

Abstract
Background
Patients with paroxysmal nocturnal hemoglobinuria harbor clonal glycosylphosphatidylinositol-anchor deficient cells arising from a multipotent hematopoietic stem cell acquiring a PIG-A mutation. Many patients with aplastic anemia and myelodysplastic syndromes also harbor small populations of glycosylphosphatidylinositol-anchor deficient cells. Patients with aplastic anemia often evolve into paroxysmal nocturnal hemoglobinuria; however, myelodysplastic syndromes seldom evolve into paroxysmal nocturnal hemoglobinuria. Here, we investigate the origin and clonality of small glycosylphosphatidylinositol-anchor deficient cell populations in aplastic anemia and myelodysplastic syndromes.
Design and Methods
We used peripheral blood flow cytometry to identify glycosylphosphatidylinositol-anchor deficient blood cells, a proaerolysin-resistant colony forming cell assay to select glycosylphosphatidylinositol-anchor deficient progenitor cells, a novel T-lymphocyte enrichment culture assay with proaerolysin selection to expand glycosylphosphatidylinositol-anchor deficient T lymphocytes, and PIG-A gene sequencing assays to identify and analyze PIG-A mutations in patients with aplastic anemia and myelodysplastic syndromes.
Results
Twelve of 15 aplastic anemia patients were found to harbor a small population of glycosylphosphatidylinositol-anchor deficient granulocytes; 11 of them were found to harbor a small population of glycosylphosphatidylinositol-anchor deficient erythrocytes, 10 patients were detected to harbor glycosylphosphatidylinositol-anchor deficient T lymphocytes, and 3 of them were detected only after T-lymphocyte enrichment in proaerolysin selection. PIG-A mutation analyses on 3 patients showed that all of them harbored a matching PIG-A mutation between CFU-GM and enriched T lymphocytes. Two of 26 myelodysplastic syndromes were found to harbor small populations of glycosylphosphatidylinositol-anchor deficient granulocytes and erythrocytes transiently. Bone marrow derived CD34+ cells from 4 patients grew proaerolysin-resistant colony forming cells bearing PIG-A mutations. No glycosylphosphatidylinositol-anchor deficient T lymphocytes were detected in myelodysplastic syndrome patients.
Conclusions
In contrast to aplastic anemia and paroxysmal nocturnal hemoglobinuria, where PIG-A mutations arise from multipotent hematopoietic stem cells, glycosylphosphatidylinositol-anchor deficient cells in myelodysplastic syndromes appear to arise from more committed progenitors.
Keywords: paroxysmal nocturnal hemoglobinuria, aplastic anemia, myelodysplastic syndrome, hematopoietic stem cell
Introduction
Myelodysplastic syndromes (MDS),1 acquired aplastic anemia (AA),2 and paroxysmal nocturnal hemoglobinuria (PNH)3 are closely related hematopoietic stem cell (HSC) diseases. MDS is a heterogeneous group of clonal, acquired HSC disorders with ineffective hematopoiesis. AA may be inherited or acquired but most adult cases are caused by an immune mediated attack of HSC that leads to bone marrow failure.4,5 PNH is caused by somatic PIG-A gene mutations that arise from a multipotent HSC. The PIG-A gene product is required for the first step in glycosylphosphatidylinositol (GPI) biosynthesis. GPI is an important glycolipid anchor that tethers a multitude of different membrane proteins to the cell surface. Thus, PNH cells have a deficiency of all GPI-anchored proteins. GPI-anchored proteins serve as the receptor for proaerolysin, an enterotoxin secreted by aeromonas hydrophila,6 and this explains why GPI anchor deficient cells are resistant to this pathogenic, channel-forming toxin. A fluorescent variant of proaerolysin (FLAER) is commonly used as a diagnostic assay for PNH.7,8 However, not all GPI-anchor protein deficient proaerolysin resistant cells harbor PIG-A mutations. Recent studies on lymphocytic cell lines, and even human embryonic stem cells, have shown that GPI anchor deficiency can result from transcriptional silencing of other genes involved in GPI anchor biosynthesis.9,10 Therefore, GPI-anchor protein deficiency is not always due to PIG-A mutations. The only way to be certain is to perform a DNA sequence analysis of the cells with GPI anchor protein deficiency.
In PNH, the majority of granulocytes are GPI-anchor protein deficient and are clonally derived from a multipotent hematopoietic stem cell that harbors a PIG-A mutation.11 Interestingly, small populations of PNH-like granulocytes can be found in up to 50% of patients with MDS (usually 0.01–5% PNH granulocytes)12 and AA (usually 0.1–10% PNH granulocytes).13,14 PIG-A mutant blood cells (~0.002%) can also been found in healthy control subjects.15,16 In contrast to PNH, most PIG-A mutations in healthy controls are transient and arise from colony forming cells that have no self-renewal capacity and no ability to differentiate into T lymphocytes.16,17
It is important to understand the relevance of small populations of PNH-like cells in AA and MDS. It is known that AA patients often, though not always, transform into classic PNH, but MDS patients seldom evolve into classic PNH.18–20 Furthermore, the presence of a small PNH-like clone in AA and MDS has been reported in some, but not all studies, to predict for responsiveness to immunosuppressive therapy.21–23 Based upon these observations, the most recent guidelines from the NCCN suggest that testing for a PNH clone should be part of the evaluation in MDS patients.24 The purpose of this study is to gain insight into the relevance of small PNH populations in AA and MDS by studying the level of hematopoiesis where PIG-A mutations arise. Our findings suggest that PIG-A mutations in AA and PNH arise from a multipotent hematopoietic stem cell; however, PIG-A mutations in MDS appear to be transient and arise from more differentiated hematopoietic cells with less self-renewal capacity.
Design and Methods
Patient selection
All patients in this study were evaluated at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University School of Medicine between July 2009 and December 2011. Acquired aplastic anemia was diagnosed as per clinical criteria.25,26 Myelodysplastic syndrome (MDS) was diagnosed as per WHO criteria.27 The bone marrow aspiration and biopsy samples were evaluated by hematopathologists at Johns Hopkins Hospital. The venous peripheral blood and bone marrow aspiration for experiment use were drawn into heparin-containing tubes after informed consent as approved by the Joint Committee Clinical Investigation of the Johns Hopkins Hospital.
