

Abstract
Aims
Despite increased understanding of the fundamental biology regulating cardiomyocyte hypertrophy and heart failure, it has been challenging to find novel chemical or genetic modifiers of these pathways. Traditional cell-based methods do not model the complexity of an intact cardiovascular system and mammalian models are not readily adaptable to chemical or genetic screens. Our objective was to create an in vivo model suitable for chemical and genetic screens for hypertrophy and heart failure modifiers
Methods and results
Using the developing zebrafish, we established that the cardiac natriuretic peptide genes (nppa and nppb), known markers of cardiomyocyte hypertrophy and heart failure, were induced in the embryonic heart by pathological cardiac stimuli. This pathological induction was distinct from the developmental regulation of these genes. We created a luciferase-based transgenic reporter line that accurately modelled the pathological induction patterns of the zebrafish nppb gene. Utilizing this reporter line, we were able to show remarkable conservation of pharmacological responses between the larval zebrafish heart and adult mammalian models.
Conclusion
By performing a focused screen of chemical agents, we were able to show a distinct response of a genetic model of hypertrophic cardiomyopathy to the histone deacetylase inhibitor, Trichostatin A, and the mitogen-activated protein kinase kinase 1/2 inhibitor, U0126. We believe this in vivo reporter line will offer a unique approach to the identification of novel chemical or genetic regulators of myocardial hypertrophy and heart failure.
Keywords: Natriuretic peptides, Hypertrophy, Heart development, Heart failure, Hypertrophic cardiomyopathy
1. Introduction
The ability of cardiomyocytes to respond to stress is critical to the preservation of myocardial function in health and disease. If the heart is unable to adapt efficiently to haemodynamic changes, myocardial function declines and ultimately leads to the development of heart failure. Despite aggressive treatment with existing therapies, the prevalence of congestive heart failure (CHF) continues to increase.1 A number of successful therapeutics have been developed using traditional target-driven screens in cell-based assays. These agents have proved effective in the later stages of the CHF syndrome, but the identification of effective agents operating earlier in the cascade, and thus capable of preventing CHF, has proved difficult. Even early in its pathogenesis, CHF is a multiorgan, multisystem disorder, and modelling this complexity is simply not feasible using existing in vitro techniques. Conversely, performing a large-scale screen for novel therapeutics using intact animal models is prohibitive. Recently, the emergence of the larval zebrafish as a tool for disease modelling and phenotype-driven small molecule screens has provided a viable in vivo alternative to traditional target-based screening techniques.2
A signalling pathway that is consistently up-regulated in both myocardial hypertrophy and CHF in humans is the cardiac natriuretic peptide axis, with two major secreted ligands; atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP). The induction of these genes serves as a robust marker of cardiomyocyte stress in mammalian models of cardiovascular disease. However, these genes are also highly expressed during cardiac development. Initial attempts at creating murine NPPA reporter lines using minimal promoter constructs were only able to recapitulate the developmental induction of these genes, but failed to mimic pathological responses.3 More recently using BAC transgenesis, a mouse model was developed that captures both the developmental and pathological expression patterns of the NPPA gene.4
The zebrafish also exhibits dynamic expression of the cardiac natriuretic peptides during cardiogenesis, but it is unclear if the pathological responses are also conserved.5 We tested whether the regulation required for pathological natriuretic peptide responses is present during the early stages of cardiogenesis. We found that expression of the natriuretic peptides is increased in the embryonic zebrafish heart in response to traditional pathological stimuli and this response is distinct from the developmental induction of these genes. In an effort to create an animal model readily adaptable to screens for heart failure modifiers, we generated transgenic zebrafish reporter lines that model the pathological transcriptional responses of the cardiac natriuretic peptides. We provide evidence that this reporter line accurately reproduces the pathological expression patterns of the endogenous natriuretic peptides and their responses to a wide range of compounds that are either clinically utilized or in preclinical testing for CHF. In addition, using a zebrafish model of a human hypertrophic cardiomyopathy mutation, we demonstrate a distinct response to the histone deactylase (HDAC) inhibitor, Trichostatin A (TSA), and mitogen-activated protein kinase kinase (MEK) inhibitor U0126. These reporter lines will be useful to efficiently screen for both chemical and genetic modifiers of myocardial hypertrophy and heart failure.
2. Methods
The investigation conforms to the ‘Guide for the Care and Use of Laboratory Animals’ published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2.1. Whole-mount in situ analysis
nppa and nppb cDNA constructs were subcloned into pDonr221 or pCR8 (Invitrogen) and then subcloned into pCSDest (kindly provided by Dr N. Lawson, University of Massachusetts). A pCS2 + eGFP plasmid (kindly provided by Dr W. Chen, Vanderbilt University) was utilized to create the YFP antisense probe. For additional details, please see Supplementary material online, Methods.
