

Abstract
Purpose
To determine whether altering the dietary content of ω-6 (n-6) and ω-3 (n-3) polyunsaturated fatty acids affects the growth of androgen-sensitive prostate cancer xenografts, tumor membrane fatty acid composition, and tumor cyclooxygenase-2 and prostaglandin E2 (PGE2) levels.
Experimental Design
Individually caged male severe combined immunodeficiency mice were fed isocaloric 20% kcal fat diets with the fat derived either primarily from n-6 fatty acids (n-6 group) or with the fat consisting of n-6 and n-3 fatty acids in a ratio of 1:1 (n-3 group), and injected s.c. with Los Angeles Prostate Cancer 4 (LAPC-4) cells. Tumor volumes and mouse weights were measured weekly, caloric intake was measured 3 days per week, and tumors and serum were harvested at 8 weeks postinjection.
Results
Tumor growth rates, final tumor volumes, and serum prostate-specific antigen levels were reduced in the n-3 group relative to the n-6 group. The n-3 group tumors had decreased proliferation (Ki67 staining) and increased apoptosis (terminal nucleotidyl transferase –mediated nick end labeling staining). In vitro proliferation of LAPC-4 cells in medium containing n-3 group serum was reduced by 22% relative to LAPC-4 cells cultured in medium containing serum from the n-6 group. The n-6/n-3 fatty acid ratios in serum and tumor membranes were lower in the n-3 group relative to the n-6 group. In addition, n-3 group tumors had decreased cyclooxygenase-2 protein and mRNA levels, an 83% reduction in PGE2 levels, and decreased vascular endothelial growth factor expression.
Conclusion
These results provide a sound basis for clinical trials evaluating the effect of dietary n-3 fatty acids from fish oil on tumor PGE2 and membrane fatty acid composition, and serum and tumor biomarkers of progression in men with prostate cancer.
Two classes of long-chain polyunsaturated fatty acids may play an important role in prostate cancer development and progression. n-6 fatty acids, i.e., linoleic acid and arachidonic acid, have been shown to exert mainly a growth-promoting effect on cell lines and xenografts, whereas n-3 fatty acids, i.e., eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), have growth-inhibitory effects (1–6). n-6 fatty acids, contained in large quantities in corn and safflower oils and in meat, are the predominant polyunsaturated fatty acids in the Western diet. In the U.S., the intake of linoleic acid (n-6) is on the order of 10 to 20 g/d; thus, constituting 85% of the total fatty acids consumed (7). In contrast, the n-3 fatty acids, EPA and DHA, are contained in fish oils and are the major fatty acids consumed in the Japanese diet. The ratio of long chain n-6 to n-3 fatty acids in the Western diet is on the order of 10:1 to 20–25:1 whereas in Japan this ratio is 4:1 (8). Epidemiologic studies support an inverse association between marine-derived n-3 fatty acid intake and prostate cancer risk, and risk of advanced prostate cancer (9–12). Other studies, however, do not confirm the inverse association between intake of marine-derived n-3 fatty acids and prostate cancer risk (13, 14). α-Linolenic acid, an n-3 fatty acid derived from plant oils, was found to increase the risk of advanced prostate cancer (12, 13). Thus, marine-derived n-3 fatty acids such as EPA and DHA may potentially inhibit prostate cancer development and progression, whereas plant oil–derived n-3 fatty acids (linolenic acid) may promote prostate cancer growth. In vitro and in vivo animal data suggests that the balance of n-6 and marine-derived n-3 fatty acids affects the growth of prostate cancer. Linoleic acid (n-6) has been shown to exert a stimulatory effect on the growth of androgen-responsive (LNCaP) and androgen-independent (PC-3) human prostate cancer cell lines, whereas DHA and EPA (n-3) inhibit in vitro growth (1, 2, 5, 6). In two prior animal feeding studies, n-3-based diets reduced the growth of s.c. DU-145 xenografts in nude mice when compared with n-6-based diets (3, 4). In one of these studies, Karmali and colleagues showed that DU-145 tumors from mice consuming a fish oil–based diet (20.52% fish oil plus 3% corn oil) had increased n-3 fatty acid levels and decreased prostaglandin E2 (PGE2) levels relative to tumors from mice consuming a corn oil–based diet (23.52% corn oil; ref. 3).
Dietary linoleic acid (n-6) is converted to arachidonic acid (n-6) and stored in membrane phospholipids (15). Arachidonic acid is released by cytosolic phospholipase A2-α and converted to prostaglandin H2 (an unstable intermediate hydroxyl-endoperoxide) by the enzymes, cyclooxygenase-1 and -2 (COX-1 and COX-2; ref. 15). Prostaglandin H2 is converted to PGE2, a proinflammatory eicosanoid implicated in promoting tumor growth and metastasis through various mechanisms including cell proliferation, inhibition of apoptosis, angiogenesis, and invasion (15–18). Increased levels of PGE2 have been observed in malignant human prostate cancers (compared with benign counterparts) and in carcinogen-induced rat and mouse prostate cancer (3, 18, 19). Whereas COX-1 is constitutively expressed, the COX-2 isoform is inducible by PGE2, inflammatory cytokines, hormones, and tumor promoters (20–23). The COX-2 isoform is overexpressed in prostate cancer, and increased mRNA and protein levels of COX-2 have been reported in malignant as compared with benign prostate tissue (24–26).
To date, animal feeding studies using androgen-sensitive prostate cancer xenografts have not evaluated if altering the dietary ratio of n-6 to marine-derived (DHA and EPA) n-3 fatty acids affects tumor growth, serum and prostate tumor membrane fatty acid levels, and COX-2 levels. This pre-clinical study was designed to evaluate the efficacy and bio-marker data related to n-6 and n-3 fatty acid intake for use in future human trials in men with androgen-sensitive prostate cancer.
Materials and Methods
Animal husbandry and feeding protocol
Thirty (15 mice/group) male CB17 beige severe combined immunodeficiency mice (8 weeks old) were obtained from the UCLA Department of Laboratory Animal Medicine facility, which is accredited by the American Association for Accreditation of Laboratory Animal Care. The mice were housed one per cage to allow for the maintenance of isocaloric intake between the diet groups. The experiments were approved by the UCLA Chancellor's Animal Research Committee, and animals were cared for in accordance with institutional guidelines.