Isolation of mononuclear cells and CD34+ cells
Bone marrow cells were obtained from posterior iliac crest aspirations and peripheral blood cells were obtained from peripheral venous phlebotomies. The mononuclear cells were recovered by Ficoll/Hypaque (density<1.077) centrifugation. To collect purified CD34+ cells, mononuclear cells were washed twice in phosphate-buffered saline (PBS; Cellgro; Mediatech, Manassas, VA, USA) incubated with anti-CD34 microbeads and processed through magnet-activated cell sorting (MACS) separation column according to the manufacturer’s instructions (Miltenyi Biotech, Auburn, CA, USA).
Further details about selection of proaerolysin-resistant CFCs, flow cytometry analysis of peripheral blood PNH cells, PNH T-lymphocyte enrichment culture and PIG-A gene sequencing assay are available in the Online Supplementary Appendix.
Statistical analysis
Fisher’s exact test was used to evaluate the significance of the prevalence difference in harboring GPI-anchor deficient T lymphocytes between MDS patients and AA patients.
Results
Origin of PIG-A mutations in aplastic anemia
PIG-A mutations in PNH arise from a multipotent hematopoietic stem cell and involve all lineages, including T lymphocytes. In order to determine whether the small population of PIG-A mutant cells (PNH cells) in patients with AA also arise from a multipotent hematopoietic stem cell, we screened 15 patients with newly diagnosed or previously treated AA for the presence of PNH cells in the peripheral blood using standard flow cytometry analysis. Patients’ characteristics are described in Table 1. A PNH granulocyte population (median 4.9%, range 0.1–46%) was found in 12 of 15 patients (80%). Out of these 12 patients, 11 were found to have a small population of PNH erythrocytes in their peripheral blood that did not express CD59 on their membranes. The median size of PNH erythrocytes was 1.0% (range 0.1–13%). Since T lymphocytes can be expanded in culture, even in patients with AA, we hypothesized that if PIG-A mutations in AA arise from a multipotent hematopoietic stem cell, then proaerolysin selection should allow for the enrichment and expansion of PIG-A mutant T lymphocytes. To test this hypothesis, we screened 13 of these 15 AA patients for GPI-anchor protein deficient T lymphocytes using FLAER staining and standard flow cytometry analyses. We identified 7 patients with a small population of PNH T lymphocytes (median PNH T-lymphocyte size: 0.3%, range 0.1–8.9%). The proportion of PNH T lymphocytes grew significantly after culturing mononuclear cells in a T-lymphocyte enrichment culture system under proaerolysin selection (Table 1, Figures 1 and 2). Interestingly, 3 patients (A2, A6 and A8) appeared to have no detectable PNH T lymphocytes by standard flow cytometry; however, after culturing their peripheral mononuclear cells in this T-lymphocyte enrichment assay, GPI-anchor protein deficient T lymphocytes were easily detected (Table 1 and Figure 2). The viability of the proaerolysin enriched T lymphocytes was higher than 87% by trypan blue staining. Of 3 AA patients with no detectable PNH cells in both granulocytes and erythrocytes (A13, A14 and A15), none had detectable PNH T lymphocytes, even after culturing their cells in proaerolysin (Table 1 and Figure 2).
Table 1.
Patients’ characteristics.
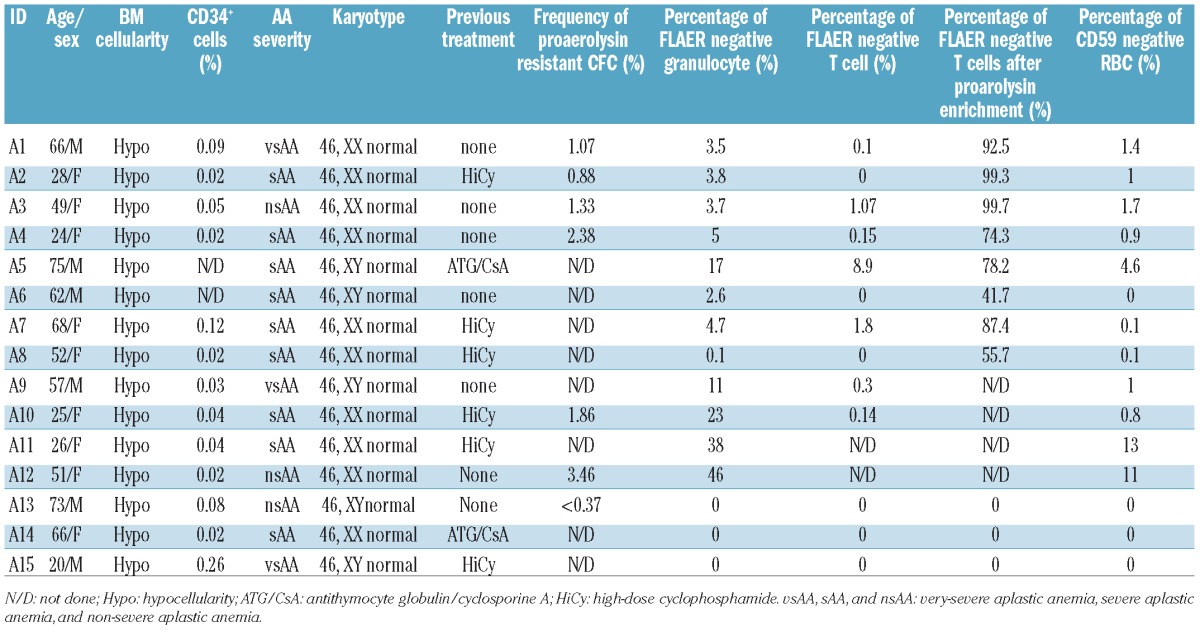
Figure 1.
