required for polymerase recognition, relies on harnessing hydrophobic and packing forces between the nucleobase analogs. This approach was also pursued in the works by Hirao and coworkers (13, 27), which developed a base pair that is amplified through PCR but with significant sequence bias.
Our efforts to develop predominantly hydrophobic unnatural base pairs have culminated in the identification of the pairs formed between d5SICS and either dMMO2 (28) or dNaM (11, 29) (Fig. 1 shows a comparison of d5SICS-dNaM with a natural dG-dC). Indeed, we have used these nucleotides to site-specifically label DNA and RNA with multiple different functional groups (2). Although both dMMO2 and dNaM are good partners for d5SICS, kinetics and preliminary PCR experiments revealed that d5SICS-dNaM is both replicated (11, 29, 30) and transcribed (12) better than d5SICS-dMMO2. Moreover, recent structural studies revealed that the efficient replication of d5SICS-dNaM results from the ability of polymerases to induce it to adopt the structure of a Watson–Crick pair, despite the absence of H-bonds (31, 32).
Fig. 1.

Unnatural (d5SICS-dNaM) and natural Watson–Crick (dC-dG) base pairs.
Here, we explore the use of d5SICS-dNaM by rigorously characterizing the sequence dependence of its replication. We found that OneTaq, a commercially available mixture of Taq and Deep Vent polymerases, simultaneously optimizes both the efficiency and fidelity of d5SICS-dNaM replication. We then show that DNA containing the unnatural base pair may be efficiently amplified in a variety of different sequence contexts, including GC- and AT-rich sequences, randomized sequences, and sequences with multiple d5SICS-dNaM pairs, with greater than 99.9% fidelity per doubling. Finally, we find through the use of a PCR selection and deep-sequencing analysis, that replication of the unnatural base pair proceeds with virtually no sequence bias. Overall, the results show that, at least for in vitro applications, d5SICS-dNaM is functionally equivalent to a natural base pair, and along with the natural base pairs, d5SICS-dNaM represents a fully functional expanded genetic alphabet.
Results and Discussion
Exploring the Scope of PCR with DNA Containing d5SICS-dNaM.
We first characterized the amplification of a DNA template containing d5SICS-dNaM flanked on each side by three nucleotides of randomized sequence with the exonuclease negative polymerases Taq, Vent (exo-), or Deep Vent (exo-) or the exonuclease positive polymerases KOD, Phusion, Vent, or Deep Vent (SI Appendix, Table S1 and Fig. S1). PCR amplification with exonuclease-deficient polymerases generally proceeded with high efficiency, allowing the use of a standard concentration of each dNTP (200 μM) but with only modest fidelity, which suggested that, just as with natural base pairs, a significant amount of fidelity is contributed by proofreading. Correspondingly, amplification with the exonuclease-proficient polymerases proceeded with higher fidelity but also required the use of high concentrations of the natural triphosphates (700 μM; likely because of inefficient primer extension past the unnatural base pair), which is associated with the error-prone amplification of natural DNA (33).
To explore conditions that might simultaneously optimize both efficiency and fidelity, we tested different combinations of Taq and an exonuclease-proficient polymerase (SI Appendix, Table S2 and Fig. S2). In general, amplification proceeded with efficiencies that were comparable with those efficiencies of Taq alone but fidelities that were characteristic of the exonuclease-proficient polymerases. This finding suggests that the ratio of the excision and extension activities of the natural exonuclease-proficient polymerases has been optimized during evolution for the natural base pairs and that efficient and high-fidelity replication of DNA containing d5SICS-dNaM requires slightly decreased exonuclease activity. Regardless, it is clear that the binary polymerase mixtures are better suited for the replication of DNA containing d5SICS-dNaM. Given its reliability as a commercial product, we chose to further explore the use of OneTaq (a mixture of Taq and Deep Vent available from New England Biolabs).
To explore the sequence dependence of amplification, the unnatural nucleotides were incorporated into a variety of DNA templates, where the flanking sequences ranged from high GC to high AT content or were randomized. With OneTaq, standard PCR conditions (e.g., 200 μM dNTPs and 1-min extension time), and 100 μM each unnatural triphosphate, all templates were efficiently amplified with fidelities ranging from 99.7% to 99.99% (Table 1 and SI Appendix, Fig. S3) (corresponding to error rates of 10−3 to 10−4 per nucleotide). These fidelities resulted in 87% to >99% retention of the unnatural base pair in the product after 1012-fold amplification. Clearly, OneTaq is able to amplify DNA containing a single d5SICS-dNaM in a variety of sequence contexts with both high efficiency and fidelity.
Table 1.
