

Abstract
Oxoiron(V) species are postulated to be involved in the mechanisms of the arene cis-dihydroxylating Rieske dioxygenases and of bioinspired nonheme iron catalysts for alkane hydroxylation, olefin cis-dihydroxylation, and water oxidation. In an effort to obtain a synthetic oxoiron(V) complex, we report herein the one-electron oxidation of the S = 1 complex [FeIV(O)(TMC)(NCCH3)]2+ (1, where TMC is tetramethylcyclam) by treatment with tert -butyl hydroperoxide and strong base in acetonitrile to generate a metastable 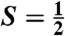 complex 2 at -44 °C, which has been characterized by UV-visible, resonance Raman, Mössbauer, and EPR methods. The defining spectroscopic characteristic of 2 is the unusual x/y anisotropy observed for the 57Fe and 17O A tensors associated with the high-valent Fe═O unit and for the 14N A tensor of a ligand derived from acetonitrile. As shown by detailed density functional theory (DFT) calculations, the unusual x/y anisotropy observed can only arise from an iron center with substantially different spin populations in the dxz and dyz orbitals, which cannot correspond to an FeIV═O unit but is fully consistent with an
complex 2 at -44 °C, which has been characterized by UV-visible, resonance Raman, Mössbauer, and EPR methods. The defining spectroscopic characteristic of 2 is the unusual x/y anisotropy observed for the 57Fe and 17O A tensors associated with the high-valent Fe═O unit and for the 14N A tensor of a ligand derived from acetonitrile. As shown by detailed density functional theory (DFT) calculations, the unusual x/y anisotropy observed can only arise from an iron center with substantially different spin populations in the dxz and dyz orbitals, which cannot correspond to an FeIV═O unit but is fully consistent with an 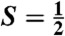 FeV center, like that found for [FeV(O)(TAML)]- (where TAML is tetraamido macrocyclic ligand), the only well-characterized oxoiron(V) complex reported. Mass spectral analysis shows that the generation of 2 entails the addition of an oxygen atom to 1 and the loss of one positive charge. Taken together, the spectroscopic data and DFT calculations support the formulation of 2 as an iron(V) complex having axial oxo and acetylimido ligands, namely [FeV(O)(TMC)(NC(O)CH3)]+.
FeV center, like that found for [FeV(O)(TAML)]- (where TAML is tetraamido macrocyclic ligand), the only well-characterized oxoiron(V) complex reported. Mass spectral analysis shows that the generation of 2 entails the addition of an oxygen atom to 1 and the loss of one positive charge. Taken together, the spectroscopic data and DFT calculations support the formulation of 2 as an iron(V) complex having axial oxo and acetylimido ligands, namely [FeV(O)(TMC)(NC(O)CH3)]+.
Keywords: high-valent iron-oxo, bioinorganic chemistry
Formally oxoiron(V) oxidants are postulated in the catalytic cycles of several iron enzymes that carry out difficult oxidations. Most prominent of these are cytochromes P450, which can hydroxylate strong C─H bonds (1–3), even those of methane (1, 2). Recent evidence has demonstrated that the active oxidant can be best described as having an oxoiron(IV) unit supported by a porphyrin cation radical (4). On the other hand, the Rieske dioxygenases activate O2 at an active site with a 2-His-1-carboxylate facial triad motif to effect the cis-dihydroxylation of C═C bonds in the biodegradation of aromatic complexes (5). For these nonheme iron enzymes, the proposed FeV═O oxidant is as yet unobserved. However, unlike the porphyrin ligand in cytochrome P450, none of the ligands in the nonheme iron active site appears likely to undergo one-electron oxidation to stabilize the high-valent state. FeV═O oxidants are also implicated in alkane hydroxylation (6, 7), olefin epoxidation and cis-dihydroxylation (8–10), and water oxidation (11) by bioinspired nonheme iron catalysts supported by neutral tetradentate ligands, and direct evidence for the formation of oxoiron(V) oxidants has been obtained in two cases by mass spectrometry (10, 12). Despite the wealth of synthetic oxoiron(IV) complexes identified during the last decade (13), to date there is only one spectroscopically well-characterized example of an oxoiron(V) complex, [FeV(O)(TAML)]- (where TAML is tetraamido macrocyclic ligand), which is stabilized by the tetraanionic nature of the TAML ligand (14). Given the ligand environments of the Rieske dioxygenases and the synthetic iron catalysts, we have been investigating the feasibility of generating an oxoiron(V) complex supported by a polydentate ligand of lower charge. Toward this end, we have focused on the one-electron oxidation of the oxoiron(IV) complex [FeIV(O)(TMC)(NCCH3)]2+ (1, see Fig. 1), where the oxoiron(IV) moiety is supported by the neutral macrocyclic tetramethylcyclam (TMC) ligand (15). We chose complex 1 as a starting point for this effort because it is structurally well-characterized and exhibits good thermal stability. One-electron oxidation of 1 could be effected by the addition of tert-butyl hydroperoxide and strong base, and our observations are reported herein.
Fig. 1.

Proposed steps in the one-electron oxidation of 1.
Results and Discussion
The reaction of 1 in CH3CN at -44 °C with at least three equivalents tBuOOH followed by the addition of 1–2 equivalents strong base (KOtBu or NBu4OH) generates an orange complex (2) that exhibits distinct UV-visible features at 410 nm (ε = 4,000 M-1 cm-1) and 780 nm (ε = 430 M-1 cm-1) (Fig. 2A). Subsequent addition of five equivalents strong acid, e.g., HBF4 or HClO4, produces a green species (2-H+) associated with spectral features at 425 nm (ε = 4,100 M-1 cm-1), 600 nm (ε = 680 M-1 cm-1), and 750 nm (ε = 530 M-1 cm-1) (Fig. 2A). (The molar extinction coefficients shown are based on samples for which Mössbauer quantification of the relevant chromophores has been carried out.) Reaction of 2-H+ with an excess of strong base readily regenerates 2, indicating that 2 and 2-H+ comprise a conjugate acid/base pair. These complexes are generated in approximately 50% yield relative to 1, as determined by Mössbauer spectroscopy (vide infra). Unlike 1, which is stable in MeCN at -44 °C, 2 and 2-H+ decay with t1/2 = 60 and 30 min, respectively. Notably, both 2 and 2-H+ are 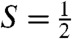 species that exhibit very similar rhombic EPR spectra (Fig. 2A, Inset) with gave ∼ 2.0, suggesting that the S = 1 species 1 has undergone one-electron oxidation to form 2.
species that exhibit very similar rhombic EPR spectra (Fig. 2A, Inset) with gave ∼ 2.0, suggesting that the S = 1 species 1 has undergone one-electron oxidation to form 2.
Fig. 2.

