

SUMMARY
In Caulobacter crescentus, the ClpXP protease degrades several crucial cell-cycle regulators, including the phosphodiesterase PdeA. Degradation of PdeA requires the response regulator CpdR and signals a morphological transition in concert with initiation of DNA replication. Here, we report the structure of a Per-Arnt-Sim (PAS) domain of PdeA and show that it is necessary for CpdR-dependent degradation in vivo and in vitro. CpdR acts as an adaptor, tethering the amino-terminal PAS domain to ClpXP and promoting recognition of the weak carboxyl-terminal degron of PdeA, a combination that ensures processive proteolysis. We identify sites on the PAS domain needed for CpdR recognition and find that one subunit of the PdeA dimer can be delivered to ClpXP by its partner. Finally, we show that improper stabilization of PdeA in vivo alters cellular behavior. These results introduce an adaptor/substrate pair for ClpXP and reveal broad diversity in adaptor-mediated proteolysis.
INTRODUCTION
Protein degradation ensures the complete and immediate removal of undesired proteins; however, because proteolysis is irreversible, target selection must be carefully regulated. In bacteria, regulated proteolysis is particularly important when cells must drastically change their proteome composition without the benefit of dilution through cell division. For example, the model bacteria Caulobacter crescentus undergoes an obligate morphological transition during cell-cycle progression. Motile swarmer cells must differentiate into nonmotile stalked cells before they begin replicating their genomes (equivalent to the G1-S transition). Rapid and controlled proteolysis of several central regulators is critical for both the developmental transition and replication initiation (Jenal, 2009; Curtis and Brun, 2010). This type of regulated degradation often uses AAA+ proteases, oligomeric enzymes that unfold and degrade specific substrates by transforming the chemical energy of ATP hydrolysis into mechanical force (Sauer and Baker, 2011). The best understood of these enzymes in Caulobacter is the essential ClpXP protease, which degrades a number of cell-cycle-regulated substrates including the master regulator CtrA (Domian et al., 1997; Jenal and Fuchs, 1998; Chien et al., 2007b).
Cyclic di-GMP (cdG) is a bacterial second messenger that signals developmental fate and fluctuates during cell-cycle progression (Hengge, 2009; Paul et al., 2008). In Caulobacter, cyclic changes in cdG regulate proteolysis of CtrA by ClpXP via the cdG effector PopA (Duerig et al., 2009). Interestingly, we have recently shown that a key regulator of cdG, the phosphodiesterase PdeA (CC3396) (Christen et al., 2005), is itself degraded by the ClpXP protease at the G1-S transition upon dephosphorylation of the response regulator CpdR (Abel et al., 2011). CpdR is a small single domain response regulator originally identified as a factor critical for degradation of CtrA in vivo that is also responsible for directing ClpX to a specific location during cell-cycle progression (Iniesta et al., 2006; Biondi et al., 2006). Thus, ClpXP causes a transient upshift in cdG levels by degrading PdeA and the resulting upshift contributes to degradation of CtrA via PopA. This feedback results in a robust switch that drives the G1-S transition as well as initiates the stalked cell specific developmental program (Abel et al., 2011).
ClpXP substrates contain specific sequence motifs (degrons) often located at the exposed termini of target proteins. Protease specificity is further tuned by the action of adaptor proteins, which are auxiliary factors that facilitate degradation of certain target substrates. Once initiated, substrate degradation is processive with cycles of ATP hydrolysis driving unfolding and translocation of the substrate through ClpX to the peptidase ClpP. Although adaptors can clearly contribute to protease specificity and substrate prioritization, there are relatively few adaptor/substrate pairs that have been biochemically characterized (Sauer and Baker, 2011; Kirstein et al., 2009). Interestingly, CpdR is necessary for degradation of a number of ClpXP substrates in vivo, such as CtrA, McpA, and KidO (Iniesta et al., 2006; Biondi et al., 2006; Radhakrishnan et al., 2010), but only PdeA has been shown to be strictly dependent on CpdR for its degradation in vitro (Abel et al., 2011). This requirement for CpdR in PdeA degradation by ClpXP suggests that it may act as a specific adaptor to deliver PdeA; however, neither PdeA nor CpdR contain motifs known to be important for other adaptor-substrate systems (Kirstein et al., 2009; Wah et al., 2002; Dougan et al., 2003; Chowdhury et al., 2010; Levchenko et al., 2000).
Here, we show that PdeA is delivered to the ClpXP protease by the CpdR response regulator mediated through an N-terminal Per-Arnt-Sim (PAS) domain in PdeA. Although degradation requires this N-terminal domain, PdeA degradation initiates at a weak C-terminal degron, a combination that ensures reengagement of potentially deleterious partially processed fragments. CpdR is needed for PdeA degradation and kinetic results suggest that CpdR not only acts as a tether to improve substrate recognition, but also improves substrate processing, especially with weakly recognized tags. We present the structure of the PAS domain and show that the dimeric PAS domain contains charged surface residues needed for CpdR-mediated delivery. Biochemical studies reveal that an adaptor insensitive monomer of PdeA can be delivered to the protease if it is partnered to a wild-type PAS domain, suggesting a role for dimerization in increasing the scope and robustness of CpdR-mediated degradation. Finally, we show that the PAS domain drives degradation of PdeA in vivo, that degradation is critical for proper cellular development, and that this domain likely performs additional functions in vivo unrelated to proteolysis. Together, our results reveal the diversity of adaptor-regulated degradation by presenting, to our knowledge, a new adaptor/substrate pair for the ClpXP protease and suggesting how substrate architecture can impact robust processive degradation.
RESULTS
PdeA Degradation Requires its N-Terminal Domain but Initiates from Its C Terminus
PdeA is composed of three domains: a C-terminal phosphodiesterase domain (EAL), a domain resembling a cdG binding domain (GEDEF), and an N-terminal domain weakly similar to a PAS domain (Figure 1A) (Christen et al., 2005). As shown in Figure 1A, degradation of PdeA by ClpXP is strongly dependent on the response regulator CpdR. Proteolysis of the full-length protein initiates from the C terminus because mutation of these residues eliminates degradation even in the presence of CpdR. Interestingly, truncating PdeA from the C terminus yields variants that are still degraded in a CpdR-dependent fashion (Figure 1B; see Figure S1 available online). The GEDEF domain is known to bind GTP (Christen et al., 2005) and GTP enhances degradation of full-length PdeA (Abel et al., 2011), therefore we tested if GTP influenced degradation of PdeA truncations lacking the GEDEF domain and found, as expected, that it did not (Figure S1). Given our original identification of the native C terminus as the site of initiation for the full-length protein, these observations collectively suggest that PdeA contains cryptic degradation tags (such as those seen in lexA; Neher et al., 2003a) that may serve as reengagement sites if the substrate becomes prematurely released during proteolysis.
Figure 1. Both Carboxyl- and Amino-Terminal Regions Are Necessary for Productive PdeA Degradation.
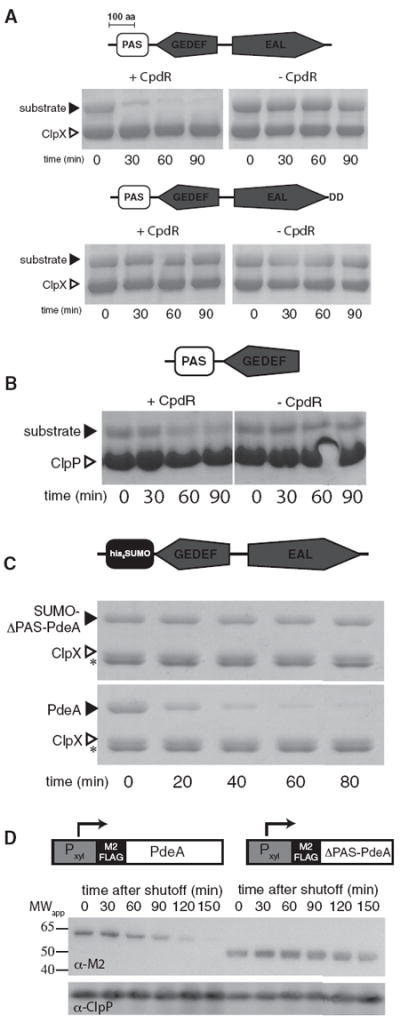
(A) PdeA consists of a C-terminal phosphodiesterase domain (EAL), a degenerate cyclic di-GMP binding domain (GEDEF), and a putative N-terminal PAS domain. In vitro degradation of PdeA requires CpdR (upper). Mutation of the C-terminal residues to Asp-Asp eliminates degradation (lower).
