

Abstract
Mitochondria have been widely studied for their critical role in cellular metabolism, energy production and cell death. New developments in research on mitochondria derived from studies in yeast have led to the discovery of entirely new mitochondrial processes that have implications for mitochondrial function in heart failure. Recent studies have identified that maintaining normal mitochondrial morphology and function depends on the dynamic balance of mitochondrial fusion and fission (division). Mitochondrial fusion and fission are constant ongoing processes, which are essential for the maintenance of normal mitochondrial function. Studies in heart failure have been very limited, but suggest a possible reduction in mitochondrial fusion. As mitochondrial fusion and fission have important links to apoptosis, a key mechanism of loss of cardiac myocytes in heart failure, there are many implications for both heart failure research and treatment.
Mitochondrial fission/fusion
The critical importance of mitochondria as a source of high energy phosphates to power contraction was recognized early in cardiovascular research, and as a result there is a long history of research on mitochondria and the heart. After a twenty year lull in cardiac mitochondrial research, there has been an upsurge in interest as a result of novel findings that change the way we perceive mitochondria and the availability of new tools to investigate mitochondrial function. Mitochondria are now recognized has having critical roles in regulating cellular metabolism, calcium handling and programmed cell death (1). In the 1990’s, scientists studying yeast began a series of groundbreaking studies that changed how we view mitochondria and demonstrated that the mitochondria are dynamic organelles that constantly undergo fission(division) and fusion. The development of targeted mitochondrial fluorescent proteins and dyes enabled real time analysis of mitochondrial shape changes and structure rearrangements in living cells from yeast to mammalian(1). Mitochondria form a highly dynamic network whose morphology is regulated by frequent fission and fusion events (figure 1). In contrast to the common view that mitochondria are static factories producing energy, mitochondria have been found to be vibrant organelles that continuously divide and fuse within the cell and have functions extending beyond energy production to cell signaling (1–2). At steady state the frequencies of fusion and fission events are balanced, maintaining the overall morphology/function of the mitochondria (2). The rate of fission and fusion in mammalian cells is thought to be much slower than in simpler organisms, such as the yeast, where fusion and fission were first described. Disruption of fission and/or fusion can lead to cellular dysfunction and as apoptosis, an important cause of cardiac myocyte death, particularly in heart failure (2–6).
Figure 1.
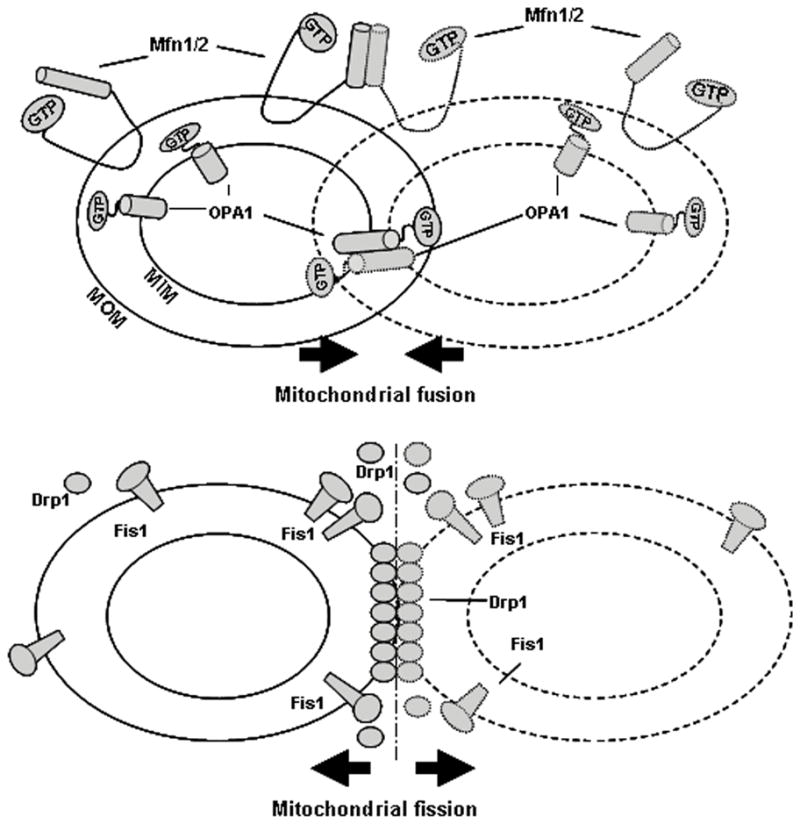
Diagram summarizes mitochondrial fusion and fission. Mfn1 and Mfn2 contribute to mitochondrial outer membrane (MOM) fusion and OPA1 is involved in the mitochondrial inner membrane (MIM) fusion. The complete fusion of two mitochondria needs both of these MOM and MIM events. On the other hand, Fis1 and Drp1 divide one mitochondria into two. When fission exceeds fusion, small fragmented mitochondria predominate.