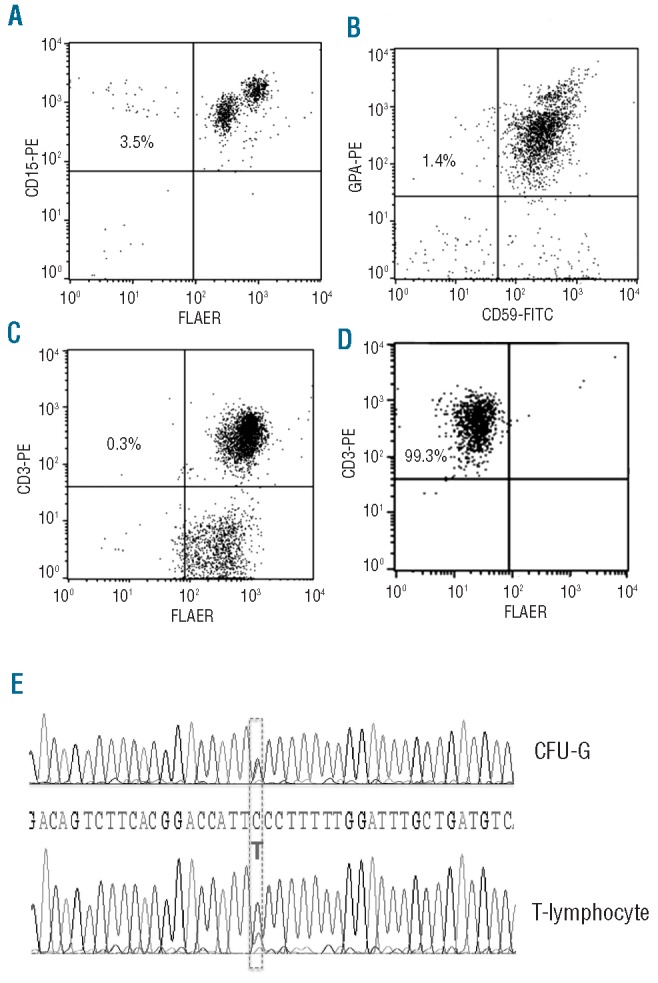
PIG-A mutations in AA arise from a multipotent HSC. Peripheral blood flow cytometry for PNH in a representative patient (A1) with AA. (A) Granulocytes stained with PE conjugated anti-CD15 and FLAER. (B) Erythrocytes stained with PE-conjugated anti-glycophorin and FITC-conjugated anti-CD59. (C) T lymphocytes stained with PE conjugated anti-CD3 and FLAER. (D) T lymphocytes stained with PE conjugated anti-CD3 and FLAER after 14 days culturing in a T-lymphocyte enrichment medium with proaerolysin selection. (E) PIG-A gene DNA sequencing analysis of a myeloid CFU-G and a T-lymphocyte colony, both grown under proaerolysin selection, showed that they both had a substitution mutation of C to T at 549bp of Exon 2. DNA was extracted from individual proaerolysin resistant CFC and T-cell colony and was PCR amplified and sequenced for PIG-A mutation analysis.
Figure 2.

T-lymphocyte enrichment assay under proaerolysin selection. Only AA patients harboring a small population of PNH cells in their peripheral blood showed enrichment of PNH T lymphocytes (P=0.00016). (A) Percentage change of GPI-AP deficient T cells before (Day 0) and after (Day 14) T-lymphocyte enrichment under proaerolysin selection in 4 MDS patients (M1, M2, M3 and M4) whose bone marrow CD34+ cells were detected to grow proaerolysin-resistant CFCs. Patient M1 and M2 harbored small populations of GPI-AP deficient granulocytes and erythrocytes but patient M3 and M4 did not harbor either. (B) Percentage change of GPI-AP deficient T-lymphocytes before (Day 0) and after (Day 14) T-lymphocyte enrichment under proaerolysin selection in 8 AA patients (A1 – A8) harboring a small population of GPI-AP deficient granulocytes and erythrocytes. (C) Percentage change of GPI-AP deficient T lymphocytes before (Day 0) and after (Day 14) T-lymphocyte enrichment under proaerolysin selection in 3 AA patients (A13 – A15) not harboring a small population of GPI-AP deficient granulocytes or erythrocytes. The images below were taken on Day 14 of the culture with 20× amplification. The red dash circle below (B) demonstrates a colony of GPI-AP deficient cells in various stages of T lymphocytopoiesis.
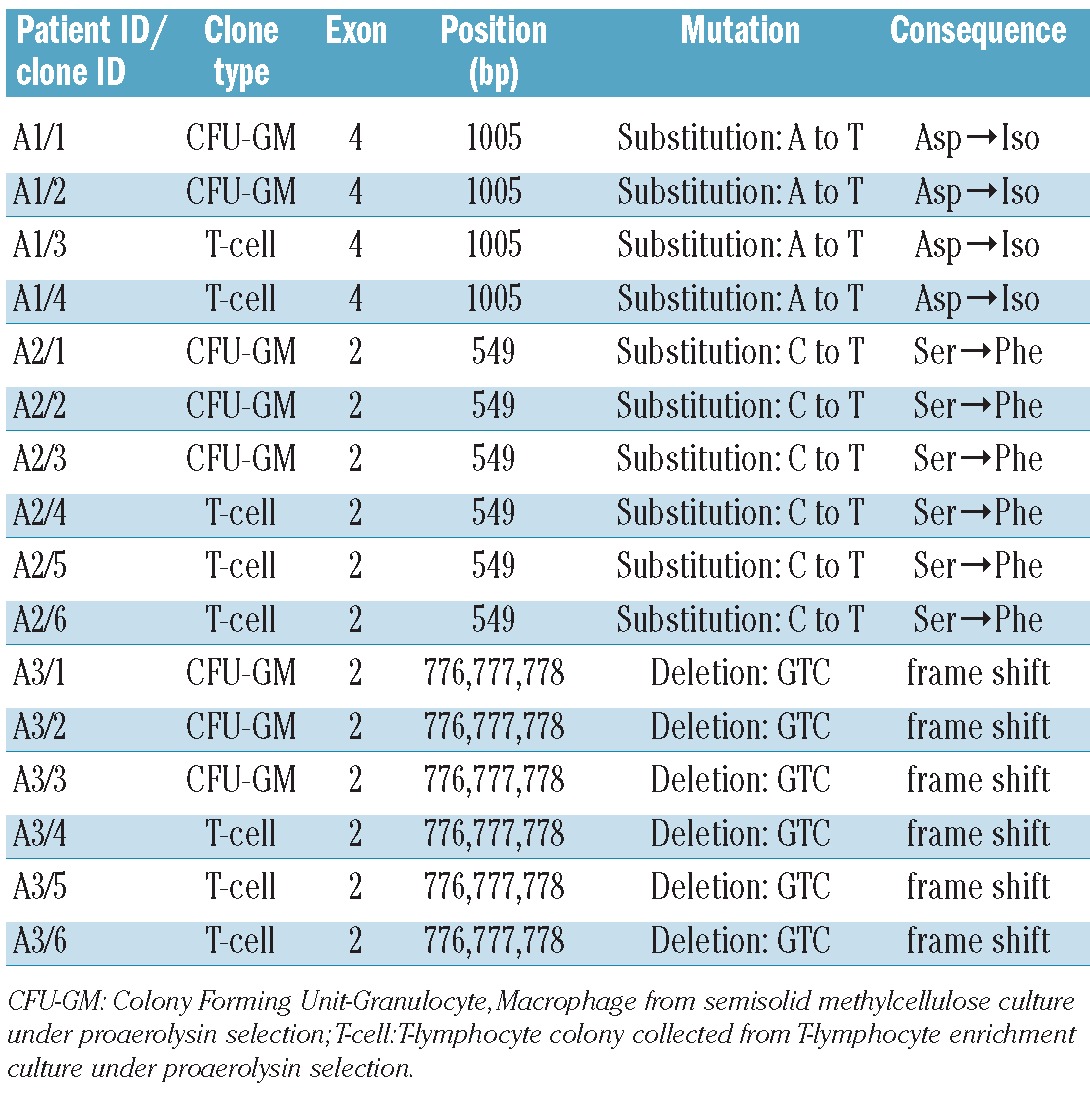
To further explore the origin and clonality of PIG-A mutations in AA patients harboring a small population of PNH granulocytes, CD34+ cells harvested from bone marrows of patients A1, A2 and A3 were plated in proaerolysin-containing semisolid methylcellulose culture as described in the Online Supplementary Appendix. Simultaneously, mononuclear cells collected from peripheral blood were cultured in the above-mentioned T-lymphocyte proaerolysin enrichment culture system. PIG-A gene sequencing and mutation studies on genomic DNA extracted from both proaerolysin-resistant CFC-GMs (2 from A1, 3 each from A2 and A3) and proaerolysin-resistant T lymphocyte colonies (2 from A1, 3 each from A2 and A3) demonstrated matching PIG-A mutations in the myeloid and lymphoid lineages in each patient (Figure 1 and Table 2). These data indicate that the PIG-A mutations in these AA patients arose in a common multipotent precursor.
