

Abstract
Myeloma is a malignancy of the antibody-producing plasma cells and, as such, these cells synthesize large quantities of unfolded or misfolded immunoglobulin. The build-up of excess protein triggers a number of downstream signal transduction cascades, including endoplasmic reticulum stress and autophagy. As a result, myeloma cells are uniquely reliant on these and other protein handling pathways for their survival. Strategies aimed at targeting this vulnerability have proved successful with the proteasome inhibitor, bortezomib, already licensed for clinical use. In addition to the proteasome, various other points within the protein handling pathways are also the subject of drug discovery projects, with some already progressing into clinical trials. These include compounds directed against heat shock proteins, the unfolded protein response and pathways both upstream and downstream of the proteasome. More recently, the role of autophagy has been recognized in myeloma. In this review, we discuss the various pathways used by myeloma cells for survival, with particular emphasis on the emerging role and conundrum of autophagy, as well as highlighting pre-clinical research on novel inhibitors targeting protein handling pathways.
Keywords: autophagy, proteasome, ER stress, heat-shock chaperones, unfolded protein response
Introduction
Myeloma is a genetically heterogeneous clonal B-cell malignancy, characterized by the accumulation of abnormal antibody-producing plasma cells in the bone marrow. It is the second most common hematologic malignancy and affects about 3,500 people in the UK each year. It tends to be a cancer of the elderly with a slightly increased incidence in men compared to woman, as well as having a higher incidence in African American populations and first degree relatives.1
Current treatment strategies include the use of immunomodulatory drugs such as thalidomide and its derivatives, proteasome inhibitors such as bortezomib, synthetic steroids like dexamethasone and prednisolone, alkylating agents and anthracycline, as well as autologous stem cell transplantation whenever possible. Despite a plethora of drugs currently used to treat myeloma, the disease remains incurable, with median survival ranging from three to six years depending on the stage of the disease.2,3
The introduction of targeted treatments such as thalidomide, bortezomib and lenalidomide has improved the median survival of myeloma patients. However, patients continue to relapse. This is in part due to the tumor cells developing resistance due to interactions with the bone marrow microenvironment.2 There is, therefore, an urgent need for novel therapies to treat this disease and this will be achieved by developing a solid understanding of tumor biology.
Although myeloma is a complex malignancy, there are underlying common characteristics that are currently being exploited to treat this disease. As a result of the large quantities of unfolded or misfolded immunoglobulin that these cells produce, they are integrally reliant on their core protein handling machinery. This idea is exemplified by the success of the proteasome inhibitor, bortezomib, which was licensed by the Food and Drug Administration in 2003 for the treatment of refractory myeloma.4 However, in addition to removal by the ubiquitin proteasome pathway, removal of excess proteins is also achieved by the autophagy pathway. This is a catabolic process whereby cells degrade portions of cytoplasm, including organelles. It has been shown to be an important part of normal development as well as playing a role in neurodegenerative diseases and cancer.5 Inhibitors of the autophagy pathway are currently being tested in myeloma in two phase I/II trials in combination with bortezomib (www.clinicaltrials.gov) underscoring the importance of these protein handling pathways in treating this malignancy.
A third pathway for removal of excess proteins is the aggresome pathway which is activated if proteasome activity is compromised. Misfolded, ubiquitinated protein aggregates are recognized by histone deacetylase 6 (HDAC6) and transported by the microtubule network to aggresomes for clearance. Recent results demonstrate synergy between an HDAC6 inhibitor and bortezomib6 and clinical trials testing this combination have begun. The role of aggresomes and HDACs has been extensively reviewed and so will not be covered further here.7,8 We aim to provide an overview of the protein handling and stress response pathways. We highlight the potential points in these pathways that can be targeted therapeutically, and review the supporting pre-clinical data.
Protein handling pathways
Under normal conditions, the synthesis, folding and degradation of cellular proteins are balanced processes. In myeloma cells, however, the protein folding capacity of the endoplasmic reticulum (ER) is overloaded by large quantities of immunoglobulin and cells must adapt to this continual stress.9 A close relationship with common signaling nodes therefore exists between the ubiquitin proteasome pathway, cellular stress pathways such as the unfolded protein response (UPR), heat shock response and autophagy.
Ubiquitin proteasome pathway
The main cellular pathway for the removal and destruction of proteins is the ubiquitin proteasome pathway. This involves the sequential enzymatic transfer of ubiquitin monomers onto an elongating polyubiquitin chain bound at the lysine 11 or lysine 48 residues of target proteins.10 In the first step of this cascade, an E1 activating enzyme, typically ubiquitin activating enzyme (UAE, also known as UBA1), binds ubiquitin. A second ubiquitin monomer binds to a different site on the E1 enzyme and the first ubiquitin is transferred to an E2 ubiquitin-conjugating enzyme. The final step involves the transfer of ubiquitin from the E2 enzyme to the lysine 48 of the target protein in a process catalyzed by an E3 ubiquitin ligase. This process is then repeated to produce a polyubiquitin chain.11 While there are only eight E1 or E1-like enzymes that have been described so far, there are around forty E2 enzymes and even more E3 enzymes.12 Therefore, specificity of target protein degradation is achieved by the action of the E2 and E3 enzymes. Tagging proteins in this manner targets them for degradation via the proteasome; proteins that are monoubiquitinated or ubiquitinated at other residues will not be degraded by this process.
The 26S proteasome consists of a 20S catalytic core particle and either one or two 19S regulatory subunits. The 20S core particle contains the active site and is responsible for the hydrolysis of peptide bonds. On the other hand, the 19S particle is important for recognizing the substrate, deubiquitination, unfolding and translocation of the substrate into the catalytic core.13 The 20S core contains four heptameric rings, the two alpha rings located at either end with the two beta rings in the middle. These rings are stacked above each other thus forming a hollow cylinder. The alpha rings act as gates that are opened upon binding of the 19S subunits whereas the beta rings contain the active sites. Of the seven subunits of the beta rings, only three are catalytically active: β1 has caspase-like activity, β2 exhibits trypsin-like activity and β5 has chymotrypsin-like activity.14 Substrate proteins are degraded into peptides that vary in length from 3 to 25 amino acids. Each substrate is cleaved at multiple locations ensuring no partially hydrolyzed proteins exit the proteasome, only short amino acid chains. These amino acid chains are then further hydrolyzed by the aminopeptidase enzymes to release free amino acids.
Another form of the proteasome, the immunoproteasome, can also be found at elevated levels in myeloma cells.15 The immunoproteasome is induced in response to interferon-gamma and contains three catalytically active β subunits together with an 11S regulatory subunit. The function of the immunoproteasome is to produce peptides for presentation to major histocompatability complex class I molecules.16 These antigenic peptides are derived from defective ribosomal products which constitute a large percentage of all newly synthesized proteins.17
The multiple enzymatic steps involved in ubiquitination, 26S proteasome activity and immunoproteasome activity result in many sites within the UPP that can be therapeutically inhibited. Inhibition of this pathway results in a build up of polyubiquitinated proteins, ER stress and resulting cell death.18,19 Importantly, targeting this pathway exploits the myeloma cells’ critical dependence on the UPP, revealing a therapeutic window between the myeloma cells and normal cells. This therapeutic potential is discussed in more detail below.
ER stress and the unfolded protein response
ER stress results from an imbalance between the amount of unfolded or misfolded protein in the ER lumen and the capacity of the ER machinery to refold these proteins. Activation of the ensuing signal transduction pathway is termed the UPR, the main functions of which are to reduce the amount of protein that enters the ER and, secondly, to increase the folding capacity of the ER. If both of these strategies fail, apoptosis is triggered.20 Additionally, if proteins cannot be folded correctly in the ER, they are retrotranslocated to the cytoplasm for degradation by the proteasome, a process termed ER-associated degradation (ERAD).21 Thus, therapeutically manipulating this pathway can interfere with the cells’ ability to deal with high protein loads, cellular stress and result in tumor cell death. Importantly, the ER stress pathway overlaps with protein removal via the proteasome, as co-administration of bortezomib with compounds that induce ER stress enhances myeloma cell apoptosis.22
The three arms of the UPR are regulated by an ER-resident transmembrane protein that detects unfolded proteins in the ER lumen and transmits the signal to the cytoplasm. These are inositol requiring enzyme 1 (IRE1), PKR-like ER kinase (PERK) and activating transcription factor 6 (ATF6) which are held in an inactive state by glucose-regulated protein, 78kDa (GRP78/BiP). In the presence of unfolded proteins, GRP78 dissociates from these proteins allowing their activation.23
IRE1 is an unusual protein in that it contains both a kinase and endoribonuclease domain, the latter of which is important for the splicing of the transcription factor X-box binding protein 1 (XBP1). Spliced XBP1 regulates the expression of genes involved in ER expansion as well as those coding for ER chaperone proteins.24 It can also activate Akt to mediate anti-apoptotic signals.25 On the other hand, the kinase domain of IRE1 is capable of autophosphorylation, with subsequent downstream activation of c-Jun N-terminal kinase (JNK).26 It is this activity of IRE1 that links it to other protein degradation pathways within the cell, such as the autophagy pathway, as JNK phosphorylates Bcl2, allowing the essential autophagy gene, Beclin1, to associate with phosphatidylinositol-3-kinase (PI3K) Class III to trigger autophagy (Figure 1).27 The role of autophagy in protein degradation and ameliorating ER stress is discussed below.
