

Abstract
Background
Chemotherapy is important in the systematic treatment of breast cancer. To enhance the response of tumours to chemotherapy, attention has been focused on agents to reverse multidrug resistance (MDR) and on the sensitivity of tumour cells to chemical drugs. Hundreds of reversal drugs have been found in vitro, but their clinical application has been limited because of their toxicity. The reversal activity of progestogen compounds has been demonstrated. However, classical agents such as progesterone and megestrol (MG) also have high toxicity. Nomegestrol (NOM) belongs to a new derivation of progestogens and shows very low toxicity. We studied the reversal activity of NOM and compared it with that of verapamil (VRP), droloxifene (DRO), tamoxifen (TAM) and MG, and investigated the reversal mechanism, i.e. effects on the expression of the MDR1, glutathione S-transferase Pi (GSTπ), MDR-related protein (MRP) and topoisomerase IIα (TopoIIα) genes, as well as the intracellular drug concentration and the cell cycle. The aim of the study was to examine the reversal effects of NOM on MDR in MCF7/ADR, an MCF7 breast cancer cell line resistant to adriamycin (ADR), and its mechanism of action.
Methods
MCF7/ADR cells and MCF7/WT, an MCF7 breast cancer cell line sensitive to ADR, were treated with NOM as the acetate ester. With an assay based on a tetrazolium dye [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; MTT], the effects of various concentrations of NOM on MDR in MCF7/ADR cells were studied. Before and after the treatment with 5 μM NOM, the expression of the MDR-related genes MDR1, GSTπ, TopoIIα and MRP were assayed with a reverse transcriptase polymerase chain reaction (RT-PCR) immunocytochemistry assay. By using flow cytometry (FCM), we observed the intracellular ADR concentration and the effects of combined treatment with NOM and ADR on the cell cycle. Results collected were analysed with Student's t test.
Results
NOM significantly reversed MDR in MCF7/ADR cells. After treatment NOM at 20, 10 and 5 μM, chemosensitivity to ADR increased 21-fold, 12-fold and 8-fold, respectively. The reversal activity of NOM was stronger than that of the precursor compound MG, and comparable to that of VRP. After treatment with 5 μM NOM, the expression of both the MDR1 and the GSTπ mRNA genes began to decline on the second day (P <0.05 and P <0.01, respectively), and reached the lowest level on the third day (both P <0.01); however, on the fifth day the expression levels began to increase again (both P <0.05). The expression of MRP and TopoIIα had no significant changes. Changes in the expression of P-glycoprotein (P-gp) and GSTπ were similar to those of their mRNA expressions, showing early declines and late increases. Two hours after treatment with 20, 10 and 5 μM NOM, the intracellular ADR concentration increased 2.7-fold, 2.3-fold and 1.5-fold respectively. However, NOM did not increase ADR accumulation in MCF7/WT cells. FCM data showed that after 48 h of combined administration of NOM (20 μM) and ADR (from low to high concentration), MCF7/ADR cells showed a gradual arrest at the G2M phase with increasing ADR dose. The arrest effect with combined drug treatment was stronger than that with the single ADR treatment.
Conclusion
MDR is the major mechanism of drug resistance in malignant tumour cells. To overcome MDR and to increase chemosensitivity, many reversal agents have been found. Most progestogen compounds have been demonstrated to have reversal effects, but we found no data on NOM, a new progestogen compound. Our results show that NOM has strong reversal activity. The reversal effects were stronger than those of the precursor compound, MG, and were comparable to that of VRP. Because NOM has low toxicity, it might have good prospects in clinical application. Using RT-PCR and immunocytochemistry assays, we studied the effects of NOM on MDR-related genes. The results were that NOM could markedly downregulate the mRNA and protein expression levels of MDR1 and GSTπ. TopoIIα and MRP gene expression showed no significant changes. It is known that P-gp induces MDR in tumour cells mainly by decreasing the intracellular drug concentration. After treatment with NOM, the intracellular drug concentration in MCF7/ADR cells increased significantly. Combined treatment with NOM and ADR induced arrest at the G2M phase. It is worth noting that NOM caused an early decrease and a late increase in the expression of some MDR-related genes in a time-dependent manner. The phenomena raise a question for the continued administration of reversal agents in clinics that merits further study. We demonstrate that NOM has strong reversal effects on MDR in MCF7/ADR cells. The reversal is via different routes, namely downregulating the mRNA and protein expression levels of MDR1 and GSTπ, increasing intracellular drug concentration and arresting cells at the G2M phase (NOM in combination with ADR). The reversal mechanism needs further study.