Table 2.
PIG-A gene analysis of proaerolysin resistant clones in 3 AA patients harboring small population of PNH cells.
Origin of PIG-A mutations in myelodysplastic syndromes
We next sought to determine whether PIG-A mutations in MDS also arise from a multipotent hematopoietic stem cell. We used standard flow cytometry analysis, proaerolysin-resistant semisolid methylcellulose CFC culture assay, T-lymphocyte enrichment culture assay under proaerolysin selection, and PIG-A gene mutation sequencing assays to study 26 MDS patients (25 of the 26 had low-grade MDS). Patients’ characteristics are described in Table 3. Using peripheral blood flow cytometry analysis, 2 patients (M1 and M2; 7.69%) were identified as bearing a small PNH granulocyte population (3.2% and 1.3%, respectively). However, using proaerolysin-resistant semisolid methylcellulose CFC culture assay, 4 patients (M1, M2, M3 and M4) were observed to grow proaerolysin-resistant CFCs from their bone marrow derived CD34+ cells. Patients M1 and M2 grew single proaerolysin resistant CFU-GM with a PIG-A mutational frequency of 0.027% and 0.035%, respectively, patient M3 grew a single proaerolysin resistant BFU-E with a PIG-A mutational frequency of 0.092%, and patient M4 grew 3 proaerolysin resistant CFCs (2 BFU-E and 1 CFU-E) with a PIG-A mutation frequency of 0.259%. PIG-A gene mutational analysis using DNA extracted from proaerolysin resistant CFCs identified a single base deletion mutation in exon 2 for M1, which caused a frame shift and premature termination of translation, and substitution mutations for M2, M3 and M4 in exons 2 and 5 (Table 4 and Figure 3). Interestingly, the presence of small populations of PNH granulocytes, initially observed in patients M1 and M2, were transient. Those populations of PNH granulocytes were not detected at subsequent follow up (Table 5 and Figure 4). Patient M1’s PNH granulocyte population became undetectable after four months of supportive care and patient M2’s PNH granulocyte population disappeared two months after 5-azacitidine therapy, even though they were not in MDS remission. Furthermore, patient M3, whose bone marrow CD34+ cells initially grew a single proaerolysin-resistant BFU-E on semisolid methylcellulose culture, persistently showed PNH phenotype negative in all lineages during five months of observation. Thus, the PIG-A mutant cells in MDS patients appear transient.
Table 3.
Characteristics of 26 MDS patients.
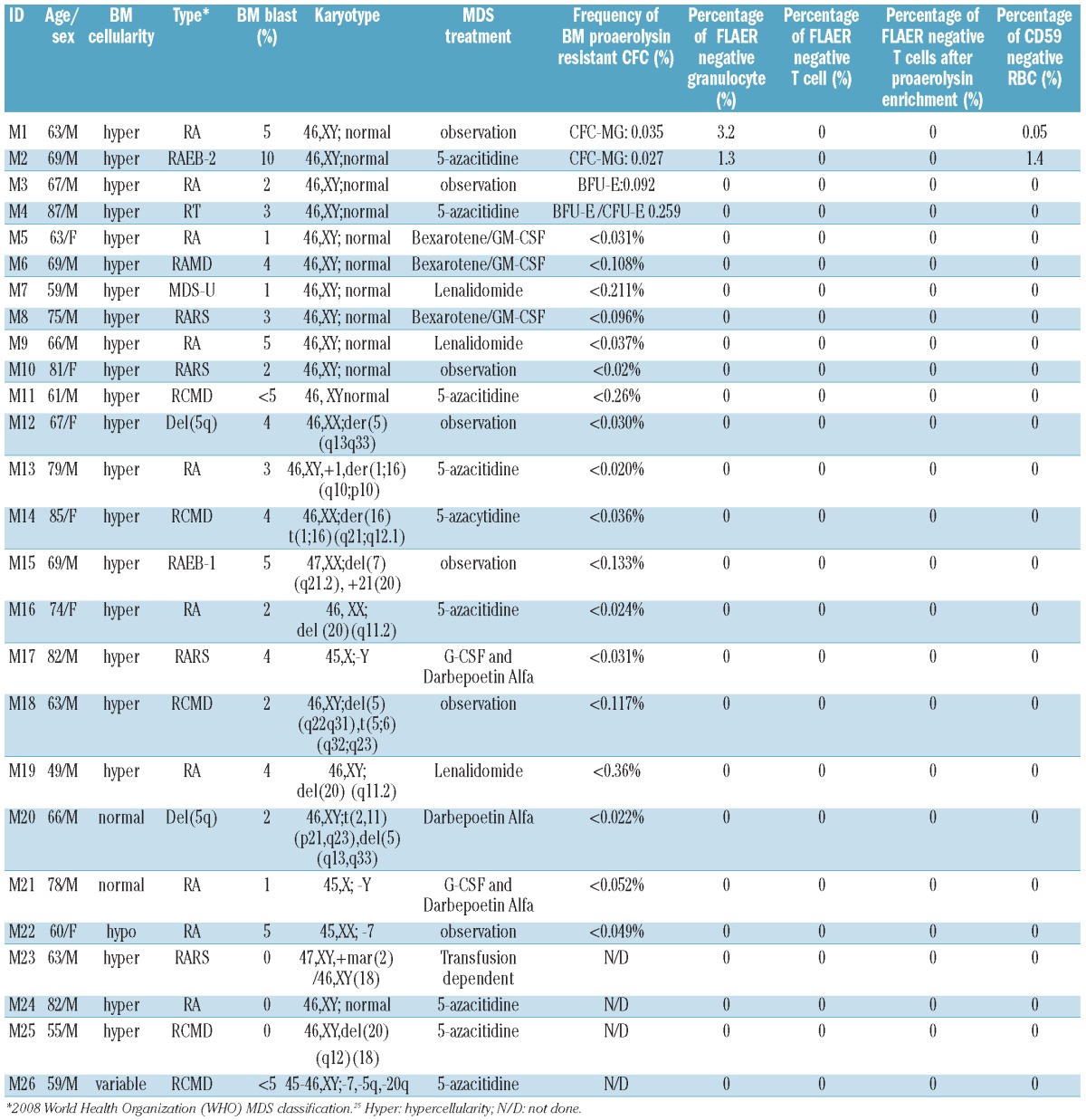
Table 4.
