

Abstract
Prophylaxis of posttraumatic epilepsy will require a detailed knowledge of the epileptogenic pathophysiological processes that follow brain injury. Results from studies of experimental models and human epilepsy highlight alterations in GABAergic interneurons and formation of excessive new excitatory synaptic connectivity as prominent targets for prophylactic therapies. Promising laboratory results suggest that it will be possible to experimentally modify these aberrant processes and interfere with epileptogenesis. However, a number of key issues must be addressed before these results can be used to frame clinical antiepileptogenic therapy.
Introduction
Posttraumatic epileptogenesis is a major neurological sequel of serious brain injury that has far-reaching impacts on affected individuals and their families, in addition to profound socioeconomic effects. The expected increased incidence in wartime and the difficulty in controlling posttraumatic seizures with anticonvulsant drugs are factors that make it important to develop strategies for prophylaxis of this disorder (Dichter [17], Garga and Lowenstein [23]). A large number of alterations in gene expression (Raghavendra et al [53]) and a variety of pathophysiological processes occur in parallel following a brain injury, making it unlikely that an intervention focused on any one of these, in isolation, will emerge as a prophylactic “silver bullet”. The situation is further complicated by the likelihood that variables such as the level of brain maturation, site and distribution of injury (focal vs multifocal vs diffuse), type of trauma (e.g. concussive vs penetrating), presence or absence of significant bleeding, genetic susceptibility factors, etc. may affect the underlying type and sequence of epileptogenic events and the optimal timing of a potentially successful intervention in a given individual. Here we briefly review essential steps for developing potential prophylactic strategies including choice of a model, identification of key pathophysiological processes underlying epileptogenesis in that model, development of experimental approaches that target these processes and recognizing unsolved issues in applying results to human posttraumatic epilepsy.
1) Choice of models
There is no perfect model of human posttraumatic epilepsy. Discussions about the merits of one model versus another are only useful in the context of the particular pathophysiological process or event to be investigated. Obviously, to determine whether a drug will be prophylactic in vivo, a model in which there is an expected high incidence of behavioral posttraumatic seizures at relatively short latency after injury (i.e. high throughput), would be desirable. However, if the goal is, for example, to determine which alterations in function of GABAergic interneurons or pyramidal cells occur at a limited site of focal neocortical epileptogenesis, and might be modified or prevented by the same drug, a model with a stereotyped restricted single epileptogenic focal injury would be necessary to facilitate detailed cellular in vitro slice experiments and avoid the complications of widespread damage and variability. The advantages of the partial cortical isolation (“undercut”) model used in our laboratories have been described in detail elsewhere (Graber and Prince [25]). Partially isolated neocortex has been associated with epileptogenesis in human EEG studies (Echlin et al [18], in human postmortem neuropathological experiments (Marin-Padilla [42]) and in vivo experiments on monkey, cat and rodents (reviewed in Graber and Prince [25]). It is interesting that areas of partially isolated cortex with underlying loss of white matter are also present in the cortical contusion model (Feeney et al [22],) and epileptogenesis following fluid percussion injury (Kharatishvili and Pitkanen [31]). It is important to note that epileptogenic mechanisms may differ in one model vs another, and may also differ from those underlying human posttraumatic epilepsy, depending on the nature of the lesion and a number of other variables, so that approaches to prophylaxis may have to be varied accordingly.
2) Identification of key targets for prophylaxis
A review of work from a number of laboratories in a variety of animal models and human material suggests that 2 prominent pathophysiological processes are of great importance in focal epileptogenesis, namely, enhanced excitatory connectivity (Tauck and Nadler [62], Cronin and Dudek [13], Molnar and Nadler [47], McKinney et al [46], Esclapez et al [20], Salin et al [58]); and alterations in GABAergic inhibitory mechanisms (Toth et al [63], Cossart et al [11], Magloczky and Freund [40], Rosen et al [56], Li and Prince [37], Prince et al [51], Kumar and Buckmaster [34], Faria and Prince [21] and many others). In this chapter we will review the evidence for alterations in these critical regulatory processes in the undercut model and summarize recent results suggesting that it may be possible to limit the ongoing pathophysiology and decrease or prevent the resulting epileptogenesis. We will also emphasize the major hurdles to be dealt with in any attempt to apply what is known in laboratory models to epileptogenesis in humans.