Keywords: MDR, MCF7/ADR, nomegestrol acetate, reversal
Introduction
In recent years, the incidence of breast cancer has become one of the most rapidly increaing among malignant tumours. With progress in understanding of the nature of breast cancer, diagnosis and treatments have been improved. Chemotherapy has become more and more important and is considered to be a major treatment to avoid the recurrence of cancer after surgery [1]. Although the remission rate is higher in previously untreated patients, relapse occurs soon. It has been reported that most initially responsive patients acquire a multidrug resistance (MDR) phenotype. Some other patients show MDR even in their first treatment. In metastatic breast cancer, the development of a MDR phenotype is primarily responsible for insensitivity to a new drug [2].
Attention has been focused recently on the study of the agents for reversing MDR. Although hundreds of compounds have been found in vitro to be able to modulate the MDR phenotype, their clinical application was limited owing to high toxicity in vivo. The key to the clinical use of reversal agents therefore lies in searching for agents with low toxicity and high reversal activity [3]. In the past, many progestogen compounds, such as MG and medroxyprogesterone, have been shown to have reversal effects in vitro [4, 5, 6, 7]. Unfortunately, the effective concentration to reverse drug resistance in vivo is very difficult to achieve [8]. Nomegestrol (NOM), a derivative of megestrol (MG), has very low toxicity. There is almost no incidence of liver lesions, pituitory-inhibiting activity, cortisol-like activity or oestrogen-like activity, which are common in other progestogen compounds [9]. NOM might therefore be suitable for clinical use.
In many previous studies on MDR reversal by compounds related to NOM, attention has been paid to the modulation of P-glycoprotein (P-gp) function. The reversal mechanism consists of, for example, binding directly to P-gp, inhibiting the transport function of drug efflux, increasing the intracellular accumulation of the drug and changing lipid mobility in the plasma membrane [4, 10, 11, 12]. However, different compounds have different mechanisms. No results on the reversal effect of NOM or on its mechanism were available. The present study was directed to the reversal activity of NOM on MDR in adriamycin (ADR)-resistant (MCF7/ADR) cells, and to compare NOM with classic reversal agents [verapamil (VRP), tamoxifen (TAM), droloxifene (DRO) and MG]. A possible reversal mechanism was explored by studying the cell cycle, intracellular drug accumulation and the expression of mRNA and proteins of MDR1, glutathione S-transferase Pi (GSTπ), MDR-related protein (MRP) and topoisomerase IIα (TopoIIα).
Materials and methods
Cell line and cell culture
The cell lines MCF7/WT (ADR-sensitive) and MCF7/ADR were used in this study; they were obtained from NCI [13] and maintained in Dulbecco's modified Eagle's medium incubated at 37°C in 5% CO2-95% air at high humidity, and passaged every 2-3 days. Cells were digested with mixture of 0.025% trypsin (Gibco BRL) and 0.01% EDTA (Sigma). Medium for MCF7/ADR cells was further supplemented with ADR (10 μM; Shanghai Hualian Pharmaceutical Co. Ltd., Shanghai, China). Before use in experiments, MCF7/ADR cells were cultured in drug-free medium for 2 weeks.