PIG-A mutations in MDS do not arise from a multipotent HSC and are transient. PIG-A gene mutation analysis of proaerolysin resistant CFCs in 4 MDS patients (M1, M2, M3 and M4).
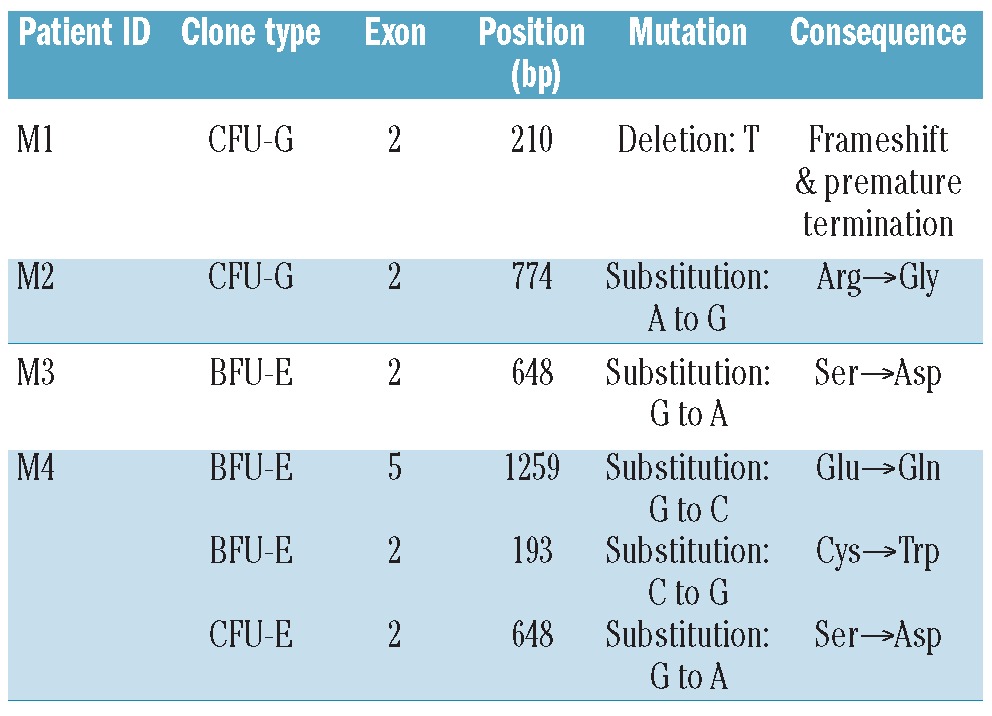
Figure 3.

PNH cell analysis in a patient (M1) with MDS. (A) Erythrocytes stained with PE conjugated anti-glycophorin A and FITC conjugated anti-CD59. (B) Granulocytes stained with PE conjugated anti-CD15 and FLAER. (C) T lymphocytes stained with PE conjugated anti-CD3 and FLAER. (D) T lymphocytes stained with PE conjugated anti-CD3 and FLAER after 14 days culturing in a T-lymphocyte enrichment medium with proaerolysin selection. (E) PIG-A DNA sequencing analysis of a single myeloid CFU-G grown under proaerolysin selection indentified a deletion mutation of “T” at 210 bp of exon 2, which caused a frameshift mutation, and a premature translation termination in the middle of exon 2.
Table 5.
Transient PNH cells in MDS patient (M1).
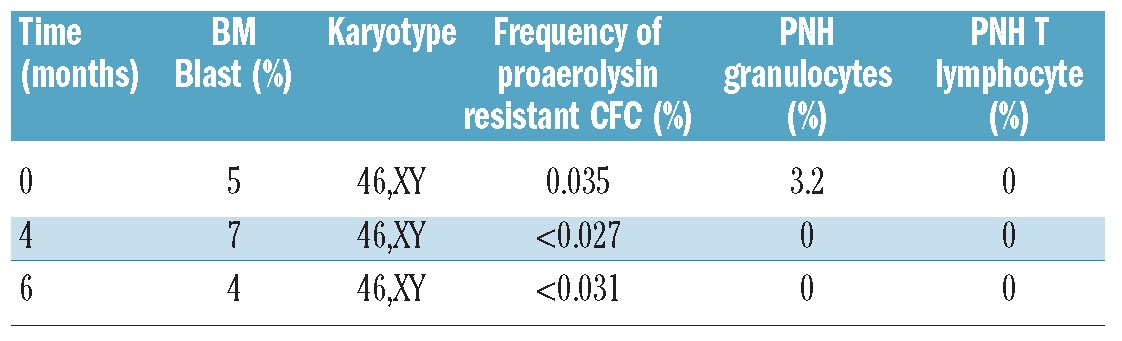
Figure 4.

PNH-like cells in patient M2 were transient: initially 1.4% M2’s RBCs and 1.3% of M2’s granulocytes were GPI-AP deficient (panel 1); however, those GPI-AP deficient cells were undetectable two months later (panel 2). Panels 1 and 2 from left to right: erythrocytes, granulocytes, T lymphocytes, and T lymphocytes from T-cell enrichment culture under proaerolysin selection.