3) Disinhibition in epileptogenesis
It has long been known that pharmacological suppression of GABAergic transmission induces acute epileptiform activity and seizures (reviewed in Prince [50]). This has occasioned a variety of experiments to determine whether disinhibition is a critical factor in lesional focal epileptogenesis. A large number of studies in both animal models and humans have emphasized alterations in anatomical or electrophysiological indices of GABAergic inhibitory function including loss of selective subtypes of inhibitory interneurons (Aronica et al [1], Buckmaster and Jongen-Relo [6], Cossart et al [12], DeFelipe [16]; reviewed in Houser [27]); alterations in postsynaptic GABAA receptors (Brooks-Kayal et al [5], Redecker et al [54], reviewed in Sperk et al [61]); and intrinsic alterations in function of interneurons such as their connectivity (Bausch [2], Magloczky and Freund [40]) and gross anatomical structure (Prince et al [51]). In the undercut model, several of these abnormalities have been demonstrated including anatomic alterations in fast-spiking (FS) parvalbumin-containing interneurons, such as decreased dendritic and axonal arbors and abnormalities in GABAergic boutons (Prince et al [51]). Electrophysiological abnormalities include decreases in the probability of GABA release (Pr) (Faria and Prince [21]; Ma and Prince, unpublished) associated with alterations in presynaptic N-type Ca++ channels (Faria and Prince unpublished). Dual whole cell recordings have confirmed the decreased Pr and also shown a markedly reduced size of unitary inhibitory postsynaptic currents (uIPSCs) from fast-spiking (FS) interneurons to excitatory cells. Decreased frequency of miniature IPSCs in pyramidal cells (Li and Prince [37]) are likely due to the decreases in Pr, together with a reduced number of inhibitory synapses. These and other results indicate that inhibitory neurons are functionally abnormal in the undercut model. Decreases in inhibitory connectivity have also been found using laser scanning photostimulation together with whole cell recordings from pyramidal neurons in undercut slices (X. Jin, J.R. Huguenard, D.A. Prince, in press).
Other data suggest that fast-spiking (FS) interneurons and their presynaptic terminals that innervate pyramidal cell somata, normally contain a high density of the sodium pump (Na+-K+ ATPase) (Chu et al, [10]). Pump activity would be essential in these terminals to prevent excessive depolarization during the high frequency spike firing that characterizes these interneurons normally and during epileptogenic discharges (McCormick et al [45]). The axonal terminal α3Na+-K+ ATPase immunoreactivity (IR) of FS interneurons is markedly decreased in epileptogenic lesions of both the undercut (L. Lee, I. Parada, D.A. Prince, unpublished) and microgyrus freeze models (Chu et al [10]), suggesting an additional potential mechanism for failure or decrease in transmitter release from inhibitory interneurons.
The remarkable anatomical changes in neocortical FS interneurons of undercuts mentioned above give them an appearance similar to that found in GABAergic neurons early in development (Jin et al [29]). One key factor influential in interneuronal development is the action of brain-derived neurotrophic factor (BDNF), released by pyramidal cells and acting at TrkB receptors (Jin et al [29], Marty [43]). BDNF is important in development and maintenance of connections of inhibitory neurons onto pyramidal cells (Kohara et al [33]). We tested the hypothesis that the undercut injury would reduce the expression of BDNF in pyramidal cells and/or its receptor, TrkB, on interneurons, as a mechanism for the observed anatomical changes. There was a significant reduction of TrkB immunoreactivity on parvalbumin-containing (FS) interneurons (Fig. 6 in Prince et al [51]) and decreased BDNF expression in pyramidal cells of UC cortex (I Parada and D.A. Prince, unpublished). These findings and others suggest that an increase in the availability of BDNF and activation of TrkB receptor in injured tissue might “rescue” inhibitory interneurons and inhibitory function. A recent report indicates that local delivery of trophic factors to hippocampi injured during status epilepticus can reduce epileptogenesis (Paradiso et al, [48]). The availability of a “small molecule” mimetic at the TrkB receptor (BD2-4), that can be administered and absorbed parenterally or intranasally (Massa et al [44]), facilitated an initial examination of the effects of BDNF on interneuronal structure in the undercut model. We found that intranasal infusion of BD2-4 for 2 wks significantly increased the ratio of phosphorylated to unphosphorylated AKT protein in brain, indicating that the drug was effective in activating TrkB receptors. Experiments thus far have dealt only with anatomical changes and results are encouraging. When BD2-4 was administered to undercut rats, there was a very substantial increase in the expression of α3Na+-K+ ATPase-IR in FS terminals versus expression of the IR in rats with undercuts treated with saline (Fig. 1). We are still a long way from knowing whether the BDNF mimetic will favorably affect other anatomical alterations in FS interneurons, the electrophysiological parameters of epileptogenesis, or behavioral seizures in this model. BDNF has many complex actions in the brain (Hu and Russek [28]) some of which may be pro-epileptogenic (e.g. Scharfman et al [59]) while others may decrease excitability and epileptiform activity (Reibel et al [55]). The timing of treatment and its duration maybe critical in effecting enhanced inhibition without increasing excitability.