(B) PdeA contains cryptic recognition sites. A truncation with the N-terminal domain, but ending in “ELAVE” rather than the native C terminus, is still degraded by ClpXP/CpdR in vitro. Additional truncations are shown in Figure S1. Black markers indicate substrates as shown, ClpX or ClpP (white markers) are shown for size comparison.
(C) Replacement of the PAS domain with a similarly sized domain (SUMO) eliminates degradation. n.b., in these electrophoresis conditions creatine kinase (asterisk) is visible.
(D) PdeA degradation in vivo requires the PAS domain. Wild-type PdeA or a mutant lacking the PAS domain was transiently induced by addition of xylose. Following removal of the inducer, wild-type or mutant PdeA levels from equal numbers of cells were determined by detection of an N-terminal M2-FLAG epitope.
The N-terminal putative PAS domain is required for CpdR-dependent degradation as replacing this region generates an otherwise full-length PdeA variant that is resistant to CpdR/ClpXP degradation in vitro (Figure 1C). PdeA stability in vivo is also dependent on this domain. Using a transient induction system, we found that an epitope tagged version of PdeA is rapidly lost after shifting to noninducing conditions, while a PdeA variant lacking the N-terminal domain is stable (Figure 1D). Together, these results demonstrate that the N-terminal domain is critical for PdeA degradation both in vivo and in vitro.
PdeA Contains a Weak C-Terminal Degron
Adaptors operate by a variety of mechanisms. For example, adaptors can increase local concentrations of poorly recognized substrates through tethering substrates to the protease (as is the case for SspB/ClpXP) (Kirstein et al., 2009; Wah et al., 2002; Dougan et al., 2003; Chowdhury et al., 2010; Levchenko et al., 2000) or they can be more actively involved in substrate delivery (as is the case for the staged delivery found in ClpS/ClpAP or MecA) (Kirstein et al., 2009; Wang et al., 2007; Román-Hernández et al., 2011; De Donatis et al., 2010). To determine how CpdR enhanced PdeA degradation, we fused GFP to the N-terminus of different PdeA constructs and assayed degradation by fluorescence (Figure 2A).
Figure 2. Degradation of PdeA Couples a Naturally Weak C-Terminal Degron with a Distal Tethering Domain.
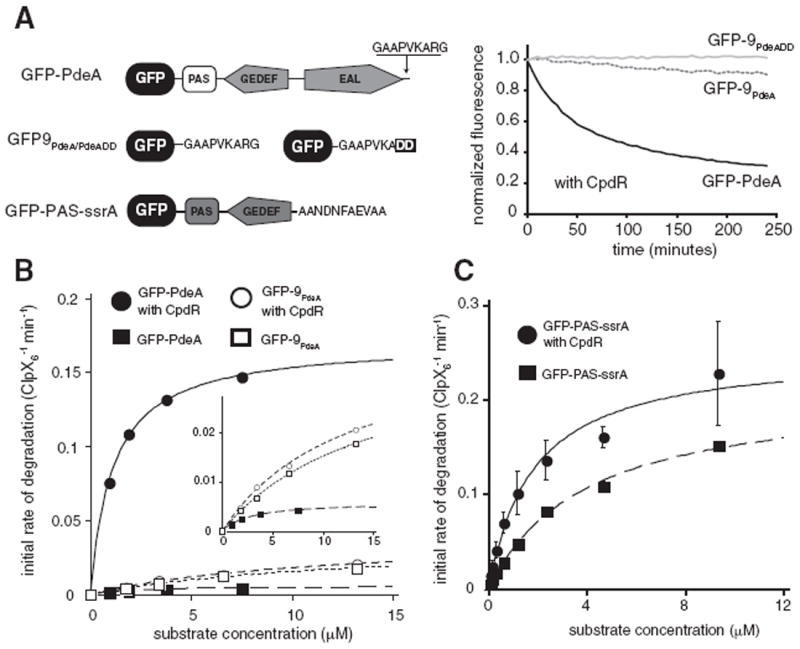
(A) A GFP reporter fusion containing only the C-terminal residues of PdeA (GFP-9PdeA) is degraded more slowly than the full-length PdeA (GFP-PdeA), but is degraded faster than GFP-9PdeADD, which contains a carboxyl-terminal Asp-Asp. Representative curves are of 1 μM substrates in the presence of 2.5 μM CpdR using standard conditions.
(B) Degradation of full-length PdeA without CpdR is inefficient but is substantially enhanced in the presence of CpdR while GFP-9PdeA degradation is unaffected by CpdR. Inset highlights the more slowly degraded substrates.
(C) CpdR can preferentially improve the KM of a readily degraded substrate. PdeA constructs containing a strong recognition tag (GFP-PAS-ssrA) show an increase in KM upon addition of CpdR, with little change in vmax relative to the degradation of this substrate in the absence of CpdR. Replacing the ssrA tag with a weaker tag also shows an improvement of KM in the presence of CpdR (Figure S2). In B and C, lines are fits to the Michaelis-Menten equation; parameters are shown in Table 1.
As expected, GFP-PdeA was robustly degraded in the presence of CpdR, but was stable in its absence (Figure 2). A fusion of the last nine residues of PdeA (GAAPVKARG) to the C terminus of GFP (GFP-9PdeA) was degraded slowly by ClpXP in a sequence-specific fashion (compare with GFP-9PdeADD) regardless of CpdR (Figures 2A, 2B). Thus, PdeA degradation appears to depend on the combination of an N-terminal adaptor-dependent site and an intrinsically weak degradation tag. With respect to degradation of the full-length GFP-PdeA fusion, addition of CpdR decreases KM by 3-fold and increases vmax by almost 30-fold (Table 1). Because addition of CpdR does not affect degradation of other ClpXP substrates such as GFP-9PdeA (Figure 2B; Table 1) this enhancement likely reflects the ability of CpdR to improve the specific efficiency of PdeA delivery rather than a global enhancement of ClpXP activity.
Table 1.
Kinetic Parameters for Substrate Degradation by CpdR/ClpXP
| KM (μM) | vmax (min−1) | vmax/KM (μM−1 min−1 × 10−3) | |
|---|---|---|---|
| GFP-PdeA | 2.8 ± 0.5 | 0.006 ± 0.0004 | 2.1 |
| GFP-PdeA with CpdR | 1.1 ± 0.1 | 0.17 ± 0.002 | 154 |
| GFP-9PdeA | 15.4 ± 1.6 | 0.04 ± 0.003 | 2.6 |
| GFP-9PdeA with CpdR | 12.6 ± 1.5 | 0.04 ± 0.002 | 3.1 |
| GFP-PAS-ssrA | 3.9 ± 0.4 | 0.21 ± 0.10 | 54 |
| GFP-PAS-ssrA with CpdR | 1.8 ± 0.4 | 0.25 ± 0.02 | 139 |
| GFP-PAS-AVAA | 19.4 ± 4.0 | 0.03 ± 0.004 | 1.6 |
| GFP-PAS-AVAA with CpdR | 4.0 ± 0.2 | 0.12 ± 0.003 | 30 |
See Experimental Procedures.
