

Abstract
As a tool for measuring the aging process, life span has been invaluable in dissecting the genes that modulate longevity. Studies over the past few decades have identified several hundred genes that can modify life span in model organisms such as yeast, worms, and flies. Yet, despite this vast amount of research, we still do not fully understand how the genes that affect life span influence how an organism ages. How does modulation of the genes that affect life span contribute to the aging process? Does life-span extension result in extension of healthy aging? Here, we will focus primarily on the insulin/IGF-1 signaling pathway in Caenorhabditis elegans because members of this pathway have been shown to be associated with extended life span across phylogeny, from worms to humans. I discuss how this connects to the aging process, age-associated disease, and the potential to increase healthy aging in addition to lengthening life span.
Keywords: Life span, Aging, Health span, C. elegans, Genetics
Defining the molecular cues that underlie the aging process has been the topic of many research studies over the last few decades. Many of the findings are derived from studies in model organisms including yeast, worms, and flies (1–4). The discovery of the first gene to modulate life span, age-1 in Caenorhabditis elegans, led to an explosion of aging research in this system (5,6). Subsequently, these analyses resulted in the finding that the insulin/IGF-1 signaling (IIS) pathway is a central regulator of life span (1,3). Downstream of the insulin/IGF-1 receptor is a conserved signaling pathway that targets the forkhead box O transcription factor (FOXO), DAF-16. Importantly, since the cloning of DAF-16/FOXO in 1997, recent data across multiple human cohorts have shown an association with a FOXO variant and life-span extension (7–10).
LIFE SPAN AS A MEASUREMENT FOR AGING RESEARCH
Over the last three decades, life span as a tool for aging research has been invaluable. Life span, defined as the time from birth until death, has been extensively used to study the aging process. This is in contrast to aging, which is a biological process that is not easily defined or measured. Definitions of aging can include what happens to an organism over time; the changes in tissues, cells, or organs over time; or an increased probability of death. The terms “life span” and “aging” tend to become intertwined, however, and, they are not equivalent.
Over the last decade, several hundred genes were found to modulate C. elegans life span. Analysis of these genes led to the finding that signaling pathways in addition to the IIS pathway also modulate life span, including target of rapamycin (TOR), sirtuins, Jun kinase (JNK), protein translation, and mitochondrial signaling (11–13). Mainly based on genetic epistasis analyses, these pathways have been shown to modulate life span either through intersection with the IIS pathway (eg, TOR signaling, dietary restriction [DR]) or as a separate pathway (DR and mitochondrial signaling) (13–15). In C. elegans, recent work has shown that six different DR regimens can extend life span (16). Genetic analysis of the different DR regimens shows that these methods seem to be regulated by either independent or additive pathways (16). In addition, TOR signaling is required for DR and intersects directly with the IIS (15,17). Therefore, primarily based on genetic analysis, the IIS and DR pathways function both in an overlapping and independent manner.
Many different types of genes (different functional classes) that modulate life span have been identified. However, thus far, mutants that show changes in life span also show changes in an additional process. For example, in addition to regulating life span, genes in the IIS pathway also modulate metabolism, development, and stress resistance (3,12). This is similar to longevity mutants in other organisms including flies in which mutations in the IIS pathway result in changes in size in addition to life span (18–20). Similarly, compared with wild-type mice, long-lived dwarf mice are smaller and have difficulty maintaining their body temperature (21). These findings suggest that regulation of life span may be molecularly linked to regulation of another biological process, such as growth or development. Does the fact that life-span mutants show pleiotropic (multiple related or unrelated phenotypic traits) effects indicate there are simply no “life-span genes?” Mutants that show changes in life span do not always show a similar secondary phenotype, although this has not been extensively studied. Alternatively, it is possible that the change in life span is simply a byproduct of another regulated process, such as reproduction or developmental stress that is coupled to life span. For example, perhaps the byproduct of a worm surviving some early developmental stress is life-span extension in the adult stage. Notably, inactivation of embryonic lethal genes in young adults can lead to life-span extension (22,23). It is also possible that the laboratory environment, in which C. elegans are grown with plenty of food, a constant temperature, and lack of crowding prevents identification of mutants that would help determine these answers. Consistent with this, daf-16 mutants are sensitive to many external stresses (24,25). It is possible that daf-16 mutants would not have ever been identified in the wild because they are stress sensitive. Alternatively, we may miss important genes because some life-extending genes would only display a longer life in the context of a “stressful” environment.
Therefore, using life span as a measurement gives us one insight into the process of how an animal ages. To date, many genes have been identified that have the ability to modulate life span when altered in dosage. But there are limited data on how these genes influence how an organism ages.
DAUER FORMATION AND LIFE SPAN ARE INTERCONNECTED
Much of our understanding of molecular pathways that regulate life span originated with studies in C. elegans. Klass (6,26) published a breakthrough study where mutants that had altered life span were identified. Subsequent studies by the Johnson lab showed that all of the mutants identified by Klass fell into a single genetic locus, named age-1 (5,27). This finding was fundamentally important as it suggested that life span, similar to other processes in development, was under genetic control. It was not until a second age mutant, daf-2 (28), was identified that aging studies in C. elegans seemed to become more popular and interesting to the public at large.