Figure 1.

Relationship between the ER stress and autophagy pathways. The three ER stress sensor molecules, PERK, IRE1 and ATF6, are held in an inactive state by GRP78. The presence of unfolded protein results in their release and subsequent activation. The PERK and IRE1 branches are the most important for ER stress-induced autophagy: in the IRE1 branch, Bcl2 is phosphorylated by JNK, resulting in the release of Bcl2 from a complex with Beclin1 thus allowing autophagy to be activated. In the PERK branch, ATF4 and CHOP both lead to the upregulation of key autophagy genes. CHOP can also inhibit Akt via Trb3, thereby allowing autophagy activation.
The PERK branch of the UPR is important for the shutdown of global protein translation. This is achieved by PERK phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α) and subsequent inhibition of the guanine nucleotide exchange factor eIF2B, blocking eIF2 activation.28 Despite global inhibition of translation, there is selective translation of specific mRNAs; one such example is ATF4. Downstream of ATF4, CHOP activates GADD34 which acts via PP1 to dephosphorylate eIF2α, thus acting as a negative feedback loop.29 Important links to the autophagy pathway are provided by ATF4 upregulation of the autophagy genes LC3B and Atg1230,31 and CHOP upregulation of Atg5.31 In addition, indirect links to autophagy are provided by inhibition of molecules in the PI3K Class I pathway that negatively regulate autophagy; ATF4 inhibits mammalian target of rapamycin (mTOR) via Redd132 and CHOP inhibits Akt via Tribbles Homolog 3 (Trb3) (Figure 1).29
ATF6 exists as 2 isoforms, α and β, but it is the α isoform that appears to be essential in the UPR.33 Full activation requires a 2-step process involving transportation to the golgi followed by cleavage to reveal a nuclear localization signal.34 In the nucleus, ATF6α transcriptionally regulates genes coding for ER chaperones and has been shown to be involved in ER expansion, enabling the ER to correct the stress and handle more protein.33 Association with XBP1 regulates the induction of ERAD components.29,35 ATF6 also plays a role in quiescence which is mediated by Akt-independent activation of Rheb and mTOR,36 further linking the UPR and autophagy pathways.
Heat shock chaperone proteins
Heat shock proteins (HSP) are a group of ubiquitously expressed chaperone proteins that are up-regulated in times of stress and aid in efficient protein folding. They exist as part of multi-protein complexes together with co-chaperone proteins, such as HSP70 and HSP40. In addition to playing important roles in ensuring the correct folding of their ‘client’ proteins, they also prevent their aggregation.37 A number of these ‘client’ proteins play vital roles in the survival of cancer cells and thus the HSP family has received a lot of attention as a potential drug target. The HSP90 family consists of the cytosolic HSP90α and HSP90β, the ER-resident Grp94 and the mitochondrial Trap-1, whereas the HSP70 family cytosolic proteins are HSP70 and heat shock cognate 70 (HSC70), the ER protein GRP78 and the mitochondrial protein mortalin.38 The interest in HSP90 is based on the finding that its ‘client’ proteins include many oncogenic proteins, including receptor tyrosine kinases, cell cycle proteins and cell surface receptors such as FGFR3, Akt, TERT and p53.39 In addition, the expression of HSP90 is regulated by pathways known to be abberantly activated in a variety of cancers. Despite a large number of client proteins regulated by HSP90 (http://www.picard.ch/downloads/Hsp90interactors.pdf), the specificity for each is guided by the association of HSP90 with numerous co-chaperones, such as Cdc37.37
The function of HSPs are integrally linked to the UPP such that proteins that cannot assume their biologically active conformation are targeted for degradation. We have also shown that inhibition of HSP90 induces the UPR40 and HSP90 is able to stabilize IRE1.41 This suggests that the chaperone systems that exist in the cytoplasm and ER are able to co-ordinate their response to alleviate ER stress, and when therapeutically targeting one pathway, there may be a paradoxical upregulation of another.
Autophagy
In addition to the UPP, the autophagy pathway also acts to remove excess protein from the cell and there seems to be a relationship between these two pathways. The last ten years has seen an explosion in the field of autophagy research and many of the genes, processes and signaling pathways have been elucidated. Three distinct types of autophagy have been recognized based on the manner in which cargo is delivered to lysosomes, but we will focus on macroautophagy, known more simply as autophagy. At basal levels, autophagy plays an important homeostatic role, maintaining a balance between the catabolism and synthesis of macromolecules and organelles.42 It is up-regulated in response to conditions of stress, such as nutrient deprivation and the presence of aggregated proteins and hypoxia, ensuring cell survival.5,42 In addition, it is also implicated in development, tissue remodeling and a number of human diseases.5 Although initially studied as a response to nutrient starvation, its role in cancer has received much attention as a potential therapeutic target. However, at the same time, its precise role is controversial as it appears to have a dual function: either inhibiting oncogenic transformation or potentially promoting cancer growth and survival. Therefore, there is uncertainty about whether to inhibit or promote autophagy for cancer treatment. The process of autophagy can be divided into four steps: induction, nucleation, vesicle expansion and closure, and autolysosome formation. Each step is regulated by the co-ordinated action of a number of autophagy related (Atg) proteins (Figure 2).
Figure 2.

Schematic overview of the autophagy pathway. Initiation of autophagy results in the formation of the ULK1 complex, consisting of ULK1, FIP200, Atg13 and Atg17, which is negatively regulated by mTOR. Atg1 also acts to recruit additional Atg proteins to the phagophore. Vps34 forms a core complex with Beclin1 and p150 and produces PI3P for membrane formation. The Atg5-Atg12-Atg16 complex is formed by the combined action of Atg7 and Atg10 while processing of LC3 is accomplished by Atg4, Atg3 and Atg7. The completed autophagosome contains cytosolic proteins and/or organelles and subsequently fuses with lysosmes. The enzymes present in lysosomes degrade the autophagosome contents and this material is then recycled back to the cell. 3-MA: 3-methyladenine.
Autophagy mechanism and processes
Autophagy induction requires the activity of ULK1, the mammalian homolog of the yeast Atg1. It forms a complex with Atg13, FIP200 (yeast Atg17) and Atg101 and is negatively regulated by mTOR. Under autophagy permissive conditions, mTOR is inactive and, therefore, Atg13 is no longer hyperphosphorylated, relieving its inhibitory block on ULK and allowing its activation.43 Nucleation then occurs with the formation of a phagophore which envelopes cytosolic proteins and organelles. The main requirement for vesicle nucleation is the activity of PI3K Class III (hVps34), which exists in a core complex with Beclin1 (yeast Atg6) and p150 (yeast Vps15), and interacts with a number of proteins to either inhibit or promote autophagy.44–46 Additionally, the binding of Bcl-2 or Bcl-xL to Beclin1 can inhibit autophagy by preventing Beclin1 binding to hVps34.27
The phagophore expands and closes to completely sequester the material in an autophagosome, a double membrane-bound vesicle which is the characteristic feature of autophagy. The expansion of the developing autophagosome is mediated by two ubiquitin-like conjugation reactions. In the first, Atg7 and Atg10 link Atg12 to Atg5, which subsequently interacts with Atg16L and oligomerizes to form the Atg16L complex. In the second reaction, Atg8/LC3 is cleaved by Atg4 and joined to a lipid moiety, phosphatidylethanolamine (PE), by Atg3 and Atg7 to produce LC3II, which is located on the surface of autophagosomes.47,48 In addition, LC3 controls the expansion of the phagophore49 and its lipidation is required for closure of the autophagosome.50,51
The final steps in the autophagy process involve fusion between the autophagosome and lysosmes and the degradation of the autophagosome contents. While the exact details of how this happens are not completely understood, a number of proteins have been shown to be essential in this process, including LAMP1, LAMP2, UVRAG and Rab7, which are required for membrane fusion.52–54 Degradation of the autophagosome contents is carried out by numerous acid hydrolases, the major group being the cathepsins. Release of the autolysosome contents back to the cell is mediated by the action of Atg22 in yeast55 but the mammalian ortholog has yet to be identified (Figure 2).