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay of cytotoxic activity
Cells were treated with NOM (purchased as the acetate ester from Pharmaceutical College, Shanghai Medical University) and other reversal agents, including MG, TAM, DRO (a gift from Professor Xia Peng) and VRP (Shanghai Hualian Pharmaceutical Co. Ltd.). MTT assays were performed as follows [14]: MCF7/WT and MCF7/ADR cells were each harvested with 0.05% trypsin/EDTA and counted. Cell lines were seeded into 96-well plates at 104 viable cells per well and left to attach to the plate for 24 h. After 24 h, the medium was changed to one containing or lacking test reversal agents or ADR. The final volume was 200 μl per well. The medium was removed after 72 h of incubation. Other medium containing 0.5 mg/ml MTT (Sigma) was added to each well in a volume of 200 μl and incubated for 4 h. The medium was then removed and 180 μl of dimethyl sulphoxide (Sigma) was added to each well for half an hour at room temperature. A 96-well microtitre plate reader (Dynatech, Chantilly, VA, USA) was used to determine A570. The mean concentration in each set of three wells was measured. To avoid interference by the red fluorescence of ADR concentrations above 8 μM, a blank well containing the corresponding ADR concentration without MTT was set up and subtracted from the test well absorbance. The absorbance of untreated controls was taken as 100% survival, and the percentage inhibition was calculated as cell survival rate (%) = 100(T - B)/(U - B), and growth inhibition (%) = 100-cell survival rate (%), where T (treated) is the absorbance of drug-treated cells, U (untreated) is the absorbance of untreated cells and B (blank) is the absorbance in the absence of both drug and MTT.
IC50 values were determined graphically from relative survival curves.
The fold reversal was calculated as IC50 for ADR/IC50 for ADR plus reversal agents.
Semiquantitative reverse transcriptase polymerase chain reaction (RT-PCR) analysis
NOM treatment
Cells were subjected to mild treatment with trypsin and plated at a density of 105 cells/ml. NOM was tested at 5 μM; cells were passaged as usual. After 2, 3, 5 and 10 days, NOM-containing medium was removed and cells were harvested.
RNA isolation
Total RNA was extracted from the treated cell line or control samples with the TRIzol system (Gibco BRL), in accordance with the manufacturer's instructions. The concentration and purity of RNA were quantified spectrophotometrically by measuring A260 and A280; the ratio A260/A280 of pure RNA is approximately 1.8. The sample was stored at -70°C.
Reverse transcription reaction
The RNA sample was added to a sterile RNase-free micro-centrifuge tube, and nuclease-free water was added to 9.5 μl. The RNA sample was heated to 70°C for 10 min, then chilled on ice. To this, 4 μl of a 5×AMV RT reaction buffer, 2 μl of 25 mM MgCl, 2 μl of 10 mM dNTP, 0.5 μl of Rnasin (1 U/μl) (Promega), 1 μl of oligo(dT)15 (0.5 μg/μl) (Promega) and 1 μl of AMV RT (10 U/μl) (Promega) were added. The final volume was 20 μl. This mixture was centrifuged shortly, then incubated at 42°C for 1 h to allow the AMV RT enzyme to catalyse the formation of cDNA on the mRNA template. The enzyme was then inactivited by being heated to 95°C for 2 min. The cDNA was stored at -20°C until required for analysis.
PCR
PCR was set up as described previously [15]. Two sets of primers were used in all reactions to yield the amplification of an endogenous control gene (β-actin, 383 bases) and the specific target genes of interest [157 bases of MDR1 (a gift from Jian Lin, Cancer Center/Institute of Cancer Research, Golumbia University, College of Physicians and Surgeons, New York, USA), 270 bases of GSTπ, 203 bases of MRP and 139 bases of TopoIIα independently (Table 1) [15]]. PCR amplification was performed on 1 μl of RT product (25 μM) incubated with 0.5 U of Taq DNA polymease (Promega) in a 25 μl reaction mixture containing 0.5 μM 10 mM dNTP, 1.5 μl of 25 mM MgCl, 2.5 μl of 10× Taq DNA polymerase buffer from Promega, 10 pmol of internal standard gene upstream and downstream primers to minimize variations in amplification efficiency between tubes.