Sequence dependence of PCR amplification
| Template (Y = 5SICS)* | Amplification | Efficiency (%)† | Retention (%)‡ | Fidelity (%)§ |
| ACTYGTG | 2.5 × 1012 | 97 | 97.76 ± 0.62 | 99.945 ± 0.016 |
| GTCYGGT | 1.5 × 1012 | 95 | 95.0 ± 1.4 | 99.874 ± 0.035 |
| AGCYCGT | 3.5 × 1012 | 96 | 97.11 ± 0.12 | 99.930 ± 0.003 |
| CCGYGAA | 8.1 × 1012 | >99 | 86.5 ± 1.4 | 99.664 ± 0.037 |
| NNNYNNN¶ | 4.8 × 1012 | 97 | 96.90 ± 0.33 | 99.925 ± 0.008 |
| NNNYNNN¶,‖ | 1.6 × 1013 | 91 | 99.2 ± 1.6 | 99.981 ± 0.037 |
| GTAYTGT | 3.1 × 1012 | 95 | 99.46 ± 0.85 | 99.987 ± 0.021 |
| AGAYAGT | 8.5 × 1012 | >99 | >99 | >99.98 |
| CCTYAAA | 8.4 × 1012 | >99 | 94.41 ± 0.57 | 99.866 ± 0.014 |
| GGTYTCC | 2.6 × 1012 | 94 | 98.30 ± 0.37 | 99.958 ± 0.012 |
Conditions: 1 ng DNA template, d5SICSTP/dNaMTP/dNTPs = 100/100/200 μM, 3 mM MgSO4, 0.02 U/mL OneTaq; cycling conditions: 96 °C, 10 s; 60 °C, 15 s; 68 °C, 1 min.
*Central sequence around Y = 5SICS is shown (SI Appendix, Table S1 shows full sequences).
†PCR efficiency (E) was calculated from A = (1 + E)n (34), where A is the amplification level and n is the total number of cycles.
‡Percentage of amplified product that retained the unnatural base pair calculated as weighted mean retention in both directions, except for the GGTYTCC (Y = 5SICS) template, where it was calculated only in one direction because of read through in the other direction that was also observed with the unamplified control template; thus, it is an artifact of Sanger sequencing. Errors were propagated from the 2 SDs determined from three independent sequencing reactions with each primer.
§Fidelity (f) was determined by sequencing (Materials and Methods), and it is defined as the retention of the unnatural base pair per doubling calculated as R = fn, where R is the retention of the unnatural base pair, n is the number of doublings calculated as log2(A), and A is the amplification level. Errors for f were propagated from the errors determined for R.
¶N represents a randomized natural nucleotide.
‖Extension time = 4 min.
To explore the amplification of sequences containing multiple unnatural base pairs, we characterized the amplification of templates containing either two consecutive d5SICS-dNaM pairs or two pairs separated by one or six natural base pairs (SI Appendix, SI Materials and Methods and Table S1). All of the templates were efficiently amplified, and when separated by six natural nucleotides, the two unnatural base pairs were replicated with fidelities of 99.5% and 99.6% per pair for extension times of 1 and 4 min, respectively (with conditions otherwise identical to those conditions described in Table 1). The fidelities when the unnatural pairs were adjacent to one another or separated by a single natural nucleotide were more difficult to calculate because of an artifactual oscillation in all four channels of the detector after the unnatural nucleotides that precluded accurate determination of the read-through signal (SI Appendix, Fig. S3). Nonetheless, when sequencing from either direction, the traces showed abrupt termination at the expected positions, and based on comparison with other templates, we estimate that the fidelities are similar. Overall, the data show that DNA containing d5SICS-dNaM in a variety of sequence contexts, including those contexts with multiple and even contiguous unnatural base pairs, may be efficiently replicated with high fidelity.
PCR Selection and Bioinformatic Analysis.
To rigorously explore the effect of sequence context, we performed a PCR selection (Fig. 2A). To account for edge effects introduced by the primers, three sublibraries were designed that incorporate d5SICS-dNaM at three different positions within a region of 40 randomized nucleotides (Fig. 2B). Sublibrary-specific two-nucleotide barcodes were included to identify the position of the unnatural base pair during the analysis of the sequencing data. The combined sublibraries, totaling ∼2 × 1010 members, were amplified by OneTaq, and aliquots were taken for analysis after 103-, 106-, 1012-, 1018-, and 1024-fold amplification (SI Appendix, Fig. S4). To vary the selection pressure for preferentially replicated sequences, we performed two sets of amplifications in parallel that varied only in extension time (1 or 4 min).
Fig. 2.

(A) PCR selection scheme. X = NaM (or when biotinylated, its analog MMO2; see Fig. S5) and Y = 5SICS. (B) Library design. The regions proximal to the unnatural base pair that were analyzed for biases are shown in red, and the distal regions used as a control are shown in green. Sublibrary-specific two-nucleotide barcodes that indicate the position of the unnatural base pair flank the randomized regions and are shown in italics. Primer binding regions are denoted as PBR (sequences in SI Appendix, Table S1).