Figure 1.
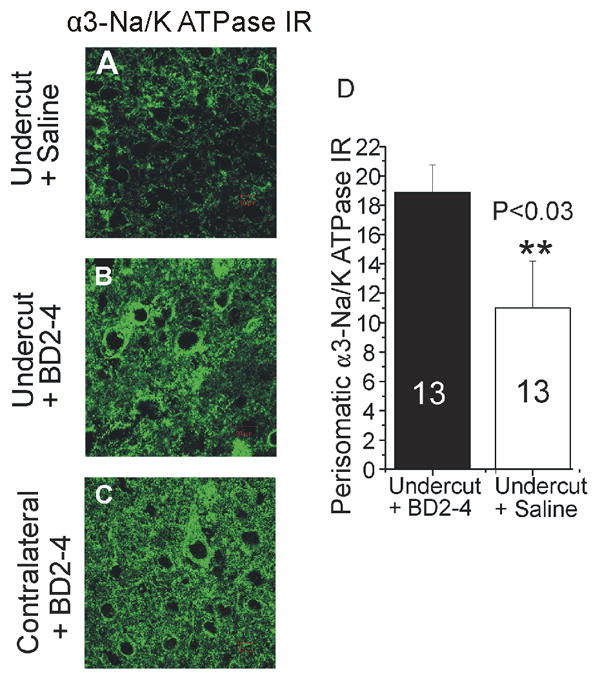
Effects of activation of TrkB receptors on perisomatic expression of α3Na/K ATPase-IR in undercut cortex.
A: Confocal image of α3-Na+/K+ ATPase immunoreactivity (IR) in undercut cortex 14 d after lesion in animal treated with intranasal saline. IR around pyramidal somata is reduced from side contralateral to isolation (C) and control naïve cortex (not shown). B: Perisomatic α3-Na+/K+ ATPase IR in undercut cortex in rat receiving intranasal BD2-4 daily × 2 wks is increased in intensity compared to saline treated control in A. D: Graph of data from 13 cells in undercut cortex of saline- vs BD2-4 treated rats shows a significant increase in α3-Na+/K+ ATPase in BD2-4 treated animals after 2 wks of treatment.
4) Enhanced excitatory connectivity/function
A large amount of work in several models of lesional epileptogenesis has shown that axonal sprouting and new excitatory synaptic connectivity are key pathophysiological mechanisms in lesional partial epilepsy (references above). In the undercut model, 2–3 wks after injury the frequency of excitatory postsynaptic currents (EPSCs) was increased significantly in the pyramidal neurons of layer V and the amplitude and input/output slope of the AMPA component of evoked EPSCs were increased (Li & Prince [37]). These findings fit with previous results (Salin et al [58]) which showed that axons of layer V pyramidal neurons have markedly increased lengths and numbers of branches, and an increased density of presynaptic boutons in layer V, where interictal epileptiform activity originates in the undercut (Prince and Tseng, [52]). To determine whether there was an activity-related mechanism involved in the sprouting response and associated epileptogenesis, we chronically applied tetrodotoxin (TTX) to the undercut cortex following the injury (Graber and Prince [24]). When the TTX application was made during a “critical period” of 3 days after the undercut (Graber and Prince [26]) or for longer periods (Graber and Prince [24]), epileptogenesis was markedly suppressed, and subsequent results showed that there was a significant decrease in anatomical markers of axonal and terminal sprouting (Fig. 1 in Prince et al [51]).
Recent experiments performed during the “critical period” after undercut have shown that enhanced excitatory postsynaptic currents (EPSCs) are present in layer V pyramidal cells 3 days after injury, and that aberrantly sprouting excitatory terminals make presumptive excitatory synapses on somata of these cells (D.K. Takahashi and D.A. Prince, unpublished). These results have provided proof in principle that, in this model, posttraumatic epileptogenesis can be prevented when therapy is administered over a limited period. They have also led to a search for more practical therapeutic interventions that would prevent formation of new excitatory connections (see below). Other experiments showed that there was an increased probability of glutamate release at excitatory synapses in epileptogenic cortical slices, indicating altered function of excitatory axonal synaptic terminals (Li et al [36]). Experiments using laser scanning and uncaging of caged glutamate have confirmed that there is increased glutamatergic excitatory connectivity in areas of the cortical undercut onto both pyramidal cells (Jin et al [30]) and interneurons (Jin, et al, 2010).