We were unable to detect GPI-anchor protein deficient T lymphocytes using either standard flow cytometry analysis or T-lymphocyte enrichment culture assay in any of the 26 MDS patients, including M1, M2, M3 and M4 (Table 3 and Figure 2). Since there were no matching mutations and there were no detectable PIG-A mutations in their T lymphocytes, the data suggest that the PIG-A mutations in MDS may have arisen from CFC rather than a multipotent HSC.
Fisher’s exact test was used to evaluate the significance of the prevalence difference in harboring GPI-anchor deficient T lymphocytes between these 2 groups of patients and demonstrated a significant difference in harboring GPI-anchor deficient T cells (P=0.001) between AA and MDS.
Discussion
The documentation of small populations of GPI-anchor deficient granulocytes by flow cytometry has led to considerable confusion about diagnosis and prognosis in bone marrow failure disorders. Both AA and MDS patients harboring a small population of PNH cells could fit in either category 2 or 3 of the IPIG classification depending on whether they have clinical or laboratory evidence of hemolysis.28 In this study, we used peripheral blood flow cytometry, proaerolysin selection of bone marrow derived CFC, a novel proaerolysin-selection T-lymphocyte enrichment assay, and PIG-A DNA sequencing to determine the relevance of small populations of GPI-anchor deficient cells in AA and MDS. We found that PIG-A mutations in AA are common, arise from multipotent HSC, and can be readily detected in T lymphocytes. In contrast, PIG-A mutations in MDS are less common, are transient, are not readily detected in T lymphocytes, and appear to arise from more differentiated and committed progenitor cells such as CFCs.
GPI-anchor deficient cells in AA have previously been shown to be caused by PIG-A mutations.29 The persistence of GPI-anchor deficient blood cells in AA and their potential to expand and to lead to classic PNH has been documented for years.12,13,30,31 Therefore, we hypothesized that minor PNH populations in AA arise from a multipotent HSC. Furthermore, since T lymphocytes are readily expanded in AA patients, we hypothesized that proaerolysin enrichment of peripheral blood T lymphocytes may be a highly sensitive and specific surrogate assay to demonstrate that PIG-A mutations are derived from multipotent HSC. Indeed, of the 8 AA patients bearing PNH granulocytes that we assayed using our T-lymphocyte enrichment culture, all of them showed enrichment of PNH T lymphocytes under proaerolysin selection (Figure 2). Interestingly, GPI-anchor deficient T lymphocytes were detected via standard flow cytometry in just 5 of these 8 patients without proaerolysin enrichment (Table 1). PIG-A DNA sequencing of the proaerolysin resistant T lymphocytes confirmed that they had identical mutations to proaerolysin resistant bone marrow derived CFC (Figure 1 and Table 2). Therefore, GPI-anchor deficient cells in patients with AA, similar to PNH patients, arise from multipotent HSC. Preferential immune attack on normal HSC that spares the PIG-A mutant HSC is the most attractive hypothesis to explain the close relationship between AA and PNH.32,33 However, other hypotheses such as secondary mutations,34 resistance to apoptosis35.36 or even neutral evolution have been proposed.37 Regardless of the mechanism, our data suggest that PIG-A mutations arise from a multi-potent HSC in both PNH and AA. We were unable to detect GPI-anchor deficient T lymphocytes or CFC using proaerolysin selective enrichment in any of the 3 patients (A13, A14, and A15) who had no detectable GPI-anchor deficient granulocytes (Figure 2). Whether or not these patients are less likely to develop PNH over time will require a prospective study and the proaerolysin T-lymphocyte enrichment culture may be particularly useful for this.
The relevance of small PNH-like populations in MDS is more controversial. Several groups have reported the presence of small PNH-like populations in patients with MDS. However, these studies have not routinely performed serial analysis of the PNH populations, neither have they determined whether the mutations occur in multiple lineages, nor performed PIG-A gene sequencing studies on the GPI-anchor deficient cells. Furthermore, the threshold for detecting PNH cells in MDS has varied from 0.003% to 0.01%. We chose a cut off of 0.01% PNH granulocytes on our flow cytometric screening assay since we and others have shown that healthy controls have roughly 0.002% PNH granulocytes that arise from mutated CFC.15,16 Thus, 0.01% granulocytes represent an expansion of greater than 1 logarithm. Several small studies have suggested that harboring PNH cells in AA and MDS might be a predictor for response to immunosuppressive therapy.12,14,21,22,38 However, a multivariate analysis on MDS patients by Sloand et al. demonstrated that finding PNH-like cells was not associated with a response to immunosuppressive therapy or with a survival advantage.23 These divergent conclusions may be due to differences in cohort composition and the difficulty in discriminating aplastic anemia and hypocellular MDS. Our data support the findings of Sloand et al. and may help to explain why the presence of PNH-like cells in MDS patients is not associated with a greater likelihood of responding to immunosuppressive therapy or with a survival advantage. In our study, we recruited patients with a strict diagnosis of MDS and most of them suffered low-grade MDS; only one MDS patient (monosomy 7) had a hypocellular bone marrow (Table 3). We found that GPI-anchor deficient blood cells occur in just 7.69% of those patients (95% CI 0.95–25.13%), which is in agreement with Wang et al. who performed sensitive PNH flow cytometry on 110 MDS patients and found that just 12% (8% of those with <5% blasts) had a small PNH population.39 We detected small populations of PNH granulocytes and PNH erythrocytes, and a single proaerolysin resistant CFC that contained a PIG-A mutation simultaneously in 2 of these patients (M1 and M2). We also detected 2 other patients (M3 and M4) growing proaerolysin resistant BFU-E/CFU-E without detectable PNH phenotype positive cells from their peripheral blood. Interestingly, the PIG-A gene mutation frequency in MDS patients (median mf=0.0635%; range 0.027–0.259%) was 10 to 100-fold higher than healthy controls. The reason for this is unclear but could be due to reduced cell number or possibly genomic instability resulting from either MDS or treatment for MDS.