Two sides to every story
The role of autophagy in cancer is unclear, with some lines of evidence indicating a tumor suppressor role whereas others suggest it may promote tumors. In support of a tumor suppressive role, a number of key autophagy genes have been demonstrated to be tumor suppressor genes. For example, Beclin-1, the mammalian homolog of the yeast autophagy-related gene 6 (Atg6), has been shown to be monoallelically deleted in 40–75% of cancers of the breast, ovaries and prostate,56 and UVRAG is monoallelically deleted in colon cancer cells.57 In addition, mice lacking Beclin1 or another autophagy protein, Bif-1, have a higher incidence of spontaneous tumors.56,58 Autophagy has also been shown to be essential for establishing senescence,59 a state of cell cycle arrest, preventing tumor establishment and/or growth.
On the other hand, the finding that autophagy is up-regulated in certain cancers argues for a role in promoting tumors. For instance, at the centre of solid tumors, where the nutrient supply is limited and cells are often faced with a hypoxic environment, autophagy acts as a survival mechanism by supplying energy and essential nutrients, and allowing cells to adapt to the low oxygen conditions. Autophagy is also up-regulated in response to radiation and certain anti-cancer drugs, such as temozolomide and etoposide, and is the likely cause of resistance to such treatments.60 Finally, autophagy may play a role in cancer metastasis by promoting the survival of cells detached from the extracellular matrix that would normally undergo apoptosis.61
Therefore, the role autophagy plays in cancer is the subject of some controversy. Is it a protective mechanism leading to tumor cell survival, or does activation of this pathway result in tumor cell death? The reality is that autophagy probably plays a role in both processes. Its precise function may depend on the nature of the activating signal, the impact of other signaling pathways, the stage of the disease, as well as the genetic make-up of the cell and the extent of cell damage. However, the prevailing view is one of autophagy being important early on to suppress tumor formation. But later, once the tumor is established, it functions to promote tumor survival.62 In the case of myeloma, high levels of autophagy have been noted in cell lines and patient samples63,64 and this was associated with shorter overall and progression-free survival.63
The relationship between autophagy and apoptosis further confounds the situation. Does autophagy activate or repress apoptosis, or can both processes be activated independently and simultaneously?65 The nature of the interplay between these pathways is crucial for us to further our knowledge of cell death in relation to cancer therapy. Clearly, many questions remain unanswered but if targeting autophagy is to be a viable option for cancer, and more specifically myeloma treatment, these questions to be addressed. The development of more specific autophagy inhibitors, and unraveling of pathways regulating autophagy will go far in helping tackle these issues.
Targeting protein handling pathways in myeloma
Although the development of proteasome inhibitors has led the way in targeting protein handling pathways in myeloma, there are many other potential targets both within the UPP as well as in the stress response and autophagy pathways. Some of these targets are early in vitro proof-of-principle whereas other examples have progressed to phase III clinical trials.
Proteasome inhibitors
Bortezomib is currently the only proteasome inhibitor licensed for clinical use. It binds reversibly to the chymotrypsin-like β5 subunit of the 20S proteasome core particle and inhibits its function.66 A number of other reversible inhibitors are under development, including MLN9708 and CEP-18770. MLN9708 has been demonstrated to have good pharmacokinetic and pharmacodynamic properties and, importantly, showed activity in xenograft models.67 CEP-18770 has shown activity in myeloma cell lines and primary patient cells and resulted in complete tumor regression in mouse models.68 In addition, combinations with melphalan and bortezomib prevented, or at the very least, delayed tumor progression in vivo.69 Interestingly, bortezomib, CEP-18770 and MLN9708 were also shown to inhibit the caspase-like activity of the proteasome, and inhibitors of the trypsin-like activity are also under development.70
The development of resistance to bortezomib has prompted the need for inhibitors with properties distinct from that of bortezomib. This has led to the development of irreversible inhibitors such as carfilzomib, NPI-0052 and ONX0912, which target both the proteasome and immunoproteasome. Carfilzomib was found to be a strong inhibitor of the chymotrypsin-like activity of the proteasome both in vitro and in vivo71 and, importantly, demonstrated activity in cells resistant to bortezomib, melphalan and dexamethasone.72 NPI-0052, in contrast to bortezomib and carfilzomib, appears to have activity against all three enzymatic activities of the proteasome.73 In addition, its non-peptidic structure ensures it is not degraded by intracellular proteases to which bortezomib and carfilzomib would be subject.14 It is able to induce apoptosis in primary myeloma cells refractory to treatment with conventional myeloma therapies73 and combines synergistically with bortezomib and lenalidomide in vivo.74–76 ONX0912 is the only irreversible proteasome inhibitor that is orally bioavailable. It too has shown activity in myeloma cells resistant to conventional therapy and enhances tumor regression in vivo. Importantly, it combines either synergistically or additively with lenalidomide, bortezomib, dexamethasone and a histone deacetylase inhibitor.75
In addition to the competitive inhibitors described above, some work has been carried out looking into allosteric proteasome inhibitors in the hope that these may be used to overcome resistance to the competitive inhibitors.77 Allosteric inhibitors work by interacting with the regulatory α subunits of the 20S proteasome, thereby blocking proteasome function non-competitively.77–79 Some of these are structurally similar to the anti-malarial drug chloroquine and their prior approval for clinical use will hopefully speed up their testing. Finally, while most research has concentrated on targeting the 20S proteasome core, the 19S regulatory subunit has recently emerged as a potential drug target80 but is yet to be tested in myeloma.
Clearly, the aim behind developing reversible/irreversible and competitive/allosteric inhibitors is to increase the anti-proteasome effect and induce more potent myeloma cell apoptosis while decreasing the clinical side-effect profile but maintaining patient tolerance and ease of administration. Pre-clinical data look promising for all the above mentioned approaches, but further clinical trials will be required to determine which approach is the best.
Inhibitors of the ubiquitin proteasome pathway
In addition to directly targeting the proteasome, it may be possible to target pathways both upstream and downstream of it. Ubiquitin E3 ligases are responsible for the ubiquitination of a variety of substrate molecules, leading to their degradation by the 26S proteasome. Skp1-Cullin-F box protein (SCF) constitutes the largest family of E3 ligases and acts as a scaffold ensuring the correct positioning of the substrate and E2 enzyme for ubiquitin transfer. An inhibitor of SCFSkp2 identified by a high-throughput screen and referred to as Compound A has been tested in vitro against myeloma.81 Compound A was able to overcome resistance to bortezomib, dexamethasone, doxorubicin and melphalan, and was synergistic with bortezomib. It also induced autophagy.81 HDM2, the human homolog of murine double minute 2 (MDM2), acts as the E3 ligase for p53 and is often over-expressed in a variety of cancers. It is a direct downstream target of p53 and marks it for degradation. Nutlin-3, which disrupts the interaction between p53 and HDM2, was tested in combination with bortezomib and was shown to have additive cytotoxicity. But in co-culture experiments with bone marrow stromal cells, the effect of Nutlin-3 on apoptosis was reduced.82 In addition to the proteasome, cullin-ring ligases are also involved in protein degradation, and NEDD8- activating enzymes (NAE) catalyze the initial step in the NEDDylation cascade in a similar manner to ubiquitin E1 enzymes.83 The NAE inhibitor, MLN4924, was recently tested in myeloma and showed activity against cells both sensitive and resistant to bortezomib, as well as primary patient cells. Importantly, it showed activity in vivo with significant inhibition of tumor growth and a reduction in tumor burden in two different models.84
Additionally, it is possible to target processes that occur after proteins exit the proteasome. Aminopeptidases catalyze the hydrolysis of amino acids from the N terminal of proteins. Tosedostat (CHR-2797), a novel metalloenzyme inhibitor, was demonstrated to induce cell cycle arrest and apoptosis of myeloma cell lines. Synergy with dexamethasone was observed while it was additive with bortezomib and melphalan. In addition, tosedostat up-regulated the UPR and induced autophagy.85
Heat-shock chaperone inhibitors
To date, most research has concentrated on developing inhibitors for HSP90, given that it sits at the intersection of numerous pathways that are aberrantly activated in cancer. Initial drug development efforts were centered around two natural products, radicicol and geldanamycin; research into geldanamycin is more pronounced. The geldanamycin-based inhibitors have undergone a number of iterations to develop drugs with improved toxicology and solubility. These are now being tested in the clinic.