Age-1 and daf-2 were originally isolated in a genetic screen that Don Riddle and coworkers conducted in C. elegans to identify mutations that altered development (29,30). Under normal growth conditions, worms progress from egg through four larval stages and then into a self-fertilizing hermaphrodite. However, under unfavorable growth conditions (particularly crowding) rather than form a reproducing adult hermaphrodite, worms enter an alternative larval stage and form a dauer larva at the third larval stage (L3). Dauers (for “enduring”) are sealed from the environment and are basically a survival stage, where worms have a thick cuticle, do not feed, and show changes in their nervous system (similar to a spore-like state), and wait until growth conditions become more favorable, primarily for healthy progeny production (29,30).
Mutations in age-1 (originally identified as daf-23) and daf-2 were found to cause a temperature-sensitive dauer formation constitutive phenotype (daf-c [29,30]). A mutant that has a daf-c phenotype will form dauers even under favorable growth conditions, and most of these mutants are also temperature sensitive. Mutants that fail to form dauers even under unfavorable growth conditions were also isolated and were termed dauer formation defective, daf-d. In these original studies, daf-16 was isolated as a daf-d mutant (29,30).
Interestingly, analysis of the genes that modulate dauer formation, which include both daf-c and daf-d mutants, placed daf-2, daf-16, daf-18, and age-1/daf-23 in a separate linear pathway from the other genes involved in dauer formation (31,32). Further studies showed that these genes were in a pathway that modulated life span and stress resistance in addition to dauer formation and that was a separate genetic pathway from other genes that modulate dauer formation (31,33,34).
From 1996 to 2001, tremendous strides were made in our understanding of the molecular nature of the daf-2/age-1/daf-16 pathway (3). Molecular cloning revealed that daf-2 encoded for a receptor that bore sequence similarity to both insulin receptor and insulin-like growth factor-1 receptor (35), and age-1 was the catalytic subunit of phosphatidylinositol 3-kinase (36). To date, analysis of the insulin-like mutants has shown that disruption of a single insulin gene cannot mimic loss of the daf-2 receptor (37–39).
Following daf-2 is a well-conserved phosphatidylinositol 3-kinase signaling cascade that ends in the direct modulation of DAF-16/FOXO (3,12,37–39). The IIS pathway regulates many biological processes including dauer/development, longevity, metabolism, and stress resistance. Therefore, DAF-16 is the target of the IIS pathway, and changes in the dosage or activity of DAF-16 result in changes in many biological functions including life span, dauer formation/development, stress resistance, and metabolism (3,12,37–39).
A large number of genes and signaling pathways that intersect with the IIS pathway or DAF-16 have been identified within the last decade (12,17,37). These include signaling pathways that intersect with the IIS such as TOR signaling or pathways that intersect directly with daf-16 such as JNK, 5’ adenosine monophosphate-activated protein kinase (AMPK), and 14-3-3, sirtuins, protein translation, and mitochondrial signaling (37–39). In addition, recent studies using null mutants and methods to uncouple egg-laying problems from life span have led to a Transforming Growth Factor-β (TGF-b) signaling pathway intersecting with the IIS pathway for dauer formation and regulation of life span (40,41). Interestingly, microarray data from long-lived daf-2 mutants and dauer larvae show significant overlap (42–44). Since worms do not age when in the dauer state (29), it is still possible in these long-lived mutants, that one is partially activating the dauer program. Therefore, in light of these findings, connections between the regulation of life span and the regulation of dauer formation need to be further examined.
AGE-ASSOCIATED CHANGES IN C. elegans
Many mutants have been isolated that can extend life span in different model organisms. However, little attention has been paid to whether or not these mutations also extend healthy life or health span (defined herein as the healthy productive time before the onset of age-associated decline). With the large numbers of genes that can alter life span, one wonders which, if any, will lead to additional healthy time. Given the health costs associated with years of decrepitude, extending life span without also extending the duration of healthy time is not a goal of aging research. But how do we define health span in such disparate species as yeast, worms, flies, mice, monkeys, and humans? Will this be a similar measurement in the different model systems? What are the measurements that one can use to define health span in a laboratory setting? Can we model aspects of human health and human age-associated disease in C. elegans? To date, there have been limited studies on health span.
Studying health span in the laboratory creates several challenges. First, how does one define health span (45) or healthy aging? Second, do we study how a given laboratory animal ages and then define health (45)? Third, do we study aging traits that are common to both the lab animal and humans? To address any or all of these questions, one first needs to define normal patterns of age-related changes. To date, the most used measure for aging research is life span: a measurement of the length of time. However, is this the best measure? If one is studying the aging process in the laboratory to benefit humans, then surely measuring life span is just a small part of understanding the aging process to ultimately extend healthy aging.
Health-span studies are largely limited and have focused on age-associated decline characteristics that resemble human aging. C. elegans has been at the forefront of studies on healthy aging due to several factors including a short life cycle, transparency of the cuticle, and the ability to observe many changes under the dissecting microscope. Therefore, the normal aging process in wild-type worms can be easily dissected and observed. This has allowed documentation of age-related changes that occur at all levels—molecular, cellular, tissue, and organ. Examining the nature of the age-related changes in different tissues allows one to examine how a particular tissue changes with age and whether the changes are on the same time scale as changes in other tissues. These changes occur in muscular, neuronal, and reproductive tissues (46–50).