2.2. Morpholino injections
Morpholinos (Gene Tools, LLC, Philomath, OR, USA) were resuspended in 1x Danieu's solution and 500 µM (2 nL drop size, 7.6 ng) of morpholino was injected into fertilized eggs from TuAB fish at the single cell stage. Tnnt2 splice targeting morpholino (tnnt2sp MO) and tnnt2 translation targeting morpholino (tnnt2 MO) were both utilized.6,7
2.3. Quantitative RT–PCR
Quantitative RT–PCR was performed using RNA extracted from whole embryos using Trizol reagent (Invitrogen) and reverse transcribed with the QuantiTect Reverse Transcription Kit (Qiagen). Gene-specific primers and Sybr Green reagent (Applied Biosystems) were utilized to perform the PCR portion of the experiment. The 2−ΔΔCt method was used to normalize the gene of interest to the endogenous housekeeping gene rpl13a and determine the fold change relative to control. For additional details, please see Supplementary material online, Methods.
2.4. Reporter construction
Natriuretic peptide reporter constructs were created using the zebrafish nppa and nppb promoters placed upstream of either YFP or Firefly luciferase. Stable transgenics were created using Tol2 transposase-mediated transgenesis. These lines were outbred for at least two generations. For additional details, please see Supplementary material online, Methods.
2.5. Whole-mount immunocytochemistry
Control or tnnt2 morpholino-injected embryos were fixed in phosphate buffered saline (PBS) with 4% paraformaldehyde overnight at 4°C. The embryos were permeabilized with 0.5% Triton-X 100 in PBS (PBS-T) and then blocked with 2 mg/mL BSA, 2% goat serum, 1% dimethyl sulfoxide (DMSO) in PBS-T. Embryos were then incubated overnight at 4°C with a monoclonal antibody directed against Firefly luciferase protein (L2164, Sigma). The embryos were then washed with PBS-T and incubated with the secondary antibody (A10667, Invitrogen) for 3 h at 25°C. They were washed again in PBS-T and then imaged with a fluorescent dissecting microscope equipped with a digital camera.
2.6. Luciferase assay
Morpholino injected or uninjected nppa:F-Luc or nppb:F-Luc embryos were placed into a 96-well microtiter plate with buffered embryo water (E3) and the chemical that was being tested. The total volume of the embryos and the E3 was 100 μL. These plates were then incubated at 28°C. At a specified time point (72 hpf, 96 hpf, etc.), the plate was removed from the incubator and 50 μL of long half-life firefly luciferase reagent (Perkin Elmer, Steady-Glo) was added to the well. The plate was then incubated in the dark for 60 min. After 60 min, the plate was read in a high-sensitivity luminescence plate reader (PE Elmer Victor X, or Biotek Synergy HL-1). The luminescence values obtained between different readers were calibrated to use the same scale.
2.7. Drug exposure
All chemicals were stored in DMSO stocks and diluted into working concentrations for specific experiments. The final DMSO concentration was kept below 1% for all experiments. All chemicals were applied at 24 hpf unless otherwise noted. The following concentrations were utilized: Isoproterenol (ISO) 250 µM, Carvedilol 25 µM, TSA 25 nM, Milrinone (MIL) 10 µM, PD98059 10 µM, U0126 10 µM, SB203580 10 µM, SB202190 10 µM, SP600125 500 nM, LY294002 10 µM. If additional drug concentrations are utilized, it is noted from the figure.
2.8. Statistics
To compare two continuous variables, a Student's t-test was used. Data are expressed as mean ± SEM, unless a box plot is utilized. The box plot data show sample minimum (lowest point), lower quartile (bottom of shaded box), median (central black square), upper quartile (top of shaded box), and largest observation (highest point).
3. Results
3.1. The cardiac natriuretic peptides are induced secondary to pathological stimuli during zebrafish development
There is a robust expression of both natriuretic peptides during zebrafish cardiogenesis, but the pathological responses of the natriuretic peptide system during cardiogenesis are unknown. We performed whole mount in situ analysis of nppa and nppb expression in the intact embryo and found that there was a qualitative increase in the expression of both nppa and nppb in response to two distinct genetic stressors of cardiomyocyte physiology: abnormal splicing of cardiac troponin T or homozygous mutation of the gene gridlock (grl,hey2) (Figure 1A). The tnnt2sp MO embryos are injected with a morpholino that causes missplicing in the zebrafish cardiac troponin T (tnnt2) gene directly paralleling a known human mutation that causes hypertrophic cardiomyopathy.6 The gridlock−/− embryos are genetic mutant embryos that are homozygous, a point mutation in the hey2 gene that causes luminal obstruction of the proximal aorta in the developing embryo.8 Notably, these embryos did not exhibit any extra-cardiac expression of the natriuretic peptides (Figure 1B).
Figure 1.

The cardiac natriuretic peptides are induced secondary to pathological stimuli in zebrafish embryos. (A) Whole mount in situ of nppa and nppb in control, tnnt2sp morphants, and gridlock (grl) mutant embryos at 96 hpf shows a qualitative increase in expression of nppa and nppb in zebrafish morphants and mutants secondary to pathological stimuli. Arrowhead points to zebrafish cardiac region. (B) This increased expression was limited to the heart in the zebrafish embryo. (C) Quantitative RT–PCR in the same treatment groups confirms a robust induction of both nppa and nppb, with a stronger induction of nppb compared with nppa. Asterisks indicate fold induction relative to control; hash symbols indicate fold induction relative to grl ± (D). A time course experiment using quantitative RT–PCR shows that the pathological expression of nppb (red line) is distinct from the developmental expression (blue line) with a clear separation by 96 hpf. All data expressed as mean ± SEM. Scale bar = 200 µm.