Table 1.
Primer sequences and fragment lengths
| Gene | Primer sequence | Fragment length (base pairs) |
| MDR1 | 5'-GTT GCC ATT GAC TGA AAG AAC-3' | 120 |
| 5'-ACA GGA GAT AGG CTG GTT TGA-3' | ||
| GSTπ | 5'-ATG CTG CTG GCA GAT CAG-3' | 270 |
| 5'-GTA GAT GAG GGA GAT GTA TTT GCA-3' | ||
| MRP | 5'-GTA CAT TAA CAT GAT CTG GTC-3' | 256 |
| 5'-CGT TCA TCA GCT TGA TCC GAT-3' | ||
| TopoIIα | 5'-ATG CTA GTC CAC CTA AGA CCA-3' | 139 |
| 5'-TGT GTA GCA GGA GGG CTT GAA GAC AG-3' | ||
| β-Actin | 5'-GAA ATC GTG CGT GAC ATT AAG GAG AAG CT-3' | 383 |
| 5'-TCA GGA GGA GCA ATG ATC TTG A-3' |
GSTπ, glutathione S-transferase PI; MRP, MDR-related protein; TopoIIα, topoisomerase IIα.
PCR was performed with the use of Gene Amp PCR Systems 9700 (Perkin Elmer, Watsonville, CA, USA). For MDR1, GSTπ and TopoIIα genes, after an initial denaturation at 94°C for 1.5 min, 30 cycles of PCR amplification were performed, each cycle consisting of a denaturing step of 94°C for 1.5 min, annealing at 54°C for 1 min, and extension at 72°C for 3 min, followed by a final step at 72°C for 10 min. For the MRP gene, after an initial denaturation step of 95°C for 4 min, 35 cycles of PCR amplification were performed, each cycle consisting of a denaturing step of 95°C for 25 s, annealing at 54°C for 30 s, and extension at 73°C for 60 s, followed by a final step at 72°C for 8 min. The amplified fragments were detected by 2% (w/v) agarose gel electrophoresis and staining with 0.3 μg/ml ethidium bromide (Sigma). Each band was analysed on image analysis system IS1000 (Alpha Innotech, San Leandro, CA, USA). The specific gene expression level was determined semiquantitatively by calculating the ratio of density metric value from specific genes expressed in relation to the internal standard (specific gene expression/β-actin expression).
Immunocytochemistry staining
Treatment with 5 μM NOM was performed as above (see RT-PCR). A smear was made from each cell sample; after the smear had dried in air, cells were fixed in acetone for 10 min. Immunocytochemistry was performed with an avidin-biotin complex immunoperoxidase method as described by Zhou [16], with some modifications. Monoclonal antibody against P-gp (JSB-1) was purchased from Boehringer Mannheim Biochemica; monoclonal antibodies against GSTπ and TopoIIα were purchased from Dako Corporation and Neomarker, respectively. In brief, after three washes with phosphate-buffered saline (PBS), fixed cells in each smear were layered with the primary antibody against P-gp (1:20 dilution), GSTπ (1:25 dilution) and TopoIIα (1:50 dilution) and incubated overnight (a minimum of 18 h). After three washes with PBS, the cells were incubated with secondary antibody mixture of anti-mouse Ig (1:200 dilution; Huamei BG Co Ltd, Shanghai, China) for 2 h at room temperature. The cells were washed as before and then incubated with ABC complex (1:100 dilution; Huamei BG Co Ltd) for 1 h at room temperature. After being washed, cells were developed in 0.04% 3,3-diaminobenzidine tetrahydrochloride dihydrate (Sigma) with 0.02% H2O2 in PBS for 15-30 min. Cells were washed again with PBS and counterstained with haematoxylin.