The mitochondria are thought to have arisen from the incorporation of unicellular organisms by other unicellular organisms millions of years ago. Thus the mitochondrion has a lipid bilayer as a membrane and contains a small amount of DNA coding for a subset of mitochondrial proteins. Complete mitochondrial fusion requires two-steps, where the outer and inner mitochondria fuse in separate events (2). The proteins involved in mammalian mitochondrial fusion show distinct mitochondrial sublocalization (figure 1). Mitofusin 1 and 2 (MFN1/MFN2, Mgm1 in yeast see Table I), are located on the outer mitochondrial membrane and contribute to outer membrane fusion (1–2). Optic atrophy 1 (OPA1, Fzo1 in yeast) is located in the inner membrane and intermembrane space, and is thought to be key for inner membrane fusion. MFN1/2 are large GTPases with two membrane-spanning domains that orient both the N- and C-termini in the cytosol (7). Both the cytosolic C-terminal coiled-coil and GTPase domains are required for fusion (8). Although the outer and inner mitochondria fusion events are distinct processes, it is likely that there are proteins synchronizing the steps required for fusion, so that both the outer and inner membrane are fused. In yeast, Ugo1 is a likely candidate for this role. It has been shown to be essential for fusion and to bind both Mgm1 and Fzo1 (9). There is no identifiable ortholog of Ugo1 in mammalian cells, but it is predicted that some protein fills this role. Reduction in either MFN1 or 2 can be partially compensated by the other MFN protein, suggesting some overlap in function (10).
Table 1.
Mammalian mitochondrial fusion/fission associated proteins
| Mammals | Yeast | Location | Links to diseases | |
|---|---|---|---|---|
| Fusion Proteins | MFN1/MFN2 | Fzo1p | Mitochondrial outer membrane | Decreased in Alzheimer’s disease (AD); MFN2 mutated in CMT type 2A disease; MFN2 is repressed in obesity |
| OPA1 | Mgm1p | Mitochondrial inner membrane | Mutated in Autosomal dominant optic atrophy (ADOA); decreased in heart failure; decreased in AD; Mutated in CMT type 2 | |
|
| ||||
| Fission Proteins | Drp1 | Dnm1p | Cytosol/mitochondri al association | Increased in heart failure; decreased AD |
| Fis1 | Fis1p | Mitochondrial outer membrane | Increased in AD | |
Proteins controlling mitochondrial fission in mammalian cells include Drp1 (DNM1 in yeast) (11) and Fis1(12). Drp1 is primarily in the cytoplasm, but also forms complexes on the outer surface of mitochondria at fission sites (13). Human Fis1 (hFis1) circumscribes the outer surface of mitochondria and is not localized specifically to mitochondrial scission sites (12, 14). hFis1 contains a tetratricopeptide repeat motif, which forms a scaffold to mediate protein – protein interactions and promote the assembly of multi-protein complexes on the cytoplasmic side of the mitochondrial membrane (14). hFis1’s c-terminal hydrophobic tail anchors the protein to the outer mitochondrial membrane (12). Interestingly, these two proteins that regulate mitochondrial fission also regulate peroxisomal fission (15).
Cellular function of mitochondrial fission/fusion
Apoptosis
Apoptosis is a key mechanism of cardiac myocyte loss in heart failure. Mitochondrial fission and fusion have important roles in regulating cell apoptosis. During apoptosis, the mitochondrial network fragments, resulting in smaller and more numerous mitochondria (3, 16–17). In the cell fission and fusion are symmetric events with equal amounts of fusion and fission occurring. Loss of this equality leads to activation of apoptotic pathways leading to cell death. Increased fission, decreased fusion, or both can induce caspase activation (16), Bax translocation tomitochondria and cytochrome c release (16). Bak may regulate apoptosis by interacting with the mitofusins (4). These studies suggest a critical mechanistic link between the dynamic process of fission/fusion and apoptosis.