We performed quantitative RT–PCR to confirm the whole mount in situ results (Figure 1C). We found that at 96 hpf, the tnnt2sp MO embryos exhibited a 2.6-fold induction of nppa and a 5.2-fold induction of nppb when compared with control embryos. The grl homozygote mutants exhibited a 2.3-fold induction of nppa and a 12.32-fold induction of nppb compared with grl heterozygote mutants (no phenotype). Since the cardiac natriuretic peptides are also induced during cardiogenesis, we sought to determine whether the pathological induction was an exaggeration of the developmental expression pattern or if unique regulators were controlling the pathological induction. Therefore, we measured the induction of nppb from 24 hpf until 96 hpf in both control and mutant embryos and we found that both groups had an early induction of nppb expression at 48 hpf, with the morphant embryos exhibiting a higher level of expression (Figure 1D) (control 25 ± 7.8 vs. TNNT2sp 35 ± 4.3-fold induction, P = N.S.). However, by 96 hpf, the control embryos had a dramatic decrease of nppb expression. In contrast, the tnnt2sp MO embryos revealed persistent elevation of nppb expression (control 4 ± 1.5 vs. tnnt2sp 39 ± 12-fold induction, P < 0.01). Thus, both control and mutant embryos responded to developmental stimuli at 48 hpf, but by 96 hpf, elevated nppb expression was maintained only in the context of the pathological stimuli in the tnnt2sp morphant embryos. These data suggest that cardiac hypertrophic stimuli do not appear to perturb the initial developmental induction of the natriuretic peptides, but markedly dysregulate subsequent control of natriuretic peptide expression and implicate discrete regulation in the control of developmental and pathological induction.
3.2. Transgenic natriuretic peptide reporter lines phenocopy endogenous gene expression patterns
The small size and translucency of the zebrafish embryo make it well suited for the real-time observation of fluorescent protein expression. We developed stable transgenic zebrafish lines that expressed fluorescent proteins under the control of the nppa and nppb minimal promoters to serve as tools in the investigation of natriuretic peptide regulation during normal development and in the face of pathological stimuli. We first created transgenic lines using minimal promoters for the nppa or nppb gene controlling the expression of yellow fluorescent protein (Figure 2). For the nppa gene, we tested multiple different upstream genomic fragments before identifying fragments that recapitulated endogenous gene expression. We found that both 1 and 2 kb upstream genomic fragments had similar but not identical expression patterns compared with endogenous nppa expression (Figure 2A; Supplementary material online, Figure S1). Specifically, there was less atrial expression of YFP in the transgenic reporter lines compared with the endogenous nppa expression. Attempts at using larger genomic fragments (>2 kb) for nppa were not successful. This may reflect the location of the zebrafish nppb gene 2.8 kb upstream of the nppa gene with inclusion of this gene in the promoter fragment preventing efficient translation of the YFP.
Figure 2.
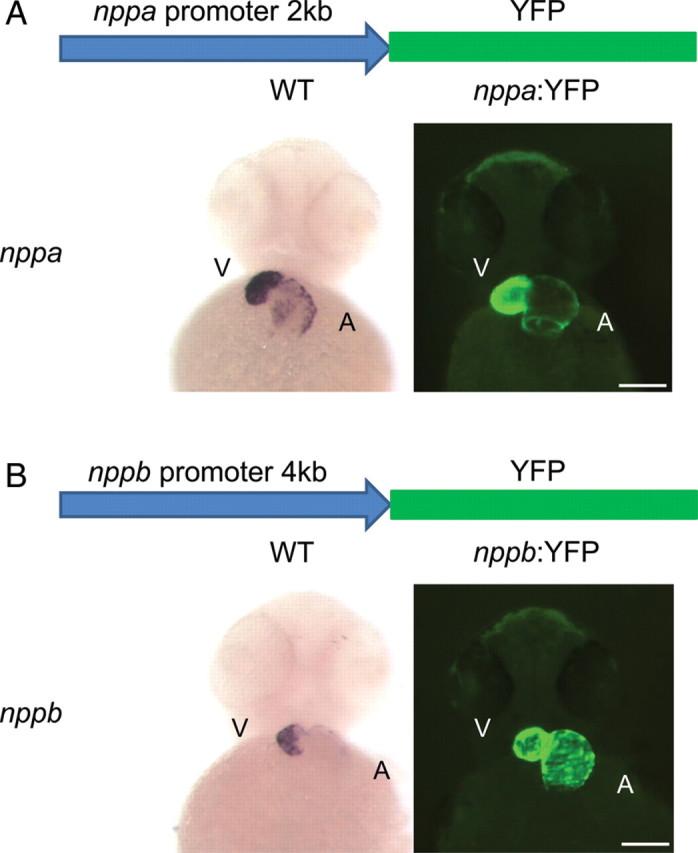
The cardiac natriuretic peptide minimal promoters can model the developmental expression pattern of nppa and nppb. (A) The nppa:YFP transgenic (right) has a similar pattern of expression compared with the endogenous nppa expression (left). (B) The nppb:YFP transgenic (right) also has localized cardiac expression of YFP but appears to have stronger atrial expression of YFP compared with the endogenous nppb expression pattern (left). V, ventricle; A, atria. Scale bar = 100 µm.
