

Abstract
Mitochondrial DNA depletion syndromes (MDSs) are autosomal recessive diseases characterized by a severe decrease in mitochondrial DNA content leading to dysfunction of the affected organ. Autosomal recessive mutations in MPV17 have been identified in the hepatocerebral form of MDS. We describe the clinical features, biochemical and molecular results of a Saudi infant with a new mutation of MPV17 and compared the features to those of previously reported cases. We stress the importance of such rare cases particularly in countries with high consanguineous marriage rate.
Keywords: Case report, mitochondrial depletion, mitochondrial, MPV17
Profound reduction of the mitochondrial DNA (mtDNA) copy number is the molecular hallmark of the mtDNA depletion syndromes (MDSs), a heterogeneous group of severe mitochondrial disorders of infancy and childhood. These syndromes are inherited as autosomal recessive traits, while no mutations were identified in the mtDNA molecule, which suggests that MDS results from defects in nucleus-encoded factors, involved in mtDNA maintenance.[1]
Clinically, MDS are usually classified as 1 of 3 forms: a myopathic form associated with mutations in the thymidine kinase 2 (TK2) gene[2] or the p53-induced ribonucleotide reductase B subunit (RRM2B) gene,[3] an encephalomyopathic form associated with mutations in the ATP-dependent succinyl CoA synthase gene (SUCLA2)[4] or the GTP-dependent succinyl CoA synthase gene (SUCLG1),[5] and a hepatocerebral form associated with mutations in either the Twinkle (PEO1) gene,[6] the polymerase-gammaA (POLG1) gene,[7] the deoxyguanosine kinase (DGUOK) gene,[8] or, more recently, with mutations in the MPV17 gene.[9]
MPV17 gene encodes a small hydrophobic protein of unknown function, located in the mitochondrial inner membrane. One hypothesis is that MPV17 could play a role in the structural preservation of the inner mitochondrial membrane, which could in turn control mtDNA maintenance and stability.[10]
Mutations in the MPV17 gene were initially identified in 3 families with infantile hepatic mtDNA depletion.[9] The phenotype in these patients was characterized by recurrent episodes of severe hypoglycemia, associated with signs of hepatopathy evolving towards liver failure and cirrhosis, and growth retardation, during the first year of life.[9] Frequent, corn-starch-based meals were proven to be effective in preventing severe hypoglycemic crisis in some of these patients, with a trend towards the stabilization of the hepatic insufficiency. However, surviving patients do develop neurologic abnormalities, including both sensory–motor peripheral neuropathy and lesions of the central nervous system, particularly in the cerebellum and cerebral cortex. A founder MPV17 mutation associated with a longer-surviving, predominantly neurologic, was later identified in patients with Navajo neurohepatopathy (NNH; MIM 256810), an autosomal recessive disorder prevalent in the Navajo population of the southwestern United States.[11]
The MPV17 gene maps to chromosome 2p23.3 and contains 8 exons. To date, 20 different mutations in 29 patients have been reported. These include 9 mis-sense mutations, 2 nonsense mutations, 3 in-frame deletions, 2 splicing site mutations, 2 insertion mutations, 1 frame-shift deletion, and 1 large deletion encompassing the entire gene.[9,11–17]
We describe here the clinical features, and the biochemical and molecular results of an infant with a new mutation of MPV17 and compared his features with those of previously reported cases.
CASE REPORT
We report the case of a Saudi boy, the first and only offspring of healthy Saudi parents who happened to be first cousins. He was the product of a fullterm pregnancy, born by spontaneous vaginal delivery after an uncomplicated pregnancy. However, he was admitted on the second day of life for 22 days because of poor feeding and repeated vomiting, and was seen to have episodes of unexplained hypoglycemia. He continued to have poor feeding and occasional episodes of vomiting. He presented with failure to thrive and dehydration at the age of 12 months. The infant was also showing delay in attaining motor development milestones. The family history was insignificant for neurologic disease, liver disease, or premature deaths in infancy or childhood.
At the age of 12 months, the infant had no dysmorphic features, and was alert and appropriately attentive to different stimuli though slightly irritable. All his growth parameters were more than 3 SD below the mean for age and sex, his weight was more than –7 SD below the mean. Liver was only mildly enlarged but with sharp border and firm consistency. There was generalized muscle wasting but with no fasciculations or abnormal movements. He was markedly hypotonic; nevertheless, he could move his limbs freely and at least against gravity. Deep tendon reflexes could not be elicited. Hair was brittle and sparse and skin was scaly, pruritic; showing eczematous changes and reticular pigmentation. [Figures 1 a–b].