Elimination of any of the mitochondrial fusion proteins, MFN1, MFN 2 or OPA1, induces mitochondrial fragmentation, as expected, as there is a loss of the symmetry between fusion and fission (3, 17). Down-regulation of Opa1 expression in cells by RNA interference(RNAi) results in spontaneous apoptosis(17). Recently we found that overexpression of Opa1 increased mitochondrial elongation, but could not prevent ischemia-induced apoptosis, suggesting that OPA1 levels are tightly controlled in the cell, and perturbation of these levels is not protective(5). Thus, the balance of fission and fusion proteins is critical to the maintenance of cell homeostasis and the prevention of apoptosis.
The fission protein, Drp1’s, binding to mitochondria increases during apoptosis (18). Provocatively, Drp1 co-localizes with Bax at mitochondrial scission sites (16). Inhibition of Drp1’s GTPase activity with a dominant negative protein defective in GTP binding (Drp1K38A) results in elongated (loss of fission) mitochondria (16, 18) and delays the process of cell death (16) in mammalian cells, suggesting that the GTPase activity is essential for Drp’s role in fission as well as in apoptosis. Down-regulating mammalian Fis1 expression by RNAi leads to mitochondrial elongation (19). Similarly, overexpression of hFis1 has been reported to induce apoptosis (20), again implicating the involvement of mitochondrial fission in apoptosis.
Mitochondrial DNA deletion
One theory is that mitochondrial fusion/fission serves as a means of distribution of mitochondrial DNA (mtDNA) to the progeny of mammalian cells (21). This function of the fission/fusion processes is supported by experiments demonstrating that the loss of these proteins leads to loss of mtDNA (22–23) The mammalian DRP1 and MFN1/2 were found to be indirect contributors to mtDNA integrity and distribution within the mitochondrial network in neuronal (24) and muscle cells (22). Chen et. al. has demonstrated that loss of MFN1 and MFN2 caused severe mitochondrial dysfunction, compensatory mitochondrial proliferation, and muscle atrophy. Mice without MFN1 and 2 have marked mtDNA depletion in muscle that precedes physiological abnormalities. Furthermore, the mitochondrial genome of the mutant muscle rapidly accumulates point mutations and deletions (22). Recently, Elachouri et. al. reported that silencing of OPA1 exon 4b leads to mtDNA depletion, secondary to inhibition of mtDNA replication, and to marked alteration of mtDNA distribution throughout the mitochondrial network (23). In a series of clinical studies, it has been found that specific OPA1 mutations induce the accumulation of mtDNA deletions in skeletal muscle of patients (25). Altogether these observations suggested that altering mitochondrial fusion proteins are essential for the maintenance of mtDNA integrity.
Mitochondrial respiration
The mitochondrial fission and fusion proteins have a key role in mitochondrial energetics and function (1). A number of reports demonstrate a direct correlation between mitochondrial fusion and the oxidative phosphorylation capacity (26). Inhibition of mitochondrial fusion results in reduced oxygen consumption (22). Silencing of MFN2 expression substantially impairs pyruvate, glucose, and fatty acid oxidation; consistent with this, there is a marked reduction in MFN2 levels in skeletal muscle from both obese humans and animal models of obesity (27). Stably transfected fibroblasts with an MFN2 antisense sequence show reduced glucose oxidation and oxygen consumption (28). These results were recently confirmed by studies in MEFs with targeted null mutations of both MFN1 and MFN2 (10). These cells show loss of mitochondrial membrane potential, reduced endogenous respiration, and an incapacity to increase respiration upon the addition of the ionophore 2,4-dinitrophenol. Respiration was restored by overexpression of MFN2 (10). On the other hand, MFN2 over-expression leads to increased respiratory complex activity, glycolysis and mitochondrial biogenesis (27) Interestingly, MFN2 expression is up-regulated in conditions of high energy demand (i.e. exercise or cold exposure) or in response to pro-apoptotic stimuli, which elicit a rapid stress-induced mitochondrial hyperfusion coupled with a transient increase in mitochondrial ATP production (29).