In each assay, five categories of staining were observed as defined previously [17]. In brief, we first determined the staining grade I: 0, no staining; 1, pale yellow; 2, brown yellow; 3, brownish. Then we calculated the grade II according to the ratio of positive staining cells to total tumour cells: 0, no staining; 1, less than 10%; 2, 11-50%; 3, 51-75%; 4, more than 75%. Finally, according to total grade (total grade = grade I × grade II), the immunocytochemistry assay standard was determined as follows: total grade 0, negative; 1-3, feeble positive; 4 or 5, weak positive; 6 or 7, moderate positive; more than 7, strong positive.
ADR accumulation
Accumulation of ADR was monitored using a standard procedure by incubating MCF7/ADR cells (5 × 105/ml) for 2 h at 37°C in the presence of ADR (10 μM) alone or in combination with NOM (20, 10 or 5 μM). Cells were then harvested and washed twice with cold (0°C) PBS, then placed in ice-water to block the reaction until analysis. After half an hour, the fluorescence intensity of cells (104/ml) was determined by flow cytometry (FCM) (FAC-SCalibar; Becton Dickinson, San Jose, CA, USA).
Cell cycle analysis
Cells were plated at 2 × 105/ml in specific medium supplemented as above. After 24 h the medium was replaced with fresh medium containing ADR alone or in combination with NOM (20 μM). The cells were harvested after 48 h and washed twice with PBS, then fixed with 70% (v/v) ethanol. The sample was concentrated by removing ethanol and treated with 1% (v/v) Triton X-100 and 0.01% RNase for 10 min. Staining of cellular DNA was performed with 0.05% propidium iodide for 20 min at 4°C. FCM analysis was performed by acquiring a minimum of 2 × 105 nuclei. Cell cycle analysis was performed with the MultiCycle software package (Phoenix, San Diego, CA, USA).
Statistical analysis
Levels of statistical significance were evaluated with data from at least three independent experiments by using Student's t test. P <0.05 was considered statistically significant.
Results
The ADR IC50 values for MCF7/ADR and MCF7/WT cells were 30 and 0.4 μM respectively: MCF7/ADR cells were therefore 75-fold more resistant to the effects of ADR that MCF7/WT cells (Fig. 1a).
Figure 1.
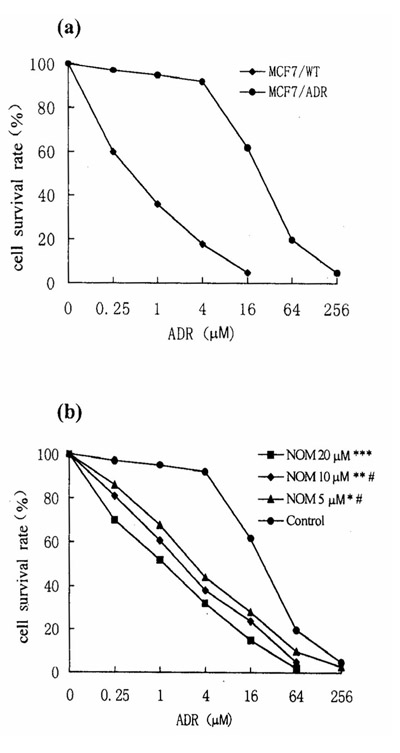
Cytotoxicity of ADR in MCF7/WT, MCF7/ADR and NOM-pretreated MCF7/ADR cells. (a) ADR cytotoxicity in MCF7/ADR (??) and MCF7/WT (??) cell lines. (b) Reversal effects of NOM on drug resistance in MCF7/ADR cells. The NOM concentrations were 20, 10 and 5 μM. Statistical significance compared with control: *, P <0.05; **, P < 0.01; ***, P < 0.005; compared with 20 μM NOM: #, P <0.05.