Spectroscopic data for 2 and 2-H+. (A) UV-visible spectra of 1 (black dashed line), 2 (thick red line), and 2-H+ (blue solid line) collected at -44 °C. (Inset) EPR spectra of 2 in CD3CN and 2-H+ in 1∶9 CH3CN∶ CH2Cl2. (B) Resonance Raman spectra of 2 (Left) and 2-H+ (Right) collected at 77 K using 413-nm laser excitation for frozen solution samples prepared with 16O (black line) and 18O (red line). Acetonitrile solvent peaks are marked as “s”.
The resonance Raman (rR) spectra of 2 and 2-H+ obtained with 413-nm excitation show features associated with an Fe═O unit. Complex 2 exhibits an intense vibrational feature at 798 cm-1 with an associated overtone at 1,587 cm-1 (Fig. 2B); corresponding features for 2-H+ are observed at 811 and 1,613 cm-1. All four features downshift when 18O-labeled 1 is used as the precursor; the observed downshifts are in accord with values calculated for an Fe═O diatomic oscillator using Hooke’s law (e.g., Δν = -35 and -36 cm-1, respectively). The observed vibrational frequencies of 798 and 811 cm-1 fall at the low end of the range of νFe═O values previously observed for 1 (839 cm-1) and its axial-ligand-substituted derivatives [FeIV(O)(TMC)(N3)]+ (814 cm-1) and [FeIV(O)(TMC)(CF3CO2)]+ (854 cm-1) (16). Thus the lower νFe═O’s of 2 and 2-H+ suggest the presence of a highly basic trans anionic ligand that weakens the Fe═O bond.
To obtain further insight into the nature of the 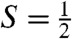 species, we have studied samples of 2 and 2-H+ with Mössbauer spectroscopy. In a typical sample, 2 and 2-H+ represented 40–55% of the total Fe in a sample. Fig. 3A shows a 4.2 K Mössbauer spectrum of 2 recorded in a 50 mT field applied parallel to the observed γ rays. In addition to 2, which accounts for approximately 55% of Fe, the sample contained several S = 1 FeIV species yielding quadrupole doublets for 1 (green, ΔEQ = 1.23 mm/s, δ = 0.17 mm/s, approximately 8%), [FeIV(O)(TMC)(OH)]+ (1-OH: blue, ΔEQ = 0.16 mm/s, δ = 0.15 mm/s, 16%), and an unidentified FeIV species (ΔEQ ∼ 0.6 mm/s, δ ∼ 0.15 mm/s, ≈10%). There is also a high-spin FeIII species (g ∼ 6.9, 5.1, ≈10%), which, fortunately, absorbs mostly outside the spectral region of 2. Similarly, the sample of 2-H+ studied here contained the following species:
species, we have studied samples of 2 and 2-H+ with Mössbauer spectroscopy. In a typical sample, 2 and 2-H+ represented 40–55% of the total Fe in a sample. Fig. 3A shows a 4.2 K Mössbauer spectrum of 2 recorded in a 50 mT field applied parallel to the observed γ rays. In addition to 2, which accounts for approximately 55% of Fe, the sample contained several S = 1 FeIV species yielding quadrupole doublets for 1 (green, ΔEQ = 1.23 mm/s, δ = 0.17 mm/s, approximately 8%), [FeIV(O)(TMC)(OH)]+ (1-OH: blue, ΔEQ = 0.16 mm/s, δ = 0.15 mm/s, 16%), and an unidentified FeIV species (ΔEQ ∼ 0.6 mm/s, δ ∼ 0.15 mm/s, ≈10%). There is also a high-spin FeIII species (g ∼ 6.9, 5.1, ≈10%), which, fortunately, absorbs mostly outside the spectral region of 2. Similarly, the sample of 2-H+ studied here contained the following species: 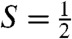 complex 2-H+ (41%), two S = 1 complexes 1 (27%) and 1-OH (2%), and S = 5/2 FeIII (30%, see SI Appendix).
complex 2-H+ (41%), two S = 1 complexes 1 (27%) and 1-OH (2%), and S = 5/2 FeIII (30%, see SI Appendix).
Fig. 3.

A 4.2 K Mössbauer spectra of 2 and 2-H+. Mössbauer spectra of samples containing 2 (A) and 2-H+ (C) recorded in a 50 mT field applied parallel to the observed γ rays. (B) Difference spectrum parallel minus perpendicular, representing 2, obtained with 50 mT applied fields. Red lines are simulations for 2 and 2-H+ based on Eq. 1, using the parameters listed below and in Table 1. The major FeIV═O contaminants are shown by the green (1) and blue (1-OH) lines. The black solid line in A is a spectral simulation for the sum of 2 (55% of Fe), 1 (8%), and 1-OH (16%). The doublets in C represent 27% of 1 and 2% of 1-OH. The arrows in C point to absorption due to a high-spin FeIII contaminant (30%). The δ, ΔEQ, and η values used in the simulations are +0.10(4) mm/s, -0.5 mm/s, and -3, respectively, for 2 and +0.10(4) mm/s, -0.2 mm/s, and -3, respectively, for 2-H+.
The presence of multiple contaminants in the sample for 2 complicates the analysis of the 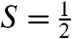 species of interest. However, two Mössbauer attributes allow us to uniquely associate a spectral component with the
species of interest. However, two Mössbauer attributes allow us to uniquely associate a spectral component with the 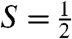 species, and to eliminate the S = 1 and
species, and to eliminate the S = 1 and 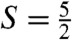 contaminants. First, we can record spectra by applying a weak (e.g., 50 mT) magnetic field either parallel or perpendicular to the observed γ rays. If the intensities of the absorption lines are different between parallel and perpendicular field, the species must be associated with an EPR active species; in this way a unique association between an
contaminants. First, we can record spectra by applying a weak (e.g., 50 mT) magnetic field either parallel or perpendicular to the observed γ rays. If the intensities of the absorption lines are different between parallel and perpendicular field, the species must be associated with an EPR active species; in this way a unique association between an 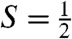 EPR signal and a Mössbauer spectral component is established. Second, for a difference spectrum thus taken, all contributions from species yielding quadrupole doublets cancel, leaving us with the analysis of a pure
EPR signal and a Mössbauer spectral component is established. Second, for a difference spectrum thus taken, all contributions from species yielding quadrupole doublets cancel, leaving us with the analysis of a pure 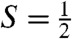 paramagnetic species. This procedure yielded the difference spectrum of Fig. 3B, which represents 2.
paramagnetic species. This procedure yielded the difference spectrum of Fig. 3B, which represents 2.