Next, we created a transgenic line with the nppb promoter controlling the expression of YFP. Again, we tested numerous genomic fragments and found that a 4 kb fragment most closely recapitulated the expression of the endogenous nppb gene (Figure 2B; Supplementary material online, Figure S1). The nppb:YFP transgenic appeared to have stronger YFP fluorescence in the atria compared with the endogenous nppb expression. However, YFP RNA expression more closely matched the endogenous gene expression. This could reflect the ≈12 h half-life of YFP protein.
We compared the expression of these fluorescent reporters over the course of cardiac development in the zebrafish to fully characterize the expression of the reporters and to detect any non-cardiac expression (Figure 3). Both reporters were initially visible at 24 hpf paralleling the onset of endogenous gene expression. Over the next 48 hpf as the embryonic heart underwent looping and further growth, the expression of the reporters remained quite high, although the intensity qualitatively decreased at 72 hpf in the nppb:YFP transgenics (Figure 3A). Importantly, there was no evidence of expression of the fluorescent reporters in non-cardiac regions of the embryo (Figure 3B).
Figure 3.
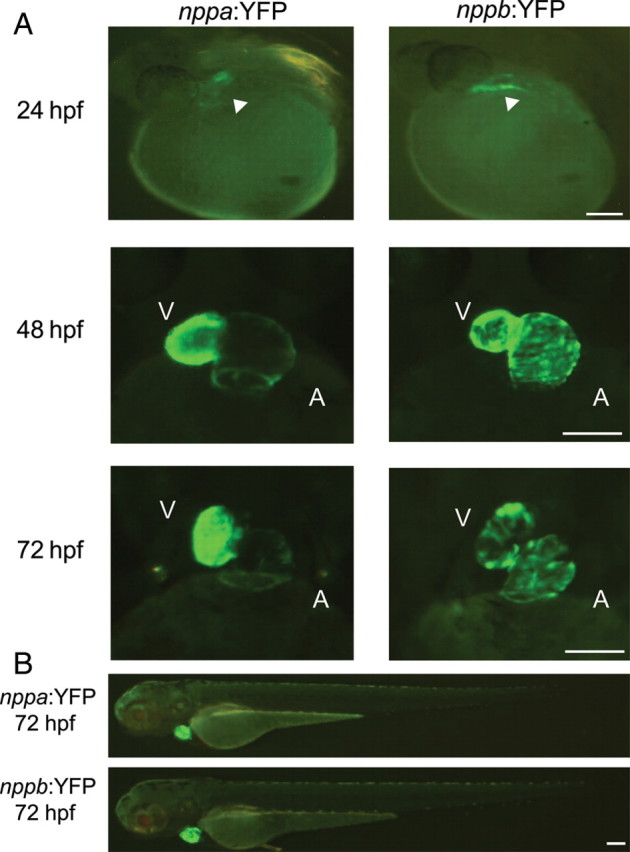
The cardiac natriuretic peptide fluorescent reporters have similar patterns of expression throughout cardiac development. (A) The earliest time point of YFP expression was 24 hpf at the heart tube stage, by 48 hpf there was distinct expression in both atria and ventricles, which persisted through 72 hpf. (B) At all time points studied, the YFP expression was limited to the heart and there was no non-cardiac expression detected. V, ventricle; A, atria. Scale bar = 100 µm.
3.3. Development of a luciferase-based whole embryo heart failure model
Fluorescent reporters are useful to observe the developmental expression patterns of the natriuretic peptides and to determine the optimal promoters for our reporter constructs. However, one of our primary goals in studying the natriuretic peptide expression pattern in the embryonic zebrafish was to develop an in vivo heart failure model that could be readily adapted for small molecule and genetic screens. Semi-quantitative measurement of fluorescent expression in the zebrafish embryo does not offer the rigorous assay characteristics that are necessary for high-throughput screens. Therefore, we coupled the validated promoters for both nppa and nppb with Firefly luciferase.
Once we had created stable transgenic zebrafish lines expressing Firefly luciferase downstream of the nppa or nppb promoter, we then exposed these embryos to either a chemical (ISO) or genetic pathological stimuli (tnnt2 morpholino). We found that fish transgenic for the nppa promoter driving luciferase did not recapitulate the pathological induction pattern of the native nppa gene (Figure 4B) (nppa:F-Luc, DMSO: 89 ± 7, Iso: 114 ± 10, tnnt2 MO:66 ± 7 average luciferase units/embryo, P < 0.04). In contrast, the nppb promoter luciferase lines increased luciferase expression in a manner paralleling that of the endogenous nppb gene (tnnt2 MO stimuli—nppb mRNA 5.2-fold increase, nppb:F-Luc 5.8-fold increase; ISO stimuli—nppb mRNA 3.1-fold increase, nppb:F-Luc 2.6-fold increase, Supplementary material online, Figure S2). This reporter line appeared to accurately model the pathological induction pattern of the native gene (nppb:F-Luc, DMSO: 207 ± 9, Iso: 546 ± 45, tnnt2 MO: 1197 ± 245 average luciferase units/embryo, P < 0.001).