The diets were prepared and sterilized (irradiated) by DYETS, Inc. (Bethelheim, PA). Isocaloric diets were formulated to contain 3,747 kcal/g with 20% of energy from protein (casein), 60% of energy from carbohydrates, and 20% of calories from fat. The fat in the n-6 diet was from corn oil and the n-6 to n-3 ratio was 26:1. The n-3 diet was formulated to contain n-6 and n-3 fatty acids in a ratio of 1:1 with menhaden oil and corn oil (Supplemental Table S1). Fatty acid analysis of the irradiated research diets confirmed the quantity and ratio of n-6 and n-3 fatty acids in the irradiated pellets (Table 1). Initially, a palatability study (without tumor injection) comparing ad lib intake of the n-6 and n-3 diets was done and the mice consumed an equal amount of calories per day in each diet group (data not shown). During the xenograft experiment, the mice were fed every Monday, Wednesday, and Friday. At the time of each feeding, the uneaten food was weighed and the average caloric intake of each diet group was calculated to monitor caloric intake. Diet restriction was not required since the mice in the two groups were found to have similar mean caloric intakes throughout the experiment.
Table 1.
Fatty acid levels of experimental diets, mouse serum, and tumor membranes from n-6 and n-3 diet groups
| Fatty acids | Diets |
Serum |
Membrane |
|||
|---|---|---|---|---|---|---|
| ω-6 diet, mg/g (%) | ω-3 diet, mg/g (%) | ω-6 diet, μmol/L (%) | ω-3 diet, Mmol/L (%) | ω-6 diet, Mmol/g protein (%) | ω-3 diet, Mmol/g protein (%) | |
| Myristic (C14:0) | 0.31 ± 0.04 (0.54) | 3.31 ± 0.02 (5.3) | 33.43 ± 7.60 (0.4) | 47.71 ± 17.54* (0.9) | 12.11 ± 1.04 (2.8) | 14.79 ± 1.97* (3.0) |
| Palmitic (C16:0) | 6.57 ± 0.29(11.5) | 10.89 ± 0.05 (17.5) | 1,594.64 ± 355.74 (20.8) | 1,263.75 ± 286.16* (24.4) | 108.71 ± 7.33 (24.7) | 123.58 ± 12.37* (25.4) |
| Palmitoleic (C16:1) | 0.19 ± 0.14 (0.3) | 3.98 ± 0.01 (6.4) | 83.29 ± 29.46 (1.1) | 143.89 ± 47.58* (2.8) | 9.06 ± 1.94 (2.05) | 14.57 ± 2.31* (3.0) |
| Stearic (C18:0) | 1.35 ± 0.09 (2.4) | 2.93 ± 0.11 (4.7) | 935.15 ± 126.23 (12.2) | 600.56 ± 132.11* (11.6) | 69.53 ± 4.53 (15.8) | 71.61 ± 6.00 (14.8) |
| Oleic (C18:1) | 14.80 ± 0.57 (25.9) | 9.88 ± 0.09 (15.8) | 987.15 ± 221.53 (12.9) | 707.19 ± 134.01* (13.6) | 90.98 ± 13.21 (20.6) | 97.65 ± 18.73 (19.9) |
| Linoleic (C18:2): ω-6 | 30.65 ± 0.64 (53.7) | 13.63 ± 0.06 (21.8) | 2,299.26 ± 527.57 (30.0) | 1,185.58 ± 243.70* (22.9) | 52.39 ± 8.43 (11.9) | 43.72 ± 5.73* (9.0) |
| Arachidic (C20:0) | 0.42 ± 0.05 (0.7) | 0.37 ± 0.02 (0.6) | 0.00 ± 0.00 (0.0) | 0.00 ± 0.00 (0.0) | 4.75 ± 2.47 (1.1) | 5.22 ± 0.86 (1.1) |
| Linolenic (C18:3): ω-3 | 0.80 ± 0.10 (1.4) | 1.06 ± 0.02 (1.7) | 24.59 ± 15.59 (0.3) | 13.29 ± 14.11 (0.3) | 4.23 ± 2.14 (0.9) | 4.52 ± 0.59 (0.9) |
| Eicosenoic (C20:1) | 0.23 ± 0.10 (0.4) | 0.73 ± 0.02 (1.2) | 0.00 ± 0.00 (0.0) | 0.00 ± 0.00 (0.0) | 9.06 ± 1.50 (2.1) | 5.77 ± 0.88* (1.2) |
| Eicosadienoic (C20:2): ω-6 | 0.26 ± 0.03 (0.5) | 0.27 ± 0.02 (0.4) | 10.37 ± 9.97 (0.1) | 0.08 ± 0.27* (0.0) | 10.28 ± 1.23 (2.3) | 5.10 ± 0.84* (1.1) |
| Behenic (C22:0) | 0.29 ± 0.01 (0.5) | 0.33 ± 0.02 (0.5) | 6.60 ± 10.21 (0.1) | 4.21 ± 7.48 (0.1) | 9.45 ± 1.47 (2.1) | 9.02 ± 1.73 (1.8) |
| Arachidonic acid (C20:4): ω-6 | 0.31 ± 0.01 (0.6) | 0.99 ± 0.00 (1.6) | 1,363.75 ± 558.88 (17.8) | 247.06 ± 55.43* (4.8) | 38.12 ± 3.87 (8.7) | 15.01 ± 1.64* (3.1) |
| EPA (C20:5): ω-3 | 0.18 ± 0.43 (0.3) | 6.08 ± 0.01 (9.7) | 0.00 ± 0.00 (0.0) | 411.24 ± 124.87* (7.9) | 1.06 ± 1.65 (0.3) | 20.18 ± 2.06* (4.2) |
| Lignoceric (C24:0) | 0.37 ± 0.03 (0.6) | 0.37 ± 0.01 (0.6) | 39.84 ± 10.26 (0.5) | 3.92 ± 6.67* (0.08) | 5.34 ± 1.35 (1.2) | 4.92 ± 0.39 (1.0) |
| Nervonic (C24:1) | 0.19 ± 0.02 (0.3) | 0.47 ± 0.01 (0.8) | 77.07 ± 17.17 (1.0) | 1.56 ± 4.93* (0.03) | 6.65 ± 1.07 (1.5) | 5.46 ± 0.45* (1.1) |
| DHA (C22:6): ω-3 | 0.22 ± 0.37 (0.4) | 7.12 ± 0.01 (11.4) | 213.51 ± 33.47 (2.8) | 555.88 ± 121.77* (10.7) | 9.10 ± 1.48 (2.1) | 45.56 ± 3.41* (9.4) |
| Total fatty acid | 57.12 ± 2.73 (100.0) | 62.39 ± 0.19 (100.0) | 7,668.65 ± 1,218.74 (100.0) | 5,185.93 ± 1,053.28 (100.0) | 545.75 ± 61.47 (100.0) | 593.33 ± 79.56 (100.0) |
| Total N-6 | 31.22 ± 0.08 (54.7) | 14.88 ± 0.60 (23.9) | 3,673.38 ± 661.59 (47.9) | 1,432.72 ± 289.76 (27.6) | 100.78 ± 12.15 (22.8) | 63.83 ± 4.08 (13.1) |
| Total ω-3 | 1.19 ± 0.01 (2.1) | 14.25 ± 0.89 (22.8) | 238.10 ± 32.78 (3.1) | 980.42 ± 209.14 (18.9) | 14.39 ± 3.88 (3.25) | 70.25 ± 7.80 (14.5) |
| ω-6/ω-3 ratio | 26.16 | 1.04 | 15.43 | 1.46 | 7.01 | 0.91 |
| Arachidonic acid/EPA + DHA | 0.80 | 0.07 | 6.4 | 0.26 | 3.9 | 0.23 |
NOTE: Data is presented with ± SD. The percentage of each fatty acid compared with the total fatty acid is presented in parentheses.