CpdR Is Capable of Tethering PdeA to ClpXP
Because simple tethering is known to be sufficient for adaptor-mediated delivery (Sauer and Baker, 2011; Davis et al., 2009) we asked if CpdR could act as a simple tether if recognition of the degradation tag was not limiting. To test this, we appended a variant of the Caulobacter ssrA tag (which is sufficient for strong recognition by ClpXP; Chien et al., 2007a), to a fragment of PdeA containing the PAS domain (GFP-PAS-ssrA). As expected, this construct was rapidly degraded in the absence of CpdR. The addition of CpdR decreased KM with little effect on vmax of this same substrate in the absence of CpdR (Figure 2C; Table 1), consistent with a model where CpdR acts as a tether to increase local concentration of substrate. Replacing the ssrA tag with a weaker tag generates a construct where both KM and vmax change substantially in the presence of CpdR (Figure S2; Table 1). Therefore, at a minimum, CpdR is capable of tethering PdeA to ClpXP, but also has strong effects on vmax, especially in the presence of a weak degradation tag.
The N-Terminal Region of PdeA Is a PAS Domain
Our results point to a crucial role for the N-terminal region of PdeA in CpdR-mediated degradation by ClpXP. As mentioned previously, this region has limited identity to PAS domains, but because PAS domains tend to diverge significantly in sequence (Möglich et al., 2009; Henry and Crosson, 2011), we took a structural approach to further characterize this domain. We crystallized the first 130 residues of PdeA, the minimum region degraded by ClpXP in a CpdR-dependent fashion (Figure S1). Despite repeated attempts, molecular replacement with known PAS domains proved unsuccessful. However, we were successful in de novo phasing using single isomorphous replacement with anomalous scatter and solved the structure to 1.7 Å resolution (Figure 3; Figure S3; Table 2).
Figure 3. Crystal Structure of the N-Terminal Region of PdeA Required for CpdR-Mediated Degradation Reveals a Characteristic PAS Domain.
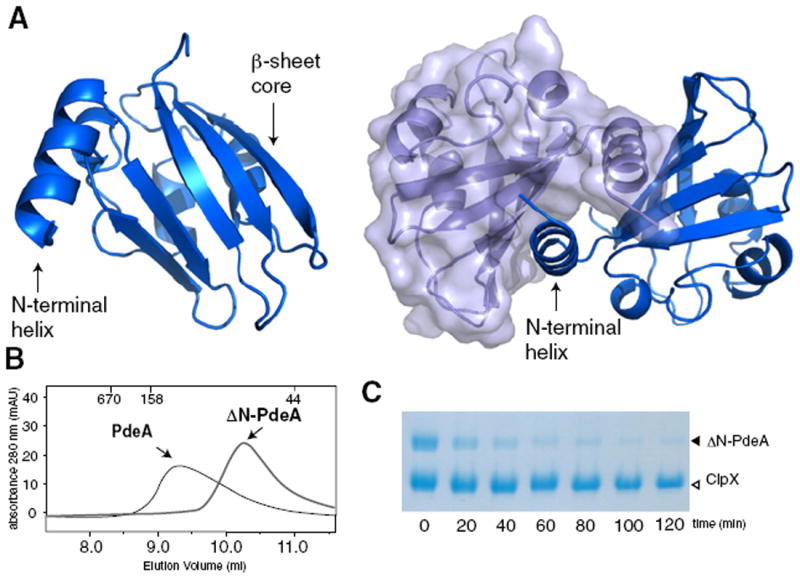
(A) Although only a monomer is present in the asymmetric unit (left), a dimer is formed across a crystallographic symmetry axis primarily through an extended N-terminal α-helix (right).
(B) Purified PdeA migrates as a dimer by size exclusion chromatography (MWapp ~110 kDa) but removal of the N-terminal helix (ΔN-PdeA) results in a monomeric PdeA (MWapp ~70 kDa). Samples were separated on a Superdex 75 10/300 gel filtration column. Numbers at the top of the trace represent molecular weights of calibration standards (in kDa).
(C) Dimerization is dispensible for PdeA degradation as monomeric ΔN-PdeA is readily degraded in the presence of CpdR/ClpXP.
Table 2.
Crystallographic Statistics
| Crystal | Native | Hg |
|---|---|---|
| Space group | C2 | C2 |
| Cell lengths, Å | 63.8, 41.9, 48.7 | 63.5, 41.4, 48.4 |
| Cell angles, ° | 90, 107.5, 90 | 90, 106.9, 90 |
| X-ray data | ||
| Resolution (last shell) | 50–1.70 (1.76–1.70) | 50–1.81 (1.87–1.81) |
| Observations | 77,322 | 25,130 |
| Unique observations | 11,481 | 9,353 |
| Completeness, % (last shell) | 84.1 (30.9) | 84.6 (41.2) |
| Redundancy (last shell) | 6.7 (4.2) | 2.7 (1.9) |
| Rsym, % (last shell) | 5.0 (27.1) | 5.5 (25.1) |
| I/σI (last shell) | 40.0 (4.0) | 28.7 (3.2) |
| Phasing | ||
| No. of sites | 3 | |
| Phasing power acentric, centric | 2.26, 1.68 | |
| Figure of merit | 0.54 | |
| Figure of merit after DM | 0.74 | |
| Refinement | ||
| Rwork / Rfree, % | 16.7 / 20.6 | |
| No. of atoms | 1037 | |
| Protein | 883 | |
| Water | 154 | |
| B-factors, Å2 | ||
| Protein | 9.4 | |
| Water | 24.0 | |
| RMS deviations | ||
| Bonds (Å) | 0.009 | |
| Angles (°) | 1.239 |
The highest resolution shell is shown in parentheses.
Rsym = ΣhΣi|h,i-〈Ih〉 |/ΣhΣi|Ih,i| where Ih,i is the ith intensity measurement of reflection h and h 〈Ih〉 is the average intensity of that reflection.
Rwork / Rfree =Σh|FP-FC|/Σh|FP|, where FC is the calculated and FP is the observed structure factor amplitude of reflection h for the working or free set, respectively.
We found that the adaptor-binding region of PdeA was a true PAS domain containing a classic five-stranded β sheet core flanked by α helices (Figure 3A). PAS domains vary significantly in the organization and orientation of these peripheral α helices, but they are generally implicated in interdomain contacts or ligand binding (Henry and Crosson, 2011; Möglich et al., 2009). Although the PAS domain crystallized as a monomer in the asymmetric unit, the crystallographic 2-fold generates a dimer with 1225 Å2 buried per monomer in an interface primarily consisting of an extended N-terminal helix (Figure 3A). Because oligomerization of some ClpXP substrates is needed for proper substrate recognition (Sharma et al., 2005; Abdelhakim et al., 2008), we speculated that PAS mediated dimerization may be important for PdeA degradation. As predicted from previous work, wild-type PdeA migrates as a dimer by size-exclusion chromatography (Christen et al., 2005) and we found that deletion of the N-terminal α-helix eliminates dimerization (ΔN-PdeA) (Figure 3B). However, unlike the previously mentioned substrates, PdeA degradation does not seem dependent on its oligomerization as the monomeric ΔN-PdeA is robustly degraded in vitro by ClpXP/CpdR (Figure 3C).
Next, we examined the PAS domain for features that might represent sites of CpdR interaction. Based on the dispensibility of PdeA dimerization for degradation, our attention was drawn to a cluster of charged residues found at opposite ends of the PAS dimer (Figure 4; Figure S3). Mutation of an arginine in this region (PdeAR69A) resulted in a variant that was degraded more slowly than wild-type PdeA (Figure 4). The mutant was deficient in interacting with CpdR (Figure S4), but retained the ability to form a dimer (see below), suggesting that dimerization and adaptor interactions are separable.
Figure 4. Arginine 69 Is Important for CpdR-Mediated PdeA Degradation.