To simulate the Mössbauer and EPR spectra, we used the 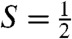 spin Hamiltonian, as follows:
spin Hamiltonian, as follows:
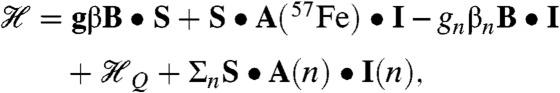 |
[1] |
where A(57Fe) is the 57Fe magnetic hyperfine tensor, I is the 57Fe nuclear spin operator, and 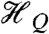 describes the 57Fe quadrupole interaction. As needed, the last term will be used to describe ligand hyperfine interactions observed by EPR; n = 14N, 15N, or 17O. From extensive simulations of the spectra of 2 and 2-H+, we obtained the parameters listed in Table 1 and SI Appendix, Fig. S6. Both 2 and 2-H+ exhibit the same isomer shifts within the uncertainties, namely δ = +0.10(4) mm/s. The δ values are smaller than those reported for S = 1 FeIV(O)(TMC) complexes (range 0.15–0.19 mm/s) (16), suggesting that the oxidation state of the iron site in 2 and 2-H+ is above FeIV. More importantly, 2 and 2-H+ exhibit unusual 57Fe A tensors with large x/y anisotropy, suggesting an electronic structure that differs significantly from that of 1.
describes the 57Fe quadrupole interaction. As needed, the last term will be used to describe ligand hyperfine interactions observed by EPR; n = 14N, 15N, or 17O. From extensive simulations of the spectra of 2 and 2-H+, we obtained the parameters listed in Table 1 and SI Appendix, Fig. S6. Both 2 and 2-H+ exhibit the same isomer shifts within the uncertainties, namely δ = +0.10(4) mm/s. The δ values are smaller than those reported for S = 1 FeIV(O)(TMC) complexes (range 0.15–0.19 mm/s) (16), suggesting that the oxidation state of the iron site in 2 and 2-H+ is above FeIV. More importantly, 2 and 2-H+ exhibit unusual 57Fe A tensors with large x/y anisotropy, suggesting an electronic structure that differs significantly from that of 1.
Table 1.
g- and A-tensors (in megahertz) experimentally observed for 2, 2-H+, and [FeV(O)(TAML)]1- and calculated for 2 and 2-H+
| Compound |
g |
A (57Fe) |
A (17O) |
A (14N) |
||||||||
| x |
y |
z |
x |
y |
z |
x |
y |
z |
x |
y |
z |
|
| HRP Cpd I | – | – | – | −26* | −26* | −8* | 35† | 36† | ND | – | – | – |
| 1 | – | – | – | −28‡ | −28‡ | −4 | −27§ | −27§ | +61§ | – | – | – |
| 2¶ | 2.053 | 2.010 | 1.971 | −47(2) | −17(2) | 0(5) | 25(15) | 128(3) | ≈20 | 29(2) | 11(3) | 11(3) |
| 2 (DFT) ∥ | 2.03 | 2.00 | 1.97 | −43.5 | −18.5 | 3.3 | −1.0 | −70.8 | 75.7 | −39.3 | 10.8 | 6.1 |
| 2-H+¶ | 2.054 | 2.011 | 1.975 | −47(2) | −17(2) | 0(5) | 25(15)** | 133(3) | 23(10) | 30(2)** | 11(3) | 10(3) |
| 2-H+ (DFT)∥ | 2.03 | 2.00 | 1.97 | −45.1 | −14.0 | −6.0 | 18.4 | −91.4 | 84.7 | −35.5∥ | −1.2 | 3.9 |
| [FeV(O)(TAML)]1-†† | 1.99 | 1.97 | 1.74 | −49.3 | −1.5 | −16.3‡‡ | ND | ND | ND | – | – | – |
ND, not determined.
*From Mössbauer data analyzed with an S = 1 Hamiltonian (34).
†From ENDOR data, S = 1 (19).
‡Refit of Mössbauer data reported in ref. 15 using g values and zero-field splitting parameters derived from more recent high-field EPR data reported in ref. 35.
§This work; B3LYP calculations yield a good match for A(57Fe).
¶This work; data analyzed with Eq. 1; 4.2 K Mössbauer spectra recorded at 2.0, 4.0, and 8.0 T show that Ax(57Fe) and Ay(57Fe) of 2 and 2-H+ are negative, see SI Appendix, Fig. S4. The z axis is along the Fe─O bond in all systems. Results for 2 apply to the majority species.
∥Values for the 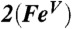 model obtained from DFT calculations using BP86 functionals (see below).
model obtained from DFT calculations using BP86 functionals (see below).
**Relative to g, A(17O) and A(14N) are rotated 8º around the z axis of the g-tensor.
††From ref. 14.
‡‡Correcting Az for the orbital contribution, which is proportional to (gz - 2), would yield Az ∼ 0.
We have conducted extensive EPR studies of 2 and 2-H+. The use of glassing solvent mixtures (1∶3 CH3CN∶CH2Cl2 or 1∶3 CH3CN∶butyronitrile) sharpened the spectra considerably (4 G linewidth). The higher resolution provided decisive clues about the ligand structure of 2 and 2-H+, as well as important magnetic hyperfine structure data. Selected spectra are displayed in Fig. 4. We found that 2 reproducibly displayed two 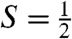 species in essentially constant proportions (ca. 2∶1 in all preparations studied); the minority species is recognized by a shoulder on the high-field feature in Fig. 4A. Our studies revealed that these two species did not differ in any significant way. The minor species, 2m, has g-values at 2.05, 1.99, and 1.96. As 2-H+ was found to be free of EPR-active contaminants in the g = 2.0 region, we focus here on this species. (EPR analysis of 2, summarized in Table 1 and detailed in the SI Appendix, afforded similar conclusions.) For 2-H+ (Fig. 4B), the signal at gx = 2.054 exhibits a 1∶1∶1 hyperfine triplet, corresponding to a single 14N nucleus with |Ax(14N)| = 28.5 MHz and |Ay,z(14N)| ≅ 11 MHz (the signs of Ax,y,z cannot be determined by EPR). A nearly identical pattern can be discerned in the spectrum of 2 shown in Fig. 4A. This splitting pattern changed to a 1∶1 doublet with |Ax(15N)| = 40 MHz (Fig. 4C) when 2-H+ was prepared using 1∶3
species in essentially constant proportions (ca. 2∶1 in all preparations studied); the minority species is recognized by a shoulder on the high-field feature in Fig. 4A. Our studies revealed that these two species did not differ in any significant way. The minor species, 2m, has g-values at 2.05, 1.99, and 1.96. As 2-H+ was found to be free of EPR-active contaminants in the g = 2.0 region, we focus here on this species. (EPR analysis of 2, summarized in Table 1 and detailed in the SI Appendix, afforded similar conclusions.) For 2-H+ (Fig. 4B), the signal at gx = 2.054 exhibits a 1∶1∶1 hyperfine triplet, corresponding to a single 14N nucleus with |Ax(14N)| = 28.5 MHz and |Ay,z(14N)| ≅ 11 MHz (the signs of Ax,y,z cannot be determined by EPR). A nearly identical pattern can be discerned in the spectrum of 2 shown in Fig. 4A. This splitting pattern changed to a 1∶1 doublet with |Ax(15N)| = 40 MHz (Fig. 4C) when 2-H+ was prepared using 1∶3 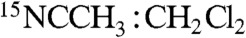 as solvent, unequivocally demonstrating that the observed hyperfine structure arises from a nitrogen atom derived from acetonitrile. This procedure yielded the parameters listed in Table 1.
as solvent, unequivocally demonstrating that the observed hyperfine structure arises from a nitrogen atom derived from acetonitrile. This procedure yielded the parameters listed in Table 1.