Figure 1.

(a-b) Image of the patient showing brittle and sparse hair; scaly and pruritic skin with eczematoid changes and lichenification
A panel of investigations was carried out under the hypothesis of a neurometabolic disorder. The following values were all normal: arterial blood gases, urea, creatinine, electrolytes, blood sugar, ammonia, creatine kinase, serum amylase, thyroid stimulating hormone, free T4, glucose, cortisol and adrenocorticotropic hormone.
Transaminases were mildly elevated [alanine aminotransferase 119 U/L (normal range 30–65 U/L), aspartate aminotransferase 214 U/L (normal range 15–37 U/L)], alkaline phosphatase level was 270 U/L (normal range 50–136 U/L)], plasma albumin 25 g/L (N 34–40); the APTT the Activated Partial Thromboplastin Time (APTT) was prolonged to 49.17 s (N, 26–4) and the international normalized ratio (INR) 1.57. Total bilirubin was 51.9 μmol/L (direct 44.0 μmol/L).
Quantitative plasma amino acids showed moderately elevated plasma tyrosine (309 μmol), while there was generalized increase in all aminoacids in urine. Succinylacetone was not increased in urine and results of very long chain fatty acids were unremarkable but serum lactate repeatedly showed moderate elevation, ranging between 2.9 and 9.7 mmol/L (N, 1.1–2.2).
Pelviabdominal renal ultrasound showed left kidney multiple stones and hydronephrotic changes.
Brain magnetic resonance imaging (MRI) showed increased signal intensity in T2-weighted images involving the cortical and subcortical white matter and extending to the subcortical U fibers with fair hyperintensity involving the cerebellar white matter and hili of the dentate nuclei, a picture suggestive of leukodystrophy with centripetal progression [Figures 2 a–b].
Figure 2.
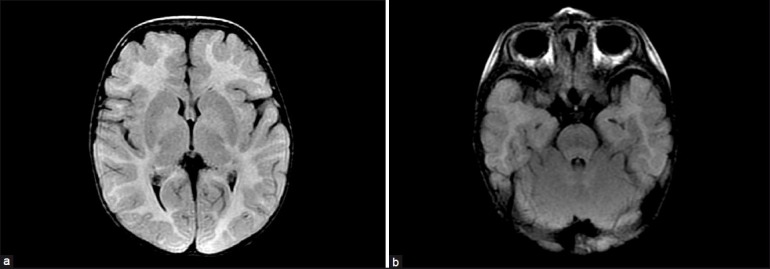
(a-b) MRI brain of the patient (axial FLAIR) showing increased signal intensity in T2 weighted images- involving the cortical and subcortical white matter
Quantitative real-time polymerase chain reaction[18] carried out on DNA extracted from muscle and liver of the patient revealed normal mtDNA amount in the former and less than 10% in the latter tissue, compared with age-matched control tissues.
For MPV17 sequence analysis, the 7 coding exons and the exon-intron boundaries of the gene were amplified from muscle DNA and sequenced directly as previously described.[9] A homozygous transversion G to T at position +1 (IVS3+1G>T) was found in intron 3. The mutation destroys the donor splice site of exon 2, therefore producing an aberrant mRNA and protein.
The boy continued to have vomiting and poor weight gain and had another episode of hypoglycemia that a gastrostomy tube was placed. However, 1 month after admission, he developed marked ascites but with no further increase in the transaminases. Doppler ultrasound scan showed normal flow of both hepatic and portal veins. Echocardiogram examination was normal. The patient's condition deteriorated further and he died in a matter of few weeks due to complications of liver failure.