Indicates P < 0.001 (two-tailed Student's t test).
Los Angeles Prostate Cancer 4 xenografts
The Los Angeles Prostate Cancer 4 (LAPC-4) cell line (a generous gift from Drs. Robert Reiter and Charles Sawyers) was developed at UCLA by direct transfer of cancer cells from a patient with advanced adenocarcinoma of the prostate into the s.c. tissue of severe combined immunodeficiency mice (27). LAPC-4 produces prostate-specific antigen (PSA), has a wild-type androgen receptor, and shows features of hormone-dependent growth and metastasis (27). Two weeks after starting their experimental diet, mice were injected s.c. in the lateral flank with 105 LAPC-4 tumor cells in 0.1 mL of Matrigel (Collaborative Biomedical Products, Bedford, MA). The LAPC-4 tumor cells used for injection were harvested from xenograft tumors propagated in severe combined immunodeficiency mice as previously described (28). Throughout the experiment, mice were weighed and tumors examined weekly. The tumor dimensions were measured using a caliper. Tumor volumes were calculated using the formula: length × width × height × 0.5236.
Serum and tumor collection
Eight weeks after the LAPC-4 cells were implanted, the mice in each group were euthanized. Serum was collected and stored at −70°C. Tumor tissue was weighed and rinsed with saline. Half of the tumor was snap-frozen in liquid nitrogen, and the other half fixed for 4 to 8 hours in 10% neutral buffered formalin and then embedded in paraffin blocks for histologic sectioning.
Serum PSA measurement
Human PSA levels in the mouse serum were measured by ELISA (Diagnostic Systems Laboratories, Inc., Webster, TX).
Ex vivo bioassay
The effect of mouse serum on LAPC-4 proliferation was examined as previously described using the CellTiter 96AQ Assay (Promega Corporation, Madison, WI; ref. 29). This method has been shown to correlate (<5% difference) with [3H]thymidine incorporation and was used to determine the viable cells in each well.
Fatty acid analysis
LAPC-4 tumor membrane was separated from the cytosolic fraction before the fatty acid analysis. LAPC-4 tumor cells (200 mg) were disrupted by douncing in 5 mL of lysis buffer [0.5 mmol/L CaCl2, 1 mmol/L NaHCO3 (pH 7.5)] by a glass homogenizer fitted with a Teflon pestle on ice. The homogenate was centrifuged at 2,000 × g for 15 minutes at 4°C. The supernatant was recentrifuged at 20,000 × g for 30 minutes at 4°C to remove the cytosol. The membrane pellet was resuspended in 1 mL of PBS. The fatty acid analysis of diet, serum, and the plasma membrane was done as previously described (30). Duplicate samples were extracted and converted to fatty acid methyl esters. Quantification was based on the recovery of a known quantity of the internal standard (tridecanoic acid; NuChek Preparation, Inc., Elysian, MN) and on the response ratio of fatty acid standards purchased from NuChek Preparation. The protein concentration of the membrane fraction was determined by bicinchoninic acid protein assay kit (Pierce, Rockford, IL) and used to express fatty acid methyl ester concentration as micromoles per gram of membrane protein.
Immunohistochemistry
Sections (4 μm thick) were cut from paraffin-embedded blocks of formalin-fixed tumors using standard methods. For Ki67 staining (monoclonal antibody; Dako A/S, Denmark), the slides were deparaffinized and the endogenous peroxidase was quenched using 3% H2O2 in 70% methanol. The heat-induced epitope retrieval was done using a pressure cooker and EDTA buffer (pH 8.0) for 2 minutes. The sections were then incubated with the primary antibody (Ki67 at 1:100), diluted in calcium chloride for 2 hours, at room temperature, followed by the secondary antibody (anti-mouse, Dako Envision HRP; Dako, Carpinteria, CA) for 30 minutes. The washing steps were done with 1× PBS Tween 20. 3,3′ Diaminobenzidine tetrahydrochloride dehydrate was used to localize the peroxidase in tissue. The slides were counterstained with hematoxylin. Apoptosis (terminal nucleotidyl transferase–mediated nick end labeling) staining was done using ApopTag Peroxidase In situ Apoptosis Detection Kit (Chem-icon, Temecula, CA).
Western analysis
The tumor protein (20 μg) was subjected to PAGE and Western blotting with anti-human COX-2 monoclonal antibody (Cayman Chemical, Ann Arbor, MI) at 1:1,000 dilution. The secondary horseradish peroxidase–linked antibody was used at 1:4,000 dilution. Signal was detected using enhanced chemiluminescence detection reagent (ECL Plus; Amersham Biosciences, Piscataway, NJ). The membrane was stripped by Restore Western blot stripping buffer (Pierce) and reprobed with monoclonal anti-β-actin antibody (1:5,000 dilution; Abcam, Cambridge, MA). Personal Densitometer SI (Molecular Dynamics) and Image Quant (Amersham Biosciences) were used to quantify the Western signals.
Quantitative reverse transcriptase-PCR
Total RNA was extracted from LAPC-4 tumors using TRIzol reagent (Invitrogen, Carlsbad, CA) and purified using a RNeasy Mini column (Qiagen, Valencia, CA) according to the manufacturer's protocols. Total RNA was reverse-transcribed by the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) with the random primers. mRNA levels were measured by real-time reverse transcriptase-PCR with the gene-specific FAM-labeled probe and primers [Applied Biosystems Assay-on-Demand Gene Expression Product: assay ID for COX-2, Hs00153133_m1; vascular endothelial growth factor (VEGF), Hs00173626_m1; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Hs99999905_m1] using TaqMan Universal PCR Master Mix without AmpErase UNG (Applied Biosystems) as described by the manufacturer. The GAPDH mRNA quantitation (endogenous control) for each sample was included in every real-time reverse transcriptase-PCR reaction. The experiments were repeated twice.