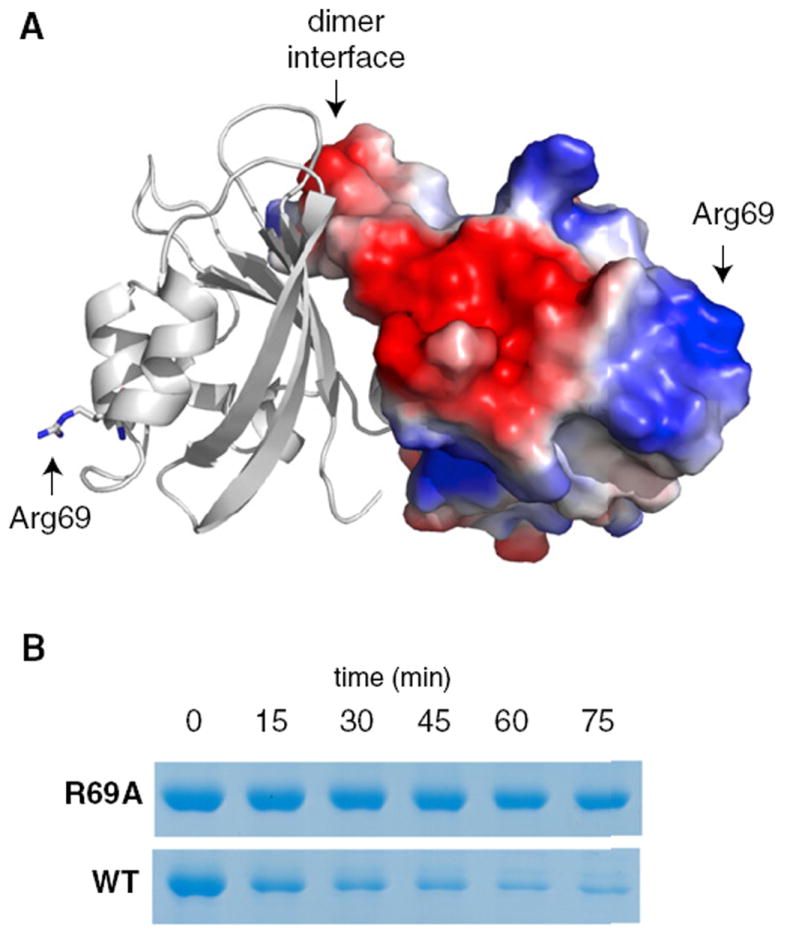
(A) Cartoon/surface representation of PAS dimer with right hand subunit surface colored by electrostatic potential (blue: positive; red: negative) using the qualitative vacuum electrostatic potential display native to Pymol (Schrödinger, LLC). Arginine 69 is modeled in stick representation on the left hand subunit and its location is marked with arrows on both subunits. See Figure S3 for the electron density map of the region surrounding this residue.
(B) Mutation of Arg69 to alanine (R69A) substantially inhibits PdeA degradation by CpdR/ClpXP, likely due to its failure to interact with CpdR (Figure S4).
A CpdR-Competent PAS Domain Can Deliver Its Partner Subunit
Based on the above results that decouple dimerization from CpdR interactions, we predicted that one PAS domain of a PdeA dimer could deliver its partner even if the partner subunit is incapable of responding to CpdR. Specifically, homodimers containing two adaptor-incompetent PAS domains (such as PdeAR69A) should not be degraded, but in a heterodimer, a wild-type PAS domain could bridge a partner PdeAR69A subunit and CpdR (Figure 5A). To test this hypothesis, we mixed PdeAR69A with either the isolated wild-type PAS domain or the dimerization defective ΔN-PAS domain under conditions that promote subunit exchange. We showed subunit exchange in the following manner. In isolation, PdeAR69A migrates as a dimer of ~110 kDa, wild-type PAS domain migrates as a dimer of ~30 kDa, and ΔN-PAS migrates as a monomer of ~16 kDa (gray traces in Figure 5B). After incubation at 45°C for 30 min to promote exchange, the PdeAR69A/PAS mixture shows a shifted elution profile in agreement with a heterodimer of PdeAR69A and the PAS domain (major elution peak at ~77 kDa; Figure 5B). In contrast, the elution profile of the PdeAR69A/ΔN-PAS mixture overlaps with the individual components, consistent with the monomeric nature of ΔN-PAS (Figure 5B).
Figure 5. PdeA(R69A) Can Be Delivered to ClpXP If Partnered with a Wild-Type PAS Domain.
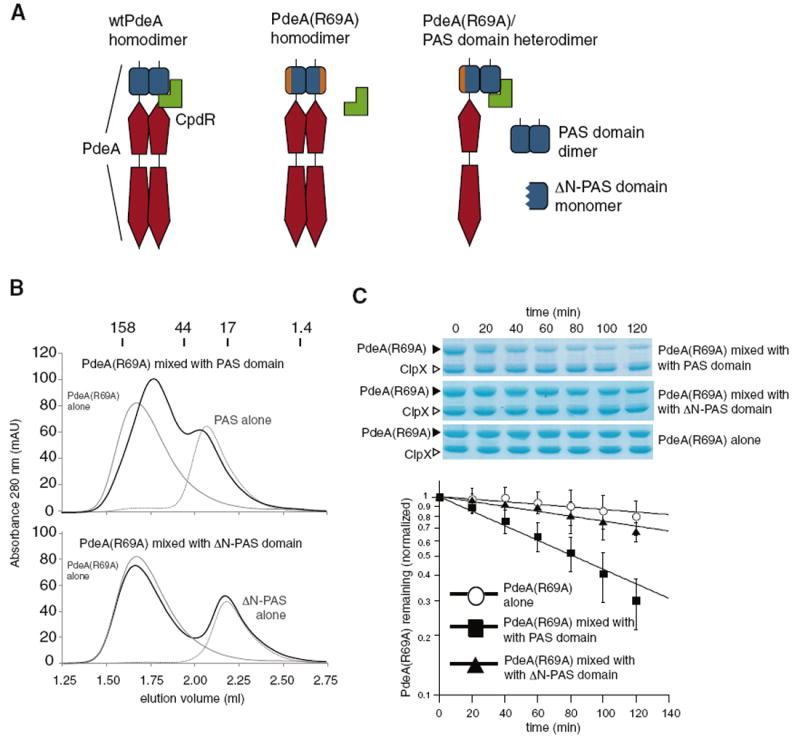
(A) Cartoon illustrating how a PdeA(R69A) protein that fails to interact with CpdR as a dimer could be targeted for degradation if it forms a heterodimer with a wild-type PAS domain.
(B) PdeA(R69A) forms a heterodimer with the wild-type PAS domain but not a monomeric PAS domain (ΔN-PAS). Dark lines show the elution profile of PdeA(R69A) mixed with either PAS or ΔN-PAS after a 30 min incubation at 45°C to promote subunit exchange. The elution profiles of the indicated proteins in isolation are gray. Gel filtration was performed on a Superdex 200 5/150 GL with size standards (in kDa) as marked above.
(C) The PdeA(R69A) subunit is degraded more rapidly when partnered with wild-type PAS. PdeA(R69A) was incubated with wild-type PAS, ΔN-PAS or alone as shown in B, then subjected to degradation by CpdR/ClpXP. The graph represents quantification of triplicate measurements, error bars represent standard errors, and solid lines are fits to single exponential decays. Enhancement of degradation requires heterodimer formation as addition of PAS domain without incubation in mixing conditions was unable to strongly promote PdeA(R69A) degradation nor was the PAS domain able to accelerate degradation of a non-tethered substrate (Figure S4).
Having generated heterodimers of both wild-type PAS domain and mutant full-length PdeA, we tested if the mutant PdeA was degraded in a CpdR-dependent fashion. As shown in Figure 5C, PdeAR69A mixed with ΔN-PAS was still degraded slowly (t1/2 = 243 ± 16 min), similar to that of pure PdeAR69A homodimers (t1/2 = 460 ± 50 min). Importantly, heterodimers of PdeAR69A mixed with the wild-type PAS domain were degraded much faster (t1/2 = 82 ± 2 min). These results suggest that CpdR can bind one subunit of a PdeA dimer and deliver the other subunit to the ClpXP protease. Thus, dimerization of the PAS domain is not necessary for degradation, but can facilitate degradation of the full-length PdeA protein in cases where one subunit is prevented from interacting with CpdR.