DISCUSSION
Liver complications are typically considered a late feature of multisystem mitochondrial disorder in which neuromuscular disease is prominent. By contrast, in MPV17-associated hepatocerebral MDS, liver involvement appears early in the course of the disease[12] and is seen in nearly all reported cases. Similar to ours, in many other reported cases liver dysfunction progressed to liver failure, leading the patient to death in infancy or early childhood, if liver transplantation was not performed. However, MPV17 mutations that are compatible with longer survival have been reported. For instance compound heterozygosities p.G94R and p.P98L,[17] or p.S170F and c.451insC[15] were found in patients surviving for as long as 30 and 47 months, respectively, without liver transplantation. The longest survival has been associated with the p.R50Q mutation, associated with NNH and identified in an Italian family, with cases surviving to their late teens.[11]
Expectedly, failure to thrive is a common manifestation and it was the main cause of parental concern when our patient first presented to our hospital. Only 3 cases in the literature were reported to have a normal growth pattern.[9,17] Common neurologic manifestations of the MPV17-associated MDS include microcephaly, developmental delay, muscle weakness, and hypotonia, all seen in our case. Other neurologic manifestations reported in the literature include seizures,[12,14] cerebrovascular infarct,[12] and dystonia.[13] By contrast, no neurologic manifestations were reported in only 2 cases.[12,17] Unfortunately, electrophysiologic studies were not done to our patient so we cannot comment on peripheral neuropathy reported in several cases,[9,11,14] whereas leukoencephalopathic lesions similar to those in our patient have also been detected by brain MRI in previous cases.[9,11,13]
Lactic acidosis and hypoglycemia are possibly the most frequent signs at onset, and our patient was no exception. Prominent vomiting as seen in our patient in a 4-month-old infant, and gastroesophageal reflux in another 2 patients were reported by El-Hattab et al.[17] The generalized aminoaciduria may be due to proximal tubulopathy as reported in other mitochondrial diseases.[19] Only 1 patient, an Iraqi girl with a homozygous nonsense mutation p.W120X[13] was reported to develop nephrolithiasis as the present case. The underlying etiology is not clear; it is possible that frequent episodes of severe dehydration and metabolic acidosis may play a role in its development. Hair abnormalities and skin changes described in our patient are among the broad spectrum of presenting symptoms of mitochondrial diseases. These are often reported to be puzzling and unexpected manifestations of mitochondrial disease, which we are describing in this patient, although we have no clear explanation.[20]
The treatment of acute liver failure and progressive liver disease in mitochondrial hepatopathies remains unsatisfactory. Present treatments involve the use of various vitamins, cofactors, and respiratory substrates, none of which have proven effective. Supportive treatments may include infusion of sodium bicarbonate for acute metabolic acidosis, and intravenous administration of glucose.[21,22] Frequent feeding every 3h may help slow down the disease progression and may be lifesaving in case of potentially fatal hypoglycemia.[17] Liver transplantation is a treatment option for end-stage liver disease associated with MDS, but the role of liver transplantation in mitochondrial hepatopathy is controversial. In some patients neurologic involvement can be non-recognizable or ensue later than the initial presentation of liver failure. Therefore, the absence of extrahepatic features of mitochondrial disease at the time of liver transplantation does not warrant a good outcome. When one is faced with a mitochondrial disorder, clinical deterioration due to the stress of transplantation, infections, and bleeding should also be emphasized and anticipated if transplantation is considered.[12]
CONCLUSION
In conclusion, the prevalence of MDS disorders is probably underestimated due to lack of awareness of such rare diseases, their clinical heterogeneity, and the difficulties in establishing the diagnosis. MDS cases are rarely reported from the Middle East. We aim to alert the clinicians, especially in areas with a high rate of consanguineous marriage as is the case in our region,[23] to such rare medical disorders. The identification of the genetic defect makes prenatal diagnosis possible in at-risk families and is particularly more important in cases with seizures where administration of valproate may have a deleterious effect.
ACKNOWLEDGMENTS
We express our appreciation to Dr. Ali Alasmari, MD, pediatric metabolic consultant at KFMC, for referring the case to pediatric neurology service.
This work was supported by Telethon grants GGP07019 and GPP10005 to Massimo Zeviani.
Footnotes
Source of Support: This work was supported by Telethon grants GGP07019 and GPP10005 to Massimo Zeviani
Conflict of Interest: None declared.