PGE2 analysis
PGE2 extraction and analysis were conducted by the method of Yang et al. under low light to avoid photooxidation (31). The extracted prostaglandins were separated by a Luna 3-μm, phenyl-hexyl, 2 × 150 mm analytic column (Phenomenex, Torrance, CA) and analyzed on a Surveyor high-performance liquid chromatography system equipped with a diode array absorbance detector. The mobile phase consisted of a gradient system of 10 mmol/L ammonium acetate (pH 8.5) and methanol. Sample injection volume was 20 μL and were kept at 4°C during the analysis. Flow rate was 0.12 μL/min with column temperature at 35°C. Quantification was done by comparing the sample peak areas to a PGE2 standard curve which was constructed using the purified PGE2 (Cayman Chemical). The protein concentration of the tumor homogenate was used to express the PGE2 levels as picograms of PGE2 per milligram of protein.
Statistical analysis
Statistical comparisons between the groups (Splus 2000; Insightful Corp, Seattle, WA) were done by Student's t test or the Wilcoxon rank sum test if the variable failed a test for normality. Correlations between outcome variables were computed using the Spearman correlation coefficient. The coefficient of variation was calculated by dividing the SD by the mean (expressed as a percentage). Tumor growth rates were computed by fitting a linear regression between time and tumor volume for each animal and then compared between the groups with a two-sample t test. Data are expressed as mean ± SE.
Results
Effect of n-3 and n-6 diets on LAPC-4 tumor growth, proliferation, and apoptosis
There was no significant difference in mean caloric intake between mice in the n-6 and n-3 diet groups, with the mice consuming an average of 0.5 kcal/g body weight/d (Fig. 1A). In addition, there was no difference in mouse body weights between the two groups (Fig. 1B). Tumor implantation and growth occurred in all animals injected, and there was no difference in time from tumor injection to the development of palpable tumor between the groups. The LAPC-4 tumors in the n-3 group had significantly slower growth rates (P < 0.01) and smaller mean final tumor volumes (68% reduction, 0.65 ± 0.14 versus 0.21 ± 0.05; P < 0.05) compared with the tumors in the n-6 group (Fig. 1C). Mean serum PSA levels at the time of euthanasia were 77% lower in the n-3 group relative to the n-6 group (21.1 ± 7.0 versus 90.2 ± 14.7 ng/mL; P < 0.01; Fig. 2A). LAPC-4 tumors in the n-3 and n-6 groups were histologically similar and revealed polygonal cells with pleomorphic vesicular nuclei and prominent nucleoli. There were large areas of necrosis with no evidence of tubule formation consistent with a poorly differentiated adenocarcinoma of the prostate. The mean Ki67 labeling index of the n-3 tumors was 64.5% lower than the labeling index in the n-6 tumors (9.4 ± 1.8% versus 26.5 ± 3.9%; P < 0.01; Fig. 2B). Apoptosis as measured by terminal nucleotidyl transferase–mediated nick end labeling staining was increased by 67% in the n-3 group relative to the n-6 group (3.1 ± 0.47% versus 2.0 ± 0.26%; P = 0.05; Fig. 2C).
Fig. 1.

Severe combined immunodeficiency mouse energy intake, weight, and LAPC-4 xenograft growth. Eight-week-old male severe combined immunodeficiency mice were fed either n-6 diet or n-3 diet (n = 15 per group) for 2 weeks prior to injection of 1×105 LAPC-4 in the lateral flank. A, caloric intake was measured for each mouse thrice per week by subtracting the weight of uneaten food from the weight of the food given at the beginning of each feeding period. B, mice were weighed weekly. C, tumor volumes were measured weekly. Points, means; bars, ±SE; two-tailed Student's t test (*, P < 0.05).
Fig. 2.

Effects of n-3 diet on serum PSA, tumor cell proliferation, and apoptosis. A, decreased serum PSA in the n-3 diet group. Human PSA levels in mouse serum collected at the time of euthanasia was measured by ELISA. n = 13 for n-6 diet group; n = 11 for n-3 diet group. Horizontal line, mean value of each group (*, P < 0.001). B, decreased cell proliferation in LAPC-4 xenografts from the n-3 diet group. Ki67-positive cells (arrows, proliferative index) decreased by 65% in the n-3 diet group compared with the n-6 group. A total of 100 cells were counted for each sample; n = 13 for n-6 diet; n = 14 for n-3 diet (*, P < 0.001). C, apoptosis was examined by terminal nucleotidyl transferase – mediated nick end labeling assay. The percentage of apoptotic cells (arrows) increased in the n-3 diet group by 67%. A total of 200 cells per sample were counted (n = 12 for n-6 diet group; n = 13 for n-3 diet; P = 0.05).
Ex vivo LAPC-4 proliferation
The proliferative effects of mouse serum were evaluated by culturing LAPC-4 cells in vitro in medium containing nonpooled n-3 or n-6 mouse serum (obtained at euthanasia). After a 48-hour incubation in medium containing mouse serum from the n-3 diet group, LAPC-4 cell proliferation was reduced by 22% compared with cells grown in medium containing n-6 mouse serum (75 ± 4.0% versus 97.9 ± 9.5% of fetal bovine serum control; P < 0.05; Fig. 3).
Fig. 3.
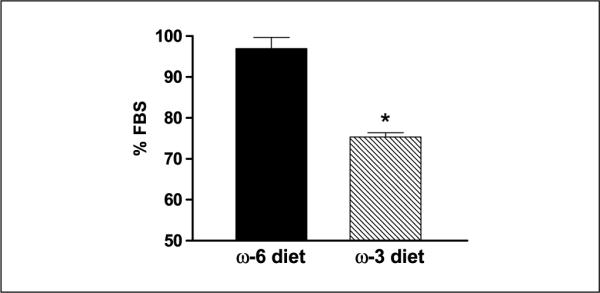
Ex vivo LAPC-4 proliferation in mouse serum. LAPC-4 cells were plated at 5 × 103 cells/well in 96-well plates for 24 hours, then incubated for 48 hours with the fresh medium containing 10% mouse serum from individual mice (serum was not pooled) from the n-6 or n-3 diet group. Cell proliferation was determined by measuring formazan product formation by dehydrogenase activity of viable cells. The doubling time of LAPC-4 cells in medium containing 10% fetal bovine serum was ~48 hours under these experimental conditions. All experiments were done in duplicate. Data is expressed as a percentage of LAPC-4 growth in medium containing 10% fetal bovine serum. Columns, means; bars, ±SE; n = 12 for n-6 diet; n = 15 for n-3 diet (*, P < 0.05). In a separate set of experiments, there was no difference in apoptosis using an ELISA assay (Cell Death Detection ELISA Plus, Roche Applied Science, Indianapolis, IN) of LAPC-4 cells incubated for 48 hours in medium containing nonpooled n-6 and n-3 mouse serum (data not shown).
