

Abstract
Endocrine therapy resistance is a primary cause of clinical breast cancer treatment failure. The p38 mitogen activated protein kinase (MAPK) signaling pathway is known to promote ligand independent tumor growth and resistance to endocrine therapy. In this study, we investigated the therapeutic potential of the p38 inhibitor RWJ67657 in the treatment of tamoxifen resistant MDA-MB-361 cells. RWJ67657 dose-dependently decreased both basal and stimulated activation of p38 MAPK signaling in this drug resistant cell system. Decreased activation of p38 by RWJ67657 resulted in inhibition of the downstream p38 targets hsp27 and MAPKAPK. Diminished p38 signaling resulted in inhibition of p38-medated gene transcription. Furthermore, pharmacological inhibition of p38 by RWJ67657 decreased biological effects of p38, including ER-mediated gene expression and clonogenic survival in a dose-dependent manner. Animal studies revealed significantly decreased p38 signaling in vivo following exposure to RWJ67657. Treatment with the inhibitor markedly decreased phosphorylation of p38 in MDA-MB-361 tumors, leading to decreased transcription of both Fra-1 and progesterone receptor. Utilizing well-established xenograft tumor models, we demonstrated that RWJ67657 exhibits potent anti-tumor properties. Treatment with RWJ67657 markedly decreased tamoxifen resistant tumor growth, both in the presence and absence of estrogen. Taken together, our findings demonstrate the therapeutic potential of targeting the p38-MAPK signaling cascade in the treatment of endocrine resistant breast cancer.
Keywords: p38, mitogen-activated protein kinase, endocrine resistance, breast cancer, drug discovery, cancer biology, hormone independence, kinase inhibitors, estrogen receptor, gene transcription
Introduction
Endocrine therapeutics, such as tamoxifen or aromatase inhibitors (AIs), represent a principle treatment for estrogen receptor positive (ER (+)) breast carcinoma in the adjuvant and metastatic setting [1]. Tamoxifen is a selective estrogen receptor modulator (SERM) and functions as an ER antagonist in the breast, but in the uterus and endometrium it acts as an agonist [2]. Despite the effectiveness of treatment with tamoxifen and AIs, de novo or acquired resistance represents a major obstacle [3,4]. While the mechanisms within breast cells leading to tamoxifen resistance are currently incompletely defined, recent research suggests a role exists for altered cellular signaling pathways [4-10]. Progression to a tamoxifen resistant phenotype may depend upon and occur through crosstalk with the ER. Anti-estrogen resistance can also be induced by growth factor mediated activation of the MAPK cascade or overexpression of components of this pathway [4,11-15]. Indeed, overexpression of growth factor receptors such as EGFR/HER2 augment both genomic and non-genomic ER actions in breast cancer [16-18]. For example, the ER can directly interact with the human epidermal growth factor receptor-2 (HER2) in the membrane, and this interaction is crucial for protecting HER2-overexpressing breast cancer cells from tamoxifen-induced apoptosis [16]. This suggests that rapid cell signaling events initiated through either ER or growth factor receptors can influence a cell’s response to endocrine therapy.
Recent studies have noted a positive correlation between activated p38 levels in breast cancer and tamoxifen resistance [19]. p38 is a member of the MAPK family which includes the extracellular signal-regulated kinase (ERK1/2, ERK5, ERK8), the c-Jun N-terminal kinase (JNK1/2/3), and p38 kinase [20-23]. A complex role exists for p38 in the regulation of cell fate, with the effects being dependent upon both duration and magnitude of p38 activation and differences in cell type [24,25]. In breast epithelial cells, we along with others have demonstrated the importance of p38 signaling in regulation of apoptosis and cell survival [26,27]. p38 MAPK can potentiate the ER in part through increased phosphorylation of ER at the Thr311 activation site, and our laboratory has described a role for p38 enhancement of ER signaling through coactivator regulation [26,28]. Increased p38 activity has been implicated in the initiation and progression of carcinogenesis and has been associated with breast cancer drug resistance and invasion, suggesting that increased p38 activation may be associated with a more malignant, therapeutically resistant and metastatic phenotype [27,29-34].
The ability to disrupt p38 signaling through small molecule inhibitors represents a potential strategy for therapeutic intervention in advanced and endocrine resistant breast cancer. There are several p38 inhibitors currently in evaluation as potential chemotherapeutic agents [35]. The most well studied p38 inhibitor is SB203580, a pyridinyl imidazole that specifically inhibits p38 in vitro and suppresses the downstream effectors of p38 [36-38]. Another pyrindinyl imidazole specific p38 MAPK inhibitor, RWJ67657, has recently been used clinically to treat chronic obstructive pulmonary disease (COPD) through inhibition of plasma level increases of interleukins 6 and 8 as well as tumor necrosis factor alpha (TNF-α) [39-41]. Our laboratory has shown that RWJ67657 can partially suppress estrogen stimulated proliferation and gene expression [26]. Yet, the efficacy of RWJ67657 as a breast cancer therapeutic has yet to be thoroughly studied.
Based on p38’s putative role in breast cancer progression to tamoxifen resistance, we investigated the role of p38 targeting and the effectiveness of RWJ67657 in pre-clinical models of tamoxifen resistant breast carcinoma. In this study, we demonstrate that RWJ67657 effectively inhibits the p38 pathway in MDA-MB-361 breast cancer cells, which are ER(+) and resistant to tamoxifen. Our results indicate RWJ67657 targets the ER as well as other factors involved in breast cancer growth and proliferation, suggesting it may be a potential therapeutic that can be used to treat tamoxifen resistant breast carcinoma.
Materials and methods
Reagents
RWJ67657 was a generous gift from Johnson and Johnson Pharmaceutical Research & Development, L.L.C. (Raritan, NJ). ICI 182780 was purchased from TOCRIS (Ballwin, MO). Estradiol and tamoxifen were purchased from Sigma (St. Louis, MO).
Cell culture
Cells were cultured as previously described [42,43]. Briefly, MDA-MB-361 human metastatic breast cancer cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS), MEM essential amino acids, MEM non-essential amino acids, sodium pyruvate, insulin, and penicillin-streptomycin. Cell were incubated at 37°C in 5% humidified CO2. Prior to experiments, media containing 10% FBS was changed to phenol red free DMEM, supplemented with 5% charcoal-stripped FBS (DCC-FBS), MEM essential amino acids, MEM non-essential amino acids, sodium pyruvate, glutamine, and penicillin-streptomycin. Before transfection the MDA-MB-361 cells were plated at an approximate density of 5 x 105 cells/well (approximately 80% confluency) in 24-well plates and were sustained for 24 hours in DMEM with 5% DCC-FBS.
Western blots
As previously described, MDA-MB-361 cells were incubated in DMEM with 5% DCC-FBS for two days and were then transferred into 10 cm2 plates [42,44]. Plates were incubated for an additional 24 hours and then vehicle (DMSO or ethanol) or increasing amounts of RWJ67657 were added. After two hours, media was removed from the plates and 100 uL of MPER containing phosphatase inhibitors (Sigma, St. Louis, MO) and protease inhibitors (Calbiochem, San Diego, CA) was added. Cells were agitated manually in the lysis buffer for 5 minutes and were then centrifuged at 12000 x g for 5 min. The supernatant was removed, sample buffer and reducing agent were added, and the samples were boiled and loaded onto a 4-12% SDS-PAGE gel (Invitrogen, Carlsbad, CA). Gels were then transferred to nitrocellulose membranes. Rabbit anti-phospho-p38, anti-p38, anti-phosho-hsp27, anti-hsp27 at a 1:1000 dilution were used as primary antibodies (Cell Signaling, Danvers, MA) followed by incubation with IR-tagged secondary antibodies (LiCor Biosciences, Lincoln, NE). The blots were analyzed using the Odyssey Infrared Imaging System (LiCor Biosciences, Lincoln, NE).
