

Abstract
The androgen insensitivity (testicular feminization) syndrome was described by Morris in phenotypic females with 46XY karyotype, presenting with primary amenorrhea, adequate breast development, and absent or scanty pubic or axillary hair. Gonads consist usually of seminiferous tubules without spermatogenesis. These patients have a 5–10% risk of developing germ cell tumors, usually after the complete development of secondary female sexual characteristics. We hereby report a case considered as a female with married life of 15 years, who was operated for severe abdominal pain. Phenotype characters were that of female. Microscopic examination of the tumor from the abdomen revealed germinoma and yolk sac tumor with adjacent seminiferous tubules. Karyotyping showed 46XY. Final diagnosis of malignant mixed germ cell tumor in androgen insensitivity syndrome was made. Surveillance may be the most appropriate option when these conditions are initially diagnosed in adulthood to prevent development of germ cell tumors.
KEY WORDS: Androgen insensitivity, germ cell tumors, karyotype
INTRODUCTION
Complete androgen insensitivity syndrome (AIS), previously called testicular feminization,[1] is a rare X-linked condition with an incidence of between 1:13,158 and 1:64,200live births.[2] An affected woman has a 46XY karyotype that leads to normal differentiation of testes in utero, but a defect in the gene coding for the androgen receptor (AR) results in complete insensitivity to circulating androgens, resulting in phenotypic female development.[3] Psychosexual orientation is, in every respect, female. There is, however, no uterus and only a partially formed vagina, and pubic and axillary hair is scant or absent.[1,3] We hereby report a case considered as a female with malignant mixed germ cell tumor in an AIS.
CASE REPORT
A middle-aged person of 35 years, said to be female, presented with history of sudden and severe acute abdominal pain. She had normal onset of puberty with normal breast development. She had not attained menarche and never had menstrual cycles. She was infertile with married life of 15 years. There was no history for inguinal hernia operation. Her family history was not relevant. She had a normal body habitus with well-developed breasts and female external genitalia. She had sparse axillary and pubic hairs [Figure 1]. Routine investigations were within normal limits. A pelvic ultrasound examination showed mass in the left iliac fossa, no uterus, and shortened vagina. Clinically, a diagnosis of ?ovarian torsion syndrome was made.
Figure 1.
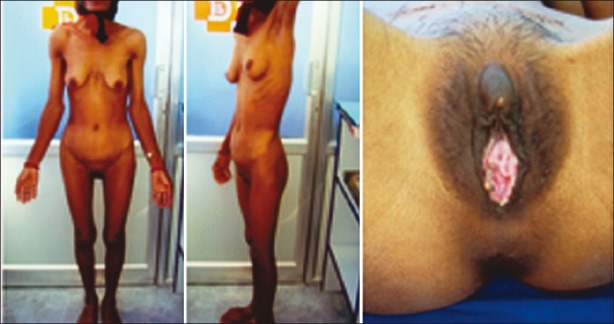
Phenotypic features of the patient with well-developed breasts and sparse axillary and pubic hairs. The external genitalia are that of female, with enlarged clitoris
On laparotomy, a tumor was seen in the left iliac fossa measuring 15cm × 12cm × 10cm. Uterus was not found. Right iliac fossa showed a fibrosed area (testis-like structure) measuring 3cm × 2cm × 1 cm. Metastatic deposits were seen in the peritoneal cavity. Gross examination of the tumor surface was irregular and showed nodules of varying sizes with small areas of tumor rupture in the capsule. Cut section showed variegated appearance [Figure 2]. Histopathology sections from the tumor area showed features of mixed germ cell tumor comprising germinoma [Figure 3] and yolk sac tumor [Figure 4], with the latter being the predominant component. The surrounding area showed testicular tissue comprising hyalinized seminiferous tubules. Ovarian tissue and stroma was not seen.
Figure 2.
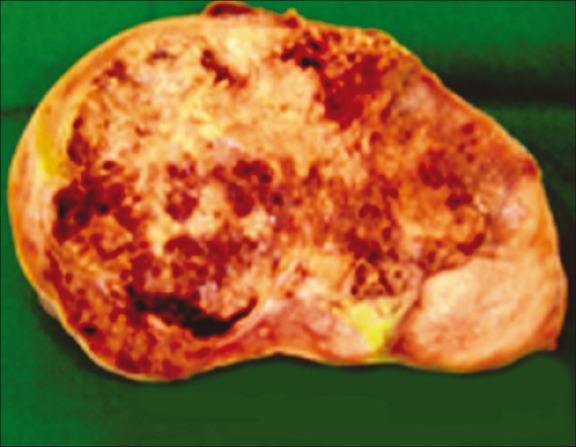
Cut section of tumor showing variegated appearance
Figure 3.