The DNA from the aliquots taken during each amplification was separated into two populations based on whether it had retained or lost the unnatural base pair by performing an additional six cycles of PCR; during PCR, dNaMTP was replaced with biotinylated dMMO2TP (2) (SI Appendix, Fig. S5) followed by passage over streptavidin solid support (Fig. 2A). To prepare the DNA for sequencing, another 10 cycles of PCR amplification were performed using Illumina primers with population-specific barcodes (SI Appendix, Table S3) and only natural dNTPs (to replace the unnatural nucleotides with natural nucleotides). Chemically synthesized (unamplified) sublibraries were subjected to the same procedure, with and without biotinylation to control for any biases introduced during analysis. In total, 23 populations were analyzed by deep sequencing on an Illumina HiSeq2000 sequencing system (SI Appendix, Table S3). From this analysis, a total of 58 million raw reads were generated and filtered by quality score and length, resulting in an average of 1.6 × 106 reads per population (∼37 million processed reads in total).
Initial analysis revealed that there were no significant differences between the sublibraries, suggesting that any biases introduced by the unnatural base pair did not depend on its position within the template. Thus, the data from the sublibraries were combined, and we focused on the 20 nt flanking the unnatural base pair (Fig. 2B, red). As a first measure of sequence bias, we quantified the diversity of each population by calculating the fraction of single-copy sequences detected. In addition, we calculated the normalized Shannon entropy (35, 36) (Eq. 1),
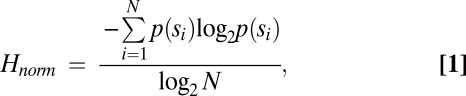 |
which is a measure of the total diversity of the population, where p(si) is the copy number of sequence si divided by the total number of sequences (N) (Fig. 3). Thus, the numerator corresponds to the sum of entropies of individual sequences (corresponding to states with standard thermodynamic entropy) weighted by their abundance, and the denominator normalizes the entropy for the size of the population (in this case, the number of sequencing reads). A population that has converged to a single sequence will have an Hnorm value of zero, whereas a population of all unique sequences will have a value of one. As expected, for any population where the diversity of templates is significantly larger than the number of sequencing reads, both of these metrics of diversity remained constant during the initial rounds of PCR but began to decrease after 106-fold amplification. Also, as expected, diversity decreased faster in the smaller populations that lost the unnatural base pair than the larger ones that retained it, simply because of population size. In addition, diversity was lost somewhat faster with 1-min extension times than with 4-min extension times, indicating the presence of difficult to amplify sequences. However, in all cases, the diversity in the analyzed region (red sequence in Fig. 2B) was lost at nearly the same rate as a more distal control region containing only natural nucleotides (green sequence in Fig. 2B). Thus, both the Shannon entropy and the fraction of unique sequences in the population suggest that the introduction of d5SICS-dNaM does not cause an increased loss of diversity during amplification.
Fig. 3.

Fraction of single-copy sequences (Upper) and normalized Shannon entropy (Lower) for amplification with 1- (Left) or 4-min (Right) extension times. The red lines correspond to the regions proximal to the unnatural base pair, and the green lines correspond to the distal control regions (Fig. 2B). Populations that retained or lost the unnatural base pair are represented with solid or dotted lines, respectively. Error bars were determined from the independent analysis of each of the three sublibraries.
To further explore sequence bias, we calculated the relative nucleotide frequency at each position, 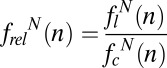 , where flN(n) is the frequency of nucleotide N at position n in the amplified library and fcN(n) is frequency of the same nucleotide at position n in a control library. Here and throughout, the position of a nucleotide n is defined relative to the position of dNaM, which is defined as zero. The 103-fold amplified libraries were used as a control to focus on the biases introduced during amplification and not during chemical synthesis [for example, because of incomplete nucleotide deprotection (37); thus, the 1024-fold amplified libraries are referred to as having been amplified 1021-fold]. Values of frelN(n) − 1 reflect the amplification bias for or against nucleotide N at position n, with zero indicating no bias and positive and negative deviations indicating that, during amplification, the nucleotide is enriched or depleted, respectively. For the population amplified with a 1-min extension time (Fig. 4A), frelN(n) − 1 deviated significantly from zero only at the positions immediately flanking the unnatural base pair (e.g., at positions 1 and −1). However, after the full 1021-fold amplification, even at these positions, |frelN(n) − 1| exceeded 0.08 in only three cases: frelC(−1) − 1 = 0.32 in the population of sequences that retained the unnatural base pair and frelA(1) − 1 = −0.19 and frelT(1) − 1 = 0.16 in the population that lost the unnatural base pair. With 4-min extension, the same general trends were apparent (SI Appendix, Fig. S6A), but the biases were even smaller (the largest was again frelC(−1) − 1 for the population that retained the unnatural base pair, which had a value of 0.20 after the full 1021-fold amplification).