Real time PCR
Real time RT-PCR was performed similar to previously reported studies [45,46]. In brief, total cellular RNA was extracted using the RNeasy® mini column (Qiagen, Valencia, CA), following the manufacturer’s instructions. The concentration of RNA was determined using an ultraviolet spectrophotometer. Reverse transcription (RT) was performed using the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA). The level of SDF-1 and PgR transcripts was determined using the iQ5 real-time quantitative PCR detection system (BioRad Inc., Hercules, CA). Primers for PCR were designed to span intron/exon junctions to minimize amplification of residual genomic DNA. The primer sequences for PgR is (sense and anti-sense, respectively): PgR (5’-TACCCGCCCTATCTCAACTACC-3’; 5’-TGCTTCATCCCCACAGATTAAACA-3’) PCR mix contained optimal concentrations of primers, cDNA and SYBR Green PCR Master Mix (BioRad Lab.). Quantification and relative gene expression were calculated with internal controls. The ratio between these values obtained provided the relative gene expression levels.
ERE-luciferase assay
Reporter gene assays were performed according to previously published protcols [42,47]. MDA-MB-361 cells were transfected with an ERE-luciferase reporter construct (Panomics, Freemont, CA) (0.4 ug/well) using Effectene (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Approximately 4-6 hours post-transfection, vehicle (DMSO or ethanol), E2 (10 pM), or E2 plus increasing concentrations of RWJ67657 was added and allowed to incubate overnight. The following day, cells were lysed using 150 uL of MPER (mammalian protein extraction reagent, Pierce Chemical, Rockford, IL) and were incubated at room temperature for 5 minutes. Samples were then read in a Berthold luminometer using Bright-Glo luminescence reagent (Promega, Madison, WI) as a substrate. Raw data was analyzed using Origin software (OriginLab Corporation, Northampton, MA).
Colony assays
Colony assays were performed as previously described [42,43]. MDA-MB-361 cells were plated in 6-well plates at a density of 500 cells/well in 3 mL DMEM with 5% DCC-FBS. Twenty-four hours later ethanol (EtOH), E2 (1.0 nM) alone or E2 + increasing concentrations of RWJ67657 and then monitored for colony growth. Ten days later the cells were fixed with 3% glutaraldehyde for 15 min. Following fixation, the plates were washed and stained with a 0.4% solution of crystal violet in 20% methanol for 30 min, washed with PBS, and dried. Colonies of ≥ 30 cells were counted as positive. Results were normalized to DMSO vehicle treated control cells.
Animal studies
Xenograft tumor studies were conducted as previously described [44,48]. Nu/nu immunecompromised female ovariectomized mice (29-32 days old) were obtained from Charles River Laboratories (Wilmington, MA). The animals were allowed a period of adaptation in a sterile and pathogen-free environment with phytoestrogen-free food and water ad libitum. Placebo or estradiol pellets (0.72 mg, 60-day release; Innovative Research of America, Sarasota, FL) were implanted s.c. in the lateral area of the neck in the middle point between the ear and shoulder using a precision trochar (10 gauge). MDA-MB-361 cells were harvested and viable cells mixed with Matrigel Reduced Factors (BD Biosciences, San Jose, CA) or injected alone in PBS. Injections (5x106 cells/injection) were made bilaterally into the mammary fat pad. All the procedures in animals were carried out under anesthesia using a mix of isofluorane and oxygen delivered by mask. Tumor size was measured every 2 days using a digital caliper. The volume of the tumor was calculated using the following formula: 4/3pLM2, where L is the larger radius, and M is the smaller radius. At necropsy on day 21, animals were euthanized by decapitation after exposure to a CO2 chamber. Tumors were removed and either frozen in liquid nitrogen or fixed in 10% formalin for further analysis. All procedures involving these animals were conducted in compliance with State and Federal laws, standards of the U.S. Department of Health and Human Services, and guidelines established by the Tulane University Animal Care and Use Committee. The facilities and laboratory animal program of Tulane University are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Results
RWJ67657 Inhibits p38 MAPK Signaling in Endocrine Therapy Resistant Breast Cancer
RWJ67657 is known to bind p38 and inhibit p38 phosphorylation and the ability of p38 to activate downstream targets in noncancerous cell systems [41]. Clinical studies have demonstrated high basal levels of phosphorylated p38 in tamoxifen resistant tumors [49]. Therefore, we set out to determine the ability of RWJ67657 to inhibit p38 MAPK activity in MDA-MB-361 breast carcinoma cells, which are ER(+) and tamoxifen resistant. Cells were treated with vehicle or increasing amounts of RWJ67657 and analyzed for protein levels of activated and total p38. Results demonstrated that these cells contain high steady-state levels of activated p38, which could not be substantially reduced by the addition of RWJ67657 (Figure 1A). Analysis of downstream p38 signaling, however, revealed decreased downstream p38 signaling following treatment with the inhibitor. Treatment with RWJ67657 resulted in decreased phosphorylation of hsp27, a known downstream target of p38 and (Figure 1A) [50]. Quantitative analysis of the western blot revealed that the 10 μM dose of RWJ67657 reduced activated levels of hsp27 by approximately 60%, without affecting phosphorylated levels of p38 (Figure 1B). These findings suggest that in vitro, RWJ67657 blocks p38 signaling through inhibition of p38 activation of downstream targets without affecting direct phosphorylation of p38.
Figure 1.
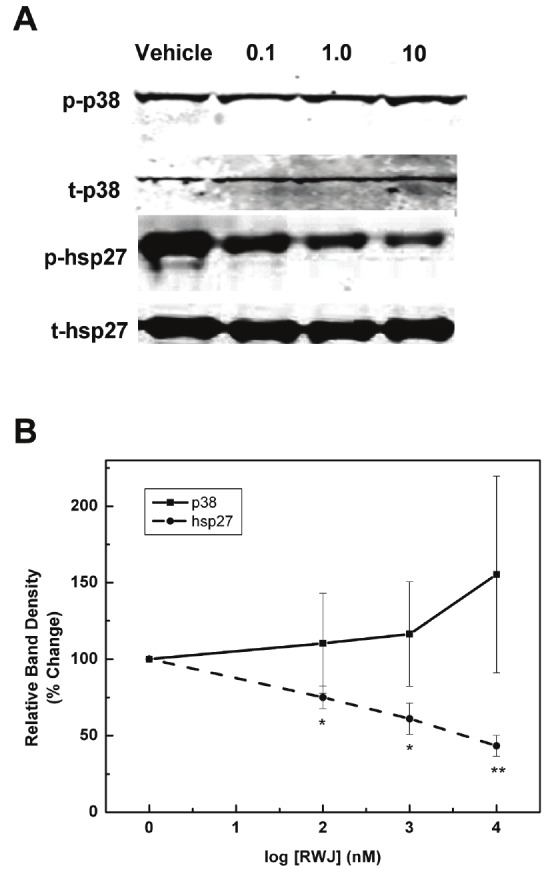
RWJ67657 inhibits basal p38 signaling pathway in tamoxifen resistant breast Cancer. (A) MDA-MB-361 cells were treated with increasing concentrations of RWJ67657 (0.1 μM, 1.0 μM, or 10 μM). Lysates were assayed by western blot using antibodies directed against phosphorylated p38 (pp38), total p38 (t-p38), phosphorylated hsp27 (phsp27), or total hsp27 (t-hsp27). The blots shown are representative of three independent experiments. (B) Densitometry analysis of protein levels. Quantified phosphorylated protein bands were divided by total protein bands within each lane and total protein load was normalized to Rho-GDI levels. The vehicle lane of each protein was set to 100% for comparative purposes. Data points and error bars represent the mean ± S.E.M. of three independent experiments in duplicate (*** p < 0.001, ** p < 0.01, *p < 0.05).