17-AAG was the first to be developed and showed single agent activity in vitro against myeloma cell lines and primary patient samples. It sensitized primary cells to treatment with conventional and novel therapies, including doxorubicin and bortezomib.86,87 This was related to the loss of cell surface expression of IGF-1R and IL-6R, with subsequent inhibition of numerous signaling pathways. Activity was mirrored in vivo where, in addition, it significantly prolonged survival.87 However, 17-AAG also induces the UPR,40 and HSP70 is up-regulated in response to HSP90 inhibition,88 suggesting that in order to achieve an optimal clinical response with HSP90 inhibitors, they should be used in a combination approach. To this end, 17-AAG or its analog 17-DMAG, in combination with either of the PI3K pathway inhibitors, perifosine or rapamycin, demonstrated synergistic cytotoxicity in myeloma cells, inhibited angiogenesis, and was able to overcome the protective effects of the bone marrow microenvironment.89,90 Similar effects on myeloma cell death were demonstrated by the combination of 17-DMAG and the autophagy inhibitor 3-MA.91 The next iteration of 17-AAG is IPI-504. This compound has improved solubility and tissue retention compared to 17-AAG, and showed similar synergy with bortezomib.92 However, in contrast to 17-AAG, IPI-504 was shown to block the UPR,93 not activate it.40 However, the reason for this discrepancy is not clear.
A number of synthetic small molecule HSP90 inhibitors with various molecular scaffolds are also available. One of the first to be described was NVP-AUY922 which showed activity at sub-micromolar concentrations in a range of cell lines and, importantly, activity in vivo.94 When tested in myeloma, NVP-AUY922 showed low nanomolar sensitivity in both cell lines and primary patient samples, even in the presence of bone marrow stromal cells. This was associated with strong induction of apoptosis and downregulation of important survival pathways.95 It also demonstrated synergy with histone deacetylase inhibitors, melphalan and doxorubicin in primary patient samples refractory to conventional therapies.96 Similarly, the orally available NVP-BEP800 was able to induce apoptosis in myeloma cell lines and primary patient samples with pronounced inhibition of the STAT3, ERK and Akt pathways. Weak synergy was demonstrated with melphalan whereas combinations with bortezomib, doxorubicin or SAHA were weakly antagonistic.97
Two additional oral HSP90 inhibitors have been tested in myeloma. NVP-HSP990 induces apoptosis and cell cycle arrest in myeloma cell lines and primary samples. This was associated with downregulation of ‘client’ proteins (IL-6R, MEK and Akt) and subsequent upregulation of HSP70 and HSP27.98 SNX-2112, which exists as a pro-drug SNX-5422, was able to induce cytotoxicity and cell cycle arrest in myeloma cell lines and patient samples. Downregulation of Akt and ERK pathways was demonstrated and, importantly, maintained in the presence of extracellular cytokines. SNX-2112 inhibited angiogenesis and osteoclastogenesis and, when tested in vivo, significantly prolonged survival. In addition, it was able to induce apoptosis, down-regulate ERK and block angiogenesis in vivo.99
The novel purine scaffold HSP90 inhibitor, PU-H71, demonstrated activity in myeloma cells both sensitive and resistant to conventional therapies, and was synergistic with thalidomide, bortezomib, dexamethasone and melphalan. It induced cell cycle arrest, apoptosis and the UPR. Interestingly, PU-H71 appeared to work by targeting HSP90 and the ER HSP90 paralog, GRP94.100 The non-ansamycin, non-purine inhibitor, KW-2478, also down-regulated levels of HSP90 ‘client’ proteins and IgH translocation products (FGFR3, c-Maf and cyclin D1). These effects on ‘client’ proteins were mirrored in vivo where, in addition, it significantly inhibited tumor growth.101
More recently, there has been a move to developing inhibitors of HSP70, given that HSP90 inhibitors induce HSP70 expression. The aim would, therefore, be combination treatment with both inhibitors.88 MAL3-101 is a non-ATP site inhibitor of HSP70 that induced apoptosis and cell cycle arrest in myeloma cell lines, and demonstrated synergy with both proteasome and HSP90 inhibitors. Synergy with proteasome inhibitors was confirmed in vivo.102 Finally, in addition to directly targeting HSP90 or HSP70, it is possible to inhibit heat shock factor 1 (HSF-1), the transcription factor that induces their expression. While many of the compounds currently used as HSF-1 inhibitors are non-specific, and their mechanism of action is not fully understood, efforts are underway to design more potent and specific inhibitors of this transcription factor.103
Unfolded protein response modulators
The potential for targeting the UPR to achieve cell death has been recognized, with the three UPR sensors, IRE1, PERK and ATF6, being the obvious candidates: they are all kinases, with two crystal structures being solved104,105 and siRNA knockdown of each kinase resulting in apoptosis in myeloma cells.106 Recently, specific inhibitors of the endoribonuclease domain of IRE1 that inhibit the splicing of XBP1 have been described107,108 and have shown activity in vivo in combination with bortezomib.107 Inhibition of the kinase domain of IRE1 has also been shown to be effective104 and more specific inhibitors are under clinical development (Walter et al., patent n. WO 2010/031056 A2, 2010; Korennykh et al., patent n. WO 2011/047384 A2, 2011), as are inhibitors of PERK (Cardozo et al., patent n.WO 2011-146748 A1, 2011). Although the cleavage of ATF6 has been targeted, the compound tested was a general serine protease inhibitor109 and, to date, no clinical grade inhibitors have been reported.
In a similar manner, many inhibitors of GRP78, the protein which holds IRE1, PERK and ATF6 in their inactive form, are available. Most are natural products but lack true specificity. The one most commonly used as a chemical tool is versipelostatin, which inhibits GRP78 transcription, but it only appears to inhibit the UPR under conditions of glucose deprivation.110 Drugs that target protein transport have also been tested in myeloma. Brefeldin A inhibits the transport of proteins from the ER to the Golgi111 while Eeyarestatin I blocks ERAD by interfering with p97, an AAA-ATPase that transfers proteins from the ER to the cytosol.112 Protein disulfide isomerases (PDI), enzymes responsible for oxidative protein folding in the ER, have also been examined as potential drug targets. Compounds targeting PDI are metabolized by glutathione S-transferases, releasing nitric oxide in cells.113 In myeloma cells, they were able to enhance cytotoxicity mediated by bortezomib and prolong survival in vivo.114 As this is a relatively new area of research, to date all inhibitors are tool compounds for use in in vitro models, although clinical grade inhibitors are under development for use in future clinical trials.
Autophagy modulators
Although much is known about the regulation of autophagy by the PI3K pathway, studies exploring the role of other autophagy regulatory pathways are in their infancy.115–117 Importantly, we know that UPR activation, removal of proteins by the proteasome and autophagy are interlinked. While inhibition of the proteasome leads to a compensatory upregulation of autophagy,64,118 blocking autophagy inhibits the proteasome (Figure 3).119 It is clear, therefore, that these interactions need to be better understood if we are to target these intracellular protein handling pathways effectively.
Figure 3.
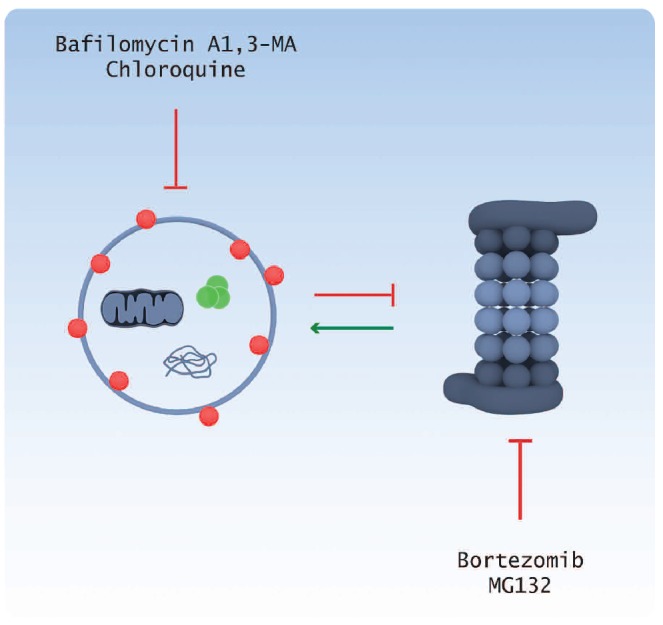
Relationship between the autophagy and proteasome pathways. Proteasome inhibition by bortezomib or MG132 results in the compensatory upregulation of the autophagy pathway, whereas inhibition of the autophagy pathway by bafilomycin A1, 3-MA or chloroquine inhibits the proteasome.