, where flN(n) is the frequency of nucleotide N at position n in the amplified library and fcN(n) is frequency of the same nucleotide at position n in a control library. Here and throughout, the position of a nucleotide n is defined relative to the position of dNaM, which is defined as zero. The 103-fold amplified libraries were used as a control to focus on the biases introduced during amplification and not during chemical synthesis [for example, because of incomplete nucleotide deprotection (37); thus, the 1024-fold amplified libraries are referred to as having been amplified 1021-fold]. Values of frelN(n) − 1 reflect the amplification bias for or against nucleotide N at position n, with zero indicating no bias and positive and negative deviations indicating that, during amplification, the nucleotide is enriched or depleted, respectively. For the population amplified with a 1-min extension time (Fig. 4A), frelN(n) − 1 deviated significantly from zero only at the positions immediately flanking the unnatural base pair (e.g., at positions 1 and −1). However, after the full 1021-fold amplification, even at these positions, |frelN(n) − 1| exceeded 0.08 in only three cases: frelC(−1) − 1 = 0.32 in the population of sequences that retained the unnatural base pair and frelA(1) − 1 = −0.19 and frelT(1) − 1 = 0.16 in the population that lost the unnatural base pair. With 4-min extension, the same general trends were apparent (SI Appendix, Fig. S6A), but the biases were even smaller (the largest was again frelC(−1) − 1 for the population that retained the unnatural base pair, which had a value of 0.20 after the full 1021-fold amplification).
Fig. 4.

Analysis of amplification bias with 1-min extension time. In all cases, retained and lost refer to the populations that retained the unnatural base pair during amplification and the populations that lost it, respectively. (A) Single nucleotide bias. frelN(n) − 1 values are shown for each natural nucleotide (indicated along the top) as a function of position relative to dNaM in the amplified library. Amplification level is shown along the far right edge. (B) Normalized pairwise correlations C(n,n′). Only positive values of C(n,n′), which indicate amplification-dependent biases, are shown. For visualization, the discrete data are represented with continuous functions (surfaces); (C) 5′- and 3′-dinucleotide biases (frelNN′(n,n′) − 1) are shown on the left and right, respectively, and are represented in a circular format, where the sequences read from the middle out, with X representing dNaM. For example, for each dinucleotide distribution, the upper-right quadrant corresponds to either 5′-NAX or XAN-3′, where N is (clockwise) A, C, G, or T. Correspondingly, the bottom-right quadrant corresponds to either 5′-NCX or XCN-3′, the bottom-left quadrant corresponds to either 5′-NGX or XGN-3′, and the top-left quadrant corresponds to either 5′-NTX or XTN-3′. Amplification level is indicated by gray shading, which is shown at the bottom.
Sequence biases may also arise from nucleotide correlations that are not apparent at the single-nucleotide level (for example, from the equal representation of only 4 of the possible 16 dinucleotides at a given site). Thus, we calculated all pairwise correlations between sequence positions using the normalized mutual information (35, 38) (Eq. 2):
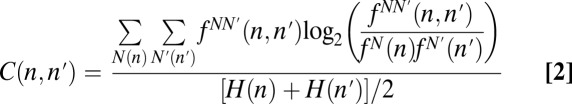 |
and (Eq. 3)
 |
where the summations are over all four natural nucleotides N at position n, fN(n) is the independent frequency of the nucleotide at position n, fNN′(n,n′) is the joint frequency of nucleotides N and N′ at positions n and n′, respectively, and finally, H(n) is the entropy of position n. C(n,n′) measures how much knowing the identity of the nucleotide at position n reduces the uncertainty of knowing the identity of the nucleotide at position n′. A value of C(n,n′) = 0 indicates that the nucleotide identity at position n does not affect that at position n′, whereas a value of C(n,n′) = 1 indicates that the nucleotide identity at position n is sufficient to determine the identity at position n′. To focus again only on amplification biases, C(n,n′) elements of the 1024-fold amplified library were corrected by subtraction of the corresponding elements of the 103-fold amplified library. For the populations amplified with a 1-min extension time (Fig. 4B), after the full 1021-fold amplification, the only off-diagonal elements in the normalized C(n,n′) matrix with values in excess of 0.001 were C(1, 2) = 0.0014 for the population of sequences that retained the unnatural base pair and C(−1, −2) = 0.0011 and C(1, 2) = 0.0012 for the population of sequences that lost the unnatural base pair. For the population amplified with a 4-min extension time, all pairwise correlations were less than 0.0007 (SI Appendix, Fig. S6B).