Fig. 4.

X-band EPR spectra of 2 and 2-H+. Experimental data (solid black lines) obtained in 1∶3 CH3CN∶butyronitrile (A, B, D, E) or 1∶3 CH3CN∶ CH2Cl2 (C) and recorded at 40 K. The conditions are, as follows: 9.62 GHz; microwave power, 20 μW; modulation, 0.3 mT. The red lines are theoretical curves generated with the parameters listed in Table 1. The 17O enrichment in E, obtained from EPR, is ≈30%. For 2 only the simulation for the majority species is shown. F shows the spectral simulation of 2-H+ assuming 100% 17O enrichment.
Species 2-H+ obtained from 17O-labeled 1 gave rise to a spectrum for which the (middle) g = gy = 2.01 feature was split to afford satellite features at both low and high field (Fig. 4E); these features arise from 17O superhyperfine splitting with |Ay(17O)| = 133 MHz. We have studied two preparations, with 17O enrichment of 10% and 30%; a spectrum of the latter shown in Fig. 4E. To analyze this spectrum we have taken the results of the fit of Fig. 4B and added an 17O hyperfine tensor. After obtaining a sufficient understanding of the splitting pattern, we have fitted the spectrum, visually focusing on the resolved two high-field peaks and the low-field feature. The simulation of Fig. 4F (plotted for 100% 17O enrichment) indicates the complexity of the spectrum, which stems to a major extent from the fact that the 17O splitting along y does not belong to a unique direction and that the splitting covers the entire Δg range. The final fit, obtained after exploring tensor rotations, gives a good value for Ay(17O) and an upper limit for Az(17O). Although the calculated intensity for the low field feature is a bit high, the fit could be improved by increasing Ax(17O) together with a distribution of that parameter.
Lastly, the 57Fe-enriched sample (Fig. 4D) exhibits significant broadening of the gx feature corresponding to |Ax(57Fe)| = 40 MHz, a value in good agreement with the largest A-tensor component of -47 MHz observed in the Mössbauer spectra of 2 and 2-H+ (EPR can only give the magnitude of the A-values); our EPR spectral simulations show that Ay and Az are at least a factor of two smaller than Ax. Most importantly, the EPR spectra of the 57Fe-enriched sample establish that the largest 57Fe A-tensor component is along x, the same direction for which the largest 14N splitting is observed. It should be noted that the EPR spectra establish a spatial correlation between the dominant components of the 57Fe, 17O, and 14N hyperfine tensors that provides insight into the electronic structure of 2 and 2-H+.
Our accumulated spectroscopic data suggest that 2 and 2-H+ are one-electron oxidized derivatives of 1 and consist of a (TMC)Fe═O unit with an axial ligand derived from acetonitrile. In Fig. 1, we propose that 2 is formed by the attack of the t-butylperoxide anion on the bound acetonitrile to form a peroxyimidic ester intermediate (17) that then undergoes homolytic cleavage of the O─O bond. To verify that homolysis occurs in this instance, we replaced tBuOOH with 2-methyl-1-phenylpropan-2-yl hydroperoxide (MPPH), a useful mechanistic probe to distinguish between homolytic and heterolytic O─O bond cleavage (18). Although substitution of MPPH for tBuOOH did not diminish the yield of 2, product analysis by GC showed quantitative conversion of MPPH to benzaldehyde. This result demonstrates that O─O bond homolysis occurs to generate 2. The formulation of 2 as shown in Fig. 1 is supported by low temperature electrospray ionization (ESI)-MS studies of 2 that reveal a feature at m/z 385.2, consistent with the formulation of 2 as [Fe(O)(TMC)(NCCH3) + O]+ (SI Appendix, Figs. S8 and S9). This feature shifts to m/z 388.2 upon preparation of 2 in CD3CN and disappears after the sample is allowed to decay. We postulate in Fig. 1 two possible limiting electronic descriptions for 2: an oxoiron(IV) unit with an axial acetaminyl radical ligand (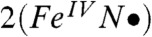 ) at one end, and an iron(V) center with trans oxo and acetylimido ligands (
) at one end, and an iron(V) center with trans oxo and acetylimido ligands (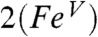 ) at the other end. Our efforts to refine electronic descriptions for 2 and 2-H+ more precisely are discussed below.
) at the other end. Our efforts to refine electronic descriptions for 2 and 2-H+ more precisely are discussed below.
The unusual A-tensor anisotropies in the xy plane listed in Table 1 for 2 and 2-H+ indicate that their electronic structures must be significantly different from that of 1. The 57Fe and 17O tensors of 1 as well as those of horseradish peroxidase compound I, an S = 1 FeIV═O species antiferromagnetically coupled to a porphyrin radical (19), are axial with respect to the Fe═O bond, the z axis in our coordinate frames. The axial anisotropy observed reflects the (dxz)1(dyz)1 electronic configuration associated with the S = 1 FeIV═O unit. In contrast, 2 and 2-H+ have drastically different A-tensor components along x and y, indicating substantially different unpaired spin densities on the dxz and dyz orbitals. This distribution can be achieved by transferring the electron from dxz to the px orbital of the aminyl ligand to create an FeV═O center with a trans imido ligand, a notion supported by the similar 57Fe A-tensor anisotropy observed for the bona fide FeV═O complex [FeV(O)(TAML)]- (Table 1) (14).