RWJ67657 inhibits stimulated p38 MAPK signaling
Given the above findings of RWJ67657-mediated inhibition of basal p38 signaling, we next determined the ability RWJ67657 to block stimulated p38 signaling in vitro. We along with others have demonstrated the ability of the organochlorine o’,p’-DDT (DDT) to induce activation of p38 in vitro [51,52]. Therefore, MDA-MB-361 cells were treated with vehicle and RWJ67657 for 24 h, in the presence and absence of DDT and analyzed the cells for total and phosphorylated levels of p38. We also determined protein activation of the downstream p38 effectors hsp27 and MAPKAPK (Figure 2A). Similar to our above findings, RWJ67657 blocked p38-induced phosphorylation of both hsp27 and MAPKAPK without affecting levels of activated p38. Quantitative analysis of protein densitometry revealed a statistically significant decrease in DDT stimulated activation of hsp27 and MAPKAPK following treatment with RWJ67657 (Figure 2B).
Figure 2.
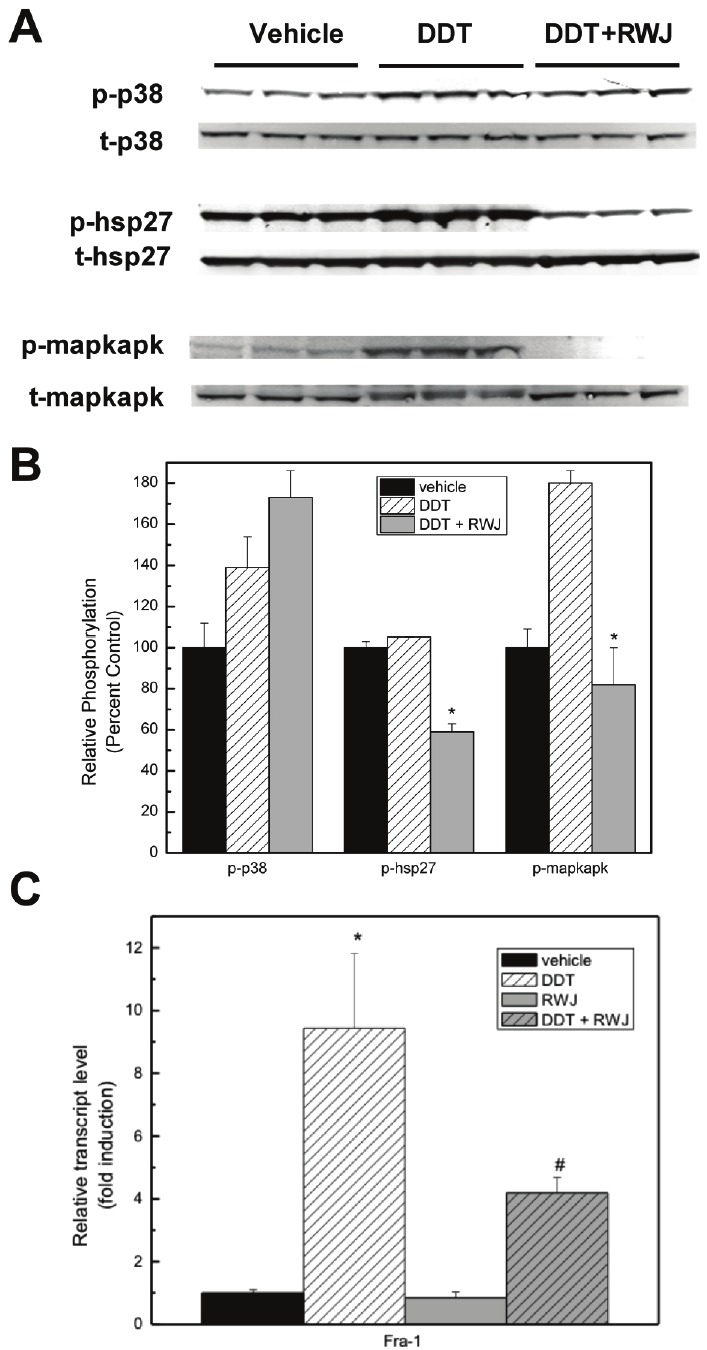
RWJ67657 inhibits stimulated p38 signaling. (A) MDA-MB-361 cells were treated with vehicle, DDT or RWJ+DDT. Lysates were assayed by western blot using antibodies directed against phosphorylated p38 (p-p38), total p38 (t-p38), phosphorylated hsp27 (p-hsp27), total hsp27 (t-hsp27), phosphorylated mapkapk (p- mapkapk), and total mapkapk (mapkapk). Each lane represents an independent experiment. (B) Densitometry analysis of the above western blots was performed. Quantified phosphorylated protein bands were divided by total protein bands within each lane and total protein was normalized to Rho-GDI levels. The vehicle lane of each protein was set to 100% for comparative purposes. Data points and error bars represent the mean ± S.E.M. of three independent experiments in duplicate (*** p < 0.001, ** p < 0.01, *p < 0.05). (C) MDA-MB-361 cells were grown in charcoal-stripped media for 48 hours, followed by treatment with vehicle, DDT (50 μM), RWJ67657 (10 μM), or DDT + RWJ67657. Samples were prepared in triplicate and analyzed using quantitative PCR for the expression of Fra-1. * indicates statistical significance compared to vehicle and # represents statistical significance compared to DDT.
We further validated the finding that RWJ67657 inhibits p38 signaling through analysis of downstream p38-mediated gene transcription. Fra-1 is a known p38 regulated gene, both in vitro and in vivo [53-56]. Therefore, we utilized quantitative RT-PCR to determine the ability of RWJ67657 to alter Fra-1 gene transcription. As seen in Figure 2C, treatment with the inhibitor markedly decreased DDT stimulated Fra-1 transcription, suggesting that the decrease in p38 activation by RWJ67657 results in diminished downstream p38 gene regulation. Taken together, these results provide strong evidence that RWJ67657 inhibits p38 MAPK signaling in endocrine resistant breast cancer cells.
Pharmacological inhibition of p38 decreases ER signaling and tamoxifen resistant breast cancer survival
We next addressed whether RWJ67657-induced inhibition of p38 signaling translated to a reduction in p38-mediated biological endpoints. The p38 MAPK pathway is known to crosstalk with the estrogen receptor to promote ER’s proliferative effects [28]. Therefore, we determined if RWJ67657 had an effect on ER transcriptional activity. MDA-MB-361 cells were transiently transfected with an ERE-luciferase reporter construct and treated with increasing concentrations of RWJ67657 in the presence of estrogen. As seen in Figure 3, treatment with RWJ67657 decreased estrogen-induced ERE transcriptional activity in a dose-dependent manner. At the highest concentration tested, RWJ67657 reduced ER transcriptional activity by approximately 70%. Taken together these results suggest that p38-mediated activation of ER can be effectively inhibited by RWJ67657.
Figure 3.