The GPI-anchor deficient blood cells in patients M1 and M2 were transient. Furthermore, all 4 patients growing proaerolysin resistant CFU-GM, BFU-E and CFU-E never had detectable GPI-anchor deficient T lymphocytes (Table 3 and Figure 2). We were unable to demonstrate whether the GPI-anchor deficient peripheral blood cells and the proaerolysin resistant CFC had matching PIG-A mutations due to the paucity of cells and the fact that the GPI-anchor deficient cells were not detected at follow up during serial analysis (Table 5 and Figure 4). However, PIG-A mutation analysis on 3 CFCs (2 BFU-Es and 1 CFU-E) growing from M4’s bone marrow derived CD34+ cells indicated that these proaerolysin resistant cells were heterogeneous and arose from different stem/progenitors (Table 4). The sensitivity of our proaerolysin enrichment assay on CFC (~0.02%) is less in MDS than in healthy controls (~0.001%) due to the fact that CFC formation, especially BFU-E formation, is markedly reduced in MDS patients. However, all patients bearing flow cytometry detectable peripheral PNH cells grew GPI-anchor deficient CFCs. Furthermore, since T lymphocytes can be readily expanded from MDS patients, the sensitivity of this assay would not be expected to differ significantly. A Fisher’s exact test comparing these 2 patient groups (MDS/PNH and AA/PNH) demonstrated a significant prevalence discrimination in harboring GPI-anchor deficient T cells between them (P=0.001). Taken together, these data suggest that small populations of GPI-anchor deficient cells (granulocytes and erythrocytes) do occur in MDS patients; however, similar to PIG-A mutations in bone marrows from healthy controls, most of these mutations seem to arise from CFC rather than HSC. An alternative interpretation of these data are that PIG-A mutations in MDS stem cells are unable to differentiate down the lymphoid lineage, or that small PIG-A mutant T lymphocytes exist in MDS but are below the level of detection of our assay. Our experiments can not definitively differentiate between these possibilities; however, the heterogeneity of PIG-A mutations in patient M4 and the transience of the PIG-A mutations, coupled with the fact that MDS seldom, if ever, evolves into PNH, suggest that those small populations of PIG-A mutant cells in MDS arise from cells more differentiated than a multipotent HSC. Classic PNH patients may evolve into MDS or even acute leukemia. These patients often (but not always) have a hypocellular marrow and a large PNH clone. Given that the GPI-AP deficient cells in classic PNH virtually always arise from a multipotent HSC-acquired PIG-A mutation, we would expect the PIG-A mutant cells in these patients to originate from a mutipotent HSC.
In summary, small populations of GPI-anchor deficient granulocytes in AA are similar to those in classic PNH in that they arise from a multipotent HSC. In contrast, small populations of GPI-anchor deficient granulocytes in MDS may be similar to those found in healthy controls in that they arise from CFC. Thus, GPI-anchor protein deficiency should be interpreted with caution in the setting of MDS, even when more than one lineage is involved. Our novel proaerolysin selection T-cell enrichment assay may be helpful in distinguishing between AA and MDS, and may also be useful in determining which AA patients are most at risk for progressing to PNH, though this will need to be confirmed in prospective studies.
Acknowledgments
We are grateful to all the patients for their contribution of their bone marrow and blood samples. We thank Steven Galkin, Sara Sakoian, RN, BSN, and Ellen Lilly-Foreman, RN, OCN for patient care and sample collection.
Footnotes
Funding: this study was supported by the National Institutes of Health (grant P01CA70970 and T32HL007525).
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Stone RM. How I treat patients with myelodysplastic syndromes. Blood. 2009;113(25):6296–303. doi: 10.1182/blood-2008-09-038935. [DOI] [PubMed] [Google Scholar]
- 2.Brodsky RA, Jones RJ. Aplastic anaemia. Lancet. 2005;365(9471):1647–56. doi: 10.1016/S0140-6736(05)66515-4. [DOI] [PubMed] [Google Scholar]
- 3.Brodsky RA. Narrative review: paroxysmal nocturnal hemoglobinuria: the physiology of complement-related hemolytic anemia. Ann Intern Med. 2008;148(8):587–95. doi: 10.7326/0003-4819-148-8-200804150-00003. [DOI] [PubMed] [Google Scholar]
- 4.Maciejewski JP, Selleri C, Sato T, Anderson S, Young NS. Increased expression of Fas antigen on bone marrow CD34+ cells of patients with aplastic anaemia. Br J Haematol. 1995;91(1):245–52. doi: 10.1111/j.1365-2141.1995.tb05277.x. [DOI] [PubMed] [Google Scholar]
- 5.Zeng W, Maciejewski JP, Chen G, Young NS. Limited heterogeneity of T cell receptor BV usage in aplastic anemia. J Clin Invest. 2001;108(5):765–73. doi: 10.1172/JCI12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brodsky RA, Mukhina GL, Nelson KL, Lawrence TS, Jones RJ, Buckley JT. Resistance of paroxysmal nocturnal hemoglobinuria cells to the glycosylphosphatidylinositol-binding toxin aerolysin. Blood. 1999;93(5):1749–56. [PubMed] [Google Scholar]
- 7.Brodsky RA, Mukhina GL, Li S, Nelson KL, Chiurazzi PL, Buckley JT, Borowitz MJ. Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am J Clin Pathol. 2000;114(3):459–66. doi: 10.1093/ajcp/114.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borowitz MJ, Craig FE, DiGiuseppe JA, Illingworth AJ, Rosse W, Sutherland DR, et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom. 2010;78(4):211–30. doi: 10.1002/cyto.b.20525. [DOI] [PubMed] [Google Scholar]
- 9.Hu R, Mukhina GL, Lee SH, Jones RJ, Englund PT, Brown P, et al. Silencing of genes required for glycosylphosphatidylinositol anchor biosynthesis in Burkitt lymphoma. Exp Hematol. 2009;37(4):423–34. doi: 10.1016/j.exphem.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen G, Ye Z, Yu X, Zou J, Mali P, Brodsky RA, Cheng L. Trophoblast differentiation defect in human embryonic stem cells lacking PIG-A and GPI-anchored cell-surface proteins. Cell Stem Cell. 2008;2(4):345–55. doi: 10.1016/j.stem.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bessler M, Mason PJ, Hillmen P, Miyata T, Yamada N, Takeda J, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994;13(1):110–7. doi: 10.1002/j.1460-2075.1994.tb06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunn DE, Tanawattanacharoen P, Boccuni P, Nagakura S, Green SW, Kirby MR, et al. Paroxysmal nocturnal hemoglobinuria cells in patients with bone marrow failure syndromes. Ann Intern Med. 1999;131(6):401–8. doi: 10.7326/0003-4819-131-6-199909210-00002. [DOI] [PubMed] [Google Scholar]