The PdeA PAS Domain Is Necessary for Regulation of Protein Levels and Function In Vivo
Finally, we examined the biological consequences of modulating PdeA stability in vivo. PdeA is part of an intricate network that governs cell-cycle progression, cellular development, and chemotaxis in Caulobacter (Abel et al., 2011). Because of the dimorphic nature of Caulobacter, changes in cell cycle or development often manifest as changes in motility. For example, defects in the swarmer-to-stalk transition ultimately result in production of fewer motile swarmer cells, which translates to a reduction in colony size when cells are inoculated into semi-solid media. Consistent with its role as a central regulator of development and the cell cycle, cells lacking PdeA (ΔPdeA) are smaller in colony size when compared to wild-type cells (Abel et al., 2011; Figure 6).
Figure 6. The PAS Domain Is Important for PdeA Activity In Vivo.
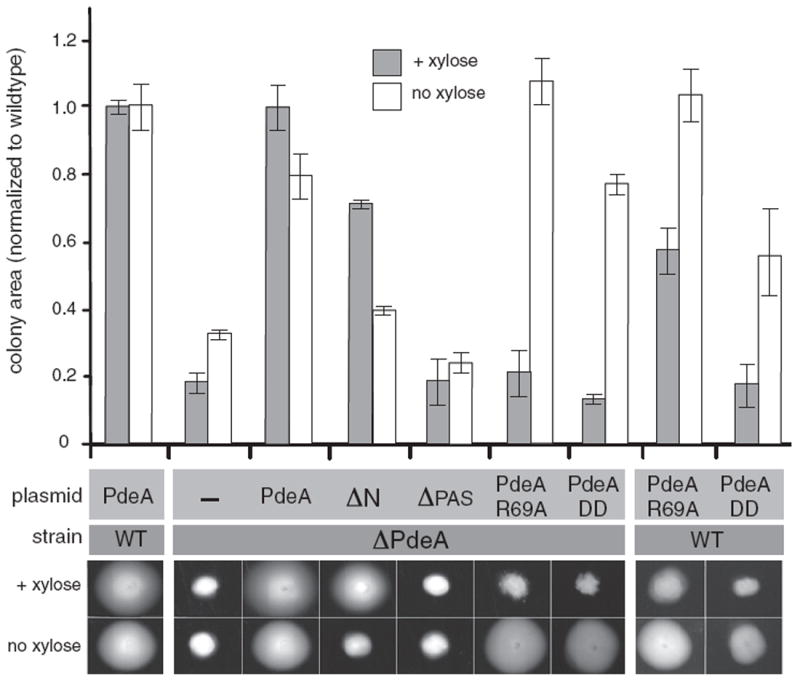
Plasmids expressing PdeA variants under control of a xylose-inducible promoter were transformed into wild-type cells (WT) or cells lacking chromosomal PdeA (ΔPdeA). Strains were inoculated into semi-solid PYE agar containing 0.2% xylose to induce high expression or without xylose for low basal expression of the PdeA variants. Control plasmid (-) expresses GFP instead of a PdeA variant. ΔN, PdeA lacking N-terminal dimerization helix of PAS domain; ΔPAS, PdeA lacking the entire PAS domain; PdeAR69A, PdeA with the R69A mutation in its PAS domain; PdeADD, PdeA with carboxyl-terminal residues replaced by Asp-Asp. Representative images are shown below, graph represents average colony area (n = 3) normalized to WT strains carrying WT PdeA (error bars represent standard errors of these measurements). Levels of PdeA protein mirrored the predicted stability as shown by western blotting (Figure S5).
In the experiments shown in Figure 6, medium copy plasmids express PdeA variants under control of a promoter that is strongly induced in the presence of xylose and weakly induced in its absence. Wild-type strains show normal motility, forming large colonies even when overexpressing wild-type PdeA. The motility defect of ΔPdeA cells is complemented by expression of wild-type PdeA in either weak or strongly inducing conditions. Plasmids containing a non-degradable full-length PdeA variant (PdeADD) complement the deletion phenotype under weak induction, but not under high induction conditions. Previous reports have shown that excess phosphodiesterase activity affects cell cycle, development, and motility due to reduced cdG levels (Duerig et al., 2009). Because PdeADD is not degraded, it is present at a higher level than PdeA in inducing conditions (Figure S5). Thus, we attribute the inability of PdeADD to complement the deletion in inducing conditions to its unregulated, prolific phosphodiesterase activity that depletes the cdG pool needed for proper motility. Consistent with this interpretation, overexpression of PdeADD reduces motility even in wild-type cells (Figure 6).
We tested the various mutants and truncations of PdeA for their ability to restore wild-type motility (Figure 6). Plasmids expressing PdeAR69A show similar phenotypic effects as those expressing PdeADD. Specifically, overexpression of PdeAR69A in wild-type cells results in a motility defect and only complements the ΔPdeA strain when expressed at low levels. In contrast, plasmids expressing monomeric PdeA (ΔN) do not complement the deletion phenotype when expression is low; however, high expression restores wild-type-like motility (Figure 6). Interestingly, the PdeA variant lacking the N-terminal PAS domain (ΔPAS) cannot complement the deletion phenotype in either weak or strongly inducing conditions even though it is present at the same level as other stabilized PdeA variants (Figures 6 and S5). Like the other stabilized PdeA variants, overexpression of ΔPAS reduces motility of wild-type cells, suggesting that it is active as a phosphodiesterase (Figure S5). Collectively, these results support a model where the PAS domain is needed for degradation of PdeA via its ability to act as an adaptor-binding site and has an additional role for PdeA function in vivo. Because PAS domains are known to act as signaling sensors (Möglich et al., 2009), it is tempting to speculate that this domain may regulate PdeA activity in response to specific cues during cell cycle or stressful conditions.
DISCUSSION
Proteolysis of cell-cycle regulators is critical for the appropriate timing of replication and developmental transitions in C. crescentus (Jenal, 2009; Curtis and Brun, 2010). Along with fluctuating protein levels, concentrations of second messengers also fluctuate during cell cycle progression (Jenal, 2009; Hengge, 2009; Paul et al., 2008). These processes have recently been shown to be linked through the phosphodiesterase PdeA. Degradation of PdeA by the essential AAA+ protease ClpXP generates an upshift in the cyclic-di-GMP pool that in turn promotes differentiation and cell cycle progression. Proteolysis of PdeA requires the response regulator CpdR both in vivo and in vitro (Abel et al., 2011), but neither PdeA nor CpdR have high similarity to known ClpX substrates or adaptors. Here, we characterize the regions of PdeA required for CpdR-mediated ClpXP recognition and structurally identify the adaptor-dependent region of PdeA as a PAS domain.
PAS domains are found in all kingdoms of life and are involved in diverse functions but have an especially strong presence in signaling networks (Möglich et al., 2009; Henry and Crosson, 2011). PAS domains often serve as protein-protein interaction modules and can generate oligomeric specificity such as that seen with the Arnt receptor (Pongratz et al., 1998). In bacteria signaling networks, these domains are found coupled to a wide range of effector domains in proteins such as sensor histidine kinases, chemoreceptors, and cdG regulating domains (Möglich et al., 2009; Henry and Crosson, 2011). PAS domains can also serve as direct sensors, as seen with the Escherichia coli cdG phosphodiesterase DosP, where oxygen binding to an N-terminal heme-bound PAS domain increases cdG breakdown (Tuckerman et al., 2009). To our knowledge, our work here points to a novel role for a PAS domain serving as an adaptor recognition site needed for regulated proteolysis by the ClpXP protease. In addition, our in vivo results suggest that the PAS domain of PdeA is necessary for functions outside of proteolysis. Although no ligand such as heme has been found yet for the PdeA PAS domain, it will be interesting to address if this domain regulates phosphodiesterase activity in response to signals similar to that seen in DosP.