Enhancement of chemosensitivity in treated cells
Cytotoxicity was expressed as the percentage growth inhibition compared with untreated control cells. MTT assays showed that 1.5% ethanol or 20 μM NOM and other various reversal agents showed no toxicity towards the two cell lines. In MCF7/ADR cells treated with NOM (5 μM) for 10 days the cell growth rate was normal compared with control cells, as judged by staining with trypan blue. The results showed that 20, 10 and 5 μM NOM significantly increased the sensitivity of MCF7/ADR cells to ADR, the degrees of reversal being 21-fold, 12-fold and 8-fold, respectively (Fig. 1b). The reversal activity of 20, 10 and 5 μM NOM was markedly stronger than that of the same concentration of DRO and MG (Fig. 2) and was similar to that of the classic agents VRP and TAM. In MCF7/WT cells neither NOM nor other reversal compounds increased chemosensitivity to ADR (Table 2).
Figure 2.
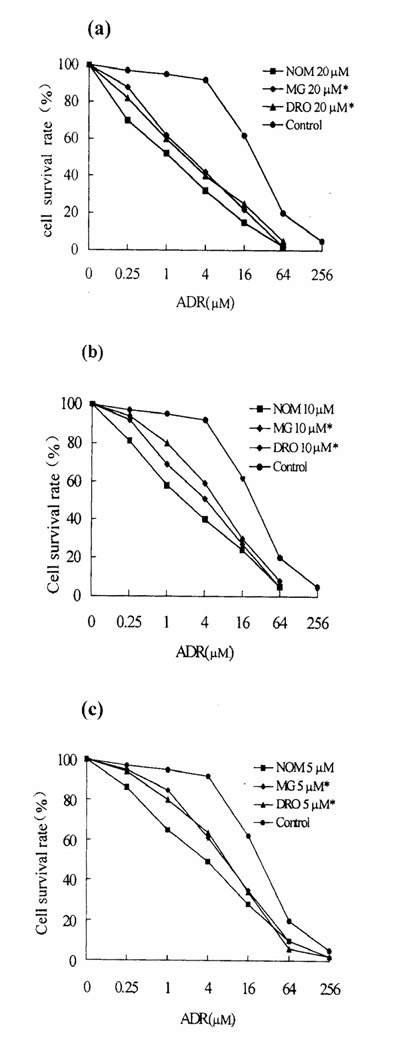
Comparison of reversal effects on drug resistance in MCF-7/ADR cells between NOM and MG or DRO, each at 20 μM (a), 10 μM (b) or 5 μM (c). Statistical significance compared with NOM: *, P <0.05. The control shows the response in the absence of treatment with reversal agent.
Table 2.
Reversal effects of nomegestrol and other reversal agents on drug resistance in MCF7/WT and MCF7/ADR cells (MTT assay)
| MCF7/WT | MSF7/ADR | ||||
| Drug | Concentration (μM) | IC50 | Reversal (fold) | IC50 | Reversal (fold) |
| ADM | 0.40 ± 0.12 | 30.0 ± 6.24 | |||
| NOM | 20 | 0.38 ± 0.09 | 1 | 1.4 ± 0.32*** | 21 |
| 10 | 0.40 ± 0.08 | 1 | 2.5 ± 0.45**† | 12 | |
| 5 | 0.41 ± 0.07 | 1 | 3.8 ± 0.63*† | 8 | |
| MG | 20 | 0.41 ± 0.09 | 1 | 3.0 ± 0.84**‡ | 10 |
| 10 | 0.39 ± 0.11 | 1 | 4.3 ± 1.01*‡ | 7 | |
| 5 | 0.39 ± 0.12 | 1 | 10.0 ± 1.42*‡ | 3 | |
| TAM | 20 | 0.36 ± 0.07 | 0.9 | 1.7 ± 0.37** | 18 |
| 10 | 0.38 ± 0.05 | 1 | 3.0 ± 0.64** | 10 | |
| 5 | 0.41 ± 0.06 | 1 | 6.0 ± 1.14* | 5 | |
| DRO | 20 | 0.40 ± 0.12 | 1 | 2.7 ± 0.81**‡ | 11 |
| 10 | 0.42 ± 0.14 | 1 | 5.0 ± 0.87*‡ | 6 | |
| 5 | 0.39 ± 0.08 | 1 | 10.0 ± 2.04*‡ | 3 | |
| VRP | 20 | 0.36 ± 0.10 | 0.9 | 1.4 ± 0.43*** | 22 |
| 10 | 0.38 ± 0.07 | 1 | 2.1 ± 0.61** | 14 | |
| 5 | 0.39 ± 0.06 | 1 | 4.3 ± 0.91* | 7 | |
Results are means ± standard deviation. Statistical significance compared with control: ***, P < 0.005; **, P < 0.01; *, P <0.05; compared with 20 μM NOM: † P <0.05; compared with responsive concentration of NOM, ‡ P <0.05. ADM, adriamycin; DRO, droloxifene; MDR, multidrug resistance; MG, megestrol; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; NOM, nomegestrol; TAM, tamoxifen; VRP, verapamil.