Figure 4.

Development of a luciferase-based reporter for rapid quantitative assessment of cardiac natriuretic peptide expression. (A) The nppa or nppb minimal promoter was placed upstream of the Firefly luciferase construct (F-Luc). (B) The nppa:F-Luc transgenic line did not consistently increase expression of the luciferase transgene secondary to pathological stimuli. However, the nppb:F-Luc transgenic line showed a robust induction secondary to the same pathological stimuli and was quite similar to the endogenous nppb gene expression. *P < 0.04 compared with nppa:F:Luc/DMSO, **P < 0.001 compared with nppb:F:Luc/DMSO, All data expressed as mean ± SEM. (C) Whole mount immunocytochemistry using a Firefly luciferase monoclonal antibody and Alexa 488 secondary antibody qualitatively confirmed that pathological stimuli increase expression of the Firelfy luciferase protein. In addition, there was no evidence of non-cardiac expression of Firefly luciferase protein. V, ventricle; A, atria. Scale bar = 100 µm.
In order to verify that the luciferase protein expression in the nppb:F-Luc transgenic was restricted to the heart, we performed immunocytochemistry using a monoclonal antibody specific for Firefly luciferase protein (Figure 4C). At baseline, we could barely detect expression of the Firefly luciferase protein. However, with the tnnt2 morpholino stimulus, there was a large increase of Firefly luciferase protein expression that was restricted to the heart.
Next, we performed a time course experiment comparing the control (DMSO) and ISO-treated nppb:F-Luc embryos exposed from 24 to 120 hpf (Supplementary material online, Figure S3A). Similar to the endogenous nppb gene expression, we found a clear divergence between control embryos and embryos exposed to ISO (48 hpf DMSO 4 ± 0.6 vs. ISO 5 ± 1, P = N.S., 72 hpf DMSO 6 ± 0.5 vs. ISO 19 ± 1.5, P < 0.001, 96 hpf DMSO 7 ± 0.5 vs. ISO 19 ± 1.7, P < 0.001, 120 hpf DMSO 3 ± 0.3 vs. ISO 16 ± 1.0-fold units, P < 0.001). However, the nppb:F-Luc embryos reached peak luciferase expression at a later time point (96 hpf) compared with the endogenous nppb gene expression (48 hpf).
We then shifted the interval of exposure of the nppb:F-Luc embryos to a later time point in order to further separate the developmental induction pattern from the pathological induction pattern (Supplementary material online, Figure S3B). We found that older embryos had a lower absolute level of luminescence when treated with either DMSO or ISO. However, since the control embryo luminescence levels were lower, the ISO-treated embryos had a greater fold induction when treated from 96 to 168 hpf (6.0-fold induction) compared with embryos treated from 24 to 96 hpf (2.8-fold induction) (24–96 hpf exposure: DMSO 205 ± 16, ISO 582 ± 54, 96–168 hpf: DMSO 62 ± 4, ISO 377 ± 40 average luciferase units/embryo, P < 0.001 for both). These data suggest that this transgenic line accurately models multiple aspects of native nppb gene expression and may be useful as a model to study pathways regulating cardiac hypertrophy and heart failure.
3.4. Development of a method to rapidly measure luminescence in the intact zebrafish embryo
In order to maximize the utility of our transgenic luciferase line, we needed to develop an assay that would be readily adaptable to large-scale screening. Luciferase measurement in zebrafish after manual homogenization is feasible for small numbers of embryos, but is not readily adaptable to a 96 or 384 well microtiter plate format. Therefore, we incorporated the use of a long half-life luciferase reagent that contained a lysis buffer (Steady-Glo, Perkin Elmer). By incubating the intact zebrafish embryo in the luciferase reagent, we were able to obtain consistent luminescence measurements even from single embryos using a standard 96-well microtiter plate and a high-sensitivity luminescence reader equipped with an agitation function (Supplementary material online, Figure S4).
3.5. Validation of pathological natriuretic peptide responses using known agents
After we developed methods to consistently measure luciferase expression in high-throughput, we began to test our nppb:F-Luc reporter lines responses to agents commonly used in human heart failure (Figure 5A). We first tested the non-selective beta-receptor agonist ISO, which mimics the elevated catecholaminergic state seen in late stage human heart failure patients. The application of this agent from 24 to 96 hpf consistently induced the reporter gene expression. Next, we co-administered the ISO with carvedilol, a non-selective beta and alpha-adrenergic blocker. This agent is one of the most effective agents in the treatment of human heart failure. When the agents were co-administered, the induction of the reporter expression was blocked. These data suggest that even at this stage of development, the embryonic zebrafish heart has an intact adrenergic signalling pathway. Next, we utilized the class I and II histone deacetylase (HDAC) inhibitor TSA, which has been shown to be effective in reducing or preventing myocardial hypertrophy in pre-clinical animal models.9 TSA has not been shown to interfere with the stimulation of the beta-adrenergic receptors, but instead modulates the expression of numerous genes by inhibition of HDACs. We found that TSA could also block the ISO induction of the nppb reporter gene (control 198 ± 6, ISO 421 ± 20, ISO/Carvedilol 213 ± 8, ISO/TSA 250 ± 12 average luciferase units/embryo, Ctl vs. ISO, P < 0.0001, ISO vs. ISO/Carvedilol, or ISO/TSA, P < 0.0001, n = 20–33 embryos/group). The final agent that we tested was milrinone (MIL), a type 3 phosphodiesterase inhibitor that increases cardiomyocyte cyclic adenosine monophosphate (cAMP) levels and has a positive ionotropic response in mammals. When this agent is administered alone to the zebrafish embryo, there is no significant change in nppb reporter expression. However, when ISO, an agent that increases intracellular cAMP levels is coadministered, there is a synergistic effect between both agents in the induction of the reporter gene. Notably, the addition of carvedilol can block this synergistic induction (MIL 219 ± 8, MIL/ISO 640 ± 52, MIL/ISO/Carvedilol 193 ± 13 average luciferase units/embryo, MIL vs. MIL/ISO, P < 0.0001, MIL/ISO vs. MIL/ISO/Carvedilol, P < 0.0001, n = 11–12 embryos/group). These data suggest that a broad selection of adult mammalian pharmacological responses is conserved in the larval zebrafish.