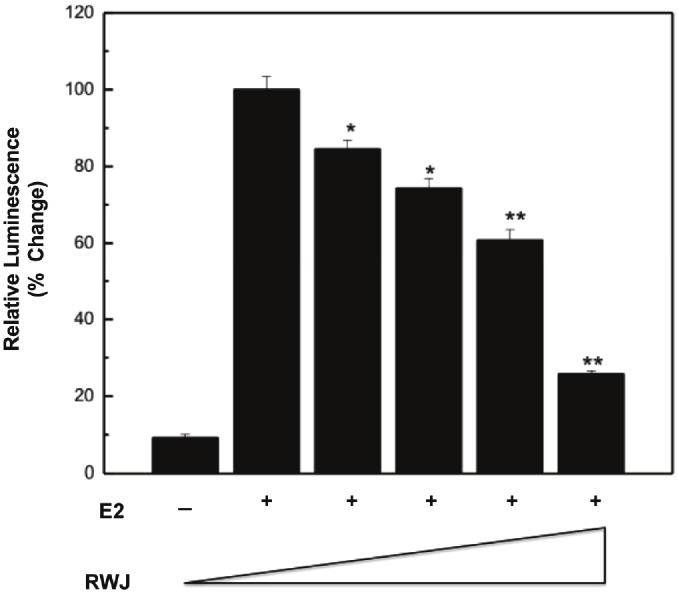
Pharmacological inhibition of p38 blocks ER transcription activity in vitro. MDA-MB-361 cells were grown in charcoal stripped media for 48 hours. The cells were then transfected with an ERE-luciferase reporter construct, followed by the addition of vehicle, 10 pM estradiol (E2), or E2 plus increasing concentrations of RWJ67657 (0, 1, 2, 4, and 10 μM). Assays were performed in triplicate. Results were normalized to E2 control. Data points and error bars represent the mean ± S.E.M. of three independent experiments in duplicate (*** p < 0.001, ** p < 0.01, *p < 0.05).
Because the p38 pathway is known to promote breast cancer cell survival and proliferation as a means of mediating tumorigenesis, [57,58], we determined whether the inhibition of p38 by RWJ67657 resulted in decreased downstream clonogenic survival. MDA-MB361 cells were incubated with vehicle, estrogen, or estrogen plus increasing concentrations of RWJ67657 and analyzed for colony formation as a measure of cancer clonogenic survival. As seen in Figure 4, treatment with RWJ67657 suppressed estrogen-stimulated clonogenic survival of these tamoxifen resistant cells, suggesting that RWJ67657 decreases downstream biological effects of p38. These results provide proof of principle that RWJ67657 blocks downstream p38 signaling activity and biological effects in vitro.
Figure 4.
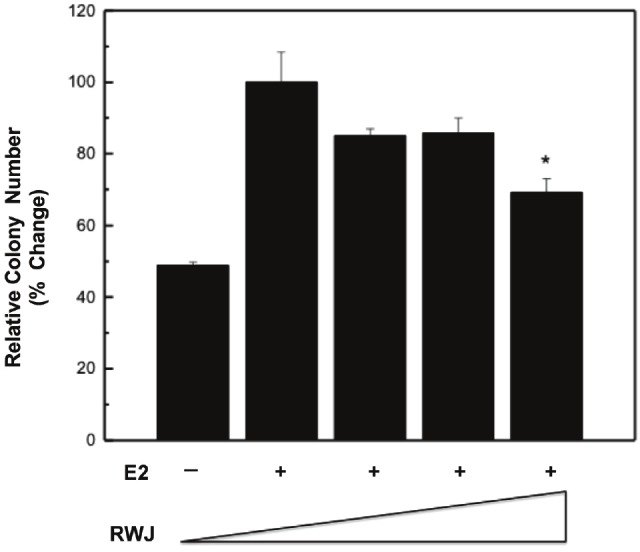
RWJ67657 inhibits tamoxifen resistant breast cancer survival. Cells were grown in charcoalstripped media for 48 hours, followed by incubation with vehicle, 1 nM estradiol or estradiol plus increasing concentrations of RWJ67657 (0.1 μM, 1 μM, 10 μM). Data points and error bars represent the mean ± S.E.M. of three independent experiments in duplicate (*** p < 0.001, ** p < 0.01, *p < 0.05).
RWJ67657 inhibits p38 MAPK signaling in vivo
Given the above findings that RWJ67657 blocks p38 MAPK signaling in vitro, we further validated the ability of RWJ67657 to inhibit p38 signaling in vivo using a murine xenograft model. We hypothesized that prolonged exposure of endocrine resistant cells to RWJ67657 may directly decrease activation of phosphorylation of p38. Female, ovariectomized SCID mice were injected subcutaneously with MDA-MB-361 cells. After tumor formation on day 10, mice were treated twice daily for two days with vehicle (DMSO/PBS) or RWJ67657 (20 mg/kg/day). Homogenized tumor samples at necroscopy on day 12 were analyzed for phosphorylated p38 protein levels using Western blot analysis (Figure 5A). Results demonstrate significantly decreased activation of p38 in RWJ67657 treated tumors compared to vehicle control tumors. Quantitative analysis of the western blot demonstrate RWJ67657 treated tumors contain a roughly 40% decrease in p38 phosphorylation compared to control tumors (Figure 5B). These in vivo results correlated with our in vitro findings that RWJ67657 reduces p38 signaling in endocrine resistant breast cancer.
Figure 5.
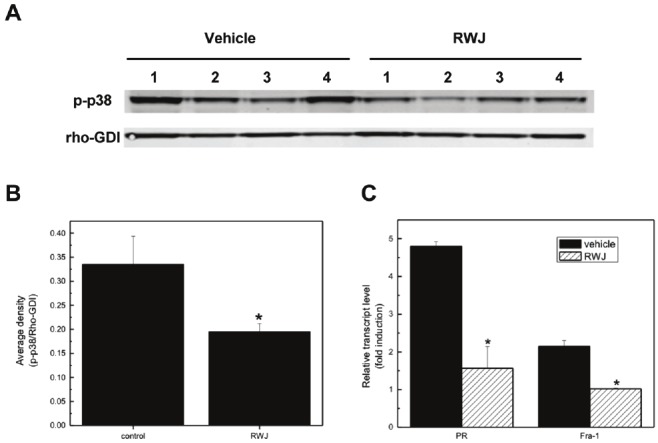
RWJ67657 inhibits p38 signaling in vivo. (A) MDA-MB-361 cells were injected into immunocompromised mice s.c. as described in the methods section. After tumor formation, mice were treated twice daily for two days with RWJ67657 (20mg/kg). At necroscopy, tumors were homogenized and lysates were assays for phosphorylated p38 levels using western blot analysis. Four tumors from each group were analyzed. Total protein load was normalized using the Rho-GDI antibody. (B) Bands from the western blot were quantified and phosphorylated p38 levels were normalized to Rho-GDI band densities (*** p < 0.001, ** p < 0.01, *p < 0.05). (B) The above tumors from each group were analyzed for expression of Fra-1 and progesterone receptor (PR) using qRT-PCR (*** p < 0.001, ** p < 0.01, *p < 0.05).
To further validate the ability of RWJ67657 to inhibit p38 MAPK activation, we investigated whether the decrease in p38 activation in the above tumor samples translated into decreased p38-mediated downstream effects. We measured relative gene expression levels of the p38-regulated gene Fra-1in RWJ67657 treated tumors and compared results to levels in vehicle treated tumors. Quantitative RT-PCR analysis of tumor samples from the above xenograft tumor experiment revealed that, similar to our in vitro findings, RWJ67657 blocked Fra-1 mRNA levels in vivo (Figure 5C). Given that MDA-MB-361 cells are ER(+) and the ability of p38 to crosstalk with the ER, we also examined the ability of RWJ67657 to inhibit transcription of the ER-regulated progesterone receptor gene (PR). MDA-MB-361 tumors treated with vehicle and RWJ65657 were analyzed for mRNA expression of Fra-1 and PR. Results demonstrated that tumors from the RWJ67657 treated animals contain decreased transcript levels of both Fra-1 and PR compared to vehicle (Figure 5C). These findings provide proof of principle that RWJ67657 blocks downstream p38 genomic signaling in vitro and in vivo. Furthermore, these results indicate a possible link between RWJ67657-mediated inhibition of p38 and ER signaling.