Density functional theory (DFT) studies of 2 and 2-H+ lend substantial support to the above conclusion (see SI Appendix, Tables S1–S7 for full computational details). For comparison, DFT calculations were also carried out for the experimentally characterized complexes 1 and [FeV(O)(TAML)]- as well as the hypothetical iron(V) species 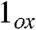 (a computational model obtained by removing one electron from 1; italics denote purely computational models). The use of the hybrid functional B3LYP led to the
(a computational model obtained by removing one electron from 1; italics denote purely computational models). The use of the hybrid functional B3LYP led to the 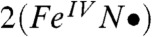 description, which is similar to that reported for HRP Cpd I, but with the radical trans rather than cis to the oxo group. This FeIV-aminyl radical description with an iron (dxz)1(dyz)1 electron configuration predicts 2 and 2-H+ to have essentially axial 57Fe and 17O A-tensors, inconsistent with the experimental data. In stark contrast, the use of the pure functional BP86 generated the
description, which is similar to that reported for HRP Cpd I, but with the radical trans rather than cis to the oxo group. This FeIV-aminyl radical description with an iron (dxz)1(dyz)1 electron configuration predicts 2 and 2-H+ to have essentially axial 57Fe and 17O A-tensors, inconsistent with the experimental data. In stark contrast, the use of the pure functional BP86 generated the 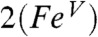 limiting description with an iron (dyz)1(dxz)0 electron configuration and yielded solutions that reproduce the observed anisotropies of the 57Fe A-tensor and rationalize the essential features of the entire dataset. In this model, the acetaminyl radical becomes an acetylimido ligand, and 2 and 2-H+ are best formulated as complexes having [O═FeV═NR]+ units (see Fig. 1). The dependence of the calculated ground state on whether a hybrid (B3LYP) or pure (BP86) functional is used may be related to the propensity of Hartree-Fock exchange to stabilize high spin states in the case of the former functional. A similar dependence on the type of functionals used has been reported for the description of electronic structures of {FeNO}7 and {FeO2}8 complexes, systems involving a noninnocent ligand radical (see for example refs. 20 and 21). In all cases, the functional chosen for further investigation most closely reproduced the experimentally observed spectroscopic parameters to generate an experimentally validated computational model.
limiting description with an iron (dyz)1(dxz)0 electron configuration and yielded solutions that reproduce the observed anisotropies of the 57Fe A-tensor and rationalize the essential features of the entire dataset. In this model, the acetaminyl radical becomes an acetylimido ligand, and 2 and 2-H+ are best formulated as complexes having [O═FeV═NR]+ units (see Fig. 1). The dependence of the calculated ground state on whether a hybrid (B3LYP) or pure (BP86) functional is used may be related to the propensity of Hartree-Fock exchange to stabilize high spin states in the case of the former functional. A similar dependence on the type of functionals used has been reported for the description of electronic structures of {FeNO}7 and {FeO2}8 complexes, systems involving a noninnocent ligand radical (see for example refs. 20 and 21). In all cases, the functional chosen for further investigation most closely reproduced the experimentally observed spectroscopic parameters to generate an experimentally validated computational model.
Table 2 compares the unpaired spin populations calculated for key orbitals of several complexes. Interestingly, although the dyz spin population remains more or less constant at about 0.58 across the series, the dxz spin population is equal to the dyz spin population for 1 and 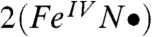 but decreases progressively for
but decreases progressively for 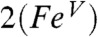 ,
, 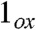 , and [FeV(O)(TAML)]-. As viewed from the progression of dxz spin density listed in Table 2, the electronic configuration of 2 more closely resembles that of
, and [FeV(O)(TAML)]-. As viewed from the progression of dxz spin density listed in Table 2, the electronic configuration of 2 more closely resembles that of 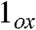 and [FeV(O)(TAML)]- than of 1 and
and [FeV(O)(TAML)]- than of 1 and 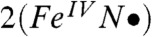 . Because the transfer of electron density from the metal dxz orbital to the singly occupied px orbital of the acetaminyl radical is not complete, the axial nitrogen retains a net β spin population in px (n = -0.30), which is much smaller than the value (n = -0.85) calculated for
. Because the transfer of electron density from the metal dxz orbital to the singly occupied px orbital of the acetaminyl radical is not complete, the axial nitrogen retains a net β spin population in px (n = -0.30), which is much smaller than the value (n = -0.85) calculated for 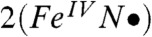 . Consistent with these ideas, the Mulliken Fe spin populations for
. Consistent with these ideas, the Mulliken Fe spin populations for 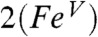 and
and 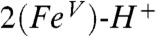 are calculated to be 0.86 and 0.77, respectively, which is lower than the 1.30 value calculated for 1. The expectation value of the operator S2 provides quantitative insight into the oxidation state predicted by DFT. The
are calculated to be 0.86 and 0.77, respectively, which is lower than the 1.30 value calculated for 1. The expectation value of the operator S2 provides quantitative insight into the oxidation state predicted by DFT. The 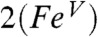 model was found to have
model was found to have 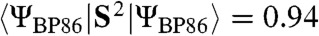 , which is close to the ideal value of S(S + 1) = 0.75 for
, which is close to the ideal value of S(S + 1) = 0.75 for 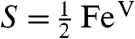 but considerably smaller than the value of 1.75 anticipated for the broken symmetry state (BS) of
but considerably smaller than the value of 1.75 anticipated for the broken symmetry state (BS) of 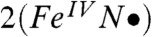 . Using the expansion |ΨBP86 > = c|FeV,
. Using the expansion |ΨBP86 > = c|FeV, 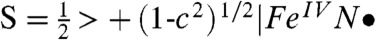 , BS > and the expectation values
, BS > and the expectation values 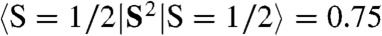 and
and 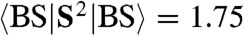 , we obtain c2 = 0.81 from
, we obtain c2 = 0.81 from 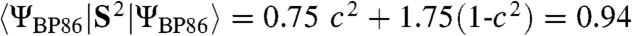 . As spin unrestricted calculations show always some degree of spin contamination, the FeV character of 2 (and 2-H+) is greater than 80%.
. As spin unrestricted calculations show always some degree of spin contamination, the FeV character of 2 (and 2-H+) is greater than 80%.
Table 2.
Calculated unpaired spin populations n = nα-nβ of Fe-dxz, Fe-dyz, and Naxial-px orbitals
| FeV(O)(TAML)* |
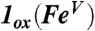 † †
|
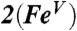 ‡ ‡
|
1 | 2 (FeIVN•)§ | |
| Fe-dxz | 0.07 | 0.15 | 0.23 | 0.58 | 0.58 |
| Fe-dyz | 0.57 | 0.63 | 0.53 | 0.58 | 0.58 |
| Naxial-px | – | ∼0 | −0.30 | 0 | −0.85 |
*Tiago de Oliveira et al. (14).
†Computational model obtained by removing one electron from 1.
‡BP86 solution.
§B3LYP solution, S = 1 FeIV═O coupled to S = 1/2 radical on N of axial ligand.
If we view 2 and 2-H+ as FeV complexes, the spin densities at the axial ligand atoms must originate from covalent spin polarization (22). Interaction of the oxo O(py) electron pair with the Fe(dyz) orbital results in partial transfer of a β oxo electron to pair off some of the α spin density associated with the unpaired Fe(dyz) electron, leaving a net α spin population in O(py) (blue in Fig. 5). The imido N(px) electron pair transfers net α spin density to the empty Fe(dxz) orbital, guided by Hund’s rule, leaving a net β spin population in N(px) (red in Fig. 5). The electron pair in the out-of-plane N(py) orbital of the imido ligand is calculated by DFT to be 10,000 cm-1 lower in energy than the redox-active in-plane N(px) orbital and thus is much less involved in covalent delocalization to the metal.