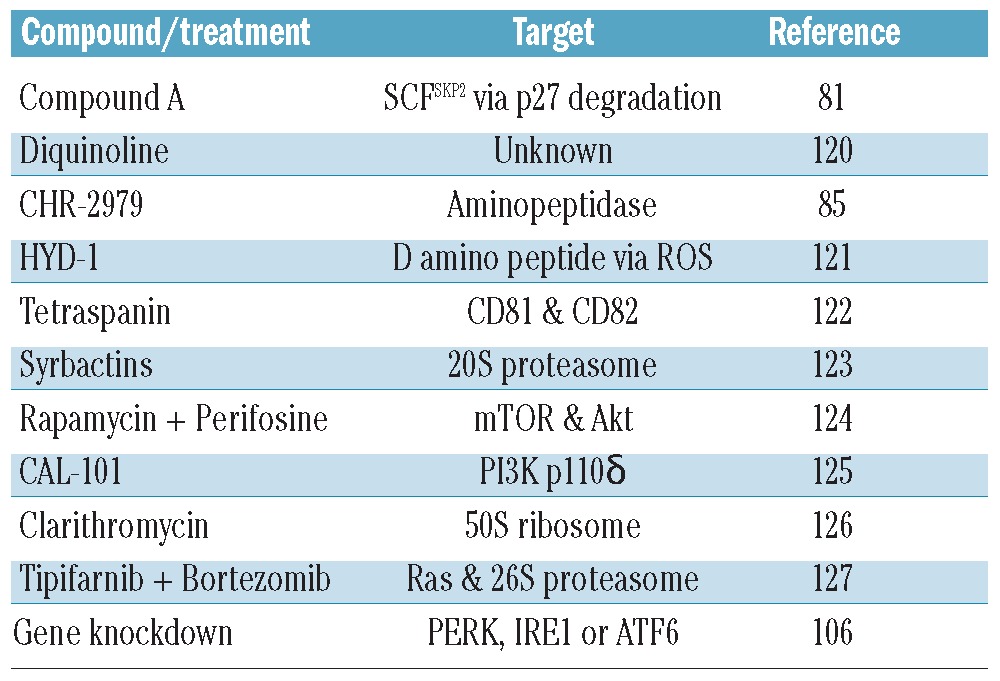
A number of researchers have begun to look at the role of autophagy in myeloma. Most of these papers have shown that a particular drug or drug combination induces autophagy (Table 1). But, as can be seen from the varied list of drug targets, no clear pattern has emerged. However, there are some familiar faces among those drugs that up-regulate autophagy. These include the proteasome inhibitor bortezomib, the PI3K/Akt/mTOR inhibitors rapamycin and perifosine, and inhibitors of the three UPR sensor molecules, PERK, IRE1 and ATF6. As mentioned above, bortezomib is already in clinical use but agents targeting Akt and mTOR are still in clinical trials. However, as a result of a negative feedback loop within the PI3K/Akt/mTOR pathway, they have had limited success, and combinations of rapamycin and bortezomib were shown to be antagonistic.128 There has, therefore, been a move to develop dual PI3K/mTOR inhibitors in order to circumvent this problem.
Table 1.
Studies in myeloma that demonstrate the upregulation of autophagy by a particular compound or treatment.
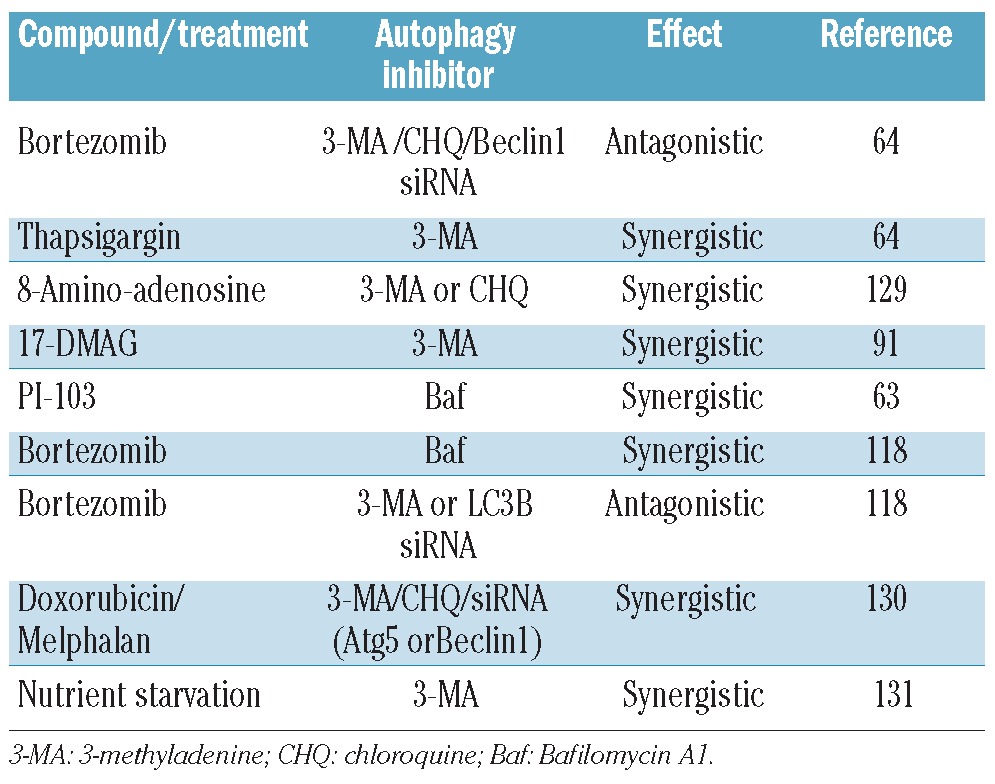
Perhaps of greater interest are the papers that look at a drug that induces autophagy in combination with autophagy inhibitors as a potential mechanism for eliciting apoptosis (Table 2). While most of the combinations are synergistic, there are two striking exceptions. In the work of Hoang et al.64 and Kawaguchi et al.118 the combination of bortezomib with 3-methyladenine (3-MA), or siRNA against LC3B or Beclin1, was antagonistic. However, Kawaguchi et al. also demonstrate that bortezomib in combination with bafilomycin A1 (Baf) is synergistic, whereas Hoang et al. show that combining bortezomib and chloroquine (CHQ) is antagonistic. 3-MA inhibits autophagy at the level of PI3K Class III, therefore, acting early on in the pathway. Bafilomycin and CHQ inhibit the fusion between the autophagosome and lysosome, therefore acting as late stage inhibitors (Figure 2). So, is the stage at which the autophagy inhibitor acts important? And why is there a difference in cells treated with bortezomib/Baf versus bortezomib/CHQ? In trying to understand these results, it is important to note that 3-MA has been shown to be a more specific inhibitor of PI3K Class I and not PI3K Class III132 and chloroquine, in addition to its effect on autophagy, is known to inhibit the proteasome.79 This is further complicated by the cross-talk between these two protein degradation systems. The main effect of proteasome inhibition seems to be induction of ER-stress,133 which in turn up-regulates autophagy.29 But we still do not know exactly how the reciprocol regulation of these pathways occurs in response to drug treatment. The different responses observed with the combination of bortezomib/Baf versus bortezomib/CHQ really underscores the urgent need for more specific autophagy inhibitors to delineate autophagy on-target, as opposed to off-target, effects.
Table 2.
Studies in myeloma examining the effect of a compound or treatment that induces autophagy, in combination with autophagy inhibitors.
Conclusions
Significant progress has been made in the development of targeted molecules for the treatment of myeloma. By understanding the basic biology of this tumor, particularly its dependence on protein handling pathways such as the ubiquitin proteasome pathway, ER stress and the unfolded protein response, heat shock proteins and autophagy, scientists have been able to exploit its weaknesses to promote its destruction. This, together with intuitive drug design, will lead to the development of more specific inhibitors with, hopefully, fewer side-effects for patients. This will be particularly important in the case of autophagy, given its important role during development. The initial excitement with bortezomib has been superseded by the development of more specific irreversible proteasome inhibitors, and inhibitors of other aspects of protein degradation are beginning to emerge. While some of these drugs are still only in early pre-clinical testing stages, especially modulators of the UPR, testing of other compounds has reached clinical trials and the initial results seem promising. Additionally, the field of autophagy research holds great promise for the identification of further therapeutic targets. However, the complex interactions between pathways and the possible upregulation of redundant pathways mean that further study is needed. As has been shown with HSP90 inhibitors, for example, there is upregulation of other HSPs and induction of the UPR.40,87,88 Indeed, HSP90 inhibitors have, in general, performed poorly as single agents for the treatment of multiple myeloma in the clinic.134 highlighting the need to look at these compensatory mechanisms in order to enhance apoptosis of myeloma cells. Additionally, the bone marrow microenvironment plays an important role in protecting cells from apoptosis, both by the direct binding of myeloma and stromal cells, and due to the secretion of numerous cytokines that influence cell growth.135 Testing of new drugs and drug combinations in the laboratory should, therefore, always be tested in a situation that mimics this environment.