Age-related changes assays in C. elegans can often be scored longitudinally: The same worms can be monitored throughout their life span. In addition, some assays are done on populations of aged worms because the assay itself results in lethality. Important information on age-related changes can be garnered in both types of assays. For a subset of these age-related changes, analysis has been done on both wild-type worms and on longevity mutants as they age, including worms with mutations in the IIS pathway (daf-2, age-1, and daf-16), the mitochondrial timing gene clk-1, and the eat-2 gene (which causes mutants to mimic DR). Here, we will focus on a subset of age-associated changes that have been studied in both wild-type worms and long-lived mutants (46,47,51,52). The reasoning is that if any of the age-related changes can be related to the causes of aging, then the physical, biochemical, and cellular change will occur either earlier than in wild-type worms (such as short-lived daf-16 mutant) or later than in wild-type worms (such as long-lived mutant daf-2, age-1, clk-1, and eat-2).
A number of neuromuscular changes have been examined in wild-type worms as well as short-lived and long-lived mutants (46). It has been suggested that the C. elegans decline in movement with age is similar to human frailty in aged individuals, and similar to humans, worms show signs of sarcopenia (muscle wasting) as they age (53). These studies measure the rates of body movements and pharyngeal pumping (the pharynx is the organ through which a worm gets its food) chronologically. Body movements can be measured on solid media, where distance and movement patterns can be tracked, or in liquid media, where movements (thrashing/swimming) are measured. In both solid and liquid media, one can detect the ability of the worms to move in a coordinated fashion as they age. Worms move in a beautiful repetitive sinusoidal wave when they are young. As worms age, the shape of the wave on solid media changes and the patterns of body bends change in liquid media as well. These age-related changes correlate with deterioration of the body muscle wall, similar to mammalian sarcopenia and frailty. Importantly, compared with wild-type worms, long-lived mutants (eg, daf-2, age-1) have a delay in age-related deterioration of body movement and muscle deterioration (46).
Pharyngeal pumping has also been studied as a phenotype that can be easily measured and shows age-related changes. In C. elegans, the pharynx undergoes rhythmic contractions that facilitate feeding. This feeding rate can be easily measured and counted under a dissecting microscope, and pharyngeal pumping shows age-related decline (46,51,54–56). Therefore, these are age-related processes that are delayed in long-lived mutants (eg, daf-2, age-1).
Another age-related change is the accumulation of fluorescent compounds, which include lipofuscin and advanced glycosylation end products (AGEs). Lipofuscin is fluorescent and consists of the membrane-bound cellular waste. Because it cannot be degraded, it accumulates in postmitotic cells and therefore is often referred to as an “aging pigment” in both invertebrates and vertebrates (57,58). A second “age pigment” also conserved from worms to humans are AGEs, formed by a nonenzymatic addition of sugar to the free amino group of proteins which is followed by cross-linking by autocatalysis (59,60). Both of these pigments are modulated by the age of the organism from worms to humans (46). Therefore, in this case, similar effects of aging are seen across phylogeny. However, it is not known if the age pigments are a causative part of the aging process or simply a consequence.
In C. elegans, both age pigments have been shown to accumulate with age. In addition, several longevity mutants have been examined for effects on the accumulation of these pigments. The long-lived IIS mutants age-1 and daf-2 show lower levels of fluorescence in young animals and a slower rate of accumulation (46,58,61). Consistent with this, short-lived IIS daf-16 mutants show the opposite effects including increased fluorescence and an accelerated accumulation of age pigments (46,57,58,61).
To further dissect the connections between age-related phenotypes, Gerstbrein and coworkers (58) examined the connections between body movement and age pigments as worms aged. Using spectrofluorimetry and microscopy, they performed a longitudinal study where they assessed both body movement and fluorescence. They found that in wild-type worms, there was a positive correlation between the decline in body movement and the accumulation of fluorescent material (58). Worms under conditions of dietary restriction as well as two long-lived mutants displayed both reduced levels of fluorescence and a slower rate of accumulation (58). For conditions that shorten life span, results have not been entirely correlated. The short-lived IIS mutant daf-16 shows an increase in accumulation of age pigments, whereas the short-lived mitochondrial mutant mev-1 has been shown to have either faster rate of accumulation of fluorescence (57) or decreased levels of age pigment (58). Therefore, accumulation of age pigments correlates with life span in wild-type and long-lived mutants.
The understanding of age-related changes in the nervous system of C. elegans is continuing to evolve. Interestingly, early studies on aging C. elegans where anatomy was examined by electron microscopy showed little change in the nervous system with age, especially when compared with muscle (53). In addition, genetic mosaic analysis suggested that the nervous system provided only a small contribution to the aging process (62). However, recent studies with newer and more specific examinations have shown evidence of neuronal aging in C. elegans. Pan and coworkers (63) examined both the touch receptor neurons and cholinergic neurons in aging worms. They observed changes in the electrical activity of the neurons, suggesting that neuronal integrity is compromised as worms age. They also found that the IIS pathway modulated neuronal integrity. Studies by Tank and coworkers (64) showed that as worms age, neurons frequently develop ectopic branches. They showed that these branches were seen in the mechanosensory neurons and suggest that the decreased mobility and decreased response to touch observed as worms age are due to these age-associated branching defects. Importantly, the age-associated ectopic branching phenotype was also modulated by the IIS pathway and the DAF-16 interactor JNK (64,65). Future studies on neuronal aging in C. elegans should examine long-lived mutants as a percentage of mean life span and determine if the neuronal changes observed correlate with associated phenotypic changes.