Alterations in OPA1 also affect mitochondrial metabolism. Depletion of OPA1 by RNAi in MEFs leads to reduction in basal respiration and inabilityto enhance oxygen consumption in the presence of the uncoupler 2,4-dinitrophenol(10). Studies of fibroblasts from patients with specific OPA1 mutations (c.2708delTTAG, c.1705 + 1G4T, c.1516 + 1G4, c.2819-2A4C, c.1346_1347insC) show impaired ATP synthesis driven by complex I substrates and decreased rates of mitochondrial fusion (26). In contrast other OPA1 mutations (c1410_144314del38, c.239A>G, c.2883A>C, c.2522A>G, c.2780T>A, c.1654delT, c.1929delC, c.2708delTTAG) did not affect mitochondrial activity and bioenergetics (30). These results suggest that there are key domains in OPA1 for mitochondrial function, and that further knowledge of post-translational modifications, protein folding and proteins that interact with OPA1 should shed light on the mechanism by which the ADOA OPA1 mutations cause disease without effecting measured mitochondrial function.
Current evidence indicates that alterations in mitochondrial fission proteins can also affect mitochondrial metabolism. Knockdown of DRP1 with RNAi in HeLa cells reduced the basal rate of oxygen consumption, decreased coupled respiration, and lowered the rate of ATP synthesis (31). Expression of a dominant negative mutant form of DRP1 also caused a marked reduction in the respiratory capacity in rat insulinoma (INS1) cells (32). Hyperglycemia induced fission, and inhibition of this by a dominant negative form of DRP1 markedly impaired the mitochondria’s ability to increase respiratory rate (33). RNAi-mediated Fis1 depletion also reduced the maximal respiratory activity of INS1 cells, and overexpression of Fis1 rescued the phenotype (32).
Fission and Fusion in Heart Failure
There are a number of inherited neuropathies, such as Charcot-Marie-Tooth disease, that are associated with mutation of genes encoding mitochondrial proteins. Two genes involved in mitochondrial fusion, OPA1 and MFN2, have been implicated in inherited optic neuropathies (1–2). Abnormal expression of MFN2 has been reported in Parkinson’s disease and type 2 diabetes (1). However, few studies have addressed whether mitochondrial fusion/fission is involved in heart failure.
Several groups have recently published data supporting the presence of dynamic mitochondria in both neonatal cardiac myocytes and cultured cardiac-like cells lines (34–35). In cultured neonatal ventricular myocytes, inhibition of mitochondrial fission by over-expression of a dominant negative mutant form of Drp1, Drp1-K38A, prevents over-production of ROS, mitochondrial permeability transition pore formation and subsequent cell death under sustained high glucose conditions (34). Cytosolic Ca2+ overload induced by thapsigargin (Tg) or potassium chloride (KCl) causes a rapid, transient mitochondrial fragmentation in neonatal and adult cardiomyocytes (35). Calcium overload is a common feature in HF, and this may increase mitochondrial fission, thus further contributing to the decrease in energetics in the failing heart. (36) Dorn et. al. have demonstrated that Drosophila heart tube-specific silencing of OPA1 and mitochondrial assembly regulatory factor (MARF) increases mitochondrial morphometric heterogeneity and induces heart tube dilation with profound contractile impairment. In this model human MFN1/2 rescued MARF RNAi induced cardiomyopathy (37).
Unlike the cardiac like cell types mentioned above, in adult mammalian cardiomyocytes mitochondria are highly organized and compacted between contractile filaments (interfibrillar) or adjacent to the sarcolemma (subsarcolemmal). During heart failure, interfibrillar mitochondria may lose their normal organization (5). There is also a reduction in size and density of interfibrillar mitochondria in rodent models of heart failure (38) We have recently reported that OPA1 (5) is decreased in both human and rat HF. Furthermore, reduction in OPA1 increased apoptosis both at baseline and after simulated ischemia, via cytochrome c release from mitochondria in H9c2 cells. Electron microscopic data showed increased number and decreased size of the mitochondria in a coronary artery ligation rat heart failure model (Figure 2). In another study, Javadov found decreased MFN2, increased Fis1 and no change in OPA1 expression in rat hearts 12–18 weeks after myocardial infarction (39). More recently, it has been reported that cardiac myocyte mitochondria lacking MFN-2 are pleiomorphic and apt to be larger. MFN-2-deficient mice have mild cardiac hypertrophy and mild depression of cardiac function (40).