The key questions now will be how best to combine these agents to achieve even better responses. For example, should a proteasome inhibitor be combined with an autophagy inhibitor, and can this be enhanced by adding an ER stress or heat shock protein inhibitor? This research needs to be accompanied by studies examining the interactions between the various protein handling pathways highlighted in this review. By understanding how these various pathways intersect and overlap, we will be able to achieve the greatest clinical benefit for our patients.
Footnotes
Funding: this work was supported by grants from Cancer Research UK and Myeloma UK. FED is a Cancer Research UK Senior Fellow, grant n. C20826/A12103.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Boyd KD, Ross FM, Chiecchio L, Dagrada G, Konn ZJ, Tapper WJ, et al. Gender disparities in the tumor genetics and clinical outcome of multiple myeloma. Cancer Epidemiol Biomarkers Prev. 2011;20(8):1703–7. doi: 10.1158/1055-9965.EPI-11-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan GJ, Davies FE, Gregory WM, Cocks K, Bell SE, Szubert AJ, et al. First-line treatment with zoledronic acid as compared with clodronic acid in multiple myeloma (MRC Myeloma IX): a randomised controlled trial. Lancet. 2010;376(9757):1989–99. doi: 10.1016/S0140-6736(10)62051-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan GJ, Gregory WM, Davies FE, Bell SE, Szubert AJ, Brown JM, et al. The role of maintenance thalidomide therapy in multiple myeloma: MRC Myeloma IX results and meta-analysis. Blood. 2012;119(1):7–15. doi: 10.1182/blood-2011-06-357038. [DOI] [PubMed] [Google Scholar]
- 4.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 5.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 6.Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012 Jan 19; doi: 10.1182/blood-2011-10-387365. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ocio EM, San Miguel JF. The DAC system and associations with multiple myeloma. Investigational new drugs. 2010;28(Suppl 1):S28–35. doi: 10.1007/s10637-010-9589-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith EM, Boyd K, Davies FE. The potential role of epigenetic therapy in multiple myeloma. Br J Haematol. 2010;148(5):702–13. doi: 10.1111/j.1365-2141.2009.07976.x. [DOI] [PubMed] [Google Scholar]
- 9.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–16. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137(1):133–45. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weissman AM, Shabek N, Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat Rev Mol Cell Biol. 2011;12(9):605–20. doi: 10.1038/nrm3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–9. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 13.Besche HC, Peth A, Goldberg AL. Getting to first base in proteasome assembly. Cell. 2009;138(1):25–8. doi: 10.1016/j.cell.2009.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruschak AM, Slassi M, Kay LE, Schimmer AD. Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst. 2011;103(13):1007–17. doi: 10.1093/jnci/djr160. [DOI] [PubMed] [Google Scholar]
- 15.Altun M, Galardy PJ, Shringarpure R, Hideshima T, LeBlanc R, Anderson KC, et al. Effects of PS-341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer Res. 2005;65(17):7896–901. doi: 10.1158/0008-5472.CAN-05-0506. [DOI] [PubMed] [Google Scholar]
- 16.Kincaid EZ, Che JW, York I, Escobar H, Reyes-Vargas E, Delgado JC, et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol. 2011;13(2):129–35. doi: 10.1038/ni.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404(6779):770–4. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 18.Bianchi G, Oliva L, Cascio P, Pengo N, Fontana F, Cerruti F, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood. 2009;113(13):3040–9. doi: 10.1182/blood-2008-08-172734. [DOI] [PubMed] [Google Scholar]
- 19.Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007;67(4):1783–92. doi: 10.1158/0008-5472.CAN-06-2258. [DOI] [PubMed] [Google Scholar]
- 20.Workman P, Davies FE. A stressful life (or death): combinatorial proteotoxic approaches to cancer-selective therapeutic vulnerability. Oncotarget. 2011;2(4):277–80. doi: 10.18632/oncotarget.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walter P, Ron D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science. 2011;334(6059):1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 22.Kelly KR, Espitia CM, Mahalingam D, Oyajobi BO, Coffey M, Giles FJ, et al. Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene. 2012;31(25):3023–38. doi: 10.1038/onc.2011.478. [DOI] [PubMed] [Google Scholar]
- 23.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–32. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 24.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23(21):7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu MC, Gong HY, Lin GH, Hu SY, Chen MH, Huang SJ, et al. XBP-1, a key regulator of unfolded protein response, activates transcription of IGF1 and Akt phosphorylation in zebrafish embryonic cell line. Biochem Biophys Res Commun. 2007;359(3):778–83. doi: 10.1016/j.bbrc.2007.05.183. [DOI] [PubMed] [Google Scholar]
- 26.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–6. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 27.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Davenport EL, Aronson LI, Davies FE. Starving to succeed. Autophagy. 2009;5(7):1052–4. doi: 10.4161/auto.5.7.9510. [DOI] [PubMed] [Google Scholar]
- 29.Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int J Cell Biol. 2010;2010:930509. doi: 10.1155/2010/930509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14(2):230–9. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 31.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120(1):127–41. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin HO, Seo SK, Woo SH, Kim ES, Lee HC, Yoo DH, et al. Activating transcription factor 4 and CCAAT/enhancer-binding protein-beta negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radic Biol Med. 2009;46(8):1158–67. doi: 10.1016/j.freeradbiomed.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 33.Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci. 2009;122(Pt 10):1626–36. doi: 10.1242/jcs.045625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3(1):99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13(3):365–76. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 36.Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA. 2008;105(30):10519–24. doi: 10.1073/pnas.0800939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J. 2008;410(3):439–53. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 38.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11(7):515–28. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 39.Davenport EL, Morgan GJ, Davies FE. Untangling the unfolded protein response. Cell Cycle. 2008;7(7):865–9. doi: 10.4161/cc.7.7.5615. [DOI] [PubMed] [Google Scholar]
- 40.Davenport EL, Moore HE, Dunlop AS, Sharp SY, Workman P, Morgan GJ, et al. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110(7):2641–9. doi: 10.1182/blood-2006-11-053728. [DOI] [PubMed] [Google Scholar]
- 41.Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, Neckers L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol. 2002;22(24):8506–13. doi: 10.1128/MCB.22.24.8506-8513.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan W, Nassiri A, Zhong Q. Auto-phagosome targeting and membrane curvature sensing by Barkor/Atg14(L) Proc Natl Acad Sci USA. 2011;108(19):7769–74. doi: 10.1073/pnas.1016472108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447(7148):1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 46.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11(4):385–96. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 47.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408(6811):488–92. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 48.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature. 1998;395(6700):395–8. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 49.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130(1):165–78. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 50.Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19(11):4651–9. doi: 10.1091/mbc.E08-03-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19(11):4762–75. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hickey CM, Wickner W. HOPS initiates vacuole docking by tethering membranes before trans-SNARE complex assembly. Mol Biol Cell. 2010;21(13):2297–305. doi: 10.1091/mbc.E10-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26(2):313–24. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117(Pt 20):4837–48. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 55.Yang Z, Huang J, Geng J, Nair U, Klionsky DJ. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006;17(12):5094–104. doi: 10.1091/mbc.E06-06-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK. Manipulation of nonsense mediated decay identifies gene mutations in colon cancer Cells with microsatellite instability. Oncogene. 2004;23(3):639–45. doi: 10.1038/sj.onc.1207178. [DOI] [PubMed] [Google Scholar]