Delivery of PdeA by CpdR requires residues present at opposite ends of the PAS dimer interface, but dimerization is not essential for PdeA degradation. However, dimerization may play an important role in improving the robustness of PdeA degradation in physiologically relevant conditions, because PdeA mutants defective in CpdR mediated degradation are still delivered to ClpXP if their partner subunit is capable of interacting with CpdR. While this situation is unlikely to occur in vivo (given that mutagenesis would impact both subunits in a homodimer), occlusion of the CpdR binding surface of one subunit may arise from binding of an inhibitory protein or through selective damage of one of the subunits. In these cases, the ability of the other “CpdR-competent” PdeA subunit to deliver its partner would support proper degradation of the entire dimer (Figure 7A). Because PAS domains are known to influence heterodimer specificity (Pongratz et al., 1998) an intriguing possibility is that the PAS domain of PdeA could also serve as a bridge between CpdR and other PAS domain containing proteins, resulting in CpdR being able to deliver substrates beyond those it interacts with directly (Figure 7). This type of mechanism has precedence in both Clp family proteases and in the eukaryotic proteasome, where degradation of multisubunit complexes can employ one subunit for targeting and another subunit for initiation (Neher et al., 2003b; Sharma et al., 2005; Abdelhakim et al., 2008; Prakash et al., 2009).
Figure 7. CpdR-Competent PdeA Subunit Supports Proper Degradation of the Entire Dimer.
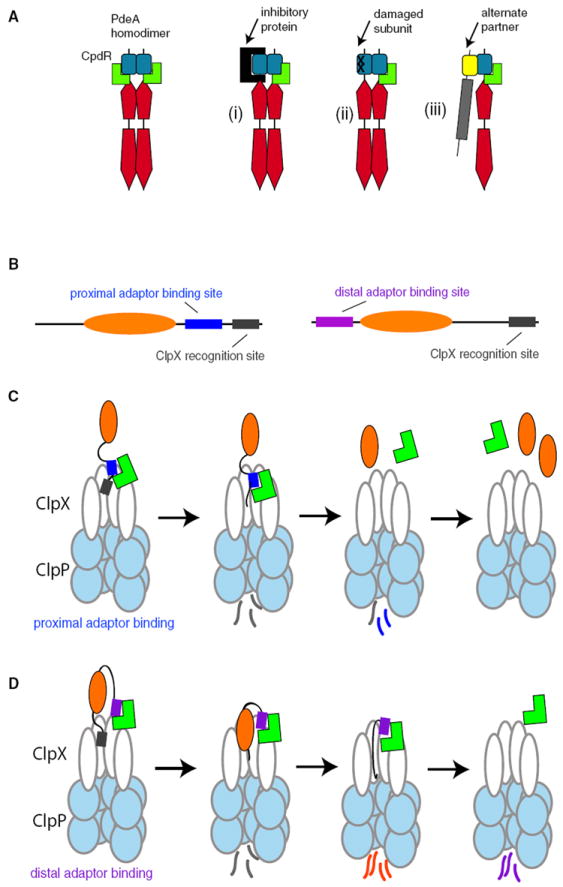
(A) Dimerization of PdeA via the PAS domain may improve degradation in vivo if one subunit is prevented from CpdR binding by inhibitory protein binding (i) or damage to the subunit (ii). Alternatively, PdeA may facilitate CpdR-dependent degradation by bridging CpdR and an alternate binding partner (iii).
(B) Cartoon illustrating organization of adaptor-binding (blue or purple) versus protease-recognition sites (gray) for different substrate architectures.
(C) Binding of adaptor (green) to sites proximal to or overlapping with protease recognition sites in a substrate improves recognition. Because both the recognition site and the adaptor site are the first to be degraded, any release of adaptor would result in a buildup of partially processed substrates.
(D) Adaptor binding to sites distal to the recognition tag promotes tight association between the protease and substrate even after proteolysis has begun, potentially leading to substrate reengagement and complete degradation.
Adaptor-mediated substrate degradation can arise from tethering interactions with the protease, decreasing the amount of substrate necessary to saturate the enzyme (a decrease in KM). Adaptors can also alter the processing or engagement of the substrate, changing both the KM and the substrate turnover rate (vmax). CpdR reduces the KM and increases the turnover rate for wild-type PdeA degradation, suggesting that it may be altering substrate processing. It is unlikely that CpdR is affecting ClpXP by increasing its ability to degrade all substrates, because substrates containing only the PdeA degradation tag are degraded independent of CpdR. Furthermore, degradation of a PAS domain-containing fragment appended with an ssrA tag shows a decrease in KM without substantial changes to the turnover rate upon addition of CpdR (Figure 2). Together, these results suggest that CpdR, at a minimum, can act as a simple tether but also improves the turnover rate of substrates with weak tags. The natural combination of adaptor-binding regions and weak degrons likely reflects a general feature of ClpXP-mediated proteolysis, similar to previous observations that artificially weakening recognition tags of adaptor-dependent substrates improves substrate selectivity (McGinness et al., 2006).
The relative locations of the adaptor binding site and degradation initiation site within PdeA are on opposite ends of the polypeptide warrant specific consideration (Figure 7B). In many characterized adaptor-substrate systems (such as SspB/ssrA and ClpS/N-end rule substrates), the adaptor-binding sites on the substrates are immediately adjacent to or overlap with the degradation tag (Levchenko et al., 2003; Song and Eck, 2003; Wang et al., 2008; Schuenemann et al., 2009). In these cases, the proximal architecture promotes a close geometry of the tag to the pore of the protease and ensures rapid engagement. However, because proteolysis is processive, the tag and the adaptor-binding site are the first elements to be degraded. If the protease fails to completely degrade the target and releases the substrate, reengagement is unlikely because the substrate has lost both the adaptor-binding regions and the recognition tag (Figure 7C). In PdeA, the location of the adaptor-binding region (the N-terminal PAS domain) relative to the site of degradation initiation (the C terminus) results in a distal architecture where partially processed substrates can still be delivered to ClpXP by the CpdR adaptor (Figure 7D). A similar separation of adaptor-binding and protease recognition sites likely explains the extended requirements for σS degradation by RssB/ClpXP in E. coli (Stüdemann et al., 2003). This architecture is well suited to ensure the complete degradation of multi-domain substrates that are susceptible to incomplete processing by ClpXP (Kenniston et al., 2005) or other proteases (Tian et al., 2005), especially when combined with cryptic degradation tags that are revealed upon partial processing of substrates as seen in PdeA (Figures 1 and S1).
The identification of the PAS domain of PdeA as the adaptor-binding site for CpdR resolves an outstanding issue that while CpdR is found in many α–proteobacteria (Brilli et al., 2010), only Caulobacter and closely related species have obvious PdeA orthologs. In particular, Sinorhizobium meliloti has a functional CpdR, but no clear PdeA ortholog (Kobayashi et al., 2009). In vitro, the S. meliloti CpdR can deliver the Caulobacter PdeA to ClpXP (unpublished observation), leading us to speculate that similar substrates exist in S. meliloti but they are not strict orthologs of PdeA. Because PAS domains have high sequence diversity (Henry and Crosson, 2011; Möglich et al., 2009), it may be difficult to identify these substrates computationally but a systematic degradation profiling of putative PAS domains would reveal these CpdR-dependent targets.