REFERENCES
- 1.Alberio S, Mineri R, Tiranti V, Zeviani M. Depletion of mtDNA: syndromes and genes. Mitochondrion. 2007;7:6–12. doi: 10.1016/j.mito.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342–4. doi: 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- 3.Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–80. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 4.Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, et al. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76:1081–6. doi: 10.1086/430843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostergaard E, Christensen E, Kristensen E, Mogensen B, Duno M, Shoubridge EA, et al. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007;81:383–7. doi: 10.1086/519222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goffart S, Cooper HM, Tyynismaa H, Wanrooij S, Suomalainen A, Spelbrink JN. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum Mol Genet. 2009;18:328–40. doi: 10.1093/hmg/ddn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naviaux RK, Nguyen KV. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706–12. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- 8.Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–41. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 9.Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D’Adamo P, Calvo S, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–5. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 10.Dallabona C, Marsano RM, Arzuffi P, Ghezzi D, Mancini P, Zeviani M, et al. Sym1, the yeast ortholog of the MPV17 human disease protein, is a stress-induced bioenergetic and morphogenetic mitochondrial modulator. Hum Mol Genet. 2010;19:1098–107. doi: 10.1093/hmg/ddp581. [DOI] [PubMed] [Google Scholar]
- 11.Karadimas CL, Vu TH, Holve SA, Chronopoulou P, Quinzii C, Johnsen SD, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet. 2006;79:544–8. doi: 10.1086/506913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong LJ, Brunetti-Pierri N, Zhang Q, Yazigi N, Bove KE, Dahms BB, et al. Mutations in the MPV17 gene are responsible for rapidly progressive liver failure in infancy. Hepatology. 2007;46:1218–27. doi: 10.1002/hep.21799. [DOI] [PubMed] [Google Scholar]
- 13.Spinazzola A, Santer R, Akman OH, Tsiakas K, Schaefer H, Ding X, et al. Hepatocerebral form of mitochondrial DNA depletion syndrome: novel MPV17 mutations. Arch Neurol. 2008;65:1108–13. doi: 10.1001/archneur.65.8.1108. [DOI] [PubMed] [Google Scholar]
- 14.Navarro-Sastre A, Martín-Hernández E, Campos Y, Quintana E, Medina E, de Las Heras RS, et al. Lethal hepatopathy and leukodystrophy caused by a novel mutation in MPV17 gene: Description of an alternative MPV17 spliced form. Mol Genet Metab. 2008;94:234–9. doi: 10.1016/j.ymgme.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Kaji S, Murayama K, Nagata I, Nagasaka H, Takayanagi M, Ohtake A, et al. Fluctuating liver functions in siblings with MPV17 mutations and possible improvement associated with dietary and pharmaceutical treatments targeting respiratory chain complex II. Mol Genet Metab. 2009;97:292–6. doi: 10.1016/j.ymgme.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 16.Parini R, Furlan F, Notarangelo L, Spinazzola A, Uziel G, Strisciuglio P, et al. Glucose metabolism and diet-based prevention of liver dysfunction in MPV17 mutant patients. J Hepatol. 2009;50:215–21. doi: 10.1016/j.jhep.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 17.El-Hattab AW, Li FY, Schmitt E, Zhang S, Craigen WJ, Wong LJ. MPV17-associated hepatocerebral mitochondrial DNA depletion syndrome: New patients and novel mutations. Mol Genet Metab. 2010;99:300–8. doi: 10.1016/j.ymgme.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 18.He L, Chinnery PF, Durham SE, Blakely EL, Wardell TM, Borthwick GM, et al. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002;30:e68. doi: 10.1093/nar/gnf067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bornstein B, Area E, Flanigan KM, Ganesh J, Jayakar P, Swoboda KJ, et al. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul Disord. 2008;18:453–9. doi: 10.1016/j.nmd.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodemer C, Rötig A, Rustin P, Cormier V, Niaudet P, Saudubray JM, et al. Hair and skin disorders as signs of mitochondrial disease. Pediatrics. 1999;103:428–33. doi: 10.1542/peds.103.2.428. [DOI] [PubMed] [Google Scholar]
- 21.Sokol RJ, Treem WR. Mitochondria and childhood liver diseases. J Pediatr Gastroenterol Nutr. 1999;28:4–16. doi: 10.1097/00005176-199901000-00005. [DOI] [PubMed] [Google Scholar]
- 22.Gillis LA, Sokol RJ. Gastrointestinal manifestations of mitochondrial disease. (v).Gastroenterol Clin North Am. 2003;32:789–817. doi: 10.1016/s0889-8553(03)00052-9. [DOI] [PubMed] [Google Scholar]
- 23.el-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Consanguinity among the Saudi Arabian population. J Med Genet. 1995;32:623–6. doi: 10.1136/jmg.32.8.623. [DOI] [PMC free article] [PubMed] [Google Scholar]