Figure 5.

Validation of the nppb:F-Luciferase transgenic reveals novel responses to chemical agents. (A) When ISO was applied to the embryos, there was a robust induction of the nppb:F-Luciferase transgene which could be blocked by either carvedilol (alpha- and beta-adrenergic antagonist) or TSA (HDAC inhibitor), *P < 0.0001 compared with DMSO, **P < 0.0001 compared with ISO alone. The combination of MIL, a phosphodiesterase type 3 inhibitor, with isoproterenol showed a significantly larger induction of the luciferase transgene. This induction could be completely blocked by administration of carvedilol, #P < 0.0001 compared with ISO alone, ##P < 0.0001 compared with MIL/ISO. (B) A targeted kinase inhibitor screen shows inhibition of reporter induction. **P < 0.04 compared with ISO alone.
To further validate our reporter line, we performed a targeted screen with a variety of kinase inhibitors (Figure 5B). We found significant reductions in reporter activity with U0126 (Mek1/2 inhibitor), SB202190 (p38 MAPK inhibitor), and SP600125 (JNK inhibitor) (Control 206 ± 9, ISO 581 ± 32, ISO/U0126 237 ± 38, ISO/SB202190 368 ± 43, and ISO/SP600125 439 ± 25 average luciferase units/embryo, ISO vs. ISO/U0126, P < 0.0001, ISO vs. ISO/SB202190, P < 0.01, ISO vs. ISO/SP600125, P < 0.04, n = 6–15 embryos/group). Upon further testing, we found that U0126 was the most effective agent at reducing ISO-induced activation of the nppb:F-Luc reporter (Figure 6A). At higher doses, SB202190 and SP600125 did not show any increased efficacy in reducing reporter activation (Supplementary material online, Figure S5).
Figure 6.

HDAC inhibition and MEK1/2 inhibition are able to blunt reporter gene induction in a zebrafish model of a hypertrophic cardiomyopathy TNNT2 mutation. (A) Dual MEK1/2 inhibition with U0126 was effective at reducing reporter gene induction. In contrast, PD98059 (MEK1 selective inhibitor) or U0124 (inactive structural analogue of U0126) were both ineffective in reducing reporter gene expression. *P < 0.01 compared with ISO alone (B) TSA and U0126 were both effective in blunting the reporter gene induction secondary to a cardiac Troponin T splice targeting morpholino (tnnt2sp). However, carvedilol and MIL were ineffective in altering the reporter gene induction. *P < 0.01 compared with tnnt2sp alone.
Finally, we tested a genetic form of hypertrophic signalling, tnnt2sp morpholino (Figure 6B). We found that the tnnt2sp morpholino caused a significant induction of the reporter gene, and co-administration of MIL showed no ability to block this induction. Interestingly, we also found that carvedilol was ineffective in reducing the induction of the reporter gene in the tnnt2sp morphant embryos. In contrast, TSA and U0126 were both effective in reducing the induction of the reporter by at least 60% (control 230 ± 10, tnnt2sp MO 1366 ± 110, tnnt2sp MO/MIL 1473 ± 235, tnnt2sp MO/Carvedilol 1705 ± 508, tnnt2sp MO/TSA 528 ± 61, and tnnt2sp MO/U0126 462 ± 78 average luciferase units/embryo, tnnt2sp MO vs. tnnt2sp MO/TSA, P < 0.01 or tnnt2sp MO/U0126, P < 0.01, n = 7–15 embryos/group).
4. Discussion
We have established that the genetic pathways controlling the pathological induction of the cardiac natriuretic peptides are intact during cardiogenesis in the zebrafish embryo and are distinct from the pathways that regulate the developmental induction of these peptides. We have generated transgenic reporter lines that express fluorescent peptides and firefly luciferase under the control of cardiac natriuretic peptides promoters. Utilizing these reporter lines, we were able to develop a rapid and reliable method to screen for genetic and pharmacological modifiers of cardiac natriuretic peptide expression, a highly conserved biomarker of cardiomyocyte dysfunction and hypertrophy. We have validated this in vivo system across a range of pathological and genetic stimuli. Finally, a targeted screen identified the HDAC inhibitor, TSA, and the MEK1/2 inhibitor, U0126, as potent inhibitors of the pathological reporter induction in a zebrafish model of hypertrophic cardiomyopathy.