Fig. 5.

Spin density plots of geometry optimized BP86 solutions for 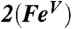 ,
, 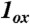 , and [Fe(O)(TAML)]-. The plot for
, and [Fe(O)(TAML)]-. The plot for 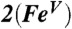 , shown in two views, reveals the contours of the orbitals carrying spin density. From top to bottom, px (Nam), dyz (Fe), and py (17O). For
, shown in two views, reveals the contours of the orbitals carrying spin density. From top to bottom, px (Nam), dyz (Fe), and py (17O). For 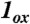 , the TMC ligand has been rotated by approximately 90° around the Fe═O bond relative to the orientation shown for
, the TMC ligand has been rotated by approximately 90° around the Fe═O bond relative to the orientation shown for 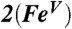 . Majority spin α in blue; minority spin β in red.
. Majority spin α in blue; minority spin β in red.
The resulting spin densities shown in Fig. 5 rationalize the spatial pattern observed in the A-tensors for 17O, 57Fe, and 14N. Thus, the net β spin population of the N(px) gives a magnetic hyperfine tensor with a large component along x and two smaller components along y and z for 2 (and 2-H+), with calculated Ax,y,z values of (-39, 11, 6) MHz for 14N in good agreement with the data. The net α spin population of Fe(dyz) produces a spin-dipolar hyperfine contribution with a large negative component along x, which together with the negative Fermi contact term yields a magnetic hyperfine tensor of which the dominant component is along x; the calculated Ax,y,z values of (-44, -19, 3) MHz (Table 1) are also in excellent agreement with the results obtained for 2 and 2-H+. Finally, the net α spin population of O(py) gives an 17O A-tensor that has a large component along y, with Ax,y,z values of (-1, -71, +76) MHz. The agreement for 17O between experiment and calculations is not as good as that for 57Fe and 14N; Ay is still too small and Az comes out too large but, importantly, the calculated tensor lacks axial symmetry around z and exhibits the x/y anisotropy observed experimentally. Finally, the g-values calculated for 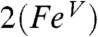 and reported in Table 1 match both in magnitude and direction the experimental data for 2 and 2-H+. Thus the congruence of the experimental EPR results with the
and reported in Table 1 match both in magnitude and direction the experimental data for 2 and 2-H+. Thus the congruence of the experimental EPR results with the 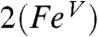 model provides a strong rationale for the description of 2 and 2-H+ as FeV complexes.
model provides a strong rationale for the description of 2 and 2-H+ as FeV complexes.
In contrast, the νFe═O frequencies hint at an FeIV oxidation state for 2 and 2-H+ and therefore may at first glance favor 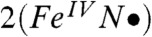 over
over 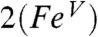 . It should be noted, however, that νFe═O is a spectroscopic parameter that reflects the length of the Fe-oxo bond (23). The Fe─O bond in the DFT model
. It should be noted, however, that νFe═O is a spectroscopic parameter that reflects the length of the Fe-oxo bond (23). The Fe─O bond in the DFT model 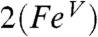 (1.70 Å) is longer than the oxoiron(V) bond in
(1.70 Å) is longer than the oxoiron(V) bond in 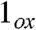 (1.60 Å) and the oxoiron(IV) bond in 1 (1.66 Å) due to delocalization of electron density over the oxoironimido moiety* (see Fig. 6) and is therefore predicted to exhibit a lower νFe═O than
(1.60 Å) and the oxoiron(IV) bond in 1 (1.66 Å) due to delocalization of electron density over the oxoironimido moiety* (see Fig. 6) and is therefore predicted to exhibit a lower νFe═O than 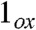 . The
. The 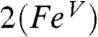 model predicts νFe═O values of 785 cm-1 for
model predicts νFe═O values of 785 cm-1 for 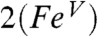 (νexp = 798 cm-1) and of 829 cm-1 for
(νexp = 798 cm-1) and of 829 cm-1 for 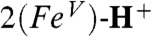 (νexp = 811 cm-1) in excellent agreement with the trend observed in our experimental data, indicating that the imido ligand exerts a sizable trans influence on the Fe─O unit.
(νexp = 811 cm-1) in excellent agreement with the trend observed in our experimental data, indicating that the imido ligand exerts a sizable trans influence on the Fe─O unit.
Fig. 6.

Calculated geometric and spectral parameters for the energy-minimized BP86 models 1ox, 2, and 2-H+. Bond lengths are reported in angstroms “L” denotes the TMC ligand.
Similarly, the weaker Fe═O bonds of 2 and 2-H+ are expected to give rise to higher Mössbauer isomer shifts, as this spectroscopic parameter reflects the 3 s electron density at the 57Fe nucleus and the extent to which the 3 s electrons are shielded by 3d electrons. Thus, we expect the δ value of 2 to be more positive than that of an FeV complex with an O═Fe─NCCH3 unit. As a further test of the trans influence of the O═Fe═N─C(O)─R unit on the value of δ, we calculated the properties for the hypothetical 1-electron reduced 2 (denoted  ) and obtained an Fe─O bond length of 1.767 Å, which is 0.10 Å longer than the Fe─O distance 1.664 Å calculated for 1. The increase in bond length is indeed accompanied by an increase of the δ value by 0.09 mm/s and a 164 cm-1 decrease in νFe═O. The significant trans influence of the acetylimido ligand is also manifested in the comparison of the calculated values for
) and obtained an Fe─O bond length of 1.767 Å, which is 0.10 Å longer than the Fe─O distance 1.664 Å calculated for 1. The increase in bond length is indeed accompanied by an increase of the δ value by 0.09 mm/s and a 164 cm-1 decrease in νFe═O. The significant trans influence of the acetylimido ligand is also manifested in the comparison of the calculated values for 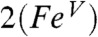 (rFe─O = 1.701 Å, δ = 0.03 mm/s, νFe═O = 785 cm-1) and
(rFe─O = 1.701 Å, δ = 0.03 mm/s, νFe═O = 785 cm-1) and 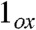 (rFe─O = 1.603 Å, δ = -0.08 mm/s, νFe═O = 944 cm-1). These calculations support our view that νFe═O and δ are poor indicators for the oxidation state of 2 and 2-H+, without properly accounting for the trans influence of the axial ligand. Judged from the full data set, 2 and 2-H+ are best described as FeV complexes.
(rFe─O = 1.603 Å, δ = -0.08 mm/s, νFe═O = 944 cm-1). These calculations support our view that νFe═O and δ are poor indicators for the oxidation state of 2 and 2-H+, without properly accounting for the trans influence of the axial ligand. Judged from the full data set, 2 and 2-H+ are best described as FeV complexes.