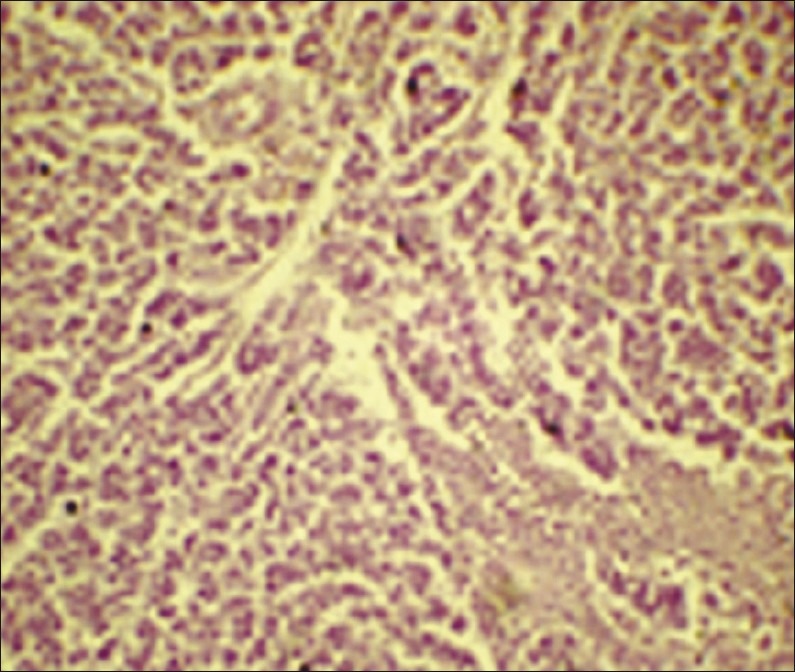
Photomicrograph of tumor tissue showing seminoma (H and E, χ10 magnification)
Figure 4.
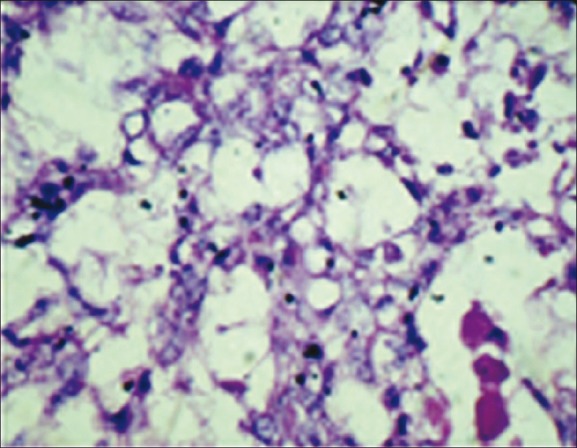
Photomicrograph of tumor tissue showing yolk sac tumor (H and E, χ40 magnification)
Buccal smear was negative for Barr body. Karyotyping revealed 46XY [Figure 5]. Final diagnosis of malignant mixed germ cell tumor in an AIS was made. Now, the patient is responding to chemotherapy.
Figure 5.
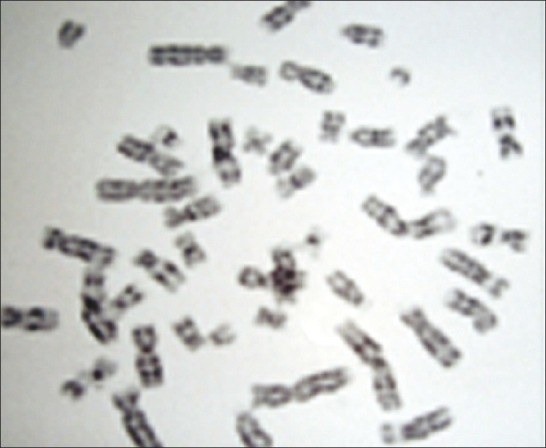
Karyotyping showing 46XY pattern
DISCUSSION
In 1953, the American gynecologist, John Morris, described the anatomical, histological, and clinical features of AIS, based on 82 cases collected from over nearly 150 years of the medical literature. He argued against disclosure of genotype and concern about the apparent high prevalence of psychiatric morbidity, casualty, and perhaps suicide, among people with intersex conditions.[1]
Development of the male genitalia is largely controlled by cells in the urogenital mesenchyme that express androgen receptors (AR).[4] AR action is a conditio sine qua non, and germline mutations of the AR gene produce a spectrum of developmental abnormalities in 46XY individuals, ranging from infertility or mild hypospadias to complete feminization, having a common denominator of loss of reproductive viability, which are collectively referred to as AIS.[5] Three main clinical phenotypes in humans define AIS: complete, partial and minimal androgen insensitivity (CAIS, PAIS, and MAIS, respectively), and they range from complete lack of virilization of the internal and external genitalia (CAIS), to intermediate virilization (PAIS), to apparently normal virilization in infertile males (MAIS).[6] In general, the degree of genital ambiguity correlates with the level of compromise of AR function: 46XY individuals with AR-inactivating mutations are completely feminized despite high levels of serum testosterone.[5,6]
Affected individuals have normal differentiation of testes in utero. They present with primary amenorrhea, adequate breast development, and absent or scanty pubic or axillary hair. The vagina is partially formed, may be shorter than normal, and ends blindly. There is, however, no uterus. The testes also show interstitial Leydig cell hyperplasia and ovarian-like stroma. Gonads consist usually of seminiferous tubules without spermatogenesis. Electron microscopic studies have shown that the gonadal ultrastructure resembles that of fetal testis with primitive germ cells and Sertoli cells lining the seminiferous tubules.[1,2]
AIS accounts for approximately 10% of cases of primary amenorrhea, ranking third after gonadal dysgenesis and congenital absence of the vagina. Patients with AIS have a 5–10% risk of developing germ cell tumor, usually after the complete development of secondary female sexual characteristics. The AIS with mixed malignant testicular germ cell tumor is very rare. Testicular biopsies in at-risk patients with intersex may aid in identifying intratubular germ cell neoplasia, seminoma, mixed germ cell tumors, and other testicular tumors at the earliest.[7] For this reason, the at least one testis should be removed after puberty and supplemental hormone therapy given.[8]