RWJ67657 inhibits endocrine resistant breast cancer tumor growth
We next determined the ability of RWJ67657 to suppress in vivo tumor growth utilizing a well-established xenograft tumor model [42,59,60]. MDA-MB-361 cells were injected into the mammary fat pad of ovariectomized and immunocompromised mice with and without concomitant estrogen pellets. After tumor formation, mice were treated with vehicle, RWJ67657 or RWJ67657 plus tamoxifen for 5 days and measured for tumor volume. In the presence of estrogen, RWJ67657 treatment resulted in an approximately 3.5 fold decrease in tumor volume compared to endpoint vehicle treated tumors (Figure 6A). This finding suggests that decreased p38 and ER signaling by RWJ67657 translates into decreased tumor proliferation. The anti-tumor effects of RWJ67657 were even more profound in the absence of estrogen. By day 40, tumors treated with RWJ67657 exhibited an approximately 4.5 fold decrease in tumor volume compared to vehicle treated tumors (Figure 6B). RWJ67657 treatment was significantly more efficacious than tamoxifen and exposure of tumors to RWJ67650 alone exhibited more anti-tumor effects than combination treatment with tamoxifen and RWJ67650. These results demonstrate that inhibition of p38 by RWJ67657 blocks downstream p38 mediated tumor growth in vivo and show the therapeutic potential of targeting p38 signaling in the treatment of endocrine resistant breast cancer.
Figure 6.
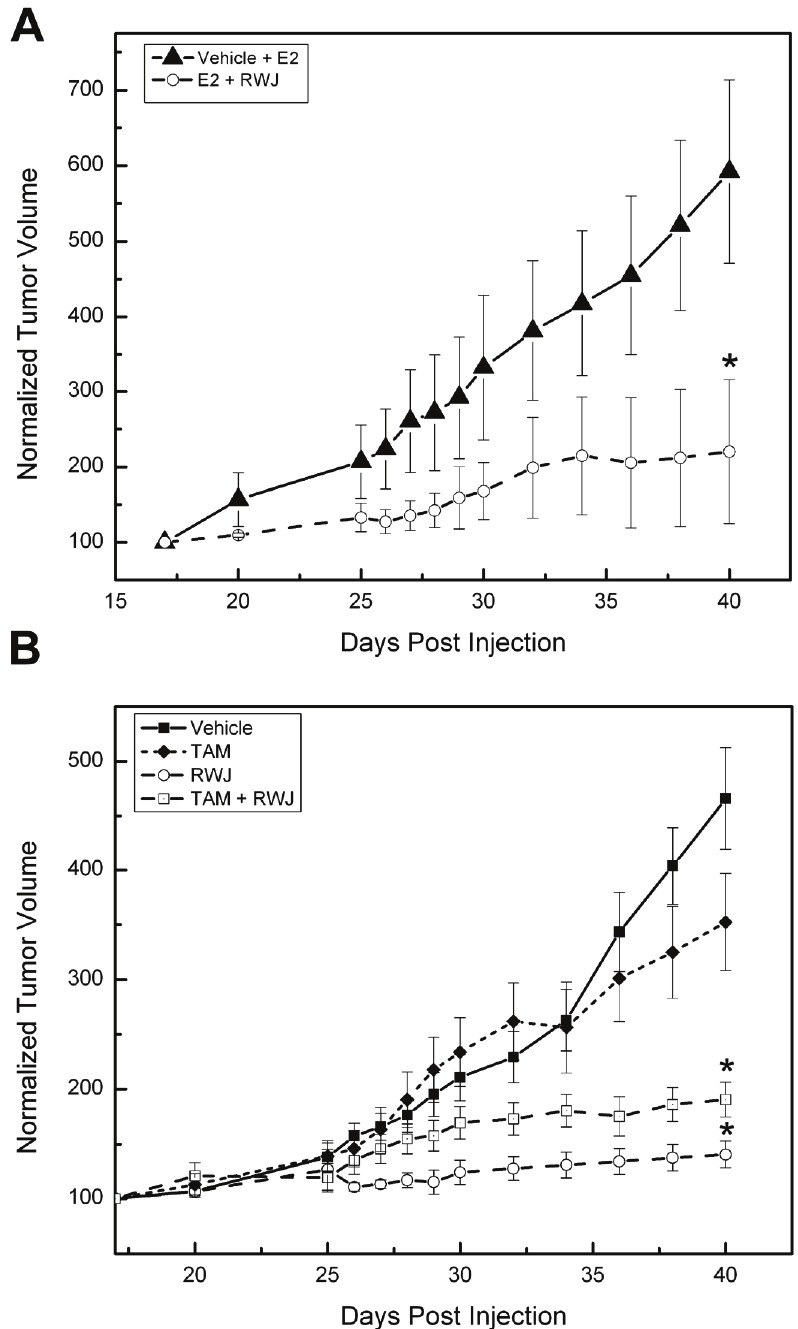
RWJ67657 inhibits endocrine resistant breast cancer tumor growth. (A) MDA-MB-361 cells were injected into mice along with slow release estradiol pellets as described in the methods section and measured for palpable tumorigenesis. Upon tumor formation, mice were treated with either vehicle or RWJ67657 and measured for tumor growth (*p < 0.05). (B) MDA-MB-361 cells were injected into mice in the absence of estrogen as described in the methods section and measured for palpable tumorigenesis. Upon tumor formation, mice were treated with either vehicle, tamoxifen (Tam), RWJ67657 or RWJ67657 + Tam and measured for tumor growth (*p < 0.05).
Discussion
Estrogen receptor-positive, tamoxifen resistant breast cancer is a major obstacle for women who have undergone therapy and subsequently developed tamoxifen resistant tumors. The mechanisms by which breast cancer cells transition from tamoxifen sensitivity to resistance remain unclear. However, both preclinical and clinical studies suggest that crosstalk between the ER and growth factor and/or stress kinase signaling cascades may play a role in driving cells to tamoxifen resistance [4,9,61]. In 2005 Guiterrez et al. showed that in paired clinical breast cancer specimens, increased phosphorylation of p38 was positively associated with acquired tamoxifen resistance [49]. Furthermore, Gauthier, et al. showed that in a premalignant variant of human mammary epithelial cells, increased basal p38 activation led to increased COX-2 expression compared to control cells [62]. COX-2 is an immediate early gene and its overexpression may be an initiating event in breast carcinogenesis [63-65]. More recently, Massarweh, et al. demonstrated in a mouse xenograft model that when ER(+) MCF-7 breast cancer cells developed a resistance to tamoxifen, the steady state levels of activated p38 were increased compared to the tamoxifen sensitive cells. Furthermore, they showed that this elevation in phosphorylated p38 could be abrogated by gefitinib, an epidermal growth factor receptor (EGFR) inhibitor [19]. These results underscore the importance of growth factor/MAPK crosstalk with p38 in the progression of breast cancer cells to tamoxifen resistance.
In this study, we tested a novel p38 inhibitor’s ability to inhibit tamoxifen resistant breast cancer growth. Several studies have demonstrated breast cancer cells that acquire tamoxifen resistance retain some level of ER expression [66-68]. To this end, we utilized the ER(+) MDA-MB-361 cell line, which is a model for acquired tamoxifen resistant and metastatic breast cancer [69,70]. Using MDA-MB-361 cells, we show in vitro that although RWJ67657 does not block phosphorylation of p38 directly, it inhibits the ability of p38 to phosphorylate and activate its downstream effector protein hsp27. RWJ67657 also exhibited potent anti-tumor properties, both in the presence and absence of estrogen, and inhibited long-term clonogenic survival. These findings demonstrate that inhibition of p38 by RWJ67657 blocks the downstream biological effects of the p38 signaling system.