Fatty acid analysesy
Table 1 shows the results of fatty acid analyses of the irradiated food pellets, fasting serum obtained at the time of euthanasia, and LAPC-4 membranes prepared from the xenografts. The n-6 food pellets contained only trace amounts of n-3 fatty acids compared with n-6 fatty acids (n-6/n-3 = 26:1), and linoleic acid was the predominant n-6 fatty acid (98% of total n-6 fatty acids). The n-3 pellets contained EPA and DHA as the predominant n-3 fatty acids (42.6% and 49.9% of total n-3 fatty acids, respectively), and linoleic acid was the predominant n-6 fatty acid (92% of total n-6 fatty acids). The n-6/n-3 ratio of the n-3 diet was 1:1.
The mean fasting-serum concentration of total n-6 fatty acids (mostly linoleic acid and arachidonic acid) was lower in mice in the n-3 compared with the n-6 diet group (n-3 group, 1,433 ± 91.7 (±SE); n-6 group, 3,676 ± 220.5 μmol/L; P < 0.001). EPA and DHA were the predominant serum n-3 fatty acids in the n-3 group (41.8 ± 2.4% and 57 ± 2.3% of total n-3 fatty acids, respectively), and serum levels of EPA plus DHA were higher in the n-3 group than the n-6 group (967.1 ± 63.5 versus 213.5 ± 11.2 μmol/L; P < 0.001). The serum n-6/n-3 ratio was 15.4:1 for the n-6 group and 1.5:1 for the n-3 group (10.6-fold difference; P < 0.001).
The ratio of n-6/n-3 fatty acids in the LAPC-4 tumor membrane was 7.8-fold higher in the n-6 diet group compared with the n-3 diet group (0.9:1 in the n-3 group and 7.0:1 in the n-6 group; P < 0.001). Arachidonic acid, eicosadienoic acid (n-6), EPA, and DHA were the only fatty acids that were significantly different between the two diet groups. Membrane arachidonic acid levels were lower in the n-3 group (15.01 ± 0.67 μmol/g protein) compared with the n-6 group (38.12 ± 1.58 μmol/g protein; P < 0.001). In addition, membrane EPA and DHA levels were higher in the n-3 group relative to the n-6 group (65.74 ± 1.49 versus 10.16 ± 0.98 μmol/g protein; P < 0.001). The arachidonic acid/EPA + DHA ratio was 17-fold higher in tumor membranes of the n-6 diet group relative to the n-3 group (P < 0.001).
Effect of dietary fatty acids on tumor COX-2, PGE2, and VEGF
The mean LAPC-4 tumor COX-2 mRNA levels (measured by quantitative reverse transcriptase-PCR) was 32% lower in the n-3 group than the n-6 group (P < 0.05; Fig. 4A). In addition, mean tumor COX-2 protein levels were 30% lower in the n-3 group relative to the n-6 group (Fig. 4B and C). Tumor PGE2 levels were significantly (83%) lower in the n-3 diet group relative to the n-6 group (0.037 ± 0.012 versus 0.218 ± 0.052 ng/mg protein; P < 0.05; Fig. 5A). The n-3 group tumors had 34% less VEGF mRNA expression compared with the n-6 group tumors (P < 0.05; Fig. 5B).
Fig. 4.

Effects of n-3 and n-6 diets on COX-2 mRNA and protein levels in LAPC-4 tumors. A, COX-2 mRNA expression was determined using real-time reverse transcriptase-PCR relative to GAPDH mRNA, a reference housekeeping gene. n = 8 for n-6 diet group and n = 10 for n-3 diet group (*, P < 0.05).The experiment was repeated twice. B, Western blot analysis of COX-2 protein. Lanes 1 to 3 are protein samples from three different tumors from the n-6 diet group and lanes 4 to 6 contain individual protein samples from three tumors from the n-3 diet group. The β-actin band was used for protein loading controls. C, the optimized bands were quantified by densitometry and shown as an average of each diet group for COX-2 protein levels normalized by β-actin band (values are relative densitomeric units with SE; n = 4 for each group, the experiment was repeated thrice.
Fig. 5.

Effects of n-3 diet on PGE2 levels and VEGF mRNA expression in LAPC-4 xenografts. A, PGE2 was extracted from tumors and quantified by high-performance liquid chromatography/diode array detection and electrospray ionization mass spectrometry analysis. PGE2 peak was clearly separated from the PGE3 peak. n = 4 for each group (*, P = 0.01). B, VEGF expression was down-regulated in the n-3 diet group. Total RNA was analyzed for VEGF mRNA relative to GAPDH mRNA using quantitative reverse transcriptase-PCR. n = 8 for n-6 diet and n = 10 for n-3 diet. The experiment was repeated thrice (*, P < 0.05).
Discussion
In the present study, we found that mice fed a 20% kcal fat diet with an n-6/n-3 fatty acid ratio of 1:1 had slower LAPC-4 tumor growth and lower serum PSA levels relative to mice fed an isocaloric 20% kcal fat diet with the majority of polyunsaturated fatty acids derived from n-6 fatty acids. In addition, LAPC-4 tumors in the n-3 diet group showed a decreased proliferative index and increased apoptosis relative to the n-6 diet group. Two prior feeding studies showed decreased s.c. growth of the androgen-insensitive human prostate cancer cell line DU-145 in mice fed predominantly n-3 fatty acids as compared with n-6 fatty acids diets (3, 4). Whereas DU-145 tumors do not produce PSA and show androgen-independent growth, LAPC-4 tumors reflect androgen-dependent growth better as LAPC-4 tumors have a wild-type androgen receptor, produce PSA, and regress following bilateral orchiectomy (27). Although total caloric intake may affect tumor growth, this potential confounding variable was carefully controlled for in the present study by housing one mouse/cage and measuring caloric intake 3 days/wk (32, 33). The mice were found to have equal caloric intake in the n-3 and n-6 diet groups, therefore, a paired feeding regimen to match caloric intake between experimental groups was not required. Our experience has been that mice may significantly alter caloric intake depending on the palatability of the research diet, and palatability studies are therefore required prior to evaluating the effect of diets and/or supplements on tumor progression in mice (29, 34).