A large number of other anatomic and electrophysiological alterations result from the epileptogenic lesion, so it is difficult to determine whether sprouting and new connectivity is a pathophysiological mechanism that is sufficient alone to induce seizure activity. However, recent studies of synapse formation and elimination have provided key information with respect to this point by showing that failure to prune excessive excitatory synapses during development, in otherwise uninjured mice, results in epilepsy associated with enhanced glutamatergic transmission, but without other obvious structural/functional abnormalities such as those affecting GABAergic inhibition (Chu et al [9]). A second line of evidence suggesting that excessive excitatory synapse formation alone might result in epileptogenesis comes from studies of normal synaptic development, and that which occurs after injury. Astrocytes secrete soluble proteins called thrombospondins (TSPs) that are key in enabling development of synapses between excitatory cells (Christoferson et al [8]). The α2δ-1 calcium channel auxiliary subunit is the receptor for both thrombospondins and the anticonvulsant drug gabapentin (GBP) (Luo et al [39], Eroglu et al [19]) and treatment of rodents with this drug early in development decreases excitatory synapse formation (Eroglu et al [19]). Further, we have recently recorded video/EEG seizures in implanted mice overexpressing the α2δ-1 receptor, as well as enhanced excitatory synaptic currents and epileptogenic activity in pyramidal neurons of in vitro slices from neocortex and hippocampus of these mice (L. Faria, B. Barres, Z.D. Luo and D.A. Prince, unpublished).
These results lead to the hypothesis that measures which interfere with development of new excitatory connections following injury could emerge as potential prophylactic approaches. We have tested the hypothesis that interference with the actions of thrombospondins at the α2δ-1 receptor would be antiepileptogenic. Field potential recordings were obtained from neocortical slices from undercut cortex of rats dosed with parenteral GBP beginning after the partial cortical isolation, using several protocols (H Li., K.D. Graber, D.A. Prince, unpublished). Results indicate that GBP can decrease the incidence of epileptogenesis in slices (Fig. 2A–C) and concurrently decrease the density of presumptive excitatory synapses in the undercut (Fig. 2D, E). There are also reductions in cell death, astrogliosis, and neurofilament staining in undercuts of GBP-treated animals (H. Li, Graber, K.D., Prince, D.A. unpublished). However, not all GBP treatment protocols were effective and further experiments are required to determine optimal dosage and timing of treatment.
Figure 2.
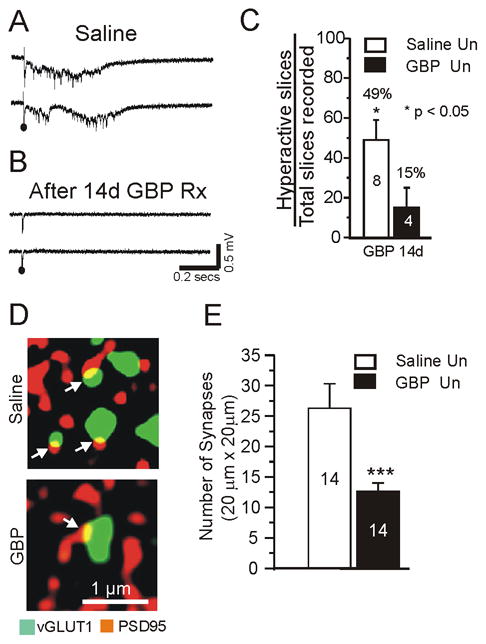
Chronic treatment with gabapentin (GBP) decreases evoked epileptiform discharges and excitatory synapse density in undercut cortex.
A: Representative field potential responses from layer V in undercut region of in vitro slice. Recordings from saline-treated rat 14 d after lesion placed. Just suprathreshold extracellular stimuli evoked epileptiform potentials. B: Same protocol as A, except rat had sc GBP infusion (10 mg/kg/d) × 14d following lesion. Recordings made on day 14 of GBP infusion. Stimuli failed to evoke epileptiform responses. C: Incidence of evoked epileptiform activity in undercut cortical slices in GBP and saline treated animals. Numbers in bars: number of animals. Average of 4.3 slices was assessed per animal. Data are expressed as percentage of slices with evoked epileptiform activity. *: p < 0.05. White bars: saline controls; black bars: GBP. D–E: Chronic GBP treatment (10 mg/kg/d sc × 7d) decreases density of excitatory synapses in undercut cortex. Data obtained from animals on 7th day of treatment following undercut.. D: Confocal images from sections of saline-treated and GBP-treated undercut rats. Double immunocytochemistry for vGlut1 (green, 1:500) and PSD95 (red, 1:200). Arrows: Sites of close apposition and presumptive synapses (yellow). E: Quantification of co-localization of pre and postsynaptic markers vGlut1 and PSD95 in undercut cortical sections from saline-treated (white bar: n =14 sections from 5 animals) and GBP-treated animals (black bar: n = 14 sections from 5 animals with 2–3 sections/rat). Three confocal images taken from each section. ***:p < 0.001. Data are expressed as mean ± SEM. Un: undercut.