- 58.Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol. 2009;3(4):366–75. doi: 10.1016/j.molonc.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14(3):548–58. doi: 10.1038/sj.cdd.4402030. [DOI] [PubMed] [Google Scholar]
- 61.Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. 2011;31(17):3616–29. doi: 10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25(19):1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aronson LI, Davenport EL, Giuntoli SG, Srikanth M, Smith E, Morgan GJ, et al. Autophagy Is a Key Myeloma Survival Pathway That Can Be Manipulated Therapeutically to Enhance Apoptosis. ASH Annual Meeting Abstracts. 2010;116(21):4083. [Google Scholar]
- 64.Hoang B, Benavides A, Shi Y, Frost P, Lichtenstein A. Effect of autophagy on multiple myeloma cell viability. Mol Cancer Ther. 2009;8(7):1974–84. doi: 10.1158/1535-7163.MCT-08-1177. [DOI] [PubMed] [Google Scholar]
- 65.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23(16):2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 66.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071–6. [PubMed] [Google Scholar]
- 67.Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, et al. Evaluation of the proteasome inhibitor MLN9708 in pre-clinical models of human cancer. Cancer Res. 2010;70(5):1970–80. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 68.Piva R, Ruggeri B, Williams M, Costa G, Tamagno I, Ferrero D, et al. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood. 2008;111(5):2765–75. doi: 10.1182/blood-2007-07-100651. [DOI] [PubMed] [Google Scholar]
- 69.Sanchez E, Li M, Steinberg JA, Wang C, Shen J, Bonavida B, et al. The proteasome inhibitor CEP-18770 enhances the anti-myeloma activity of bortezomib and melphalan. Br J Haematol. 2010;148(4):569–81. doi: 10.1111/j.1365-2141.2009.08008.x. [DOI] [PubMed] [Google Scholar]
- 70.Mirabella AC, Pletnev AA, Downey SL, Florea BI, Shabaneh TB, Britton M, et al. Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem Biol. 2011;18(5):608–18. doi: 10.1016/j.chembiol.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007;67(13):6383–91. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 72.Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007;110(9):3281–90. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8(5):407–19. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 74.Chauhan D, Singh A, Brahmandam M, Podar K, Hideshima T, Richardson P, et al. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2008;111(3):1654–64. doi: 10.1182/blood-2007-08-105601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chauhan D, Singh AV, Ciccarelli B, Richardson PG, Palladino MA, Anderson KC. Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2010;115(4):834–45. doi: 10.1182/blood-2009-03-213009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Obaidat A, Weiss J, Wahlgren B, Manam RR, Macherla VR, McArthur K, et al. Proteasome regulator marizomib (NPI-0052) exhibits prolonged inhibition, attenuated efflux, and greater cytotoxicity than its reversible analogs. J Pharmacol Exp Ther. 2011;337(2):479–86. doi: 10.1124/jpet.110.177824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li X, Wood TE, Sprangers R, Jansen G, Franke NE, Mao X, et al. Effect of noncom-petitive proteasome inhibition on bortezomib resistance. J Natl Cancer Inst. 2010;102(14):1069–82. doi: 10.1093/jnci/djq198. [DOI] [PubMed] [Google Scholar]
- 78.Gaczynska M, Osmulski PA, Gao Y, Post MJ, Simons M. Proline- and arginine-rich peptides constitute a novel class of allosteric inhibitors of proteasome activity. Biochemistry. 2003;42(29):8663–70. doi: 10.1021/bi034784f. [DOI] [PubMed] [Google Scholar]
- 79.Sprangers R, Li X, Mao X, Rubinstein JL, Schimmer AD, Kay LE. TROSY-based NMR evidence for a novel class of 20S proteasome inhibitors. Biochemistry. 2008;47(26):6727–34. doi: 10.1021/bi8005913. [DOI] [PubMed] [Google Scholar]
- 80.D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17(12):1636–40. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 81.Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27-and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood. 2008;111(9):4690–9. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ooi MG, Hayden PJ, Kotoula V, McMillin DW, Charalambous E, Daskalaki E, et al. Interactions of the Hdm2/p53 and proteasome pathways may enhance the antitumor activity of bortezomib. Clin Cancer Res. 2009;15(23):7153–60. doi: 10.1158/1078-0432.CCR-09-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watson IR, Irwin MS, Ohh M. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell. 2011;19(2):168–76. doi: 10.1016/j.ccr.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 84.McMillin DW, Jacobs HM, Delmore JE, Buon L, Hunter ZR, Monrose V, et al. Molecular and cellular effects of NEDD8 activating enzyme (NAE) inhibition in myeloma. Mol Cancer Ther. 2012;11(4):942–51. doi: 10.1158/1535-7163.MCT-11-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moore HE, Davenport EL, Smith EM, Muralikrishnan S, Dunlop AS, Walker BA, et al. Aminopeptidase inhibition as a targeted treatment strategy in myeloma. Mol Cancer Ther. 2009;8(4):762–70. doi: 10.1158/1535-7163.MCT-08-0735. [DOI] [PubMed] [Google Scholar]
- 86.Duus J, Bahar HI, Venkataraman G, Ozpuyan F, Izban KF, Al-Masri H, et al. Analysis of expression of heat shock protein-90 (HSP90) and the effects of HSP90 inhibitor (17-AAG) in multiple myeloma. Leuk Lymphoma. 2006;47(7):1369–78. doi: 10.1080/10428190500472123. [DOI] [PubMed] [Google Scholar]
- 87.Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Kung AL, Davies FE, et al. Antimyeloma activity of heat shock protein-90 inhibition. Blood. 2006;107(3):1092–100. doi: 10.1182/blood-2005-03-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Davenport EL, Zeisig A, Aronson LI, Moore HE, Hockley S, Gonzalez D, et al. Targeting heat shock protein 72 enhances Hsp90 inhibitor-induced apoptosis in myeloma. Leukemia. 2010;24(10):1804–7. doi: 10.1038/leu.2010.168. [DOI] [PubMed] [Google Scholar]
- 89.Francis LK, Alsayed Y, Leleu X, Jia X, Singha UK, Anderson J, et al. Combination mammalian target of rapamycin inhibitor rapamycin and HSP90 inhibitor 17-ally-lamino-17-demethoxygeldanamycin has synergistic activity in multiple myeloma. Clin Cancer Res. 2006;12(22):6826–35. doi: 10.1158/1078-0432.CCR-06-1331. [DOI] [PubMed] [Google Scholar]
- 90.Huston A, Leleu X, Jia X, Moreau AS, Ngo HT, Runnels J, et al. Targeting Akt and heat shock protein 90 produces synergistic multiple myeloma cell cytotoxicity in the bone marrow microenvironment. Clin Cancer Res. 2008;14(3):865–74. doi: 10.1158/1078-0432.CCR-07-1299. [DOI] [PubMed] [Google Scholar]
- 91.Palacios C, Martin-Perez R, Lopez-Perez AI, Pandiella A, Lopez-Rivas A. Autophagy inhibition sensitizes multiple myeloma cells to 17-dimethylaminoethylamino-17-demethoxygeldanamycin-induced apoptosis. Leuk Res. 2010;34(11):1533–8. doi: 10.1016/j.leukres.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 92.Sydor JR, Normant E, Pien CS, Porter JR, Ge J, Grenier L, et al. Development of 17-ally-lamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci USA. 2006;103(46):17408–13. doi: 10.1073/pnas.0608372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patterson J, Palombella VJ, Fritz C, Normant E. IPI-504, a novel and soluble HSP-90 inhibitor, blocks the unfolded protein response in multiple myeloma cells. Cancer Chemother Pharmacol. 2008;61(6):923–32. doi: 10.1007/s00280-007-0546-0. [DOI] [PubMed] [Google Scholar]
- 94.Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, et al. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68(8):2850–60. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 95.Stuhmer T, Zollinger A, Siegmund D, Chatterjee M, Grella E, Knop S, et al. Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia. 2008;22(8):1604–12. doi: 10.1038/leu.2008.111. [DOI] [PubMed] [Google Scholar]
- 96.Kaiser M, Lamottke B, Mieth M, Jensen MR, Quadt C, Garcia-Echeverria C, et al. Synergistic action of the novel HSP90 inhibitor NVP-AUY922 with histone deacetylase inhibitors, melphalan, or doxorubicin in multiple myeloma. Eur J Haematol. 2010;84(4):337–44. doi: 10.1111/j.1600-0609.2009.01403.x. [DOI] [PubMed] [Google Scholar]