To estimate the effect of these correlations, we focused on the positions with the largest values (i.e., those positions flanking the unnatural base pair). We calculated the relative frequencies of all possible dinucleotides at each flanking position, 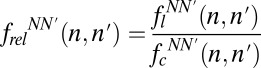 , where flNN′(n, n′) is the percentage of the sequences in the amplified library with nucleotides N and N′ at positions n = −2 and n′ = −1 or n = 1 and n′ = 2 and fcNN′(n, n′) is the same for the 103-fold amplified control library. Thus, frelNN′(n, n′) − 1 reflects any biases introduced during amplification in dinucleotides that flank the unnatural base pair, with zero indicating no bias and positive and negative deviations indicating enrichment and depletion of the dinucleotide, respectively. For the population amplified with a 1-min extension time that retained the unnatural base pair (Fig. 4C), the largest biases observed after the full 1021-fold amplification were for each of the four dinucleotides with C at the −1 position, with values of frelNC(−2, −1) − 1 that ranged from 0.21 to 0.51. The largest bias found at the n = 1, n′ = 2 position was for the AA dinucleotide, with frelAA(1, 2) − 1 = −0.23. For the populations of DNA that lost the unnatural base pair, the largest 5′- and 3′-dinucleotide biases were frelAA(−2, −1) − 1 = −0.18, frelTN(1, 2) − 1 = 0.10–0.21, and frelAN(1, 2) − 1 = −0.16 to −0.26. All of these dinucleotide biases are similar to those biases predicted from the single-nucleotide biases frelN [for example, frelGC(2, −1) = 1.51 compared with the value of 1.36 predicted from single biases without correlation; i.e., frelG(−2) × frelC(−1)], revealing that the biases associated with sequence correlations are small. For the population amplified with a 4-min extension time (SI Appendix, Fig. S6C), the general trends in the dinucleotide biases were almost identical to those trends observed with 1-min extension time, except that they were even smaller. The largest bias detected, frelGC(−2, −1), in the population that retained the unnatural base pair only reached a value of 1.36 after the full 1021-fold amplification and was again similar to the bias predicted from the single-nucleotide frelN values [frelG(−2) × frelC(−1) = 1.25]. Thus, even at the positions with the highest C(n,n′) values, the correlations contribute little to sequence bias.
, where flNN′(n, n′) is the percentage of the sequences in the amplified library with nucleotides N and N′ at positions n = −2 and n′ = −1 or n = 1 and n′ = 2 and fcNN′(n, n′) is the same for the 103-fold amplified control library. Thus, frelNN′(n, n′) − 1 reflects any biases introduced during amplification in dinucleotides that flank the unnatural base pair, with zero indicating no bias and positive and negative deviations indicating enrichment and depletion of the dinucleotide, respectively. For the population amplified with a 1-min extension time that retained the unnatural base pair (Fig. 4C), the largest biases observed after the full 1021-fold amplification were for each of the four dinucleotides with C at the −1 position, with values of frelNC(−2, −1) − 1 that ranged from 0.21 to 0.51. The largest bias found at the n = 1, n′ = 2 position was for the AA dinucleotide, with frelAA(1, 2) − 1 = −0.23. For the populations of DNA that lost the unnatural base pair, the largest 5′- and 3′-dinucleotide biases were frelAA(−2, −1) − 1 = −0.18, frelTN(1, 2) − 1 = 0.10–0.21, and frelAN(1, 2) − 1 = −0.16 to −0.26. All of these dinucleotide biases are similar to those biases predicted from the single-nucleotide biases frelN [for example, frelGC(2, −1) = 1.51 compared with the value of 1.36 predicted from single biases without correlation; i.e., frelG(−2) × frelC(−1)], revealing that the biases associated with sequence correlations are small. For the population amplified with a 4-min extension time (SI Appendix, Fig. S6C), the general trends in the dinucleotide biases were almost identical to those trends observed with 1-min extension time, except that they were even smaller. The largest bias detected, frelGC(−2, −1), in the population that retained the unnatural base pair only reached a value of 1.36 after the full 1021-fold amplification and was again similar to the bias predicted from the single-nucleotide frelN values [frelG(−2) × frelC(−1) = 1.25]. Thus, even at the positions with the highest C(n,n′) values, the correlations contribute little to sequence bias.
To evaluate the potential impact of the observed biases, it is instructive to consider their consequences. The largest single- and dinucleotide biases observed were frelC(−1) − 1 for frelGC(−1, −2) − 1 in the population that retained the unnatural base pair, which after the full 1021-fold amplification, only reached values of 0.32 and 0.51, respectively. These values correspond to an increase in the frequency of 5′-CNaM from 18.71% to 24.65%, with the subpopulation having a 5′-GCNaM sequence increasing from 2.30% to 3.48%. These biases are not larger than the biases observed among natural sequences (39), and they are unlikely to interfere with any in vitro application of DNA containing the unnatural base pair, even including those applications requiring massive amplification.
Conclusion
DNA containing d5SICS-dNaM is PCR-amplified by OneTaq polymerase with both high efficiency and fidelity. Importantly, the efficiencies and fidelities seem excellent regardless of the natural sequence context of the unnatural base pair. Moreover, the error rate with which the unnatural base pair is lost or gained, which ranges from 10−3 to 10−4 per nucleotide, overlaps with the error rate of fully natural DNA with commonly used commercial PCR systems, which ranges between 10−4 and 10−7 (40). Thus, at least for in vitro applications, d5SICS-dNaM represents a fully functional base pair, and along with the natural base pairs, it represents a fully functional, expanded genetic alphabet. It is remarkable that this unnatural base pair with proven functional equivalence to the natural base pairs relies on hydrophobic interactions for replication, without the aid of complementary H-bonding. Clearly, the natural purine and pyrimidine scaffolds that pair through complementary H-bonding are not unique solutions to the challenge of biological information storage and retrieval. With linkers added to the unnatural nucleotides (2), the unnatural base pair could be used to site-specifically modify DNA or RNA with any functionality of interest, and should find uses in different in vitro applications, even those requiring massive and sequence-independent amplification. Moreover, the efficient and high-fidelity replication and transcription of d5SICS-dNaM also suggest that it might allow for the in vivo expansion of the genetic alphabet and the creation of a semisynthetic organism with an increased potential for information storage and retrieval. Efforts toward these goals are currently underway.