The issue of adaptive vs maladaptive changes in connectivity is a key one that must be considered in approaching potential preventative treatments that decrease epileptogenic sprouting after injury. A number of reports implicate axonal sprouting and new connections as major adaptive plastic events in recovery of function after cortical lesions ((Dancause et al [15], Lee et al [35]). In recent experiments in a stroke model where middle cerebral artery occlusion induces expression of TSPs in astrocytes, TSP1-2 knock-out mice showed significant defects in the axonal sprouting and synaptic density compared to wild type animals, together with defects in functional recovery (Liauw et al [38]). The post-stroke incidence of epilepsy was not studied in these experiments; however the results, and those in the above references, provide a cautionary note.
5) Discussion and important unsolved issues
A number of pathophysiological processes occur in parallel after a serious epileptogenic brain injury. Although any one of these in isolation might not induce seizure activity, their effects on excitability would summate and epileptogenesis could result. Thus, a single prophylactic approach might be ineffective and a “prophylactic cocktail” might be required. Two key elements in developing epileptogenesis in a variety of injury models are reductions in functional GABAergic inhibition and enhanced new excitatory connectivity. However, the relationships between both excitatory and inhibitory circuit function and epileptiform activity are complex. GABAergic synchronization of cortical networks occurs in epileptogenic cortical lesions (D’Antuono et al [14], and in both acute (Marchionni and Maccaferri [41]) and genetic models of epileptiform discharge (Klaassen et al [32]). Also, depolarizing GABA responses due to altered chloride gradients occur in excitatory cells during development (Tyzio et al [64]) and after injury (Pathak et al [49], van den Pol et al [65]). These factors make the net effect of enhanced interneuronal output hard to predict. Also, decreases in excitatory circuit activities might result in decreased activation of interneurons (Sloviter [60], but see Zhang and Buckmaster [66]; Halabisky et al, [27]). As more becomes known about control of these processes during cortical development or following injury, it is possible that prophylactic therapies selectively affecting maladaptive processes might be applied. One important obstacle at this time is the unavailability of a reliable biological marker that would select for individuals who will go on to develop post-traumatic epilepsy, although it is clear that the incidence of posttraumatic epilepsy increases with the severity of brain injury (reviewed in Chen et al [7]). We know little about the temporal extent of critical periods in man when prophylactic intervention would be effective, or how to identify epileptogenesis “in progress”. The latent period may be very long between injury and expression of behavioral seizures (Salazar et al, 1985 [57]), however the critical period for intervention could closely follow injury (Benardo [3], Graber and Prince [26]). As noted above, another major problem is the difficulty in distinguishing between adaptive and maladaptive processes that follow brain injury. Finally, multiple offsetting potential effects by a given intervention are possible, such as both enhancement of excitatory connectivity together with “rescue” of inhibitory interneurons by BDNF agonists (e.g. Binder et al [4] vs Reibel et al [55]). The data presented in this issue suggest that significant progress is being made and that we may have potential prophylactic therapies available in the years to come, providing that some of the issues mentioned above are settled by detailed basic and clinical investigations.