Figure 2.

Representative EM images for mitochondria from Sham and HF rat left ventricles. In sham control myocytes, mitochondria are well organized and compacted between contractile filaments or adjacent to the sarcolemma. However, in heart failure mitochondria are disorganized and smaller compared to sham, suggesting increased mitochondrial fission vs. fusion in HF. Bar = 500nm, M = mitochondria.
Mitochondrial fusion/fission are relatively newly described processes and thus studies in heart failure are quite limited; however, mitochondrial morphologic changes have been found in other types of cardiomyopathy. With cuprizone, a copper chelator that inhibits monoamine oxidase, the increased ROS leads to numerous partitioned mitochondria, suggestive of decreased fusion (41). The high prevalence of abnormal mitochondrial morphology in cardiac disease suggests that mitochondrial fusion/fission are frequently, if not always, impaired in cardiac disease/muscle disease. Further work will be needed to fully define the role of abnormal mitochondrial fission/fusion in HF, and whether interventions can positively influence the delicate fusion/fission balance and mitochondrial function.
Implications of these changes for treatment of heart failure
The heart, like the brain, has a high demand for ATP and is dependent on mitochondrial function. Despite intensive studies of mitochondrial dynamics in other fields, much remains to be understood in the heart, where fusion and fission occur likely at a much slower rate than in yeast. Disorders in mitochondrial organization and the presence of abnormally small and fragmented mitochondria have been observed in end-stage dilated cardiomyopathy, myocardial hibernation, and ventricular-associated congenital heart disease (35), suggesting the important role of mitochondrial fusion/fission in cardiac diseases. The contribution of asymmetric mitochondrial fusion/fission in heart failure as a cause vs. a consequence of myocardial injury remains to be defined. Nevertheless, the restoration of mitochondrial fusion/fission symmetry may help rescue the failing heart. Treatment of adult murine cardiac myocytes with mitochondrial division inhibitor-1, an inhibitor of Drp1, reduced cell death and inhibited mitochondrial permeability transition pore opening after simulated ischemia/reperfusion injury(42). In vivo mitochondrial division inhibitor-1 reduced myocardial infarct size in mice (42). These results suggest a novel pharmacological strategy for cardioprotection.
Research interest in mitochondria had waned, but with the development of new tools and models, along with the discovery of fusion and fission, new avenues of investigation have opened that have the potential to provide fresh insight into cardiac diseases, such as heart failure. Thus, the next decade of mitochondrial research should be an exciting one indeed, and holds the potential for the development of new therapies for the patient with HF.
Acknowledgments
Supported by NIH grants HL077281 and HL079071 both to AAK.