A number of mechanisms may be responsible for the decreased LAPC-4 tumor growth, decreased proliferation, and increased apoptosis we observed in the n-3 group in the present study. The most likely mechanisms revolve around the effects of the diet on serum and membrane fatty acid ratios. Serum from mice consuming the n-3-based diet stimulated less LAPC-4 cell proliferation in vitro compared with serum from mice consuming the n-6-based diet. The serum factors responsible for this effect may be DHA (n-3), EPA (n-3), arachidonic acid (n-6), and linoleic acid (n-6). Mice consuming the n-3 diet had increased serum DHA and EPA levels and decreased serum arachidonic acid and linoleic acid levels relative to the n-6 diet group. Prior in vitro studies found that DHA and EPA had a growth-inhibitory effect, whereas linoleic acid and arachidonic acid had growth-promoting effects on prostate cancer cell lines (1, 2). The LAPC-4 tumor membrane arachidonic acid/EPA + DHA ratio was significantly lower in the n-3 diet group relative to the n-6 group. The membrane arachidonic acid/EPA + DHA ratio in the n-6 diet group was 3.9 compared with 0.23 in the n-3 group. In response to a variety of stimuli, arachidonic acid is released from membrane phospholipids by phospholipases and converted by cyclooxygenase enzymes to prostaglandin H2 (15). Specific prostaglandin synthases in turn convert prostaglandin H2 to bioactive eicosanoids including PGE2, PGD-2, PGF-2α, PGI-2, and thromboxane A2 (15). PGE2 has been shown to promote the growth of prostate cancer cells in tissue culture and potentially affect tumor cell invasion and metastasis through a number of signaling pathways which affect proliferation, angiogenesis, apoptosis, and immune responses (15, 18, 22, 35, 36). Conversely, EPA and DHA compete with arachidonic acid as a substrate for the cyclooxgenases (COX-2 and COX-1) resulting in the production of three-series prostanoids such as PGE-3 and five-series leukotrienes that are antiinflammatory and have no mitogenic effects (37). The net biological effect of prostaglandins may be determined by the balance in the tissues between prostaglandins derived from dietary n-6 and n-3 fatty acids. In the present study, PGE2 levels were 83% lower in LAPC-4 tumors in the n-3 diet group relative to the n-6 group. Therefore, alterations in tumor membrane fatty acid levels may have played a role in the decreased tumor growth seen in the n-3 diet group by decreasing the tumor PGE2 concentration.
A number of other mechanisms involving membrane phospholipid composition and eicosanoid biosynthesis may also explain the differences in LAPC-4 tumor growth seen in the n-6 and n-3 diet groups. Arachidonic acid (derived from dietary linoleic acid) and EPA compete for binding to lipoxygenase enzymes (38). Specific lipoxygenase enzymes convert arachidonic acid to eicosanoids that potentially affect prostate cancer progression through proliferation, apoptosis, and angiogenesis (39). For example, 5-lipoxygenase in LNCaP and PC3 cell lines converts arachidonic acid to 5-hydroxyeicosatetraenoic acid, which stimulates the proliferation and survival of prostate cancer cell lines in vitro (40, 41). Levels of 5-lipoxygenase and its product, 5-hydroxyeicosatetraenoic acid, are significantly higher in malignant as compared with benign prostate tissue, and inhibition of 5-lipoxygenase induces apoptosis in prostate cancer cell lines (42, 43). 12-Lipoxygenase converts arachidonic acid to 12(S)-hydroxyeicosatetraenoic acid. 12-Lipoxygenase levels were found to be higher in pathologically advanced and higher grade prostate cancers, and 12(S)-hydroxyeicosatetraenoic acid promotes tumor cell invasion and metastasis in a number of experimental systems (44, 45). For example, 12-lipoxygenase transfected PC-3 cells injected in athymic mice produced larger tumors with increased vascularity relative to untransfected PC-3 tumors (46). Thus, decreasing the ratio of arachidonic acid/EPA+ DHA in cell membrane phospholipids may potentially decrease the production of hydroxyeicosatetraenoic acids implicated in prostate cancer invasion and metastasis.
In the present study, the n-3 diet group had decreased COX-2 mRNA and protein levels relative to the n-6 group. A number of studies have shown that COX-2 levels are increased in prostatic intraepithelial neoplasia relative to benign prostate tissue, and is highest in malignant prostate tissue (24, 25). Numerous in vitro and animal studies incorporating COX-2 inhibitors have shown a clear role for COX-2 in promoting prostate cancer progression (38, 47). COX-2 likely exerts its proliferative, antiapoptotic, and angiogenic effects through mechanisms involving the end product PGE2 (48). Furthermore, COX-2 is inducible by a variety of inflammatory stimuli including one of its end products, PGE2 (20, 23). Thus, the reduced COX-2 levels in the n-3 diet group may potentially be explained by a reduced arachidonic acid/EPA+ DHA ratio in the membrane phospholipids, resulting in decreased PGE2 production, which in turn, affected COX-2 expression.
In vitro studies by Liu et al. showed that PGE2 increased hypoxia-induced VEGF expression by promoting hypoxiainducible factor 1-α translocation from the cytosol to the nucleus (36). Preclinical prostate cancer models incorporating COX-2 inhibitors showed decreased angiogenesis (47, 49). Likewise, focuses of inflammation in prostate cancer tissue in radical prostatectomy specimens had increased COX-2 expression and microvessel density staining (50). In the present study, the higher tumor PGE2 levels in the n-6 diet group may therefore be the mechanism for higher COX-2 mRNA and protein levels and increased VEGF expression seen in the LAPC-4 tumors.
Based on extensive literature on the tumor-promoting effects of COX-2, as well as epidemiologic studies on decreased prostate cancer risk in men with increased use of nonsteroidal antiinflammatory drugs, a great deal of interest exists for using specific and nonspecific COX-2 inhibitors for prostate cancer prevention and treatment (48). The results of the present preclinical trial further suggest that dietary intervention trials incorporating n-3 supplements (DHA and EPA) with reduced dietary n-6 fatty acid designed to decrease tissue PGE2 levels may also play an important role in primary and secondary prostate cancer prevention. An additional strategy may be the combination of n-3 supplements with COX-2 inhibitors given that the combination had additive growth-inhibitory and proapoptotic effects on prostate cancer cell lines in vitro (6). Based on data generated from the present study, the authors are conducting a prospective randomized trial in men undergoing radical prostatectomy, comparing the effects of a low-fat diet combined with fish oil supplements (achieving an n-6/n-3 ratio of 2:1) and a western diet on serum and tissue fatty acid levels and on tissue COX-2 and PGE2 levels. Further prospective clinical trials incorporating n-3-based dietary interventions and/or COX-2 inhibitors with relevant serum and tissue biomarkers are warranted.
Supplementary Material
Acknowledgments
Grant support: Department of Veterans Affairs, NIH grants: Specialized Programs of Research Excellence P50 CA92131-01A1, 1R01CA100938, and UCLA Jonsson Comprehensive Cancer Center Interdisciplinary grant (W.J. Aronson); Department of Defense, American Urological Association Foundation Astellas Rising Star in Urology Award (S.J. Freedland); and NIH grants 5P01 CA42710, AT00151 (D. Heber).