Using quantitative PCR, we show that RWJ67657 inhibits the ability of p38 to stimulate transcription of Fra-1 both in vitro and in vivo. The fos-related antigen 1 (Fra-1) is an immediate early gene encoding a member of the AP-1 family of transcription factors and is involved in cell proliferation, differentiation, apoptosis, and other biological processes [53,56]. Fra-1 transcription can be regulated by p38 [71]. The ability of RWJ67657 to inhibit p38 and decrease Fra-1 expression is particularly intriguing because Fra-1, as an immediate early gene, seems to play a pivotal role in the process of cell transformation and carcinogenesis. Suzuki et al showed that Fra-1 exhibited oncogenic potential in that its overexpression in rat fibroblasts stimulated anchorage-independent growth in the absence of clear morphological transformation [72]. The inhibition of Fra-1 protein synthesis by stable transfection with a Fra-1 antisense construct significantly reduced the malignant phenotype of transformed thyroid cells [55]. Furthermore, Fra-1 induced morphological transformation and increased in vitro invasiveness and motility of epithelioid adenocarcinoma cells [54]. High Fra-1 expression is also associated with a more malignant cancer cell phenotype, suggesting this gene may have a significant role in cancer progression, including that of the breast [53,73,74]. In light of this data, a drug that can selectively knockdown expression of Fra-1 may have therapeutic promise in treating metastatic breast cancer, especially in cells that have already transitioned to tamoxifen resistance.
Given that MDA-MB-361 cells are ER(+) and p38 is known to phosphorylate and activate the ER, we tested the ability of RWJ67657 to inhibit the transcription of the progesterone receptor (PR), a known ER-mediated gene. Using a mouse xenograft model, we showed RWJ67657 was able to significantly reduce PR expression in MDA-MB-361 tumors compared to vehicle alone. We further demonstrated that RWJ67657 decreased PR expression at least in part by altering ER signaling. RWJ67657 also inhibited ER activity in an in vitro ERE-luciferase assay. These results support the recently identified role of p38-ER crosstalk in the progression of breast carcinoma [19,26,28,75].
The findings presented here demonstrate that p38 is crucial for breast cancer proliferation and transition from tamoxifen sensitivity to resistance. We show that the novel p38 inhibitor RWJ67657, which inhibits both p38 and ER signaling to promote its effects, shows promise as a targeted anti-cancer agent. Taken together, our results demonstrate the therapeutic potential of targeting p38 in the treatment of endocrine therapy resistant breast cancer.
Acknowledgements
This work was supported by National Institutes of Health grants CA125806 (MB) and DK059389 (MB).
References
- 1.Schiff R, Chamness GC, Brown PH. Advances in breast cancer treatment and prevention: preclinical studies on aromatase inhibitors and new selective estrogen receptor modulators (SERMs) Breast Cancer Res. 2003;5:228–231. doi: 10.1186/bcr626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Agthoven T, Sieuwerts AM, Meijer-van Gelder ME, Look MP, Smid M, Veldscholte J, Sleijfer S, Foekens JA, Dorssers LC. Relevance of breast cancer antiestrogen resistance genes in human breast cancer progression and tamoxifen resistance. J. Clin. Oncol. 2009;27:542–549. doi: 10.1200/JCO.2008.17.1462. [DOI] [PubMed] [Google Scholar]
- 3.Fan M, Yan PS, Hartman-Frey C, Chen L, Paik H, Oyer SL, Salisbury JD, Cheng AS, Li L, Abbosh PH, Huang TH, Nephew KP. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 2006;66:11954–11966. doi: 10.1158/0008-5472.CAN-06-1666. [DOI] [PubMed] [Google Scholar]
- 4.Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, Skaar TC, Gomez B, O'Brien K, Wang Y, Hilakivi-Clarke LA. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22:7316–7339. doi: 10.1038/sj.onc.1206937. [DOI] [PubMed] [Google Scholar]
- 5.Ghayad SE, Vendrell JA, Larbi SB, Dumontet C, Bieche I, Cohen PA. Endocrine resistance associated with activated ErbB system in breast cancer cells is reversed by inhibiting MAPK or PI3K/Akt signaling pathways. Int J Cancer. 2010;126:545–562. doi: 10.1002/ijc.24750. [DOI] [PubMed] [Google Scholar]
- 6.Nicholson RI, Hutcheson IR, Hiscox SE, Knowlden JM, Giles M, Barrow D, Gee JM. Growth factor signalling and resistance to selective oestrogen receptor modulators and pure anti-oestrogens: the use of anti-growth factor therapies to treat or delay endocrine resistance in breast cancer. Endocr Relat Cancer. 2005;12(Suppl 1):S29–36. doi: 10.1677/erc.1.00991. [DOI] [PubMed] [Google Scholar]
- 7.Normanno N, Di Maio M, De Maio E, De Luca A, de Matteis A, Giordano A, Perrone F. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer. 2005;12:721–747. doi: 10.1677/erc.1.00857. [DOI] [PubMed] [Google Scholar]
- 8.Schiff R, Massarweh SA, Shou J, Bharwani L, Arpino G, Rimawi M, Osborne CK. Advanced concepts in estrogen receptor biology and breast cancer endocrine resistance: implicated role of growth factor signaling and estrogen receptor coregulators. Cancer Chemother Pharmacol. 2005;56(Suppl 1):10–20. doi: 10.1007/s00280-005-0108-2. [DOI] [PubMed] [Google Scholar]
- 9.Zilli M, Grassadonia A, Tinari N, Di Giacobbe A, Gildetti S, Giampietro J, Natoli C, Iacobelli S. Molecular mechanisms of endocrine resistance and their implication in the therapy of breast cancer. Biochim Biophys Acta. 2009;1795:62–81. doi: 10.1016/j.bbcan.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 11.Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2:101–112. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- 12.Johnston SR, Head J, Pancholi S, Detre S, Martin LA, Smith IE, Dowsett M. Integration of signal transduction inhibitors with endocrine therapy: an approach to overcoming hormone resistance in breast cancer. Clin Cancer Res. 2003;9:524S–532S. [PubMed] [Google Scholar]
- 13.MacGregor JI, Jordan VC. Basic guide to the mechanisms of antiestrogen action. Pharmacol Rev. 1998;50:151–196. [PubMed] [Google Scholar]
- 14.Martini PG, Katzenellenbogen BS. Modulation of estrogen receptor activity by selective coregulators. J Steroid Biochem Mol Biol. 2003;85:117–122. doi: 10.1016/s0960-0760(03)00207-3. [DOI] [PubMed] [Google Scholar]
- 15.Schiff R, Massarweh S, Shou J, Osborne CK. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin Cancer Res. 2003;9:447S–454S. [PubMed] [Google Scholar]
- 16.Chung YL, Sheu ML, Yang SC, Lin CH, Yen SH. Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int J Cancer. 2002;97:306–312. doi: 10.1002/ijc.1614. [DOI] [PubMed] [Google Scholar]
- 17.Kumar R, Wang RA, Mazumdar A, Talukder AH, Mandal M, Yang Z, Bagheri-Yarmand R, Sahin A, Hortobagyi G, Adam L, Barnes CJ, Vadlamudi RK. A naturally occurring MTA1 variant sequesters oestrogen receptor-alpha in the cytoplasm. Nature. 2002;418:654–657. doi: 10.1038/nature00889. [DOI] [PubMed] [Google Scholar]
- 18.Stoica GE, Franke TF, Moroni M, Mueller S, Morgan E, Iann MC, Winder AD, Reiter R, Wellstein A, Martin MB, Stoica A. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 2003;22:7998–8011. doi: 10.1038/sj.onc.1206769. [DOI] [PubMed] [Google Scholar]