Acknowledgments
Supported by grants NS 12151 and NS 39579 from the NINDS and a grant from Citizens United for Research in Epilepsy. The authors have no conflicts of interest to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Aronica E, Redeker S, Boer K, Spliet WG, van Rijen PC, Gorter JA, Troost D. Inhibitory networks in epilepsy-associated gangliogliomas and in the perilesional epileptic cortex. Epilepsy Res. 2007;74:33–44. doi: 10.1016/j.eplepsyres.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Bausch SB. Axonal sprouting of GABAergic interneurons in temporal lobe epilepsy. Epilepsy Behav. 2005;7:390–400. doi: 10.1016/j.yebeh.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 3.Benardo LS. Prevention of epilepsy after head trauma: do we need new drugs or a new approach? Epilepsia. 2003;44(Suppl 10):27–33. doi: 10.1046/j.1528-1157.44.s10.2.x. [DOI] [PubMed] [Google Scholar]
- 4.Binder DK, Croll SD, Gall CM, Scharfman HE. BDNF and epilepsy: too much of a good thing? Trends Neurosci. 2001;24:47–53. doi: 10.1016/s0166-2236(00)01682-9. [DOI] [PubMed] [Google Scholar]
- 5.Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABAA receptor subunit expression and function in temporal lobe epilepsy. Nature Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- 6.Buckmaster PS, Jongen-Relo AL. Highly specific neuron loss preserves lateral inhibitory circuits in the dentate gyrus of kainate-induced epileptic rats. J Neurosci. 1999;19:9519–9529. doi: 10.1523/JNEUROSCI.19-21-09519.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen JW, Ruff RL, Eavey R, Wasterlain CG. Posttraumatic epilepsy and treatment. J Rehabil Res Dev. 2009;46:685–696. doi: 10.1682/jrrd.2008.09.0130. [DOI] [PubMed] [Google Scholar]
- 8.Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 9.Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu Y, Parada I, Prince DA. Temporal and topographic alterations in expression of the alpha3 isoform of Na+, K(+)-ATPase in the rat freeze lesion model of microgyria and epileptogenesis. Neuroscience. 2009;162:339–348. doi: 10.1016/j.neuroscience.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cossart R, Bernard C, Ben Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Esclapez M, Bernard C, Ben Ari Y. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. NATURE NEUROSCIENCE. 2001;4:52–62. doi: 10.1038/82900. [DOI] [PubMed] [Google Scholar]
- 13.Cronin J, Dudek FE. Chronic seizures and collateral sprouting of dentate mossy fibers after kainic acid treatment in rats. Brain Res. 1988;474(1):181–184. doi: 10.1016/0006-8993(88)90681-6. [DOI] [PubMed] [Google Scholar]
- 14.D’Antuono M, Louvel J, Kohling R, Mattia D, Bernasconi A, Olivier A, Turak B, Devaux A, Pumain R, Avoli M. GABAA receptor-dependent synchronization leads to ictogenesis in the human dysplastic cortex. Brain. 2004;127:1626–1640. doi: 10.1093/brain/awh181. [DOI] [PubMed] [Google Scholar]
- 15.Dancause N, Barbay S, Frost SB, Plautz EJ, Chen D, Zoubina EV, Stowe AM, Nudo RJ. Extensive cortical rewiring after brain injury. J Neurosci. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeFelipe J. Chandelier cells and epilepsy. Brain. 1999;122:1807–1822. doi: 10.1093/brain/122.10.1807. [DOI] [PubMed] [Google Scholar]
- 17.Dichter MA. Posttraumatic epilepsy: the challenge of translating discoveries in the laboratory to pathways to a cure. Epilepsia. 2009;50(Suppl 2):41–45. doi: 10.1111/j.1528-1167.2008.02009.x. [DOI] [PubMed] [Google Scholar]
- 18.Echlin FA, Arnett V, Zoll J. Paroxysmal high voltage discharges from isolated and partially isolated human and animal cerebral cortex. Electroenceph clin Neurophysiol. 1952;4:147–164. doi: 10.1016/0013-4694(52)90004-7. [DOI] [PubMed] [Google Scholar]
- 19.Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esclapez M, Hirsch JC, Ben Ari Y, Bernard C. Newly formed excitatory pathways provide a substrate for hyperexcitability in experimental temporal lobe epilepsy. J Comp Neurol. 1999;408:449–460. doi: 10.1002/(sici)1096-9861(19990614)408:4<449::aid-cne1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 21.Faria LC, Prince DA. Presynaptic inhibitory terminals are functionally abnormal in a rat model of posttraumatic epilepsy. J Neurophysiol. 2010;104:280–290. doi: 10.1152/jn.00351.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to Cortical Injury: I. Methodology and Local Effects of Contusions in the Rat. Brain Res. 1981;211:67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- 23.Garga N, Lowenstein DH. Posttraumatic epilepsy: a major problem in desperate need of major advances. Epilepsy Curr. 2006;6:1–5. doi: 10.1111/j.1535-7511.2005.00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graber KD, Prince DA. Tetrodotoxin prevents posttraumatic epileptogenesis in rats. Ann Neurol. 1999;46:234–242. doi: 10.1002/1531-8249(199908)46:2<234::aid-ana13>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 25.Graber KD, Prince DA. Chronic partial cortical isolation. In: Pitkanen A, SPaMS, editors. Models of Seizures and Epilepsy. Elsevier Academic Press; San Diego: 2006. pp. 477–493. [Google Scholar]