EXPERIMENTAL PROCEDURES
Plasmid Construction, Protein Purification, Gel Filtration, and Degradation Assays
Table S1 (available online) lists strains and plasmids. Constructs for expression in Caulobacter were generated using the Gateway cloning system as previously described (Skerker et al., 2005). All untagged PdeA and CpdR constructs for biochemical assays were cloned as his6SUMO fusion constructs and purified/cleaved as described (Wang et al., 2007). When needed, proteins were further purified by size-exclusion chromatography and buffer exchanged into H-buffer (25 mM HEPES-KOH pH 7.4, 100 mM KCl, 10% Glycerol, 10 mM β-mercaptomethanol) for storage. GFP-PdeAwas purified as a his-tag fusion protein (Skerker et al., 2005). ClpX and ClpP were purified as before (Chien et al., 2007b; Levchenko et al., 2000). Final concentrations of components in degradation assays (30°C) were as follows unless noted otherwise: ClpX 0.4 μM, ClpP 0.8 μM, ATP 4 mM, 75 μg/ml creatine kinase, 5mM creatine phosphate, 1 mM GTP, substrate 2.5 μM, CpdR 2.5 μM (if present) using H-buffer as the reaction buffer. GTP improves PdeA degradation for an unknown reason (Abel et al., 2011) and is included in the reaction to standardize the experiments; however, experiments with PdeA lacking the GTP binding region (Figure S1) show that CpdR-dependent degradation does not require GTP. Degradation of GFP-PdeA was performed on a Spectramax M5 (Molecular Devices) as described (Román-Hernández et al., 2011). Protein degradation was also monitored by SDS-PAGE. Aliquots were removed at indicated times, quenched with SDS loading dye, and frozen immediately. Aliquots were heated at 65°C for 5 min, then immediately separated by SDS-PAGE, stained by Coomassie Blue G-250, scanned on a flatbed scanner, and quantified using ImageJ (NIH). Gel filtration experiments were performed at 4°C on a GE ATKA Purifier UPC 10 FPLC with a GE Superdex 75 10/300 GL or Superdex 200 column equilibrated in H-buffer. To generate heterodimers, mixtures of PdeA and PAS domains (20 μM each) were mixed in H-buffer, heated at 45°C for 30 min to induce mixing then kept at 4°C until gel filtration or degradation assays.
Motility Assays and Protein Stability In Vivo
Wild-type CB15N or NA1000 ΔPdeA (Abel et al., 2011) cells were transformed with the plasmids as described using standard techniques (Skerker et al., 2005). Motility assays were performed by inoculating three independent colonies into PYE media containing 0.3% agar with or without 0.2% xylose, and incubated for 3 days at 30°C. Colony sizes were determined automatically following thresholding using ImageJ (NIH). For measurements of protein degradation in vivo, plasmids with xylose promoters driving amino-terminal M2-FLAG tagged were expressed for one hour, then shifted to glucose containing media for the indicated times, aliquots were removed, centrifuged, and pellets frozen immediately. After the timecourse was complete, SDS loading dye was added to the pellets, suspensions were heated at 100°C for 10 min, and centrifuged at 20,000G for 10 min. Ten microliters of supernatant were resolved by SDS-PAGE, transferred to PVDF (Millipore), and probed with monoclonal M2-FLAG antibodies (Sigma-Aldrich), developed with HRP-conjugated antibodies (Millipore), and developed with Immobilon ECL substrate (Millipore).
Crystallization, Diffraction, and Refinement
The isolated PAS domain of PdeA (residues 2–130) was purified as described previously, exchanged into water, and concentrated to >5 mg/ml using centrifugal ultrafiltration (Vivaspin; Millipore). Initial screening of sitting drops used the JCSG+ Suite (QIAGEN). Final crystallization conditions were 0.1 M sodium acetate pH 4.5, 4% (w/v) PEG 4,000 (1:1 protein:well solution) with crystals forming within 4 hours at 20°C. For heavy atom derivatization, crystals were grown using 0.1 M HEPES pH 7.5, 4% (w/v) PEG 4,000, then 10 mM mercury acetate in the same solution was added and incubated for a further 24 hr. Before freezing, crystals were cryoprotected by being soaked briefly in well solution supplemented with 20% glycerol. X-ray data were collected on a Cu rotating anode source with an Raxis IV++ detector (Rigaku) at 100K. Reflection data were indexed, integrated, and scaled with HKL2000 (Collaborative Computational Project, Number 4, 1994). The crystals diffracted better than anticipated, so we integrated data to the corners of the image, resulting in low completeness in the high-resolution shells. Native and mercury datasets were scaled in SCALEIT (Collaborative Computational Project, Number 4, 1994). Isomorphous and anomalous patterns indicated a single Hg location, which was refined to 3.0 Å in MLPHARE (Cowtan, 2006) to a figure of merit of 0.40 and phasing power of 1.73 (acentric) and 1.35 (centric). Inspection of double-difference Fourier maps revealed the presence of two additional Hg sites, which were refined in MLPHARE. The resulting maps were improved in DM (Collaborative Computational Project, Number 4, 1994) by solvent flattening and histogram matching. Alpha carbons were initially located in Buccaneer (Cowtan, 2006); the model was manually built in Coot (Emsley and Cowtan, 2004), and refined with Refmac (Collaborative Computational Project, Number 4, 1994). The final model contains 94.6% and 5.4% of the residues in the most favored and additionally allowed regions of the Ramachandran plot according to Procheck.
Supplementary Material
Acknowledgments
The authors thank all members of the Chien lab for discussion and G. Roman-Hernandez, A. Olivares, and L. Francis for comments on the manuscript. Support for this work was from NIH grants R00 GM084157 to P.C. and R01 DK76877 to S.C.G. in addition to funds provided by the University of Massachusetts, Amherst to P.C. N.E.C. was supported in part by a traineeship from the NSF-sponsored Institute for Cellular Engineering IGERT program (Grant Number DGE-0654128).
K.R., P.R.S, and P.C. conceived the experiments. K.R., P.R.S., and P.C. performed experiments. K.R. crystallized all proteins. K.R. and N.E.C. collected the diffraction data. N.E.C. and S.C.G. solved and refined the crystal structure. P.C. wrote the paper.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and one table and can be found with this article online at doi:10.1016/j.str.2012.04.019.
ACCESSION NUMBERS
Coordinates and structure factors are deposited into the Protein Data Bank under accession code 3U2A.