The specific issue of revealing the genotype in CAIS at diagnosis remained contentious until the 1990s. Shah, in 1992, argued that: “The disclosure of genotype is irrelevant to care and may be confusing to patient and family.”[8] Advances in information technology have exponentially increased the chance of a woman finding out her diagnosis. It is now established practice to disclose the genotype of CAIS at diagnosis. Knowing her diagnosis may also provide options that optimize her welfare.[9] The harm associated with disclosure, conversely, is likely to be short term and minimized by a sensitive and skillful approach.[10] Despite the clinical dogma that “AIS is not treatable,” some sporadic PAIS and MAIS patients respond to endocrine management, consisting of pharmacologic doses of androgens.[9,10]
Surveillance may be the most appropriate option when these conditions are initially diagnosed in adulthood. Germ cell tumors occurring in the setting of androgen resistance are managed similarly to the testicular cancers, and attention should be given to genetic counseling for families of complete AIS patients.[9,10]
In conclusion, testicular biopsy is warranted as soon as the syndrome is diagnosed and the findings of in situ seminoma should indicate immediate orchidectomy. At least one testis should be left until the puberty to result in secondary sexual characteristics.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Morris JM. The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol. 1953;65:1192–211. doi: 10.1016/0002-9378(53)90359-7. [DOI] [PubMed] [Google Scholar]
- 2.Blackless M, Charuvastra A, Derryck A, Fausto-Sterling A, Lauzanne K, Lee E. How sexually dimorphic are we? Review and synthesis. Am J Hum Biol. 2000;12:151–66. doi: 10.1002/(SICI)1520-6300(200003/04)12:2<151::AID-AJHB1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.Weiner JS, Teague JL, Roth DR, Gonzales ET, Jr, Lamb DJ. Molecular biology and function of the androgen receptor in genital development. J Urol. 1997;157:1377–86. [PubMed] [Google Scholar]
- 4.Takeda H, Mizuno T, Lasnitzki I. Autoradiographic studies of androgen-binding sites in the rat urogenital sinus and postnatal prostate. J Endocrinol. 1985;104:87–92. doi: 10.1677/joe.0.1040087. [DOI] [PubMed] [Google Scholar]
- 5.Holterhus PM, Brüggenwirth HT, Hiort O, Kleinkauf-Houcken A, Kruse K, Sinnecker GH, et al. Mosaicism due to a somatic mutation of the androgen receptor gene determines phenotype in androgen insensitivity syndrome. J Clin Endocrinol Metab. 1997;82:3584–9. doi: 10.1210/jcem.82.11.4375. [DOI] [PubMed] [Google Scholar]
- 6.Brown TR, Maes M, Rothwell SW, Migeon CJ. Human complete androgen insensitivity with normal dihydrotestosterone receptor binding capacity in cultured genital skin fibroblasts: Evidence for a qualitative abnormality of the receptor. J Clin Endocrinol Metab. 1982;55:61–9. doi: 10.1210/jcem-55-1-61. [DOI] [PubMed] [Google Scholar]
- 7.Chen CP, Chern SR, Wang TY, Wang W, Wang KL, Jeng CJ. Androgen receptor gene mutations in 46 XY females with germ cell tumors. Hum Reprod. 1999;14:664–70. doi: 10.1093/humrep/14.3.664. [DOI] [PubMed] [Google Scholar]
- 8.Shah R, Woolley MM, Costin G. testicular feminization: The androgen insensitivity syndrome. J Pediatr Surg. 1992;27:757–60. doi: 10.1016/s0022-3468(05)80110-1. [DOI] [PubMed] [Google Scholar]
- 9.Weidemann W, Peters B, Romalo G, Spindler KD, Schweikert HU. Response to androgen treatment in a patient with partial androgen insensitivity and a mutation in the deoxyribonucleic acid-binding domain of the androgen receptor. J Clin Endocrinol Metab. 1998;83:1173–6. doi: 10.1210/jcem.83.4.4704. [DOI] [PubMed] [Google Scholar]
- 10.Kemp BD. Sex, lies and androgen insensitivity syndrome. CMAJ. 1996;154:1829. author reply 1833. [PMC free article] [PubMed] [Google Scholar]