- 26.Graber KD, Prince DA. A critical period for prevention of posttraumatic neocortical hyperexcitability in rats. Ann Neurol. 2004;55:860–870. doi: 10.1002/ana.20124. [DOI] [PubMed] [Google Scholar]
- 27.Halabisky B, Parada I, Buckmaster PS, Prince DA. Excitatory Input onto Hilar Somatostatin Interneurons is Increased in a Chronic Model of Epilepsy. J Neurophysiol. 2010;104:2214–2223. doi: 10.1152/jn.00147.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem. 2008;105:1–17. doi: 10.1111/j.1471-4159.2008.05237.x. [DOI] [PubMed] [Google Scholar]
- 29.Jin X, Hu H, Mathers PH, Agmon A. Brain-derived neurotrophic factor mediates activity-dependent dendritic growth in nonpyramidal neocortical interneurons in developing organotypic cultures. J Neurosci. 2003;23:5662–5673. doi: 10.1523/JNEUROSCI.23-13-05662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jin X, Huguenard JR, Prince DA. Reorganization of inhibitory synaptic circuits in rodent chronically injured epileptogenic neocortex. Cereb Cortex. 2010 doi: 10.1093/cercor/bhq181. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer v pyramidal neurons of chronically injured epileptogenic neocortex in rats. J Neurosci. 2006;26:4891–4900. doi: 10.1523/JNEUROSCI.4361-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kharatishvili I, Pitkanen A. Association of the severity of cortical damage with the occurrence of spontaneous seizures and hyperexcitability in an animal model of posttraumatic epilepsy. Epilepsy Res. 2010 doi: 10.1016/j.eplepsyres.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A. 2006;103:19152–19157. doi: 10.1073/pnas.0608215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohara K, Yasuda H, Huang Y, Adachi N, Sohya K, Tsumoto T. A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci. 2007;27:7234–7244. doi: 10.1523/JNEUROSCI.1943-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar SS, Buckmaster PS. Hyperexcitability, interneurons, and loss of GABAergic synapses in entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2006;26:4613–4623. doi: 10.1523/JNEUROSCI.0064-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JK, Kim JE, Sivula M, Strittmatter SM. Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J Neurosci. 2004;24:6209–6217. doi: 10.1523/JNEUROSCI.1643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Bandrowski AE, Prince DA. Cortical injury affects short-term plasticity of evoked excitatory synaptic currents. J Neurophysiol. 2005;93:146–156. doi: 10.1152/jn.00665.2004. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Prince DA. Synaptic activity in chronically injured, epileptogenic sensory-motor neocortex. J Neurophysiol. 2002;88:2–12. doi: 10.1152/jn.00507.2001. [DOI] [PubMed] [Google Scholar]
- 38.Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28:1722–1732. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- 39.Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI, Myers RR. Injury type-specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J Pharmacol Exp Ther. 2002;303:1199–1205. doi: 10.1124/jpet.102.041574. [DOI] [PubMed] [Google Scholar]
- 40.Magloczky Z, Freund TF. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005;28:334–340. doi: 10.1016/j.tins.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 41.Marchionni I, Maccaferri G. Quantitative dynamics and spatial profile of perisomatic GABAergic input during epileptiform synchronization in the CA1 hippocampus. J Physiol. 2009;587:5691–5708. doi: 10.1113/jphysiol.2009.179945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. II: white matter lesions of the neocortex. J Neuropathol Exp Neurol. 1997;56:219–235. doi: 10.1097/00005072-199703000-00001. [DOI] [PubMed] [Google Scholar]
- 43.Marty S, Berzaghi M, Berninger B. Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends Neurosci. 1997;20:198–202. doi: 10.1016/s0166-2236(96)01026-0. [DOI] [PubMed] [Google Scholar]
- 44.Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, Longo FM. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 2010;120:1774–1785. doi: 10.1172/JCI41356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCormick DA, Connors BW, Lighthall JW, Prince DA. Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J Neurophysiol. 1985;54:782–806. doi: 10.1152/jn.1985.54.4.782. [DOI] [PubMed] [Google Scholar]