References
- Abel S, Chien P, Wassmann P, Schirmer T, Kaever V, Laub MT, Baker TA, Jenal U. Regulatory cohesion of cell cycle and cell differentiation through interlinked phosphorylation and second messenger networks. Mol Cell. 2011;43:550–560. doi: 10.1016/j.molcel.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelhakim AH, Oakes EC, Sauer RT, Baker TA. Unique contacts direct high-priority recognition of the tetrameric Mu transposase-DNA complex by the AAA+ unfoldase ClpX. Mol Cell. 2008;30:39–50. doi: 10.1016/j.molcel.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi EG, Reisinger SJ, Skerker JM, Arif M, Perchuk BS, Ryan KR, Laub MT. Regulation of the bacterial cell cycle by an integrated genetic circuit. Nature. 2006;444:899–904. doi: 10.1038/nature05321. [DOI] [PubMed] [Google Scholar]
- Brilli M, Fondi M, Fani R, Mengoni A, Ferri L, Bazzicalupo M, Biondi EG. The diversity and evolution of cell cycle regulation in alpha-proteobacteria: a comparative genomic analysis. BMC Syst Biol. 2010;4:52. doi: 10.1186/1752-0509-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien P, Grant RA, Sauer RT, Baker TA. Structure and substrate specificity of an SspB ortholog: design implications for AAA+ adaptors. Structure. 2007a;15:1296–1305. doi: 10.1016/j.str.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Chien P, Perchuk BS, Laub MT, Sauer RT, Baker TA. Direct and adaptor-mediated substrate recognition by an essential AAA+ protease. Proc Natl Acad Sci USA. 2007b;104:6590–6595. doi: 10.1073/pnas.0701776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury T, Chien P, Ebrahim S, Sauer RT, Baker TA. Versatile modes of peptide recognition by the ClpX N domain mediate alternative adaptor-binding specificities in different bacterial species. Protein Sci. 2010;19:242–254. doi: 10.1002/pro.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen M, Christen B, Folcher M, Schauerte A, Jenal U. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J Biol Chem. 2005;280:30829–30837. doi: 10.1074/jbc.M504429200. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1 Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Curtis PD, Brun YV. Getting in the loop: regulation of development in Caulobacter crescentus. Microbiol Mol Biol Rev. 2010;74:13–41. doi: 10.1128/MMBR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JH, Baker TA, Sauer RT. Engineering synthetic adaptors and substrates for controlled ClpXP degradation. J Biol Chem. 2009;284:21848–21855. doi: 10.1074/jbc.M109.017624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Donatis GM, Singh SK, Viswanathan S, Maurizi MR. A single ClpS monomer is sufficient to direct the activity of the ClpA hexamer. J Biol Chem. 2010;285:8771–8781. doi: 10.1074/jbc.M109.053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan DA, Weber-Ban E, Bukau B. Targeted delivery of an ssrA-tagged substrate by the adaptor protein SspB to its cognate AAA+ protein ClpX. Mol Cell. 2003;12:373–380. doi: 10.1016/j.molcel.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Duerig A, Abel S, Folcher M, Nicollier M, Schwede T, Amiot N, Giese B, Jenal U. Second messenger-mediated spatiotemporal control of protein degradation regulates bacterial cell cycle progression. Genes Dev. 2009;23:93–104. doi: 10.1101/gad.502409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domian IJ, Quon KC, Shapiro L. Cell type-specific phosphorylation and proteolysis of a transcriptional regulator controls the G1-to-S transition in a bacterial cell cycle. Cell. 1997;90:415–424. doi: 10.1016/s0092-8674(00)80502-4. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- Henry JT, Crosson S. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu Rev Microbiol. 2011;65:261–286. doi: 10.1146/annurev-micro-121809-151631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iniesta AA, McGrath PT, Reisenauer A, McAdams HH, Shapiro L. A phospho-signaling pathway controls the localization and activity of a protease complex critical for bacterial cell cycle progression. Proc Natl Acad Sci USA. 2006;103:10935–10940. doi: 10.1073/pnas.0604554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal U, Fuchs T. An essential protease involved in bacterial cell-cycle control. EMBO J. 1998;17:5658–5669. doi: 10.1093/emboj/17.19.5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal U. The role of proteolysis in the Caulobacter crescentus cell cycle and development. Res Microbiol. 2009;160:687–695. doi: 10.1016/j.resmic.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Sauer RT. Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci USA. 2005;102:1390–1395. doi: 10.1073/pnas.0409634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J, Molière N, Dougan DA, Turgay K. Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Microbiol. 2009;7:589–599. doi: 10.1038/nrmicro2185. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, De Nisco NJ, Chien P, Simmons LA, Walker GC. Sinorhizobium meliloti CpdR1 is critical for co-ordinating cell cycle progression and the symbiotic chronic infection. Mol Microbiol. 2009;73:586–600. doi: 10.1111/j.1365-2958.2009.06794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Seidel M, Sauer RT, Baker TA. A specificity-enhancing factor for the ClpXP degradation machine. Science. 2000;289:2354–2356. doi: 10.1126/science.289.5488.2354. [DOI] [PubMed] [Google Scholar]
- Levchenko I, Grant RA, Wah DA, Sauer RT, Baker TA. Structure of a delivery protein for an AAA+ protease in complex with a peptide degradation tag. Mol Cell. 2003;12:365–372. doi: 10.1016/j.molcel.2003.08.014. [DOI] [PubMed] [Google Scholar]
- McGinness KE, Baker TA, Sauer RT. Engineering controllable protein degradation. Mol Cell. 2006;22:701–707. doi: 10.1016/j.molcel.2006.04.027. [DOI] [PubMed] [Google Scholar]
- Möglich A, Ayers RA, Moffat K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure. 2009;17:1282–1294. doi: 10.1016/j.str.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher SB, Flynn JM, Sauer RT, Baker TA. Latent ClpX-recognition signals ensure LexA destruction after DNA damage. Genes Dev. 2003a;17:1084–1089. doi: 10.1101/gad.1078003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher SB, Sauer RT, Baker TA. Distinct peptide signals in the UmuD and UmuD’ subunits of UmuD/D’ mediate tethering and substrate processing by the ClpXP protease. Proc Natl Acad Sci USA. 2003b;100:13219–13224. doi: 10.1073/pnas.2235804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul R, Jaeger T, Abel S, Wiederkehr I, Folcher M, Biondi EG, Laub MT, Jenal U. Allosteric regulation of histidine kinases by their cognate response regulator determines cell fate. Cell. 2008;133:452–461. doi: 10.1016/j.cell.2008.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongratz I, Antonsson C, Whitelaw ML, Poellinger L. Role of the PAS domain in regulation of dimerization and DNA binding specificity of the dioxin receptor. Mol Cell Biol. 1998;18:4079–4088. doi: 10.1128/mcb.18.7.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, Inobe T, Hatch AJ, Matouschek A. Substrate selection by the proteasome during degradation of protein complexes. Nat Chem Biol. 2009;5:29–36. doi: 10.1038/nchembio.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan SK, Pritchard S, Viollier PH. Coupling prokaryotic cell fate and division control with a bifunctional and oscillating oxidoreductase homolog. Dev Cell. 2010;18:90–101. doi: 10.1016/j.devcel.2009.10.024. [DOI] [PubMed] [Google Scholar]
- Román-Hernández G, Hou JY, Grant RA, Sauer RT, Baker TA. The ClpS adaptor mediates staged delivery of N-end rule substrates to the AAA+ ClpAP protease. Mol Cell. 2011;43:217–228. doi: 10.1016/j.molcel.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- Schuenemann VJ, Kralik SM, Albrecht R, Spall SK, Truscott KN, Dougan DA, Zeth K. Structural basis of N-end rule substrate recognition in Escherichia coli by the ClpAP adaptor protein ClpS. EMBO Rep. 2009;10:508–514. doi: 10.1038/embor.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Hoskins JR, Wickner S. Binding and degradation of heterodimeric substrates by ClpAP and ClpXP. J Biol Chem. 2005;280:5449–5455. doi: 10.1074/jbc.M412411200. [DOI] [PubMed] [Google Scholar]
- Skerker JM, Prasol MS, Perchuk BS, Biondi EG, Laub MT. Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a system-level analysis. PLoS Biol. 2005;3:e334. doi: 10.1371/journal.pbio.0030334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HK, Eck MJ. Structural basis of degradation signal recognition by SspB, a specificity-enhancing factor for the ClpXP proteolytic machine. Mol Cell. 2003;12:75–86. doi: 10.1016/s1097-2765(03)00271-5. [DOI] [PubMed] [Google Scholar]
- Stüdemann A, Noirclerc-Savoye M, Klauck E, Becker G, Schneider D, Hengge R. Sequential recognition of two distinct sites in sigma(S) by the proteolytic targeting factor RssB and ClpX. EMBO J. 2003;22:4111–4120. doi: 10.1093/emboj/cdg411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Holmgren RA, Matouschek A. A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-kappaB. Nat Struct Mol Biol. 2005;12:1045–1053. doi: 10.1038/nsmb1018. [DOI] [PubMed] [Google Scholar]
- Tuckerman JR, Gonzalez G, Sousa EH, Wan X, Saito JA, Alam M, Gilles-Gonzalez MA. An oxygen-sensing diguanylate cyclase and phosphodiesterase couple for c-di-GMP control. Biochemistry. 2009;48:9764–9774. doi: 10.1021/bi901409g. [DOI] [PubMed] [Google Scholar]
- Wah DA, Levchenko I, Baker TA, Sauer RT. Characterization of a specificity factor for an AAA+ ATPase: assembly of SspB dimers with ssrA-tagged proteins and the ClpX hexamer. Chem Biol. 2002;9:1237–1245. doi: 10.1016/s1074-5521(02)00268-5. [DOI] [PubMed] [Google Scholar]
- Wang KH, Sauer RT, Baker TA. ClpS modulates but is not essential for bacterial N-end rule degradation. Genes Dev. 2007;21:403–408. doi: 10.1101/gad.1511907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KH, Roman-Hernandez G, Grant RA, Sauer RT, Baker TA. The molecular basis of N-end rule recognition. Mol Cell. 2008;32:406–414. doi: 10.1016/j.molcel.2008.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.