Materials and Methods
OneTaq PCR.
PCR reactions were performed in 1× OneTaq buffer containing 3 mM Mg2+, 200 μM each natural dNTP, 100 μM d5SICSTP and dNaMTP, 1 μM each primer, 0.5× SYBR Green, 1 ng DNA template (SI Appendix, SI Materials and Methods, and Table S1), and 0.02 unit/μL OneTaq in a total of 50 μL in a MyiQ system (Bio-Rad) under the following thermal cycling conditions: 96 °C, 10 s; 60 °C, 15 s; 68 °C, 1 min. The total number of PCR cycles varied between 42 and 47 (based on the real-time monitoring of SYBR green), with 104-fold dilution after the initial 14–16 cycles and then again after an additional 18 cycles. A negative control reaction lacking template was run in parallel. On completion, an aliquot (5 μL) was analyzed on a 2% (wt/vol) agarose gel to confirm the size of the product. DNA was purified using DNA Clean & Concentrator-5 (Zymo Research Corp.), quantified by the Quant-iT dsDNA HS Assay (Invitrogen), and sequenced on 3730 DNA Analyzer (Applied Biosystems). Efficiencies of amplification were quantified by normalizing the amount of product produced by the number of PCR cycles. Fidelity was determined from the retention level of the unnatural base pair normalized by the number of doublings [log2(amplification)]. The retention was quantified by Sanger sequencing using natural triphosphates. Under these conditions, an unnatural nucleotide in the template causes abrupt termination of the sequencing reaction, making it possible to quantitatively determine its level of retention as the ratio of the normalized amplitudes of sequencing chromatogram peaks before and after termination (details in ref. 30 and SI Appendix, SI Materials and Methods). Raw Sanger sequencing traces are shown in SI Appendix, Fig. S3.
PCR Selection and Library Design.
Three sublibraries were prepared as d5SICS strands using standard automated DNA synthesis (sequences are shown in SI Appendix, Table S1, and dNaM strands are shown in Fig. 2B). A mixture of phosphoramidites for the randomized region synthesis was prepared as described previously (41). The three purified sub-libraries were quantified by UV and mixed in a 1:1:1 ratio, and 5 ng were subjected to OneTaq PCR amplification as described above. After 13 cycles, reactions were diluted by a factor of 103 and transferred to PCR tubes with fresh reagents followed by 10 cycles at 103 dilution, 2 × 20 cycles at 106 dilution, and finally, 21 cycles (84 PCR cycles in total) (SI Appendix, Fig. S4A shows quantitative PCR data). Samples at different amplification levels were purified, quantified, and analyzed on 10% nondenaturing PAGE (SI Appendix, Fig. S4B).
Duplex Biotinylation.
The amplified products (5 ng) were subjected to six additional rounds of PCR and run under conditions identical to the conditions described above, except that a biotinylated variant of dMMO2TP was used instead of dNaMTP (2) (SI Appendix, Fig. S5) and an 80-nt-long primer (Primer1-poly-dT) was used instead of the 21-nt long Primer1 to allow separation of the amplification product on a 4% agarose gel (SI Appendix, Table S1 shows full sequences). The fragment corresponding to ∼180 bp was excised and extracted from the gel, purified, and quantified. The biotinylation level of each duplex was quantified by streptavidin gel mobility assay (SI Appendix, SI Materials and Methods).
Deep-Sequencing and Bioinformatic Analysis.
Biotinylated populations of amplified libraries were separated using Streptavidin Sepharose High Performance resin (GE Healthcare); DNA that lost the unnatural base pair was purified from the supernatant, whereas DNA that retained the unnatural base pair was recovered from the resin by DTT treatment. Both the populations that retained the unnatural base pair and the populations that lost it at different levels of amplification were subjected to PCR with only natural dNTPs and Illumina primers with population-specific Multiplex TruSeq Index barcodes (sequences in SI Appendix, SI Materials and Methods and Tables S1 and S3). All populations were then sequenced on a HiSeq2000 Sequencing System (Illumina) on a single-flow cell lane. A total of 58 million raw reads were generated and filtered by quality score (Q > 30). After identification of the primer regions, all reads were binned by their population-specific barcode into 23 populations (SI Appendix, Table S3). Primer regions were trimmed, and only sequences with an exact length of 41 nt (N40 + dNaM) with two correct sublibrary-specific barcodes (Fig. 2B) were included in the analysis to avoid insertion and deletion mutations that may misrepresent the position of the unnatural base pair. Custom python scripts were used to determine nucleotide frequencies, read counts, mutual information, and Shannon entropy scores. Raw data are available for download from the National Center for Biotechnology Information Short Read Archive (http://trace.ncbi.nlm.nih.gov/Traces/sra; accession no. SRA050408.1).