References
- 1.Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89(3):799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 2.Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci. 2010;1201:21–5. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- 3.Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, et al. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. 2011;41(2):150–60. doi: 10.1016/j.molcel.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A. 2007;104(28):11649–54. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 2009;84(1):91–9. doi: 10.1093/cvr/cvp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Zhou J, Li Y, Qin D, Li P. Mitochondrial fission controls DNA fragmentation by regulating endonuclease G. Free Radic Biol Med. 2010;49(4):622–31. doi: 10.1016/j.freeradbiomed.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 7.Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. Journal of Cell Science. 2002;115(8):1663–74. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- 8.Meeusen S, McCaffery JM, Nunnari J. Mitochondrial Fusion Intermediates Revealed in Vitro. Science. 2004;305(5691):1747–52. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- 9.Sesaki H, Jensen RE. UGO1 Encodes an Outer Membrane Protein Required for Mitochondrial Fusion. The Journal of Cell Biology. 2001;152(6):1123–34. doi: 10.1083/jcb.152.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Chomyn A, Chan DC. Disruption of Fusion Results in Mitochondrial Heterogeneity and Dysfunction. Journal of Biological Chemistry. 2005;280(28):26185–92. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 11.Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, et al. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1(5):298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23(15):5409–20. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12(8):2245–56. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki M, Jeong SY, Karbowski M, Youle RJ, Tjandra N. The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR-like helix bundle. J Mol Biol. 2003;334(3):445–58. doi: 10.1016/j.jmb.2003.09.064. [DOI] [PubMed] [Google Scholar]
- 15.Koch A, Thiemann M, Grabenbauer M, Yoon Y, McNiven MA, Schrader M. Dynamin-like Protein 1 Is Involved in Peroxisomal Fission. Journal of Biological Chemistry. 2003;278(10):8597–605. doi: 10.1074/jbc.M211761200. [DOI] [PubMed] [Google Scholar]
- 16.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15(11):5001–11. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278(10):7743–6. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 18.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1(4):515–25. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 19.Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci. 2004;117(Pt 7):1201–10. doi: 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- 20.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278(38):36373–9. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 21.Margineantu DH, Gregory Cox W, Sundell L, Sherwood SW, Beechem JM, Capaldi RA. Cell cycle dependent morphology changes and associated mitochondrial DNA redistribution in mitochondria of human cell lines. Mitochondrion. 2002;1(5):425–35. doi: 10.1016/s1567-7249(02)00006-5. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141(2):280–9. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elachouri G, Vidoni S, Zanna C, Pattyn A, Boukhaddaoui H, Gaget K, et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Research. 2011;21(1):12–20. doi: 10.1101/gr.108696.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130(3):548–62. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 25.Hudson G, Amati-Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131(2):329–37. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- 26.Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. 2008;131(2):352–67. doi: 10.1093/brain/awm335. [DOI] [PubMed] [Google Scholar]
- 27.Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, et al. The Charcots–Marie–Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Human Molecular Genetics. 2005;14(11):1405–15. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 28.Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, et al. Mitofusin-2 Determines Mitochondrial Network Architecture and Mitochondrial Metabolism. Journal of Biological Chemistry. 2003;278(19):17190–7. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 29.Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, et al. Preventing Mitochondrial Fission Impairs Mitochondrial Function and Leads to Loss of Mitochondrial DNA. PLoS One. 2008;3(9):e3257. doi: 10.1371/journal.pone.0003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayorov V, Lowrey A, Biousse V, Newman N, Cline S, Brown M. Mitochondrial oxidative phosphorylation in autosomal dominant optic atrophy. BMC Biochemistry. 2008;9(1):22. doi: 10.1186/1471-2091-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, et al. Mitochondrial bioenergetics and structural network organization. Journal of Cell Science. 2007;120(5):838–48. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- 32.Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27(2):433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8):2653–8. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu T, Sheu S-S, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovascular Research. 2008;79(2):341–51. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hom J, Yu T, Yoon Y, Porter G, Sheu SS. Regulation of mitochondrial fission by intracellular Ca2+ in rat ventricular myocytes. Biochim Biophys Acta. 2010;1797(6–7):913–21. doi: 10.1016/j.bbabio.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kane LA, Youle RJ. Mitochondrial fission and fusion and their roles in the heart. J Mol Med. 2010;88(10):971–9. doi: 10.1007/s00109-010-0674-6. [DOI] [PubMed] [Google Scholar]
- 37.Dorn GW, 2nd, Clark CF, Eschenbacher WH, Kang MY, Engelhard JT, Warner SJ, et al. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ Res. 2011;108(1):12–7. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukyanenko V, Chikando A, Lederer WJ. Mitochondria in cardiomyocyte Ca2+ signaling. The International Journal of Biochemistry & Cell Biology. 2009;41(10):1957–71. doi: 10.1016/j.biocel.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Javadov S, Rajapurohitam V, Kilic A, Hunter JC, Zeidan A, Said Faruq N, et al. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res Cardiol. 2011;106(1):99–109. doi: 10.1007/s00395-010-0122-3. [DOI] [PubMed] [Google Scholar]
- 40.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31(6):1309–28. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tandler B, Hoppel CL. Possible division of cardiac mitochondria. Anat Rec. 1972;173(3):309–23. doi: 10.1002/ar.1091730306. [DOI] [PubMed] [Google Scholar]
- 42.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121(18):2012–22. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]