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Rose DP, Connolly JM. Effects of fatty acids and eicosanoid synthesis inhibitors on the growth of two human prostate cancer cell lines. Prostate. 1991;18:243–54. doi: 10.1002/pros.2990180306. [DOI] [PubMed] [Google Scholar]
- 2.Pandalai PK, Pilat MJ, Yamazaki K, Naik H, Pienta KJ. The effects of omega-3 and omega-6 fatty acids on in vitro prostate cancer growth. Anticancer Res. 1996;16:815–20. [PubMed] [Google Scholar]
- 3.Karmali RA, Reichel P, Cohen LA, et al. The effects of dietary omega-3 fatty acids on the DU-145 transplantable human prostatic tumor. Anticancer Res. 1987;7:1173–9. [PubMed] [Google Scholar]
- 4.Rose DP, Cohen LA. Effects of dietary menhaden oil and retinyl acetate on the growth of DU 145 human prostatic adenocarcinoma cells transplanted into athymic nude mice. Carcinogenesis. 1988;9:603–5. doi: 10.1093/carcin/9.4.603. [DOI] [PubMed] [Google Scholar]
- 5.Chung BH, Mitchell SH, Zhang JS, Young CY. Effects of docosahexaenoic acid and eicosapentaenoic acid on androgen-mediated cell growth and gene expression in LNCaP prostate cancer cells. Carcinogenesis. 2001;22:1201–6. doi: 10.1093/carcin/22.8.1201. [DOI] [PubMed] [Google Scholar]
- 6.Narayanan NK, Narayanan BA, Reddy BS. A combination of docosahexaenoic acid and celecoxib prevents prostate cancer cell growth in vitro and is associated with modulation of nuclear factor-κB, and steroid hormone receptors. Int J Oncol. 2005;26:785–92. [PubMed] [Google Scholar]
- 7.Kris-Etherton PM, Taylor DS, Yu-Poth S, et al. Polyunsaturated fatty acids in the food chain in the United States. Am J Clin Nutr. 2000;71:179–88S. doi: 10.1093/ajcn/71.1.179S. [DOI] [PubMed] [Google Scholar]
- 8.Sugano M, Hirahara F. Polyunsaturated fatty acids in the food chain in Japan. Am J Clin Nutr. 2000;71:189–96S. doi: 10.1093/ajcn/71.1.189S. [DOI] [PubMed] [Google Scholar]
- 9.Norrish AE, Skeaff CM, Arribas GL, Sharpe SJ, Jackson RT. Prostate cancer risk and consumption of fish oils: a dietary biomarker-based case-control study. Br J Cancer. 1999;81:1238–42. doi: 10.1038/sj.bjc.6690835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terry P, Lichtenstein P, Feychting M, Ahlbom A, Wolk A. Fatty fish consumption and risk of prostate cancer. Lancet. 2001;357:1764–6. doi: 10.1016/S0140-6736(00)04889-3. [DOI] [PubMed] [Google Scholar]
- 11.Augustsson K, Michaud DS, Rimm EB, et al. A prospective study of intake of fish and marine fatty acids and prostate cancer. Cancer Epidemiol Biomarkers Prev. 2003;12:64–7. [PubMed] [Google Scholar]
- 12.Leitzmann MF, Stampfer MJ, Michaud DS, et al. Dietary intake of n-3 and n-6 fatty acids and the risk of prostate cancer. Am J Clin Nutr. 2004;80:204–16. doi: 10.1093/ajcn/80.1.204. [DOI] [PubMed] [Google Scholar]
- 13.Giovannucci E, Rimm EB, Colditz GA, et al. A prospective study of dietary fat and risk of prostate cancer. J Natl Cancer Inst. 1993;85:1571–9. doi: 10.1093/jnci/85.19.1571. [DOI] [PubMed] [Google Scholar]
- 14.Schuurman AG, van den Brandt PA, Dorant E, Brants HA, Goldbohm RA. Association of energy and fat intake with prostate carcinoma risk: results from The Netherlands Cohort Study. Cancer. 1999;86:1019–27. [PubMed] [Google Scholar]
- 15.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–22. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen HS. Dietary essential fatty acids and in vivo prostaglandin production in mammals. World Rev Nutr Diet. 1983;42:102–34. doi: 10.1159/000408352. [DOI] [PubMed] [Google Scholar]
- 17.Levy GN. Prostaglandin H synthases, nonsteroidal anti-inflammatory drugs, and colon cancer. FASEB J. 1997;11:234–47. [PubMed] [Google Scholar]
- 18.Ablin RJ, Shaw MW. Prostaglandin modulation of prostate tumor growth and metastases. Anticancer Res. 1986;6:327–8. [PubMed] [Google Scholar]
- 19.Shaw MW, Ablin RJ, Ray P, Rubenstein M, Guinan PD, McKiel CF. Immunobiology of the Dunning R-3327 rat prostate adenocarcinoma sublines: plasma and tumor effusion prostaglandins. Am J Reprod Immunol Microbiol. 1985;8:77–9. doi: 10.1111/j.1600-0897.1985.tb00312.x. [DOI] [PubMed] [Google Scholar]
- 20.Lin AH, Bienkowski MJ, Gorman RR. Regulation of prostaglandin H synthase mRNA levels and prostaglandin biosynthesis by platelet-derived growth factor. J Biol Chem. 1989;264:17379–83. [PubMed] [Google Scholar]
- 21.Herschman HR. Prostaglandin synthase 2. Biochim Biophys Acta. 1996;1299:125–40. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 22.Tjandrawinata RR, Dahiya R, Hughes-Fulford M. Induction of cyclo-oxygenase-2 mRNA by prostaglandin E2 in human prostatic carcinoma cells. Br J Cancer. 1997;75:1111–8. doi: 10.1038/bjc.1997.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bagga D, Wang L, Farias-Eisner R, Glaspy JA, Reddy ST. Differential effects of prostaglandin derived from omega-6 and omega-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc Natl Acad Sci U S A. 2003;100:1751–6. doi: 10.1073/pnas.0334211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta S, Srivastava M, Ahmad N, Bostwick DG, Mukhtar H. Over-expression of cyclooxygenase-2 in human prostate adenocarcinoma. Prostate. 2000;42:73–8. doi: 10.1002/(sici)1097-0045(20000101)42:1<73::aid-pros9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 25.Kirschenbaum A, Klausner AP, Lee R, et al. Expression of cyclooxygenase-1 and cyclooxygenase-2 in the human prostate. Urology. 2000;56:671–6. doi: 10.1016/s0090-4295(00)00674-9. [DOI] [PubMed] [Google Scholar]
- 26.Yoshimura R, Sano H, Masuda C, et al. Expression of cyclooxygenase-2 in prostate carcinoma. Cancer. 2000;89:589–96. [PubMed] [Google Scholar]