There are two distinct settings in which cardiac natriuretic peptide production is induced, in cardiac development and in pathological states where myocardial function is impaired, such as CHF or myocardial hypertrophy. Many of the upstream transcriptional regulators of natriuretic peptide expression during heart development have been identified, and most of these transcription factors (Nkx2.5, Gata4, SRF, Tbx5) are also important for cardiomyocyte differentiation. These transcription factors not only control the developmental induction of the cardiac natriuretic peptides, but also are important in restricting the expression of NPPA and NPPB to the embryonic atria and ventricle.10
In contrast, the transcriptional control of NPPA under pathological conditions is not well defined. This is highlighted by the fact that multiple transgenic reporter mouse lines with a range of NPPA promoters failed to respond to pathological stimuli (aortic banding) despite accurate recapitulation of the endogenous developmental expression patterns.3 The only mouse transgenic NPPA reporter line that has a preserved response to pathological stimuli was constructed using BAC transgenesis incorporating a segment of chromosomal DNA that spans from −141 kb upstream to +58 kb downstream of the NPPA gene.4 In contrast, the developmental and pathological regulation of NPPB expression can be accurately reproduced with much less extensive promoter constructs.11 Importantly, we were able to demonstrate robust developmental and pathological induction of nppb in the zebrafish using relatively limited promoter fragments. In order to build a truly quantitative reporter system, we created luciferase-based reporter lines, which are able to rapidly measure single embryo luciferase expression at a scale similar to cell-based luciferase assays. Our method, unlike those previously published, does not require the manual homogenization of zebrafish embryos.12 This modification allows this assay to be readily adapted to primary genetic or chemical screens.
Modelling complex disease states such as CHF is challenging using in vitro systems, which are limited in their representation. Further, currently available cardiomyocyte cell lines either share properties of both cardiac and skeletal muscle (H9C2) or are derived from a cardiac tumour (HL-1).13,14 Using the intact zebrafish embryo with a functioning multi-chambered heart, intact endothelium, and active circulation is a better model of complex disease states such as CHF. The incorporation of a quantitative natriuretic peptide luciferase reporter enables the rapid assessment of a gene network that is conserved in most forms of myocardial hypertrophy and heart failure across multiple vertebrate species.15
We were able to validate our reporter lines against several classes of cardioactive agents. Those agents that increased luciferase expression in the nppb reporter line (Beta-receptor agonists and Phosphodiesterase Type 3 inhibitors) have also been shown to increase mortality in human heart failure patients.16,17 In contrast, the agents that decreased expression of NPPB luciferase reporter line were the beta-receptor antagonist, Carvedilol, the class I and II HDAC inhibitor TSA, and the MEK1/2 inhibitor U0126. Carvedilol is a well-established agent administered to human heart failure patients and has been shown to significantly improve mortality in these patients.18 However, TSA has only recently emerged as a promising agent to inhibit and reverse the development of left ventricular hypertrophy in murine animal models.9 Likewise, U0126 has been shown in cell culture to be effective in blunting the induction of hypertrophic signalling pathways, but has limited in vivo efficacy data.19 Of note, we also tested PD98059 a selective inhibitor of MEK1. This compound was ineffective in reducing pathological induction of the reporter gene. These results suggest that both MEK1 and MEK2 may be contributing to the pathological signalling cascade, or that MEK2 may be the primary regulator. Mouse models genetically targeting the Raf/MEK/ERK signalling cascade have shown divergent results with regards to the role of this signalling cascade in the pathological hypertrophic response.20 However, recent data from a mouse model with a Raf1 (L613V) mutation that causes Noonan syndrome in humans suggest that pharmacolgical inhibition of MEK may be a viable strategy in preventing myocardial hypertrophy.21
Finally, to further illustrate the utility of our reporter line, we injected a morpholino that causes missplicing in cardiac Troponin T paralleling a human mutation that causes hypertrophic cardiomyopathy.6 The reporter embryos showed a robust induction of the luciferase transgene, but in contrast to the ISO-treated embryos, the TNNT2sp morphants did not uniformly respond to carvedilol. However, there was a significant inhibitory response to the HDAC inhibitor TSA and MEK 1/2 inhibitor U0126. This is the first experimental evidence from an in vivo model that sarcomeric gene mutations associated with hypertrophic cardiomyopathy may respond to HDAC inhibition or MEK1/2 inhibition. Currently, the pharmacological agents available to treat this condition are limited and are unable to induce regression of established ventricular hypertrophy.22
The cardiac natriuretic peptide axis is a well-established marker of pathological cardiac signalling. Our zebrafish natriuretic peptide reporter lines enable the in vivo identification of genetic and chemical modifiers of this signalling pathway. We used this line to identify two compounds, TSA and U0126, which could potentially be useful in modifying the pathological response to sarcomeric gene mutations that cause hypertrophic cardiomyopathy. Screening chemical libraries with this reporter line will provide the ability to rapidly assess the in vivo effectiveness of agents to modulate heart failure and myocardial hypertrophic signalling pathways.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by NIH T32HL007208 and Vanderbilt Physician Scientist Development Program award to J.R.B. Massachusetts General Hospital Hypertrophic Cardiomyopathy Center and British Heart Foundation grants to C.A.M.