Supplementary Material
Acknowledgments
We thank Steven Head, Lana Schaffer, and Dennis Shpakov for assistance with deep sequencing and analysis. Funding for this work was provided by National Institutes of Health\x{2013}National Center for Research Resources Clinical and Translational Science Award Grant UL1 RR025774 (to A.T.) and National Institute of Health Grant GM060005 (to F.E.R.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The sequences reported in the paper have been deposited in the National Center of Biotechnology Information Short Read Archive (accession no. SRA050408.1).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1205176109/-/DCSupplemental.
References
- 1.Benner SA, Sismour AM. Synthetic biology. Nat Rev Genet. 2005;6:533–543. doi: 10.1038/nrg1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seo YJ, Malyshev DA, Lavergne T, Ordoukhanian P, Romesberg FE. Site-specific labeling of DNA and RNA using an efficiently replicated and transcribed class of unnatural base pairs. J Am Chem Soc. 2011;133:19878–19888. doi: 10.1021/ja207907d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawai R, et al. Site-specific fluorescent labeling of RNA molecules by specific transcription using unnatural base pairs. J Am Chem Soc. 2005;127:17286–17295. doi: 10.1021/ja0542946. [DOI] [PubMed] [Google Scholar]
- 4.Kimoto M, et al. A new unnatural base pair system between fluorophore and quencher base analogues for nucleic acid-based imaging technology. J Am Chem Soc. 2010;132:15418–15426. doi: 10.1021/ja1072383. [DOI] [PubMed] [Google Scholar]
- 5.Hollenstein M, Hipolito CJ, Lam CH, Perrin DM. A self-cleaving DNA enzyme modified with amines, guanidines and imidazoles operates independently of divalent metal cations (M2+) Nucleic Acids Res. 2009;37:1638–1649. doi: 10.1093/nar/gkn1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keefe AD, Cload ST. SELEX with modified nucleotides. Curr Opin Chem Biol. 2008;12:448–456. doi: 10.1016/j.cbpa.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Collins ML, et al. A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/ml. Nucleic Acids Res. 1997;25:2979–2984. doi: 10.1093/nar/25.15.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimoto M, Cox RS, 3rd, Hirao I. Unnatural base pair systems for sensing and diagnostic applications. Expert Rev Mol Diagn. 2011;11:321–331. doi: 10.1586/erm.11.5. [DOI] [PubMed] [Google Scholar]
- 9.Seeman NC. Nanomaterials based on DNA. Annu Rev Biochem. 2010;79:65–87. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z, Chen F, Alvarado JB, Benner SA. Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J Am Chem Soc. 2011;133:15105–15112. doi: 10.1021/ja204910n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavergne T, Malyshev DA, Romesberg FE. Major groove substituents and polymerase recognition of a class of predominantly hydrophobic unnatural base pairs. Chemistry. 2012;18:1231–1239. doi: 10.1002/chem.201102066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seo YJ, Matsuda S, Romesberg FE. Transcription of an expanded genetic alphabet. J Am Chem Soc. 2009;131:5046–5047. doi: 10.1021/ja9006996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimoto M, Kawai R, Mitsui T, Yokoyama S, Hirao I. An unnatural base pair system for efficient PCR amplification and functionalization of DNA molecules. Nucleic Acids Res. 2009;37:e14. doi: 10.1093/nar/gkn956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirao I, et al. An unnatural hydrophobic base pair system: Site-specific incorporation of nucleotide analogs into DNA and RNA. Nat Methods. 2006;3:729–735. doi: 10.1038/nmeth915. [DOI] [PubMed] [Google Scholar]
- 15.Krueger AT, Peterson LW, Chelliserry J, Kleinbaum DJ, Kool ET. Encoding phenotype in bacteria with an alternative genetic set. J Am Chem Soc. 2011;133:18447–18451. doi: 10.1021/ja208025e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaul C, Müller M, Wagner M, Schneider S, Carell T. Reversible bond formation enables the replication and amplification of a crosslinking salen complex as an orthogonal base pair. Nat Chem. 2011;3:794–800. doi: 10.1038/nchem.1117. [DOI] [PubMed] [Google Scholar]
- 17.Heuberger BD, Shin D, Switzer C. Two Watson-Crick-like metallo base-pairs. Org Lett. 2008;10:1091–1094. doi: 10.1021/ol703029d. [DOI] [PubMed] [Google Scholar]
- 18.Clever GH, Shionoya M. Alternative DNA base pairing through metal coordination. Met Ions Life Sci. 2012;10:269–294. doi: 10.1007/978-94-007-2172-2_10. [DOI] [PubMed] [Google Scholar]