- 13.Mukhina GL, Buckley JT, Barber JP, Jones RJ, Brodsky RA. Multilineage glycosylphosphatidylinositol anchor deficient hematopoiesis in untreated aplastic anemia. Br J Haematol. 2001;115(2):476–82. doi: 10.1046/j.1365-2141.2001.03127.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Chuhjo T, Yamazaki H, Shiobara S, Teramura M, Mizoguchi H, Nakao S. Relative increase of granulocytes with a paroxysmal nocturnal haemoglobinuria phenotype in aplastic anaemia patients: the high prevalence at diagnosis. Eur J Haematol. 2001;66(3):200–5. doi: 10.1034/j.1600-0609.2001.00376.x. [DOI] [PubMed] [Google Scholar]
- 15.Araten DJ, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc Natl Acad Sci USA. 1999;96(9):5209–14. doi: 10.1073/pnas.96.9.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu R, Mukhina GL, Piantadosi S, Barber JP, Jones RJ, Brodsky RA. PIG-A mutations in normal hematopoiesis. Blood. 2005;105(10):3848–54. doi: 10.1182/blood-2004-04-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Traulsen A, Pacheco JM, Dingli D. On the origin of multiple mutant clones in paroxysmal nocturnal hemoglobinuria. Stem Cells. 2007;25(12):3081–4. doi: 10.1634/stemcells.2007-0427. [DOI] [PubMed] [Google Scholar]
- 18.Pu JJ, Mukhina G, Wang H, Savage WJ, Brodsky RA. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011;87(1):37–45. doi: 10.1111/j.1600-0609.2011.01615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheinberg P, Marte M, Nunez O, Young NS. Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica. 2010;95(7):1075–80. doi: 10.3324/haematol.2009.017889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112(8):3099–106. doi: 10.1182/blood-2008-01-133918. [DOI] [PubMed] [Google Scholar]
- 21.Sugimori C, Chuhjo T, Feng X, Yamazaki H, Takami A, Teramura M, et al. Minor population of CD55-CD59- blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107(4):1308–14. doi: 10.1182/blood-2005-06-2485. [DOI] [PubMed] [Google Scholar]
- 22.Ishikawa T, Tohyama K, Nakao S, Yoshida Y, Teramura M, Motoji T, et al. A prospective study of cyclosporine A treatment of patients with low-risk myelodysplastic syndrome: presence of CD55(−)CD59(−) blood cells predicts platelet response. Int J Hematol. 2007;86(2):150–7. doi: 10.1532/IJH97.07052. [DOI] [PubMed] [Google Scholar]
- 23.Sloand EM, Wu CO, Greenberg P, Young N, Barrett J. Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J Clin Oncol. 2008;26(15):2505–11. doi: 10.1200/JCO.2007.11.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Myelodysplastic Syndrome: NCCN Clincial Practice Guideline in Oncology (NCCN GuidelinesTM)-Version 2. 2011. p. 3. [Google Scholar]
- 25.Camitta BM, Rappeport JM, Parkman R, Nathan DG. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood. 1975;45(3):355–63. [PubMed] [Google Scholar]
- 26.Bacigalupo A, Hows JM, Gluckman E, Nissen C, Marsh J, Van Lint MT, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA Working Party. Br J Haematol. 1988;70(2):177–82. doi: 10.1111/j.1365-2141.1988.tb02460.x. [DOI] [PubMed] [Google Scholar]
- 27.Swerdlow SH, Campo E, Harris NL, Stein H, Jaffe ES, Theille J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue. IARC; Lyon, France: 2008. [Google Scholar]
- 28.Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagarajan S, Brodsky R, Young NS, Medof ME. Genetic defects underlying paroxysmal nocturnal hemoglobinuria that arises out of aplastic anemia. Blood. 1995;86(12):4656–61. [PubMed] [Google Scholar]
- 30.Tichelli A, Gratwohl A, Nissen C, Speck B. Late clonal complications in severe aplastic anemia. Leuk Lymphoma. 1994;12(3–4):167–75. doi: 10.3109/10428199409059587. [DOI] [PubMed] [Google Scholar]
- 31.Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253–8. doi: 10.1056/NEJM199511093331904. [DOI] [PubMed] [Google Scholar]
- 32.Luzzatto L, Bessler M, Rotoli B. Somatic mutations in paroxysmal nocturnal hemoglobinuria: A blessing in disguise? Cell. 1997;88(1):1–4. doi: 10.1016/s0092-8674(00)81850-4. [DOI] [PubMed] [Google Scholar]
- 33.Young NS. The problem of clonality in aplastic anemia: Dr. Dameshek’s riddle, restated. Blood. 1992;79(6):1385–92. [PubMed] [Google Scholar]
- 34.Inoue N, Izui-Sarumaru T, Murakami Y, Endo Y, Nishimura J, Kurokawa K, et al. Molecular basis of clonal expansion of hematopoiesis in 2 patients with paroxysmal nocturnal hemoglobinuria (PNH) Blood. 2006;108(13):4232–6. doi: 10.1182/blood-2006-05-025148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brodsky RA, Vala MS, Barber JP, Medof ME, Jones RJ. Resistance to apoptosis caused by PIG-A gene mutations in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA. 1997;94(16):8756–60. doi: 10.1073/pnas.94.16.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savage WJ, Barber JP, Mukhina GL, Hu R, Chen G, Matsui W, et al. Glycosylphosphatidylinositol-anchored protein deficiency confers resistance to apoptosis in PNH. Exp Hematol. 2009;37(1):42–51. doi: 10.1016/j.exphem.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dingli D, Luzzatto L, Pacheco JM. Neutral evolution in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA. 2008;105(47):18496–500. doi: 10.1073/pnas.0802749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maciejewski JP, Follmann D, Nakamura R, Saunthararajah Y, Rivera CE, Simonis T, et al. Increased fraquency of HLA-DR2 in patients with paroxysmal nocturnal hemoglobinuria and the PNH/aplastic anemia syndrome. Blood. 2001;98(13):3513–9. doi: 10.1182/blood.v98.13.3513. [DOI] [PubMed] [Google Scholar]
- 39.Wang SA, Pozdnyakova O, Jorgensen JL, Medeiros LJ, Stachurski D, Anderson M, et al. Detection of paroxysmal nocturnal hemoglobinuria clones in patients with myelodysplastic syndromes and related bone marrow diseases, with emphasis on diagnostic pitfalls and caveats. Haematologica. 2009;94(1):29–37. doi: 10.3324/haematol.13601. [DOI] [PMC free article] [PubMed] [Google Scholar]