- 19.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68:826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 20.Chao TH, Hayashi M, Tapping RI, Kato Y, Lee JD. MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J Biol Chem. 1999;274:36035–36038. doi: 10.1074/jbc.274.51.36035. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Cobb MH. Regulation of stress-responsive mitogen-activated protein (MAP) kinase pathways by TAO2. J Biol Chem. 2001;276:16070–16075. doi: 10.1074/jbc.M100681200. [DOI] [PubMed] [Google Scholar]
- 22.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 23.Maemura M, Iino Y, Koibuchi Y, Yokoe T, Morishita Y. Mitogen-activated protein kinase cascade in breast cancer. Oncology. 1999;57(Suppl 2):37–44. doi: 10.1159/000055273. [DOI] [PubMed] [Google Scholar]
- 24.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 25.Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007;1773:1358–1375. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 26.Frigo DE, Basu A, Nierth-Simpson EN, Weldon CB, Dugan CM, Elliott S, Collins-Burow BM, Salvo VA, Zhu Y, Melnik LI, Lopez GN, Kushner PJ, Curiel TJ, Rowan BG, McLachlan JA, Burow ME. p38 mitogen-activated protein kinase stimulates estrogen-mediated transcription and proliferation through the phosphorylation and potentiation of the p160 coactivator glucocorticoid receptor-interacting protein 1. Mol Endocrinol. 2006;20:971–983. doi: 10.1210/me.2004-0075. [DOI] [PubMed] [Google Scholar]
- 27.Weldon CB, Parker AP, Patten D, Elliott S, Tang Y, Frigo DE, Dugan CM, Coakley EL, Butler NN, Clayton JL, Alam J, Curiel TJ, Beckman BS, Jaffe BM, Burow ME. Sensitization of apoptotic-ally-resistant breast carcinoma cells to TNF and TRAIL by inhibition of p38 mitogenactivated protein kinase signaling. Int J Oncol. 2004;24:1473–1480. [PubMed] [Google Scholar]
- 28.Lee H, Bai W. Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol Cell Biol. 2002;22:5835–5845. doi: 10.1128/MCB.22.16.5835-5845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta A, Rosenberger SF, Bowden GT. Increased ROS levels contribute to elevated transcription factor and MAP kinase activities in malignantly progressed mouse keratinocyte cell lines. Carcinogenesis. 1999;20:2063–2073. doi: 10.1093/carcin/20.11.2063. [DOI] [PubMed] [Google Scholar]
- 30.Reddy KB, Nabha SM, Atanaskova N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev. 2003;22:395–403. doi: 10.1023/a:1023781114568. [DOI] [PubMed] [Google Scholar]
- 31.Guo YL, Kang B, Han J, Williamson JR. p38beta MAP kinase protects rat mesangial cells from TNF-alpha-induced apoptosis. J Cell Biochem. 2001;82:556–565. doi: 10.1002/jcb.1180. [DOI] [PubMed] [Google Scholar]
- 32.Nemoto S, Xiang J, Huang S, Lin A. Induction of apoptosis by SB202190 through inhibition of p38beta mitogen-activated protein kinase. J Biol Chem. 1998;273:16415–16420. doi: 10.1074/jbc.273.26.16415. [DOI] [PubMed] [Google Scholar]
- 33.Chen J, Baskerville C, Han Q, Pan ZK, Huang S. Alpha(v) integrin, p38 mitogen-activated protein kinase, and urokinase plasminogen activator are functionally linked in invasive breast cancer cells. J Biol Chem. 2001;276:47901–47905. doi: 10.1074/jbc.M107574200. [DOI] [PubMed] [Google Scholar]
- 34.Kim MS, Lee EJ, Kim HR, Moon A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003;63:5454–5461. [PubMed] [Google Scholar]
- 35.Banerjee A, Koziol-White C, Panettieri R Jr. p38 MAPK inhibitors, IKK2 inhibitors, and TNFalpha inhibitors in COPD. Curr Opin Pharmacol. 2012 doi: 10.1016/j.coph.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henklova P, Vrzal R, Papouskova B, Bednar P, Jancova P, Anzenbacherova E, Ulrichova J, Maurel P, Pavek P, Dvorak Z. SB203580, a pharmacological inhibitor of p38 MAP kinase transduction pathway activates ERK and JNK MAP kinases in primary cultures of human hepatocytes. Eur J Pharmacol. 2008;593:16–23. doi: 10.1016/j.ejphar.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Clerk A, Sugden PH. The p38-MAPK inhibitor, SB203580, inhibits cardiac stress-activated protein kinases/c-Jun N-terminal kinases (SAPKs/JNKs) FEBS Lett. 1998;426:93–96. doi: 10.1016/s0014-5793(98)00324-x. [DOI] [PubMed] [Google Scholar]
- 38.Ward SG, Parry RV, Matthews J, O'Neill L. A p38 MAP kinase inhibitor SB203580 inhibits CD28-dependent T cell proliferation and IL-2 production. Biochem Soc Trans. 1997;25:304S. doi: 10.1042/bst025304s. [DOI] [PubMed] [Google Scholar]
- 39.Westra J, Doornbos-van der Meer B, de Boer P, van Leeuwen MA, van Rijswijk MH, Limburg PC. Strong inhibition of TNF-alpha production and inhibition of IL-8 and COX-2 mRNA expression in monocyte-derived macrophages by RWJ 67657, a p38 mitogen-activated protein kinase (MAPK) inhibitor. Arthritis Res Ther. 2004;6:R384–392. doi: 10.1186/ar1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J Clin Pharmacol. 2003;43:406–413. doi: 10.1177/0091270002250615. [DOI] [PubMed] [Google Scholar]
- 41.Wadsworth SA, Cavender DE, Beers SA, Lalan P, Schafer PH, Malloy EA, Wu W, Fahmy B, Olini GC, Davis JE, Pellegrino-Gensey JL, Wachter MP, Siekierka JJ. RWJ 67657, a potent, orally active inhibitor of p38 mitogen-activated protein kinase. J Pharmacol Exp Ther. 1999;291:680–687. [PubMed] [Google Scholar]
- 42.Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, Elliott S, Smith CD, Burow ME, Beckman BS. Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther. 2011;11:678–689. doi: 10.4161/cbt.11.7.14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antoon JW, Liu J, Ponnapakkam AP, Gestaut MM, Foroozesh M, Beckman BS. Novel D: -erythro N-octanoyl sphingosine analogs as chemo- and endocrine-resistant breast cancer therapeutics. Cancer Chemother Pharmacol. 2010;65:1191–1195. doi: 10.1007/s00280-009-1233-0. [DOI] [PubMed] [Google Scholar]
- 44.Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, Rhodes LV, Ashe HB, Wiese TE, Smith CD, Burow ME, Beckman BS. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. 2010;151:5124–5135. doi: 10.1210/en.2010-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhodes LV, Muir SE, Elliott S, Guillot LM, Antoon JW, Penfornis P, Tilghman SL, Salvo VA, Fonseca JP, Lacey MR, Beckman BS, McLachlan JA, Rowan BG, Pochampally R, Burow ME. Adult human mesenchymal stem cells enhance breast tumorigenesis and promote hormone independence. Breast Cancer Res Treat. 2010;121:293–300. doi: 10.1007/s10549-009-0458-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boue SM, Tilghman SL, Elliott S, Zimmerman MC, Williams KY, Payton-Stewart F, Miraflor AP, Howell MH, Shih BY, Carter-Wientjes CH, Segar C, Beckman BS, Wiese TE, Cleveland TE, McLachlan JA, Burow ME. Identification of the potent phytoestrogen glycinol in elicited soybean (Glycine max) Endocrinology. 2009;150:2446–2453. doi: 10.1210/en.2008-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antoon JW, Meacham WD, Bratton MR, Slaughter EM, Rhodes LV, Ashe HB, Wiese TE, Burow ME, Beckman BS. Pharmacological inhibition of sphingosine kinase isoforms alters estrogen receptor signaling in human breast cancer. J Mol Endocrinol. 2011;46:205–216. doi: 10.1530/JME-10-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rhodes LV, Short SP, Neel NF, Salvo VA, Zhu Y, Elliott S, Wei Y, Yu D, Sun M, Muir SE, Fonseca JP, Bratton MR, Segar C, Tilghman SL, Sobolik-Delmaire T, Horton LW, Zaja-Milatovic S, Collins-Burow BM, Wadsworth S, Beckman BS, Wood CE, Fuqua SA, Nephew KP, Dent P, Worthylake RA, Curiel TJ, Hung MC, Richmond A, Burow ME. Cytokine receptor CXCR4 mediates estrogen-independent tumorigenesis, metastasis, and resistance to endocrine therapy in human breast cancer. Cancer Res. 2011;71:603–613. doi: 10.1158/0008-5472.CAN-10-3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, Schiff R, Osborne CK, Dowsett M. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol. 2005;23:2469–2476. doi: 10.1200/JCO.2005.01.172. [DOI] [PubMed] [Google Scholar]