The characterization of 2 and 2-H+ reported in this paper increases to three the number of synthetic oxoiron(V) complexes for which detailed spectroscopic and electronic structure information is available. Such complexes demonstrate that it is possible to stabilize such a high-valent state in an iron coordination complex. For [FeV(O)(TAML)]-, the iron(V) oxidation state was achieved by the use of a tetraanionic tetraazamacrocyclic ligand that mitigates the high charge of the metal center (14). In contrast, 2 and 2-H+ are supported by a neutral tetraazamacrocycle, so the stabilization of the iron(V) state, it would seem, must derive from the presence of the charged axial oxo and imido ligands. The structure we favor for 2 is closely related to that of [MnV(O)2(TPFPP)]- [where TPFPP is meso-tetrakis(pentafluorophenyl)porphinate dianion], which is the only known example of a first row transition metal containing an [MV(O)2] moiety (24). Examples of nitridoiron(V) and nitridoiron(VI) complexes have also been reported, demonstrating the ability of the N3- ligand to allow access to high-valent iron complexes, even with supporting polydentate ligands that are neutral or monoanionic (25–29). Lastly, the characterization of 2 and 2-H+ demonstrates the possibility of a neutral nonheme ligand to support an Fe(V) center and lends credence to mechanistic proposals for FeV(O)(OH) oxidants in arene cis-dihydroxylation by Rieske dioxygenases (5) and in alkane, olefin, and water oxidation by a series of bioinspired synthetic iron catalysts (6–9, 11). For the latter, evidence for the fleeting iron(V) intermediates has been obtained from low temperature spectroscopic analysis of catalytic reactions by EPR (30, 31) and by mass spectrometry (10, 12).
Experimental Procedures
Preparation of 1, 2, and 2-H+.
All reagents and solvents were purchased from commercial sources and used as received unless otherwise noted. As previously reported (15), a solution of [FeII(TMC)(O3SCF3)] in acetonitrile was reacted with 1.2 equivalents of PhIO solubilized in methanol to obtain 1 in quantitative yield, as monitored by the appearance of its distinctive chromophore at 820 nm. Compound 2 was prepared at -44 °C in acetonitrile by the addition of at least three equivalents of tert-butyl hydroperoxide followed by the addition of 1–2 equivalents of NBu4OH or KOtBu. Subsequent addition of at least five equivalents of strong acid, HBF4 or HClO4, yielded 2-H+. Forty percent 17O-enriched H2O was used to prepare the 17O EPR samples.
Physical Methods.
A Hewlett Packard 8453A diode-array spectrometer equipped with a Unisoku Scientific Instruments cryostat to maintain a constant temperature was employed to collect electronic absorption spectrum. Resonance Raman spectra were generated via excitation with Kr+ and Ar+ lasers (Spectra Physics BeamLok 2060-RM) with a power of < 10 mW at the samples, and the spectra were collected using an ACTON AM-506M3 monochromator and a Princeton Instruments ACTON PyLoN LN/CCD-1340 × 400 detector. X band (9.28 GHz) EPR spectra were recorded on a Bruker ESR 300 spectrometer equipped with an Oxford ESR 910 liquid Helium cryostat and an Oxford temperature controller. Mössbauer spectra were recorded using Janis Research Super-Varitemp dewars that allowed studies in applied magnetic fields up to 8.0 T. Mössbauer spectral simulations were performed using the WMOSS software package (SEE Co), and EPR spectra were simulated with SpinCount, a program developed by Prof. M. P. Hendrich of Carnegie Mellon University. Isomer shifts are quoted relative to Fe metal at 298 K. ESI-MS experiments were conducted using a Bruker BioTOF II mass spectrometer. The spray chamber voltage was set to 4,000 V and the gas carrier temperature was set at 60 °C.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Professor Thomas C. Brunold at the University of Wisconsin-Madison for kindly providing access to his computer cluster. We are particularly grateful to two of the reviewers for suggesting a key improvement in our proposed mechanism in Figure 1. This work was supported by National Institutes of Health Grants GM33162 and GM38767 (to L.Q.) and EB001475 (to E.M.) and postdoctoral fellowships GM-093479 (to K.M.V.H.) and GM-079839 (to A.T.F.) and National Science Foundation Grant CHE1058248 (to L.Q.) and Grant CHE070073 (to E.L.B.) through TeraGrid resources provided by the National Center for Supercomputing Applications and Pittsburgh Supercomputing Center.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1206457109/-/DCSupplemental.
*Unfortunately, our Raman experiments have not been able to identify vibrational modes that can be associated with the Fe═NR unit. This result may not be surprising, as vibrational data are also not available for the only previously reported six-coordinate Fe═NR complex (32), although Raman data for four-coordinate Fe═NR complexes have been obtained (33).
References
- 1.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 2.Zilly FE, et al. Tuning a P450 enzyme for methane oxidation. Angew Chem Int Ed. 2011;50:2720–2724. doi: 10.1002/anie.201006587. [DOI] [PubMed] [Google Scholar]
- 3.Kawakami N, Shoji O, Watanabe Y. Use of perfluorocarboxylic acids to trick cytochrome P450BM3 into initiating the hydroxylation of gaseous alkanes. Angew Chem Int Ed. 2011;50:5315–5318. doi: 10.1002/anie.201007975. [DOI] [PubMed] [Google Scholar]
- 4.Rittle J, Green MT. Cytochrome P450 compound I: Capture, characterization, and C─H bond activation kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 5.Kovaleva EG, Lipscomb JD. Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen K, Que L., Jr Stereospecific alkane hydroxylation by nonheme iron catalysts: Mechanistic evidence for an FeV═O active species. J Am Chem Soc. 2001;123:6327–6337. doi: 10.1021/ja010310x. [DOI] [PubMed] [Google Scholar]
- 7.Company A, et al. Alkane hydroxylation by a nonheme iron catalyst that challenges the heme paradigm for oxygenase action. J Am Chem Soc. 2007;129:15766–15767. doi: 10.1021/ja077761n. [DOI] [PubMed] [Google Scholar]