MDR-related gene expression in NOM-treated cells
RNA from untreated cells served as a control. In the control MCF7/ADR cells, MDR1, GSTπ and MRP gene expression levels were all high. A positive band of TopoIIα gene expression was not detected after 30 cycles of amplification; a weak band was found after 35 cycles of amplification. In the control MCF7/WT cells, the MDR1 and GSTπ genes showed negative results, whereas the MRP and TopoIIα genes were expressed positively.
The effects of NOM on MDR-related gene mRNA levels were evaluated by calculating the ratio of the expression of the resistant gene to that of β-actin by semiquantitative analysis (at least three independent NOM treatment experiments).
MDR1 and GSTπ mRNA expression
After treatment with 5 μM NOM, the MDR1 and GSTπ expression levels of MCF7/ADR cells were modulated in a time-dependent manner. The expression of both mRNA species began to decrease on the second day (P <0.05 and P < 0.01, respectively), and reached the lowest level on the third day (both P < 0.01). The expression level began to rise as detected on the fifth day (P >0.05 and P <0.05, respectively), and reached a level close to that of untreated controls on the tenth day (Fig. 3, Table 3).
Figure 3.
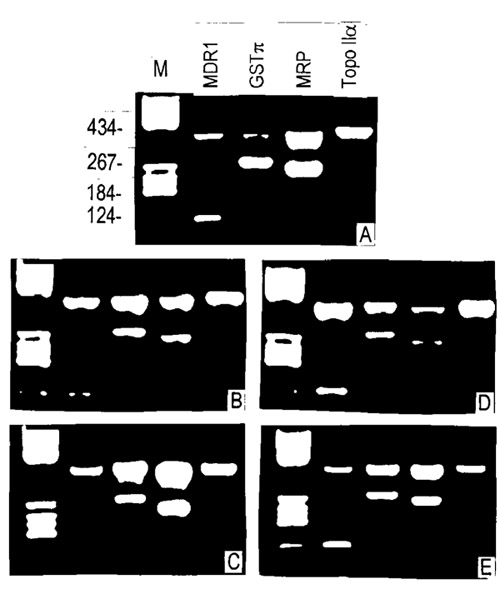
Reverse transcriptase polymerase chain reaction (RT-PCR) analysis of MDR-related gene mRNA expression in NOM-pretreated MCF-7/ADR cells, amplifying β-actin (as endogenous control) with MDR1, GSTπ, MRP and TopoIIα. Lane M, DNA molecular weight marker standards (bases). The sizes of the specific RT-PCR products were 120 base pairs (bp) for MDR, 270 bp for GSTπ, 256 bp for MRP, 139 bp for TopoIIα and 383 bp for β-actin. The cell line was treated with NOM at the start (A) (control) and after 2 days (B), 3 days (C), 5 days (D) and 10 days (E).
Table 3.