To further define the aging process in C. elegans, McGee and coworkers (47) performed extensive serial 3D reconstruction of aging worms, both wild-type and long-lived daf-2 mutants. These studies identified a number of cellular age-related changes that were previously unidentified in the aging intestine, including changes in the size and shape of intestinal nuclei, loss of intestinal nuclei, and loss of microvilli. The loss of intestinal nuclei as worms age was highly stochastic at several levels, including within individuals and intestinal cells (47). When long-lived daf-2 worms were examined, they showed a prolonged duration of time with a normal intestine, and the loss of intestinal integrity was delayed. However, it remains unclear how these age-associated changes in the C. elegans intestine contribute to any age-associated phenotypes in the worm or the death of the worm. Taken together, many C. elegans studies have shown that many age-related changes are accelerated in a short-lived mutant and delayed in a long-lived mutant.
HEALTH-SPAN STUDIES IN C. elegans
Recent studies have combined several of these changes to identify health-span regulators in C. elegans. Because there is no universal definition of health span nor is this term defined in the laboratory setting, Iwasa and coworkers (66) defined health span as “the period of midlife vigor that precedes significant functional decline.” With this definition of health span, they performed a genetic screen using an automated recording device to screen for C. elegans mutants that showed a prolonged swimming ability (movement in liquid media) when compared with wild-type (66). This screen identified two genes termed high performance in advanced age locomotory 1 and 2 (hpa-1 and hpa-2). Subsequent molecular and genetic studies showed that these genes were part of the epidermal growth factor (EGF) signaling pathway. hpa-1 and hpa-2 mutants were further characterized for other age-associated phenotypes and showed decreased lipofuscin accumulation as well as decreased advanced glycation end-product accumulation. It is noteworthy that although these animals showed prolonged swimming ability, their median and mean life span were only slightly increased, and maximum life span was unaffected. hpa-1 and hpa-2 mutants also showed similar mortality rates over adult life when compared with wild type (where the rates of mortality rise exponentially with age). Therefore, these mutants live the same time as wild type but are active longer without changing their rate of aging. Analysis of additional components of the EGF signaling pathway components revealed that other EGF signaling genes also modulate healthy aging in worms, and these effects were largely independent of the IIS pathway. Therefore, by defining health span as locomotor swimming ability at advanced ages, the EGF signaling pathway was identified as a regulator of healthy aging in C. elegans.
The study by Iwasa and colleagues (66) showed for the first time that the ability to screen for healthy aging genes has the potential to identify new genes that modulate multiple age-associated parameters. It is somewhat surprising that the genes identified in this screen, hpa-1 and hpa-2, seem to function independent of the IIS pathway (66) when multiple previous studies have suggested that the long-lived IIS mutants show a delay in many age-associated phenotypes including locomotion (46). How could this be?
One clue may come from important studies by Huang and coworkers (51) where different physiological processes were studied as worms age in wild type, IIS components (daf-2, age-1, and daf-16), clk-1, and eat-2. The life span was divided up into four different stages (I–IV) based on reproductive time, pharyngeal pumping time, and fast movement. Worms were evaluated as they aged and scored for the number of days spent in each stage. Data were then further analyzed by setting the mean life span equal to 100% and examining the fraction of the mean life span each strain occupied at the different stages. daf-2 and age-1 showed an expansion of the last stage when animals are no longer pumping and hardly moving. Therefore, examining all strains including wild type and mutants at their mean life span (51,66,67) gave additional insight into age-associated characteristics. In this way, one can also measure what proportion of life the animal is spending active. For example, if one defines health span as a disease-free state before the onset of age-associated decline, then all long-lived mutants have an increase in health span (Figure 1C). However, thus far, only one study (51) has measured a few parameters as a percentage of mean life span. Almost all studies examine age-associated changes in long-lived mutants chronologically: comparing mutants and wild type on the same day in the life span. It is also important to note that very few age-related assays have been done on older long-lived animals to examine their capabilities in their extended periods to determine if their extended life span results in extended health or decrepitude. Therefore, the connection between life span and health span needs to be further explored. It is unclear what the interplay between them will be: entirely separate (eg, EGF studies [66]), some genes in common, or completely overlapping (Figure 1).
Figure 1.