- 97.Stuhmer T, Chatterjee M, Grella E, Seggewiss R, Langer C, Muller S, et al. Antimyeloma activity of the novel 2-aminothienopyrimidine Hsp90 inhibitor NVP-BEP800. Br J Haematol. 2009;147(3):319–27. doi: 10.1111/j.1365-2141.2009.07852.x. [DOI] [PubMed] [Google Scholar]
- 98.Khong T, Spencer A. Targeting HSP 90 induces apoptosis and inhibits critical survival and proliferation pathways in multiple myeloma. Mol Cancer Ther. 2011;10(10):1909–17. doi: 10.1158/1535-7163.MCT-11-0174. [DOI] [PubMed] [Google Scholar]
- 99.Okawa Y, Hideshima T, Steed P, Vallet S, Hall S, Huang K, et al. SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell growth, angiogenesis, and osteoclastogenesis in multiple myeloma and other hematologic tumors by abrogating signaling via Akt and ERK. Blood. 2009;113(4):846–55. doi: 10.1182/blood-2008-04-151928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Usmani SZ, Bona RD, Chiosis G, Li Z. The antimyeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1. J Hematol Oncol. 2010;3:40. doi: 10.1186/1756-8722-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nakashima T, Ishii T, Tagaya H, Seike T, Nakagawa H, Kanda Y, et al. New molecular and biological mechanism of antitumor activities of KW-2478, a novel non-ansamycin heat shock protein 90 inhibitor, in multiple myeloma cells. Clin Cancer Res. 2010;16(10):2792–802. doi: 10.1158/1078-0432.CCR-09-3112. [DOI] [PubMed] [Google Scholar]
- 102.Braunstein MJ, Scott SS, Scott CM, Behrman S, Walter P, Wipf P, et al. Antimyeloma Effects of the Heat Shock Protein 70 Molecular Chaperone Inhibitor MAL3–101. J Oncol. 2011;2011:232037. doi: 10.1155/2011/232037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.de Billy E, Powers MV, Smith JR, Workman P. Drugging the heat shock factor 1 pathway: exploitation of the critical cancer cell dependence on the guardian of the proteome. Cell Cycle. 2009;8(23):3806–8. doi: 10.4161/cc.8.23.10423. [DOI] [PubMed] [Google Scholar]
- 104.Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011;30(5):894–905. doi: 10.1038/emboj.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cui W, Li J, Ron D, Sha B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 5):423–8. doi: 10.1107/S0907444911006445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Michallet AS, Mondiere P, Taillardet M, Leverrier Y, Genestier L, Defrance T. Compromising the unfolded protein response induces autophagy-mediated cell death in multiple myeloma cells. PLoS One. 2011;6(10):e25820. doi: 10.1371/journal.pone.0025820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117(4):1311–4. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Volkmann K, Lucas JL, Vuga D, Wang X, Brumm D, Stiles C, et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem. 2011;286(14):12743–55. doi: 10.1074/jbc.M110.199737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, et al. A serine pro-tease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J Biol Chem. 2003;278(33):31024–32. doi: 10.1074/jbc.M300923200. [DOI] [PubMed] [Google Scholar]
- 110.Park HR, Tomida A, Sato S, Tsukumo Y, Yun J, Yamori T, et al. Effect on tumor cells of blocking survival response to glucose deprivation. J Natl Cancer Inst. 2004;96(17):1300–10. doi: 10.1093/jnci/djh243. [DOI] [PubMed] [Google Scholar]
- 111.Carew JS, Nawrocki ST, Krupnik YV, Dunner K, Jr, McConkey DJ, Keating MJ, et al. Targeting endoplasmic reticulum protein transport: a novel strategy to kill malignant B cells and overcome fludarabine resistance in CLL. Blood. 2006;107(1):222–31. doi: 10.1182/blood-2005-05-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci USA. 2009;106(7):2200–5. doi: 10.1073/pnas.0807611106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Townsend DM, Manevich Y, He L, Xiong Y, Bowers RR, Jr, Hutchens S, et al. Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009;69(19):7626–34. doi: 10.1158/0008-5472.CAN-09-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, et al. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007;110(2):709–18. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell. 2010;18(6):1041–52. doi: 10.1016/j.devcel.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Orvedahl A, Sumpter R, Jr, Xiao G, Ng A, Zou Z, Tang Y, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480(7375):113–7. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Szyniarowski P, Corcelle-Termeau E, Farkas T, Hoyer-Hansen M, Nylandsted J, Kallunki T, et al. A comprehensive siRNA screen for kinases that suppress macroautophagy in optimal growth conditions. Autophagy. 2011;7(8):892–903. doi: 10.4161/auto.7.8.15770. [DOI] [PubMed] [Google Scholar]
- 118.Kawaguchi T, Miyazawa K, Moriya S, Ohtomo T, Che XF, Naito M, et al. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagylysosome and ER stress. Int J Oncol. 2011;38(3):643–54. doi: 10.3892/ijo.2010.882. [DOI] [PubMed] [Google Scholar]
- 119.Qiao L, Zhang J. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett. 2009;456(1):15–9. doi: 10.1016/j.neulet.2009.03.085. [DOI] [PubMed] [Google Scholar]
- 120.Hurren R, Beheshti Zavareh R, Dalili S, Wood T, Rose D, Chang H, et al. A novel diquinolonium displays preclinical anti-cancer activity and induces caspase-independent cell death. Apoptosis. 2008;13(6):748–55. doi: 10.1007/s10495-008-0209-6. [DOI] [PubMed] [Google Scholar]
- 121.Nair RR, Emmons MF, Cress AE, Argilagos RF, Lam K, Kerr WT, et al. HYD1-induced increase in reactive oxygen species leads to autophagy and necrotic cell death in multiple myeloma cells. Mol Cancer Ther. 2009;8(8):2441–51. doi: 10.1158/1535-7163.MCT-09-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zismanov V, Lishner M, Tartakover-Matalon S, Radnay J, Shapiro H, Drucker L. Tetraspanin-induced death of myeloma cell lines is autophagic and involves increased UPR signalling. Br J Cancer. 2009;101(8):1402–9. doi: 10.1038/sj.bjc.6605291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Archer CR, Koomoa DL, Mitsunaga EM, Clerc J, Shimizu M, Kaiser M, et al. Syrbactin class proteasome inhibitor-induced apoptosis and autophagy occurs in association with p53 accumulation and Akt/PKB activation in neuroblastoma. Biochem Pharmacol. 2010;80(2):170–8. doi: 10.1016/j.bcp.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 124.Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S, et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther. 2010;9(4):963–75. doi: 10.1158/1535-7163.MCT-09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ikeda H, Hideshima T, Fulciniti M, Perrone G, Miura N, Yasui H, et al. PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood. 2010;116(9):1460–8. doi: 10.1182/blood-2009-06-222943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nakamura M, Kikukawa Y, Takeya M, Mitsuya H, Hata H. Clarithromycin attenuates autophagy in myeloma cells. Int J Oncol. 2010;37(4):815–20. [PubMed] [Google Scholar]
- 127.David E, Kaufman JL, Flowers CR, Schafer-Hales K, Torre C, Chen J, et al. Tipifarnib sensitizes cells to proteasome inhibition by blocking degradation of bortezomib-induced aggresomes. Blood. 2010;116(24):5285–8. doi: 10.1182/blood-2010-03-272393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther. 2005;4(10):1533–40. doi: 10.1158/1535-7163.MCT-05-0068. [DOI] [PubMed] [Google Scholar]
- 129.Shanmugam M, McBrayer SK, Qian J, Raikoff K, Avram MJ, Singhal S, et al. Targeting glucose consumption and autophagy in myeloma with the novel nucleoside analogue 8-aminoadenosine. J Biol Chem. 2009;284(39):26816–30. doi: 10.1074/jbc.M109.000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pan Y, Gao Y, Chen L, Gao G, Dong H, Yang Y, et al. Targeting autophagy augments in vitro and in vivo antimyeloma activity of DNA-damaging chemotherapy. Clin Cancer Res. 2011;17(10):3248–58. doi: 10.1158/1078-0432.CCR-10-0890. [DOI] [PubMed] [Google Scholar]
- 131.Zeng R, He J, Peng J, Chen Y, Yi S, Zhao F, et al. The time-dependent autophagy protects against apoptosis with possible involvement of Sirt1 protein in multiple myeloma under nutrient depletion. Ann Hematol. 2012;91(3):407–17. doi: 10.1007/s00277-011-1315-z. [DOI] [PubMed] [Google Scholar]
- 132.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285(14):10850–61. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, et al. Linking of auto-phagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171(2):513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Richardson PG, Mitsiades CS, Laubach JP, Lonial S, Chanan-Khan AA, Anderson KC. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br J Haematol. 2011;152(4):367–79. doi: 10.1111/j.1365-2141.2010.08360.x. [DOI] [PubMed] [Google Scholar]
- 135.Caligaris-Cappio F, Bergui L, Gregoretti MG, Gaidano G, Gaboli M, Schena M, et al. Role of bone marrow stromal cells in the growth of human multiple myeloma. Blood. 1991;77(12):2688–93. [PubMed] [Google Scholar]