- 27.Klein KA, Reiter RE, Redula J, et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat Med. 1997;3:402–8. doi: 10.1038/nm0497-402. [DOI] [PubMed] [Google Scholar]
- 28.Craft N, Chhor C, Tran C, et al. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res. 1999;59:5030–6. [PubMed] [Google Scholar]
- 29.Ngo TH, Barnard RJ, Cohen P, et al. Effect of isocaloric low-fat diet on human LAPC-4 prostate cancer xenografts in severe combined immunodeficiency mice and the insulin-like growth factor axis. Clin Cancer Res. 2003;9:2734–43. [PubMed] [Google Scholar]
- 30.Lepage G, Roy CC. Direct transesterification of all classes of lipids in a one-step reaction. J Lipid Res. 1986;27:114–20. [PubMed] [Google Scholar]
- 31.Yang P, Felix E, Madden T, Fischer SM, Newman RA. Quantitative high-performance liquid chromatography/electrospray ionization tandem mass spectrometric analysis of 2- and 3-series prostaglandins in cultured tumor cells. Anal Biochem. 2002;308:168–77. doi: 10.1016/s0003-2697(02)00218-x. [DOI] [PubMed] [Google Scholar]
- 32.Mukherjee P, Sotnikov AV, Mangian HJ, Zhou JR, Visek WJ, Clinton SK. Energy intake and prostate tumor growth, angiogenesis, and vascular endothelial growth factor expression. J Natl Cancer Inst. 1999;91:512–23. doi: 10.1093/jnci/91.6.512. [DOI] [PubMed] [Google Scholar]
- 33.Pariza MW. Dietary fat, calorie restriction, ad libitum feeding, and cancer risk. Nutr Rev. 1987;45:1–7. doi: 10.1111/j.1753-4887.1987.tb06064.x. [DOI] [PubMed] [Google Scholar]
- 34.Aronson WJ, Tymchuk CN, Elashoff RM, et al. Decreased growth of human prostate LNCaP tumors in SCID mice fed a low-fat, soy protein diet with iso-flavones. Nutr Cancer. 1999;35:130–6. doi: 10.1207/S15327914NC352_6. [DOI] [PubMed] [Google Scholar]
- 35.Subbarayan V, Sabichi AL, Llansa N, Lippman SM, Menter DG. Differential expression of cyclooxygenase-2 and its regulation by tumor necrosis factor-α in normal and malignant prostate cells. Cancer Res. 2001;61:2720–6. [PubMed] [Google Scholar]
- 36.Liu XH, Kirschenbaum A, Lu M, et al. Prostaglandin E2 induces hypoxia-inducible factor-1α stabilization and nuclear localization in a human prostate cancer cell line. J Biol Chem. 2002;277:50081–6. doi: 10.1074/jbc.M201095200. [DOI] [PubMed] [Google Scholar]
- 37.Culp BR, Titus BG, Lands WE. Inhibition of prostaglandin biosynthesis by eicosapentaenoic acid. Prostaglandins Med. 1979;3:269–78. doi: 10.1016/0161-4630(79)90068-5. [DOI] [PubMed] [Google Scholar]
- 38.Wallace JM. Nutritional and botanical modulation of the inflammatory cascade-eicosanoids, cyclooxygenases, and lipoxygenases-as an adjunct in cancer therapy [discussion] Integr Cancer Ther. 2002;1:7–37. doi: 10.1177/153473540200100102. [DOI] [PubMed] [Google Scholar]
- 39.Nie D, Che M, Grignon D, Tang K, Honn KV. Role of eicosanoids in prostate cancer progression. Cancer Metastasis Rev. 2001;20:195–206. doi: 10.1023/a:1015579209850. [DOI] [PubMed] [Google Scholar]
- 40.Ghosh J, Myers CE. Arachidonic acid stimulates prostate cancer cell growth: critical role of 5-lipoxygenase. Biochem Biophys Res Commun. 1997;235:418–23. doi: 10.1006/bbrc.1997.6799. [DOI] [PubMed] [Google Scholar]
- 41.Anderson KM, Seed T, Vos M, et al. 5-Lipoxygenase inhibitors reduce PC-3 cell proliferation and initiate nonnecrotic cell death. Prostate. 1998;37:161–73. doi: 10.1002/(sici)1097-0045(19981101)37:3<161::aid-pros5>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 42.Gupta S, Srivastava M, Ahmad N, Sakamoto K, Bostwick DG, Mukhtar H. Lipoxygenase-5 is overexpressed in prostate adenocarcinoma. Cancer. 2001;91:737–43. doi: 10.1002/1097-0142(20010215)91:4<737::aid-cncr1059>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh J, Myers CE. Inhibition of arachidonate 5-lipoxygenase triggers massive apoptosis in human prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95:13182–7. doi: 10.1073/pnas.95.22.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao X, Grignon DJ, Chbihi T, et al. Elevated 12-lipoxygenase mRNA expression correlates with advanced stage and poor differentiation of human prostate cancer. Urology. 1995;46:227–37. doi: 10.1016/s0090-4295(99)80198-8. [DOI] [PubMed] [Google Scholar]
- 45.Nie D, Nemeth J, Qiao Y, et al. Increased metastatic potential in human prostate carcinoma cells by overexpression of arachidonate 12-lipoxygenase. Clin Exp Metastasis. 2003;20:657–63. doi: 10.1023/a:1027302408187. [DOI] [PubMed] [Google Scholar]
- 46.Nie D, Hillman GG, Geddes T, et al. Platelet-type 12-lipoxygenase in a human prostate carcinoma stimulates angiogenesis and tumor growth. Cancer Res. 1998;58:4047–51. [PubMed] [Google Scholar]
- 47.Liu XH, Kirschenbaum A, Yao S, Lee R, Holland JF, Levine AC. Inhibition of cyclooxygenase-2 suppresses angiogenesis and the growth of prostate cancer in vivo. J Urol. 2000;164:820–5. doi: 10.1097/00005392-200009010-00056. [DOI] [PubMed] [Google Scholar]
- 48.Lin DW, Nelson PS. The role of cyclooxygenase-2 inhibition for the prevention and treatment of prostate carcinoma. Clin Prostate Cancer. 2003;2:119–26. doi: 10.3816/cgc.2003.n.020. [DOI] [PubMed] [Google Scholar]
- 49.Narayanan BA, Narayanan NK, Pittman B, Reddy BS. Regression of mouse prostatic intraepithelial neoplasia by nonsteroidal anti-inflammatory drugs in the transgenic adenocarcinoma mouse prostate model. Clin Cancer Res. 2004;10:7727–37. doi: 10.1158/1078-0432.CCR-04-0732. [DOI] [PubMed] [Google Scholar]
- 50.Wang W, Bergh A, Damber JE. Cyclooxygenase-2 expression correlates with local chronic inflammation and tumor neovascularization in human prostate cancer. Clin Cancer Res. 2005;11:3250–6. doi: 10.1158/1078-0432.CCR-04-2405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.