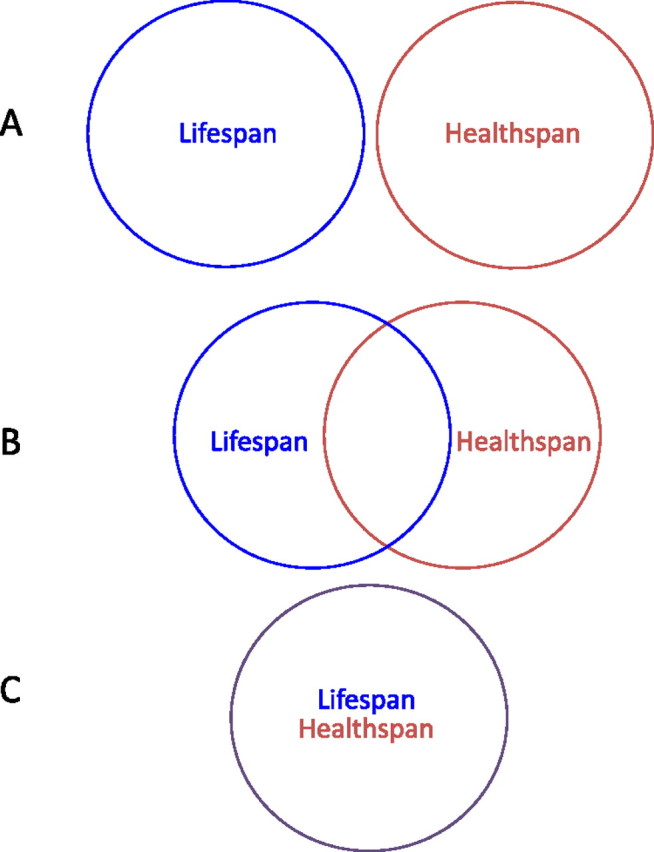
Determining the connection between life span and health span. In (A) all of the genes that regulate life span are different than the genes that modulate health span (eg, pha-1 and hpa-2). In (B), there is partial overlap, and in (C), there is complete overlap. See text for details.
MODELING HUMAN AGE-ASSOCIATED DISEASE
One possibility for extending life span and healthy aging is to prolong the time before the onset of age-associated disease. To examine this directly, several age-associated neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease have been explored using transgenic models in lower organisms. In C. elegans, this has allowed for dissecting connections between longevity and age-associated disease with a focus on whether or not life-span extension could change susceptibility to the neurodegenerative disease.
One of the attributes associated with Alzheimer’s disease is the proteotoxicity and cell death that arises from the toxic aggregation of plaques consisting of beta-amyloid (Aβ) protein. In the C. elegans transgenic model for Alzheimer’s disease, worms bear a transgene that expresses the human Aβ1–42 driven by a muscle-specific promoter (known as Aβ worms) originally described by Link and colleagues (68). These worms develop plaque deposits in the muscle, and this leads to progressive paralysis. Two life-extending paradigms have been evaluated in the transgenic Aβ C. elegans. In the first study, transgenic worms were subject to one paradigm of DR, bacterial deprivation, where food is completely removed and increases both median and maximum life span by 50% (69). Bacterial deprivation leads to a significant suppression in susceptibility to Aβ proteotoxicity (70). In the second study, reduction of IIS signaling also leads to protection against and Aβ aggregation–associated proteotoxicity (71). These data showed that when IIS is reduced during development and adulthood, animals are protected from paralysis mediated by Aβ1–42. Interestingly, neither Aβ expression levels nor the Aβ total protein amounts are affected in these worms (71). More recently, Cohen and coworkers (72) directly assessed the connection between proteotoxicity and longevity with relation to IIS. Using temporal RNAi against the IIS revealed that decreasing levels of the IIS receptor daf-2 either early or late in life protects from age onset proteotoxicity by invoking a mechanism that converts toxic aggregates into larger, less toxic high-molecular weight aggregates. Remarkably, further dissection of the temporal requirements of daf-2 and daf-16 revealed that these genes are required for protection against proteotoxicity well into adulthood, but this is independent of the life-span extension effects.
In Huntington’s disease, individuals with the disease show an expansion of a CAG repeat. In the C. elegans model, transgenic worms bear a transgene with a tract of 35 conserved glutamine residues fused to YFP that is expressed in the body muscle, resulting in age-related proteotoxicity (73). Similar to the effects of Aβ, bacterial deprivation suppressed the proteotoxicity in this polyQ C. elegans strain (70), and reduction in IIS can also protect worms from polyQ-associated proteotoxicity (73,74). A third method of life-span extension is overexpression of the sirtuin sir-2.1, which has also been shown to be neuroprotective in this model (75).
Parkinson’s disease is characterized by selective loss of dopaminergic neurons. In the C. elegans Parkinson’s disease model, the dat-1 promoter was fused to green fluorescence protein and specifically expressed in eight dopaminergic neurons. Worms were then treated with the neurotoxin 6-hydroxy dopamine, which resulted in dopaminergic neurodegeneration. Interestingly, under dietary restriction, these worms were protected from dopaminergic neurodegeneration. In addition, the sirtuin sir-2.1 was necessary for achieving the neuroprotective effect of dietary restriction (76).
Therefore, dietary restriction and IIS signaling have protective effects against age-associated proteotoxicity in neurodegenerative disease. Importantly, one study in C. elegans suggests that this effect is independent of the longevity effect. Therefore, future studies are required to determine mechanistically how different signaling pathways can regulate these parameters independently of each other.
Longevity research has made tremendous strides since the first discovery of a gene that modifies life span (age-1) in 1983 (6). To date, several hundred genes have been found to be able to modulate life span. The hope and challenge for the future lies with dissecting the connections between life-span extension and healthy aging/health span. The key to determining these connections may lie in how we define, assess, and evaluate the data and the effects of the laboratory setting.