- 19.Megger DA, Fonseca Guerra C, Bickelhaupt FM, Müller J. Silver(I)-mediated Hoogsteen-type base pairs. J Inorg Biochem. 2011;105:1398–1404. doi: 10.1016/j.jinorgbio.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Yaren O, Mosimann M, Leumann CJ. A parallel screen for the discovery of novel DNA base pairs. Angew Chem Int Ed Engl. 2011;50:1935–1938. doi: 10.1002/anie.201005300. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka K, Tasaka M, Cao H, Shionoya M. An approach to metal-assisted DNA base pairing: Novel beta-C-nucleosides with a 2-aminophenol or a catechol as the nucleobase. Eur J Pharm Sci. 2001;13:77–83. doi: 10.1016/s0928-0987(00)00210-4. [DOI] [PubMed] [Google Scholar]
- 22.Piccirilli JA, Krauch T, Moroney SE, Benner SA. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature. 1990;343:33–37. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- 23.Switzer CY, Moroney SE, Benner SA. Enzymatic recognition of the base pair between isocytidine and isoguanosine. Biochemistry. 1993;32:10489–10496. doi: 10.1021/bi00090a027. [DOI] [PubMed] [Google Scholar]
- 24.Yang Z, Chen F, Chamberlin SG, Benner SA. Expanded genetic alphabets in the polymerase chain reaction. Angew Chem Int Ed Engl. 2010;49:177–180. doi: 10.1002/anie.200905173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMinn DL, et al. Efforts toward expansion of the genetic alphabet: DNA polymerase recognition of a highly stable, self-pairing hydrophobic base. J Am Chem Soc. 1999;121:11585–11586. [Google Scholar]
- 26.Moran S, Ren RXF, Rumney S, Kool ET. Difluorotoluene, a nonpolar isostere for thymine, codes specifically and efficiently for adenine in DNA replication. J Am Chem Soc. 1997;119:2056–2057. doi: 10.1021/ja963718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashige R, et al. Highly specific unnatural base pair systems as a third base pair for PCR amplification. Nucleic Acids Res. 2012;40:2793–2806. doi: 10.1093/nar/gkr1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leconte AM, et al. Discovery, characterization, and optimization of an unnatural base pair for expansion of the genetic alphabet. J Am Chem Soc. 2008;130:2336–2343. doi: 10.1021/ja078223d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seo YJ, Hwang GT, Ordoukhanian P, Romesberg FE. Optimization of an unnatural base pair toward natural-like replication. J Am Chem Soc. 2009;131:3246–3252. doi: 10.1021/ja807853m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malyshev DA, Seo YJ, Ordoukhanian P, Romesberg FE. PCR with an expanded genetic alphabet. J Am Chem Soc. 2009;131:14620–14621. doi: 10.1021/ja906186f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malyshev DA, et al. Solution structure, mechanism of replication, and optimization of an unnatural base pair. Chemistry. 2010;16:12650–12659. doi: 10.1002/chem.201000959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Betz K, et al. Replication without H-bonds: The structure of a DNA polymerase replicating an expanding genetic alphabet. Nat Chem Biol. 2012;8:612–614. [Google Scholar]
- 33.Cline J, Braman JC, Hogrefe HH. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996;24:3546–3551. doi: 10.1093/nar/24.18.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cha RS, Thilly WG. Specificity, efficiency, and fidelity of PCR. PCR Methods Appl. 1993;3:S18–S29. doi: 10.1101/gr.3.3.s18. [DOI] [PubMed] [Google Scholar]
- 35.Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27:623–656. [Google Scholar]
- 36.Vinga S, Almeida J. Alignment-free sequence comparison—a review. Bioinformatics. 2003;19:513–523. doi: 10.1093/bioinformatics/btg005. [DOI] [PubMed] [Google Scholar]
- 37.Fu C, Smith S, Simkins SG, Agris PF. Identification and quantification of protecting groups remaining in commercial oligonucleotide products using monoclonal antibodies. Anal Biochem. 2002;306:135–143. doi: 10.1006/abio.2002.5687. [DOI] [PubMed] [Google Scholar]
- 38.Ohya M, Sato K. Use of information theory to study genome sequences. Rep Math Phys. 2000;46:419–428. [Google Scholar]
- 39.Arezi B, Xing W, Sorge JA, Hogrefe HH. Amplification efficiency of thermostable DNA polymerases. Anal Biochem. 2003;321:226–235. doi: 10.1016/s0003-2697(03)00465-2. [DOI] [PubMed] [Google Scholar]
- 40.New England Biolabs . PCR Reagents, Version 3.0. Ipswich, MA: New England Biolabs; Properties of PCR polymerases; p. 3. [Google Scholar]
- 41.Unrau PJ, Bartel DP. RNA-catalysed nucleotide synthesis. Nature. 1998;395:260–263. doi: 10.1038/26193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.