Supplementary Material
Acknowledgements
We would like to acknowledge all members of the Becker, Peterson, and MacRae laboratory for helpful discussion and comments.
Conflict of interest: None declared.
References
- 1.Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, et al. Heart disease and stroke statistics–2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–486. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- 2.Schlueter PJ, Peterson RT. Systematizing serendipity for cardiovascular drug discovery. Circulation. 2009;120:255–263. doi: 10.1161/CIRCULATIONAHA.108.824177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knowlton KU, Rockman HA, Itani M, Vovan A, Seidman CE, Chien KR. Divergent pathways mediate the induction of ANF transgenes in neonatal and hypertrophic ventricular myocardium. J Clin Invest. 1995;96:1311–1318. doi: 10.1172/JCI118166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horsthuis T, Houweling AC, Habets PE, de Lange FJ, el Azzouzi H, Clout DE, et al. Distinct regulation of developmental and heart disease-induced atrial natriuretic factor expression by two separate distal sequences. Circ Res. 2008;102:849–859. doi: 10.1161/CIRCRESAHA.107.170571. [DOI] [PubMed] [Google Scholar]
- 5.Auman HJ, Coleman H, Riley HE, Olale F, Tsai HJ, Yelon D. Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol. 2007;5:e53. doi: 10.1371/journal.pbio.0050053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker JR, Deo RC, Werdich AA, Panakova D, Coy S, MacRae CA. Human cardiomyopathy mutations induce myocyte hyperplasia and activate hypertrophic pathways during cardiogenesis in zebrafish. Dis Model Mech. 2011;4:400–410. doi: 10.1242/dmm.006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sehnert AJ, Huq A, Weinstein BM, Walker C, Fishman M, Stainier DY. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat Genet. 2002;31:106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 8.Zhong TP, Rosenberg M, Mohideen MA, Weinstein B, Fishman MC. Gridlock, an HLH gene required for assembly of the aorta in zebrafish. Science. 2000;287:1820–1824. doi: 10.1126/science.287.5459.1820. [DOI] [PubMed] [Google Scholar]
- 9.Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation. 2006;113:2579–2588. doi: 10.1161/CIRCULATIONAHA.106.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Small EM, Krieg PA. Transgenic analysis of the atrialnatriuretic factor (ANF) promoter: Nkx2–5 and GATA-4 binding sites are required for atrial specific expression of ANF. Dev Biol. 2003;261:116–131. doi: 10.1016/s0012-1606(03)00306-3. [DOI] [PubMed] [Google Scholar]
- 11.LaPointe MC. Molecular regulation of the brain natriuretic peptide gene. Peptides. 2005;26:944–956. doi: 10.1016/j.peptides.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 12.Shin JT, Priest JR, Ovcharenko I, Ronco A, Moore RK, Burns CG, et al. Human-zebrafish non-coding conserved elements act in vivo to regulate transcription. Nucleic Acids Res. 2005;33:5437–5445. doi: 10.1093/nar/gki853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimes BW, Brandt BL. Properties of a clonal muscle cell line from rat heart. Exp Cell Res. 1976;98:367–381. doi: 10.1016/0014-4827(76)90447-x. [DOI] [PubMed] [Google Scholar]
- 15.Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006;27:47–72. doi: 10.1210/er.2005-0014. [DOI] [PubMed] [Google Scholar]
- 16.Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325:1468–1475. doi: 10.1056/NEJM199111213252103. [DOI] [PubMed] [Google Scholar]
- 17.Krell MJ, Kline EM, Bates ER, Hodgson JM, Dilworth LR, Laufer N, et al. Intermittent, ambulatory dobutamine infusions in patients with severe congestive heart failure. Am Heart J. 1986;112:787–791. doi: 10.1016/0002-8703(86)90475-8. [DOI] [PubMed] [Google Scholar]
- 18.Poole-Wilson PA, Swedberg K, Cleland JG, Di Lenarda A, Hanrath P, Komajda M, et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362:7–13. doi: 10.1016/S0140-6736(03)13800-7. [DOI] [PubMed] [Google Scholar]
- 19.Yue TL, Gu JL, Wang C, Reith AD, Lee JC, Mirabile RC, et al. Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:37895–37901. doi: 10.1074/jbc.M007037200. [DOI] [PubMed] [Google Scholar]
- 20.Lorenz K, Schmitt JP, Vidal M, Lohse MJ. Cardiac hypertrophy: targeting Raf/MEK/ERK1/2-signaling. Int J Biochem Cell Biol. 2009;41:2351–2355. doi: 10.1016/j.biocel.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Simpson J, Hong JH, Kim KH, Thavarajah NK, Backx PH, et al. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest. 2011;121:1009–1025. doi: 10.1172/JCI44929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashrafian H, McKenna WJ, Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res. 2011;109:86–96. doi: 10.1161/CIRCRESAHA.111.242974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.