FUNDING
H.A.T. is supported by grants from the National Institute of Aging (AG025891 and AG031237), the Ellison Medical Foundation, and an endowment from the William Randolph Hearst Foundation.
Acknowledgments
I am grateful to Kelvin Yen and Ankita Bansal for advice and critical comments on the manuscript. H.A.T. is a William Randolph Hearst Young Investigator.
References
- 1.Barbieri M, Bonafe M, Franceschi C, et al. Insulin/IGF-I-signaling pathway: an evolutionarily conserved mechanism of longevity from yeast to humans. Am J Physiol Endocrinol Metab. 2003;285(5):E1064–E1071. doi: 10.1152/ajpendo.00296.2003. [DOI] [PubMed] [Google Scholar]
- 2.Bartke A. Single-gene mutations and healthy ageing in mammals. Philos Trans R Soc Lond B Biol Sci. 2011;366(1561):28–34. doi: 10.1098/rstb.2010.0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenyon C. The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing. Philos Trans R Soc Lond B Biol Sci. 2011;366(1561):9–16. doi: 10.1098/rstb.2010.0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith ED, Kennedy BK, Kaeberlein M. Genome-wide identification of conserved longevity genes in yeast and worms. Mech Ageing Dev. 2007;128(1):106–111. doi: 10.1016/j.mad.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Friedman DB, Johnson TE. Three mutants that extend both mean and maximum life span of the nematode, Caenorhabditis elegans, define the age-1 gene. J Gerontol. 1988;43(4):B102–B109. doi: 10.1093/geronj/43.4.b102. [DOI] [PubMed] [Google Scholar]
- 6.Klass MR. A method for the isolation of longevity mutants in the nematode Caenorhabditis elegans and initial results. Mech Ageing Dev. 1983;22:279–286. doi: 10.1016/0047-6374(83)90082-9. [DOI] [PubMed] [Google Scholar]
- 7.Flachsbart F, Caliebe A, Kleindorp R, et al. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A. 2009;106(8):2700–2705. doi: 10.1073/pnas.0809594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willcox BJ, Donlon T, He Q, et al. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A. 2008;105(37):13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anselmi CV, Malovini A, Roncarati R, et al. Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 2009;12(2):95–104. doi: 10.1089/rej.2008.0827. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Wang WJ, Cao H, et al. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum Mol Genet. 2009;18(24):4897–4904. doi: 10.1093/hmg/ddp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolff S, Dillin A. The trifecta of aging in Caenorhabditis elegans. Exp Gerontol. 2006;41(10):894–903. doi: 10.1016/j.exger.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 12.Landis JN, Murphy CT. Integration of diverse inputs in the regulation of Caenorhabditis elegans DAF-16/FOXO. Dev Dyn. 2010;239(5):1405–1412. doi: 10.1002/dvdy.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braeckman BP, Vanfleteren JR. Genetic control of longevity in C. elegans. Exp Gerontol. 2007;42(1–2):90–98. doi: 10.1016/j.exger.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Antebi A. Genetics of aging in Caenorhabditis elegans. PLoS Genet. 2007;3(9):1565–1571. doi: 10.1371/journal.pgen.0030129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mair W, Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem. 2008;77:727–754. doi: 10.1146/annurev.biochem.77.061206.171059. [DOI] [PubMed] [Google Scholar]
- 16.Greer EL, Brunet A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell. 2009;8:113–127. doi: 10.1111/j.1474-9726.2009.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narasimhan SD, Yen K, Tissenbaum HA. Converging pathways in lifespan regulation. Curr Biol. 2009;19(15):R657–R666. doi: 10.1016/j.cub.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429(6991):562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- 19.Aigaki T, Seong KH, Matsuo T. Longevity determination genes in Drosophila melanogaster. Mech Ageing Dev. 2002;123(12):1531–1541. doi: 10.1016/s0047-6374(02)00089-1. [DOI] [PubMed] [Google Scholar]
- 20.Clancy DJ, Gems D, Harshmann LG, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 21.Brown-Borg HM, Rakoczy SG, Sharma S, Bartke A. Long-living growth hormone receptor knockout mice: potential mechanisms of altered stress resistance. Exp Gerontol. 2009;44(1–2):10–19. doi: 10.1016/j.exger.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3(4):e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen D, Pan KZ, Palter JE, Kapahi P. Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging Cell. 2007;6(4):525–533. doi: 10.1111/j.1474-9726.2007.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nemoto-Sasaki Y, Kasai K. Deletion of lec-10, a galectin-encoding gene, increases susceptibility to oxidative stress in Caenorhabditis elegans. Biol Pharm Bull. 2009;32(12):1973–1977. doi: 10.1248/bpb.32.1973. [DOI] [PubMed] [Google Scholar]
- 25.Yanase S, Yasuda K, Ishii N. Adaptive responses to oxidative damage in three mutants of Caenorhabditis elegans (age-1, mev-1 and daf-16) that affect life span. Mech Ageing Dev. 2002;123(12):1579–1587. doi: 10.1016/s0047-6374(02)00093-3. [DOI] [PubMed] [Google Scholar]
- 26.Klass M, Nguyen PN, Dechavigny A. Age-correlated changes in the DNA template in the nematode Caenorhabditis elegans. Mech Ageing Dev. 1983;22(3–4):253–263. doi: 10.1016/0047-6374(83)90080-5. [DOI] [PubMed] [Google Scholar]