- 50.Xu L, Chen S, Bergan RC. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene. 2006;25:2987–2998. doi: 10.1038/sj.onc.1209337. [DOI] [PubMed] [Google Scholar]
- 51.Bratton MR, Frigo DE, Vigh-Conrad KA, Fan D, Wadsworth S, McLachlan JA, Burow ME. Organochlorine-mediated potentiation of the general coactivator p300 through p38 mitogen-activated protein kinase. Carcinogenesis. 2009;30:106–113. doi: 10.1093/carcin/bgn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frigo DE, Tang Y, Beckman BS, Scandurro AB, Alam J, Burow ME, McLachlan JA. Mechanism of AP-1-mediated gene expression by select organochlorines through the p38 MAPK pathway. Carcinogenesis. 2004;25:249–261. doi: 10.1093/carcin/bgh009. [DOI] [PubMed] [Google Scholar]
- 53.Belguise K, Kersual N, Galtier F, Chalbos D. FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene. 2005;24:1434–1444. doi: 10.1038/sj.onc.1208312. [DOI] [PubMed] [Google Scholar]
- 54.Kustikova O, Kramerov D, Grigorian M, Berezin V, Bock E, Lukanidin E, Tulchinsky E. Fra-1 induces morphological transformation and increases in vitro invasiveness and motility of epithelioid adenocarcinoma cells. Mol Cell Biol. 1998;18:7095–7105. doi: 10.1128/mcb.18.12.7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vallone D, Battista S, Pierantoni GM, Fedele M, Casalino L, Santoro M, Viglietto G, Fusco A, Verde P. Neoplastic transformation of rat thyroid cells requires the junB and fra-1 gene induction which is dependent on the HMGI-C gene product. EMBO J. 1997;16:5310–5321. doi: 10.1093/emboj/16.17.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Young MR, Colburn NH. Fra-1 a target for cancer prevention or intervention. Gene. 2006;379:1–11. doi: 10.1016/j.gene.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Ballif BA, Blenis J. Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ. 2001;12:397–408. [PubMed] [Google Scholar]
- 58.Breitwieser W, Lyons S, Flenniken AM, Ashton G, Bruder G, Willington M, Lacaud G, Kouskoff V, Jones N. Feedback regulation of p38 activity via ATF2 is essential for survival of embryonic liver cells. Genes Dev. 2007;21:2069–2082. doi: 10.1101/gad.430207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duong BN, Elliott S, Frigo DE, Melnik LI, Vanhoy L, Tomchuck S, Lebeau HP, David O, Beckman BS, Alam J, Bratton MR, McLachlan JA, Burow ME. AKT regulation of estrogen receptor beta transcriptional activity in breast cancer. Cancer Res. 2006;66:8373–8381. doi: 10.1158/0008-5472.CAN-05-3845. [DOI] [PubMed] [Google Scholar]
- 60.Rhodes LV, Muir SE, Elliott S, Guillot LM, Antoon JW, Penfornis P, Tilghman SL, Salvo VA, Fonseca JP, Lacey MR, Beckman BS, McLachlan JA, Rowan BG, Pochampally R, Burow ME. Adult human mesenchymal stem cells enhance breast tumorigenesis and promote hormone independence. Breast Cancer Res Treat. 2010;121:293–300. doi: 10.1007/s10549-009-0458-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, Osborne CK. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. 2004;10:331S–336S. doi: 10.1158/1078-0432.ccr-031212. [DOI] [PubMed] [Google Scholar]
- 62.Gauthier ML, Pickering CR, Miller CJ, Fordyce CA, Chew KL, Berman HK, Tlsty TD. p38 regulates cyclooxygenase-2 in human mammary epithelial cells and is activated in premalignant tissue. Cancer Res. 2005;65:1792–1799. doi: 10.1158/0008-5472.CAN-04-3507. [DOI] [PubMed] [Google Scholar]
- 63.Half E, Tang XM, Gwyn K, Sahin A, Wathen K, Sinicrope FA. Cyclooxygenase-2 expression in human breast cancers and adjacent ductal carcinoma in situ. Cancer Res. 2002;62:1676–1681. [PubMed] [Google Scholar]
- 64.Shim V, Gauthier ML, Sudilovsky D, Mantei K, Chew KL, Moore DH, Cha I, Tlsty TD, Esserman LJ. Cyclooxygenase-2 expression is related to nuclear grade in ductal carcinoma in situ and is increased in its normal adjacent epithelium. Cancer Res. 2003;63:2347–2350. [PubMed] [Google Scholar]
- 65.Boland GP, Butt IS, Prasad R, Knox WF, Bundred NJ. COX-2 expression is associated with an aggressive phenotype in ductal carcinoma in situ. Br J Cancer. 2004;90:423–429. doi: 10.1038/sj.bjc.6601534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Encarnacion CA, Ciocca DR, McGuire WL, Clark GM, Fuqua SA, Osborne CK. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res Treat. 1993;26:237–246. doi: 10.1007/BF00665801. [DOI] [PubMed] [Google Scholar]
- 67.Kuukasjarvi T, Kononen J, Helin H, Holli K, Isola J. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J. Clin. Oncol. 1996;14:2584–2589. doi: 10.1200/JCO.1996.14.9.2584. [DOI] [PubMed] [Google Scholar]
- 68.Bachleitner-Hofmann T, Pichler-Gebhard B, Rudas M, Gnant M, Taucher S, Kandioler D, Janschek E, Dubsky P, Roka S, Sporn E, Jakesz R. Pattern of hormone receptor status of secondary contralateral breast cancers in patients receiving adjuvant tamoxifen. Clin Cancer Res. 2002;8:3427–3432. [PubMed] [Google Scholar]
- 69.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat. 2004;83:249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- 70.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cook SJ, Aziz N, McMahon M. The repertoire of fos and jun proteins expressed during the G1 phase of the cell cycle is determined by the duration of mitogen-activated protein kinase activation. Mol Cell Biol. 1999;19:330–341. doi: 10.1128/mcb.19.1.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suzuki T, Murakami M, Onai N, Fukuda E, Hashimoto Y, Sonobe MH, Kameda T, Ichinose M, Miki K, Iba H. Analysis of AP-1 function in cellular transformation pathways. J Virol. 1994;68:3527–3535. doi: 10.1128/jvi.68.6.3527-3535.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chiappetta G, Ferraro A, Botti G, Monaco M, Pasquinelli R, Vuttariello E, Arnaldi L, Di Bonito M, D'Aiuto G, Pierantoni GM, Fusco A. FRA-1 protein overexpression is a feature of hyperplastic and neoplastic breast disorders. BMC Cancer. 2007;7:17. doi: 10.1186/1471-2407-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zajchowski DA, Bartholdi MF, Gong Y, Webster L, Liu HL, Munishkin A, Beauheim C, Harvey S, Ethier SP, Johnson PH. Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res. 2001;61:5168–5178. [PubMed] [Google Scholar]
- 75.Bartucci M, Morelli C, Mauro L, Ando S, Surmacz E. Differential insulin-like growth factor I receptor signaling and function in estrogen receptor (ER)-positive MCF-7 and ER-negative MDA-MB-231 breast cancer cells. Cancer Res. 2001;61:6747–6754. [PubMed] [Google Scholar]