- 46.McKinney RA, Debanne D, Gahwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of post-traumatic epilepsy. Nature Med. 1997;3:990–996. doi: 10.1038/nm0997-990. [DOI] [PubMed] [Google Scholar]
- 47.Molnar P, Nadler JV. Mossy fiber-granule cell synapses in the normal and epileptic rat dentate gyrus studied with minimal laser photostimulation. J Neurophysiol. 1999;82:1883–1894. doi: 10.1152/jn.1999.82.4.1883. [DOI] [PubMed] [Google Scholar]
- 48.Paradiso B, Marconi P, Zucchini S, Berto E, Binaschi A, Bozac A, Buzzi A, Mazzuferi M, Magri E, Mora GN, Rodi D, Su T, Volpi I, Zanetti L, Marzola A, Manservigi R, Fabene PF, Simonato M. Localized delivery of fibroblast growth factor-2 and brain-derived neurotrophic factor reduces spontaneous seizures in an epilepsy model. Proc Natl Acad Sci U S A. 2009;106:7191–7196. doi: 10.1073/pnas.0810710106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prince DA. Thirty years among cortical neurons. In: Gutnick MJ, Mody I, editors. The Cortical Neuron. Oxford Univ. Press; New York: 1995. pp. 3–23. [Google Scholar]
- 51.Prince DA, Parada I, Scalise K, Graber K, Jin X, Shen F. Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis. Epilepsia. 2009;50(Suppl 2):30–40. doi: 10.1111/j.1528-1167.2008.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prince DA, Tseng GF. Epileptogenesis in chronically injured cortex: in vitro studies. J Neurophysiol. 1993;69:1276–1291. doi: 10.1152/jn.1993.69.4.1276. [DOI] [PubMed] [Google Scholar]
- 53.Raghavendra R, Dhodda VVK, Song G, Bowen KK, Dempsey RJ. Traumatic brain injury-induced acute gene expression changes in rat cerebral cortex identified by GeneChip analysis. J Neurosci Res. 2003;71:208–219. doi: 10.1002/jnr.10486. [DOI] [PubMed] [Google Scholar]
- 54.Redecker C, Luhmann HJ, Hagemann G, Fritschy JM, Witte OW. Differential downregulation of GABAA receptor subunits in widespread brain regions in the freeze-lesion model of focal cortical malformations. J Neurosci. 2000;20:5045–5053. doi: 10.1523/JNEUROSCI.20-13-05045.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reibel S, Depaulis A, Larmet Y. BDNF and epilepsy--the bad could turn out to be good. Trends Neurosci. 2001;24:318–319. doi: 10.1016/s0166-2236(00)01869-5. [DOI] [PubMed] [Google Scholar]
- 56.Rosen GD, Jacobs KM, Prince DA. Effects of neonatal freeze lesions on expression of parvalbumin in rat neocortex [In Process Citation] Cereb Cortex. 1998;8:753–761. doi: 10.1093/cercor/8.8.753. [DOI] [PubMed] [Google Scholar]
- 57.Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology. 1985;35:1406–1414. doi: 10.1212/wnl.35.10.1406. [DOI] [PubMed] [Google Scholar]
- 58.Salin P, Tseng GF, Hoffman S, Parada I, Prince DA. Axonal sprouting in layer V pyramidal neurons of chronically injured cerebral cortex. J Neurosci. 1995;15:8234–8245. doi: 10.1523/JNEUROSCI.15-12-08234.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scharfman HE, Goodman JH, Sollas AL, Croll SD. Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor. Exp Neurol. 2002;174:201–214. doi: 10.1006/exnr.2002.7869. [DOI] [PubMed] [Google Scholar]
- 60.Sloviter RS, Zappone CA, Harvey BD, Bumanglag AV, Bender RA, Frotscher M. “Dormant basket cell” hypothesis revisited: relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J Comp Neurol. 2003;459:44–76. doi: 10.1002/cne.10630. [DOI] [PubMed] [Google Scholar]
- 61.Sperk G, Furtinger S, Schwarzer C, Pirker S. GABA and its receptors in epilepsy. Adv Exp Med Biol. 2004;548:92–103. doi: 10.1007/978-1-4757-6376-8_7. [DOI] [PubMed] [Google Scholar]
- 62.Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toth Z, Hollrigel GS, Gorcs T, Soltesz I. Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J Neurosci. 1997;17:8106–8117. doi: 10.1523/JNEUROSCI.17-21-08106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tyzio R, Minlebaev M, Rheims S, Ivanov A, Jorquera I, Holmes GL, Zilberter Y, Ben Ari Y, Khazipov R. Postnatal changes in somatic gamma-aminobutyric acid signalling in the rat hippocampus. Eur J Neurosci. 2008;27:2515–2528. doi: 10.1111/j.1460-9568.2008.06234.x. [DOI] [PubMed] [Google Scholar]
- 65.van den Pol AN, Obrietan K, Chen G. Excitatory actions of GABA after neuronal trauma. J Neurosci. 1996;16:4283–4292. doi: 10.1523/JNEUROSCI.16-13-04283.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang W, Buckmaster PS. Dysfunction of the dentate basket cell circuit in a rat model of temporal lobe epilepsy. J Neurosci. 2009;29:7846–7856. doi: 10.1523/JNEUROSCI.6199-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]