- 27.Friedman DB, Johnson TE. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 1988;118:75–86. doi: 10.1093/genetics/118.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366(6454):461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 29.Riddle DL. The Nematode Caenorhabditis elegans. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. pp. 393–412. The dauer larva. [Google Scholar]
- 30.Riddle DL, Albert PS. Genetic and environmental regulation of dauer larva development. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 739–768. [PubMed] [Google Scholar]
- 31.Vowels JJ, Thomas JH. Genetic analysis of chemosensory control of dauer formation in Caenorhabditis elegans. Genetics. 1992;130:105–123. doi: 10.1093/genetics/130.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gottlieb S, Ruvkun G. daf-2, daf-16 and daf-23: genetically interacting genes controlling Dauer formation in Caenorhabditis elegans. Genetics. 1994;137(1):107–120. doi: 10.1093/genetics/137.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorman JB, Albinder B, Shroyer T, Kenyon C. The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics. 1995;141(4):1399–1406. doi: 10.1093/genetics/141.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larsen PL, Albert PS, Riddle DL. Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics. 1995;139(4):1567–1583. doi: 10.1093/genetics/139.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura KD, Tissenbaum HA, Liu Y, Ruvkin G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277(5328):942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 36.Morris JZ, Tissenbaum HA, Ruvkun G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature. 1996;382(6591):536–539. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- 37.Yen K, Narasimhan SD, Tissenbaum HA. DAF-16/Forkhead box O transcription factor: many paths to a single Fork(head) in the road. Antioxid Redox Signal. 2011;14(4):623–634. doi: 10.1089/ars.2010.3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukhopadhyay A, Oh SW, Tissenbaum HA. Worming pathways to and from DAF-16/FOXO. Exp Gerontol. 2006;41(10):928–934. doi: 10.1016/j.exger.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 39.Kenyon CJ. The genetics of ageing. Nature. 2010;464(7288):504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 40.Narasimhan SD, Yen K, Bansal A, Kwon ES, Padmanabhan S, Tissenbaum HA. PDP-1 links the TGF-beta and IIS pathways to regulate longevity, development, and metabolism. PLoS Genet. 2011;7(4):e1001377. doi: 10.1371/journal.pgen.1001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaw WM, Luo S, Landis J, Ashraf J, Murphy CT. The C. elegans TGF-beta Dauer pathway regulates longevity via insulin signaling. Curr Biol. 2007;17(19):1635–1645. doi: 10.1016/j.cub.2007.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McElwee JJ, Schuster E, Blanc E, Thornton J, Gems D. Erratum to “Diapause-associated metabolic traits reiterated in long-lived daf-2 mutants in the nematode Caenorhabditis elegans” [Mech. Ageing Dev. 127(5) (2006) 458-472] Mech Ageing Dev. 2006;127(12):922–936. doi: 10.1016/j.mad.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 43.McElwee JJ, Schuster E, Blanc E, Thomas JH, Gems D. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J Biol Chem. 2004;279(43):44533–44543. doi: 10.1074/jbc.M406207200. [DOI] [PubMed] [Google Scholar]
- 44.McElwee JJ, Schuster E, Blanc E, Thornton J, Gems D. Diapause-associated metabolic traits reiterated in long-lived daf-2 mutants in the nematode Caenorhabditis elegans. Mech Ageing Dev. 2006;127(5):458–472. doi: 10.1016/j.mad.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 45.Tatar M. Can we develop genetically tractable models to assess healthspan (rather than life span) in animal models? J Gerontol A Biol Sci Med Sci. 2009;64(2):161–163. doi: 10.1093/gerona/gln067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins JJ, Huang C, Hughes S, Kornfeld K. The measurement and analysis of age-related changes in Caenorhabditis elegans. WormBook. 2008:1–21. doi: 10.1895/wormbook.1.137.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGee MD, Weber D, Day N, et al. Loss of intestinal nuclei and intestinal integrity in aging C. elegans. Aging Cell. 2011;10(4):699–710. doi: 10.1111/j.1474-9726.2011.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hughes SE, Huang C, Kornfeld K. Identification of mutations that delay somatic or reproductive aging of Caenorhabditis elegans. Genetics. 2011;189(1):341–356. doi: 10.1534/genetics.111.130450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo S, Kleemann GA, Ashraf JM, Shaw WM, Murphy CT. TGF-beta and insulin signaling regulate reproductive aging via oocyte and germline quality maintenance. Cell. 2010;143(2):299–312. doi: 10.1016/j.cell.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Golden TR, Hubbard A, Dando C, Herren MA, Melov S. Age-related behaviors have distinct transcriptional profiles in Caenorhabditis elegans. Aging Cell. 2008;7(6):850–865. doi: 10.1111/j.1474-9726.2008.00433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang C, Xiong C, Kornfeld K. Measurements of age-related changes of physiological processes that predict lifespan of Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2004;101(21):8084–8089. doi: 10.1073/pnas.0400848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hughes SE, Evason K, Xiong C, Kornfeld K. Genetic and pharmacological factors that influence reproductive aging in nematodes. PLoS Genet. 