- 8.Chen K, Costas M, Kim J, Tipton AK, Que L., Jr Olefin cis-dihydroxylation versus epoxidation by nonheme iron catalysts: Two faces of an FeIII─OOH coin. J Am Chem Soc. 2002;124:3026–3035. doi: 10.1021/ja0120025. [DOI] [PubMed] [Google Scholar]
- 9.Company A, et al. Olefin-dependent discrimination between two nonheme HO-FeV═ tautomeric species in catalytic H2O2 epoxidations. Chem Eur J. 2009;15:3359–3362. doi: 10.1002/chem.200802597. [DOI] [PubMed] [Google Scholar]
- 10.Chow TW-S, et al. Cis-dihydroxylation of alkenes with oxone catalyzed by iron complexes of a macrocyclic tetraaza ligand and reaction mechanism by ESI-MS spectrometry and DFT calculations. J Am Chem Soc. 2010;132:13229–13239. doi: 10.1021/ja100967g. [DOI] [PubMed] [Google Scholar]
- 11.Fillol JL, et al. Efficient water oxidation catalysts based on readily available iron coordination complexes. Nat Chem. 2011;3:807–813. doi: 10.1038/nchem.1140. [DOI] [PubMed] [Google Scholar]
- 12.Prat I, et al. Observation of Fe(V)═O using variable temperature mass spectrometry and its enzyme-like C─H and C═C oxidation reactions. Nat Chem. 2011;3:788–793. doi: 10.1038/nchem.1132. [DOI] [PubMed] [Google Scholar]
- 13.Que L., Jr The road to non-heme oxoferryls and beyond. Acc Chem Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- 14.Tiago de Oliveira F, et al. Chemical and spectroscopic evidence for an FeV─oxo complex. Science. 2007;315:835–838. doi: 10.1126/science.1133417. [DOI] [PubMed] [Google Scholar]
- 15.Rohde J-U, et al. Crystallographic and spectroscopic evidence for a nonheme FeIV═O complex. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 16.Jackson TA, et al. Axial ligand effects on the geometric and electronic structures of nonheme oxoiron(IV) complexes. J Am Chem Soc. 2008;130:12394–12407. doi: 10.1021/ja8022576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Payne GB, Deming PH, Williams PH. Reactions of hydrogen peroxide. VII. Alkali-catalyzed epoxidation and oxidation using a nitrile as co-reactant. J Org Chem. 1961;26:659–663. [Google Scholar]
- 18.MacFaul PA, Arends IWCE, Ingold KI, Wayner DDM. Oxygen activation by metal complexes and alkyl hydroperoxides. Applications of mechanistic probes to explore the role of alkoxyl radicals in alkane functionalization. J Chem Soc Perkin Trans. 1997;2:135–146. [Google Scholar]
- 19.Roberts JE, Hoffman BM, Rutter R, Hager LP. 17O ENDOR of horseradish peroxidase compound I. J Am Chem Soc. 1981;103:7654–7656. [PubMed] [Google Scholar]
- 20.Schenk G, Pau MYM, Solomon EI. Comparison between the geometric and electronic structures and reactivities of {FeNO}7 and {FeO2}8 complexes: A density functional theory study. J Am Chem Soc. 2004;126:505–515. doi: 10.1021/ja036715u. [DOI] [PubMed] [Google Scholar]
- 21.Hopmann KH, Conradie J, Ghosh A. Broken-symmetry DFT spin densities of iron nitrosyls, including Roussin’s red and black salts: Striking differences between pure and hybrid functionals. J Phys Chem B. 2009;113:10540–10547. doi: 10.1021/jp904135h. [DOI] [PubMed] [Google Scholar]
- 22.Drago RS. Physical Methods for Chemists. 2nd Ed. Philadelphia: Saunders; 1992. [Google Scholar]
- 23.Green MT. Application of Badger’s rule to heme and non-heme iron–oxygen bonds: An examination of ferryl protonation states. J Am Chem Soc. 2006;128:1902–1906. doi: 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- 24.Jin N, Ibrahim M, Spiro TG, Groves JT. Trans-dioxo manganese(V) porphyrins. J Am Chem Soc. 2007;129:12416–12417. doi: 10.1021/ja0761737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer K, Bill E, Mienert B, Weyhermüller T, Wieghardt K. Photolysis of cis- and trans-[FeIII(cyclam)(N3)2]+ complexes: Spectroscopic characterization of a nitridoiron(V) species. J Am Chem Soc. 1999;121:4859–4876. [Google Scholar]
- 26.Grapperhaus CA, Mienert B, Bill E, Weyhermüller T, Wieghardt K. Mononuclear (nitrido)iron(V) and (oxo)iron(IV) complexes via photolysis of [(cyclam-acetato)FeIII(N3)]+ and ozonolysis of [(cyclam-acetato)FeIII(O3SCF3)]+ in water/acetone mixtures. Inorg Chem. 2000;39:5306–5317. doi: 10.1021/ic0005238. [DOI] [PubMed] [Google Scholar]
-
27.Aliaga-Alcalde N, et al. The geometric and electronic structure of [(cyclam-acetato)Fe(N)]+: A genuine iron(V) species with a ground-state spin
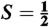 . Angew Chem Int Ed. 2005;44:2908–2912. doi: 10.1002/anie.200462368. [DOI] [PubMed] [Google Scholar]
. Angew Chem Int Ed. 2005;44:2908–2912. doi: 10.1002/anie.200462368. [DOI] [PubMed] [Google Scholar] - 28.Berry JF, et al. An octahedral coordination complex of iron(VI) Science. 2006;312:1937–1941. doi: 10.1126/science.1128506. [DOI] [PubMed] [Google Scholar]
- 29.Scepaniak JJ, et al. Synthesis, structure, and reactivity of an iron(V) nitride. Science. 2011;331:1049–1052. doi: 10.1126/science.1198315. [DOI] [PubMed] [Google Scholar]
- 30.Lyakin OY, Bryliakov KP, Britovsek GJP, Talsi EP. EPR spectroscopic trapping of the active species of nonheme iron-catalyzed oxidation. J Am Chem Soc. 2009;131:10798–10799. doi: 10.1021/ja902659c. [DOI] [PubMed] [Google Scholar]
- 31.Lyakin OY, Bryliakov KP, Talsi EP. EPR, 1H and 2H NMR, and reactivity studies of the iron-oxygen intermediates in bioinspired catalyst systems. Inorg Chem. 2011;50:5526–5538. doi: 10.1021/ic200088e. [DOI] [PubMed] [Google Scholar]
- 32.Klinker EJ, et al. A tosylimido analogue of a nonheme oxoiron(IV) complex. Angew Chem Int Ed. 2006;45:7394–7397. doi: 10.1002/anie.200602799. [DOI] [PubMed] [Google Scholar]
- 33.Mehn MP, Brown SD, Jenkins DM, Peters JC, Que L., Jr Vibrational spectroscopy and analysis of pseudo-tetrahedral complexes with metal imido bonds. Inorg Chem. 2006;45:7417–7427. doi: 10.1021/ic060670r. [DOI] [PubMed] [Google Scholar]
- 34.Schulz CE, Rutter R, Sage JT, Debrunner PG, Hager LP. Mössbauer and electron paramagnetic resonance studies of horseradish peroxidase and its catalytic intermediates. Biochemistry. 1984;23:4743–4754. doi: 10.1021/bi00315a033. [DOI] [PubMed] [Google Scholar]
- 35.Krzystek J, et al. Determination by high-frequency and -field EPR of zero-field splitting in iron(IV) oxo complexes: Implications for intermediates in nonheme iron enzymes. Inorg Chem. 2008;47:3483–3485. doi: 10.1021/ic800411c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.