2007;3(2):e25. doi: 10.1371/journal.pgen.0030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herndon LA, Schmeissner PA, Dudaronek JM. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419(6909):808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- 54.Hosono R, Sato Y, Aizawa SI, Mitsui Y. Age-dependent changes in mobility and separation of the nematode Caenorhabditis elegans. Exp Gerontol. 1980;15(4):285–289. doi: 10.1016/0531-5565(80)90032-7. [DOI] [PubMed] [Google Scholar]
- 55.Chow DK, Glenn CF, Johnston JL, Goldberg IG, Wolkov CA. Sarcopenia in the Caenorhabditis elegans pharynx correlates with muscle contraction rate over lifespan. Exp Gerontol. 2006;41(3):252–260. doi: 10.1016/j.exger.2005.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Croll NA, Smith JM, Zuckerman BM. The aging process of the nematode Caenorhabditis elegans in bacterial and axenic culture. Exp Aging Res. 1977;3(3):175–189. doi: 10.1080/03610737708257101. [DOI] [PubMed] [Google Scholar]
- 57.Hosokawa H, Ishii N, Ishida H, Ichimori K, Nakazawa H, Suzuki K. Rapid accumulation of fluorescent material with aging in an oxygen-sensitive mutant mev-1 of Caenorhabditis elegans. Mech Ageing Dev. 1994;74(3):161–170. doi: 10.1016/0047-6374(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 58.Gerstbrein B, Stamatas G, Kollias N, Driscoll M. In vivo spectrofluorimetry reveals endogenous biomarkers that report healthspan and dietary restriction in Caenorhabditis elegans. Aging Cell. 2005;4(3):127–137. doi: 10.1111/j.1474-9726.2005.00153.x. [DOI] [PubMed] [Google Scholar]
- 59.Ulrich P, Cerami A. Protein glycation, diabetes, and aging. Recent Prog Horm Res. 2001;56:1–21. doi: 10.1210/rp.56.1.1. [DOI] [PubMed] [Google Scholar]
- 60.Krautwald M, Munch G. Advanced glycation end products as biomarkers and gerontotoxins—a basis to explore methylglyoxal-lowering agents for Alzheimer’s disease? Exp Gerontol. 2010;45(10):744–751. doi: 10.1016/j.exger.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 61.Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics. 2002;161(3):1101–1112. doi: 10.1093/genetics/161.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115(4):489–502. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- 63.Pan CL, Peng CY, Chen CH, McIntire S. Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc Natl Acad Sci U S A. 2011;108(22):9274–9279. doi: 10.1073/pnas.1011711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tank EM, Rodgers KE, Kenyon C. Spontaneous age-related neurite branching in Caenorhabditis elegans. J Neurosci. 2011;31(25):9279–9288. doi: 10.1523/JNEUROSCI.6606-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci U S A. 2005;102(12):4494–4499. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iwasa H, Yu S, Xue J, Driscoll M. Novel EGF pathway regulators modulate C. elegans healthspan and lifespan via EGF receptor, PLC-gamma, and IP3R activation. Aging Cell. 2010;9(4):490–505. doi: 10.1111/j.1474-9726.2010.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edrey YH, Hanes M, Pinto M, Mele J, Buffenstein R. Successful aging and sustained good health in the naked mole rat: a long-lived mammalian model for biogerontology and biomedical research. ILAR J. 2011;52(1):41–53. doi: 10.1093/ilar.52.1.41. [DOI] [PubMed] [Google Scholar]
- 68.Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis-elegans. Proc Natl Acad Sci U S A. 1995;92(20):9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaeberlein TL, Smith ED, Tsuchiya M, et al. Lifespan extension in Caenorhabditis elegans by complete removal of food. Aging Cell. 2006;5(6):487–494. doi: 10.1111/j.1474-9726.2006.00238.x. [DOI] [PubMed] [Google Scholar]
- 70.Steinkraus KA, Smith ED, Davis C, et al. Dietary restriction suppresses proteotoxicity and enhances longevity by an hsf-1-dependent mechanism in Caenorhabditis elegans. Aging Cell. 2008;7(3):394–404. doi: 10.1111/j.1474-9726.2008.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313(5793):1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 72.Cohen E, Du D, Joyce D, Kapernick EA, Volovik Y, Kelly JW, Dillin A. Temporal requirements of insulin/IGF-1 signaling for proteotoxicity protection. Aging Cell. 2010;9(2):126–134. doi: 10.1111/j.1474-9726.2009.00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99(16):10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300(5622):1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 75.Parker JA, Arango M, Abderrahmane S, Lambert E, Tourette C, Catoire H, Néri C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet. 2005;37(4):349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- 76.Jadiya P, Chatterjee M, Sammi SR, Kaur S, Palit G, Nazir A. Sir-2.1 modulates ‘calorie-restriction-mediated’ prevention of neurodegeneration in Caenorhabditis elegans: implications for Parkinson’s disease. Biochem Biophys Res Commun. 2011;413(2):306–310. doi: 10.1016/j.bbrc.2011.08.092. [DOI] [PubMed] [Google Scholar]