Expression of MDR-related gene mRNA species in nomegestrol-pretreated MCF7/ADR cells
| Comparison | Duration of treatment (days) | MDR1 | GSTπ | MRP | TopoIIα |
| MA | 0 | 1.7 ± 0.61 | 2.0 ± 0.82 | 0.9 ± 0.30 | 0.4 ± 0.10 |
| 2 | 0.5 ± 0.22* | 0.5 ± 0.24** | 0.7 ± 0.19 | 0.4 ± 0.15 | |
| 3 | 0.1 ± 0.04** | 0.5 ± 0.15** | 0.7 ± 0.34 | 0.5 ± 0.21 | |
| 5 | 0.5 ± 0.21* | 0.7 ± 0.30* | 0.8 ± 0.17 | 0.5 ± 0.18 | |
| 10 | 1.5 ± 0.60 | 0.9 ± 0.80* | 0.7 ± 0.25 | 0.4 ± 0.09 | |
| MW | 0 | - | - | 0.6 ± 0.24 | 0.8 ± 0.31 |
| 2 | - | - | 0.7 ± 0.27 | 0.8 ± 0.38 | |
| 3 | - | - | 0.5 ± 0.30 | 0.7 ± 0.19 | |
| 5 | - | - | 0.7 ± 0.31 | 0.7 ± 0.32 | |
| 10 | - | - | 0.6 ± 0.18 | 0.7 ± 0.26 |
Values are ratios of β-actin to MDR-related genes: MA, MCF7/ADR; MW, MCF7/WT. Statistical significance compared with control: *, P <0.05; **, P < 0.01. Data are means ± standard deviation from three experiments. GSTπ, glutathione S-transferase Pi; MDR, multidrug resistance; MRP, MDR-related protein; TopoIIα, topoisomerase IIα.
MRP and TopoIIα mRNA expression
After treatment with NOM, the MRP mRNA expression level of MCF7/ADR cells tended to decrease on the second day, with no statistical significance. No significant change was found in TopoIIα expression level when measured on the second, third, fifth and tenth days after treatment in comparison with the control. No significant changes in MRP and TopoIIα mRNA expression levels were found in MCF7/WT cells (Table 3).
Expression of P-gp, GSTπ and TopoIIα proteins
MCF7/ADR cells had high expression levels of P-gp and GSTπ proteins and a moderate expression level of TopoIIα protein. MCF7/WT cells expressed a high level of TopoIIα only: P-gp and GSTπ protein expression were undetectable. After treatment with NOM, the expression levels of P-gp and GSTπ in MCF7/ADR cells gradually declined, the lowest level being on the third day; on the fifth day their expression levels began to rise. No marked difference whose TopoIIα protein expression was found before and after NOM treatment in either MCF7/ADR or MCF7/WT cells. The effects of NOM on the expression of drug resistance protein were similar to those of NOM on mRNA expression (Figs 4 and 5).
Figure 4.
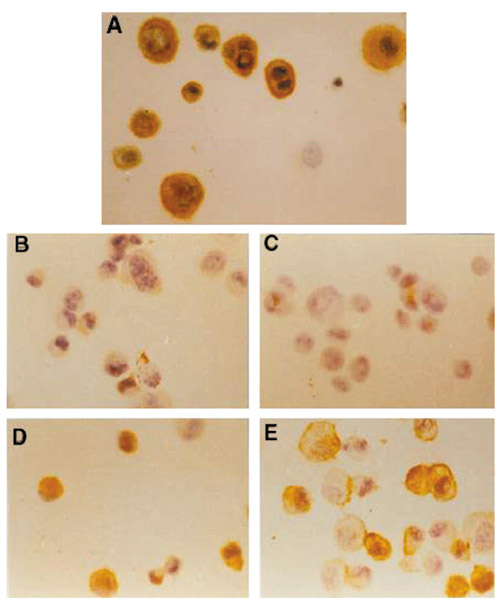
Expression of P-glycoprotein in NOM-pretreated MCF-7/ADR cells, detected with monoclonal antibody. The cell line was treated with NOM at the start (A) (control) and after 2 days (B), 3 days (C), 5 days (D) and 10 days (E). Magnification ×200.
Figure 5.
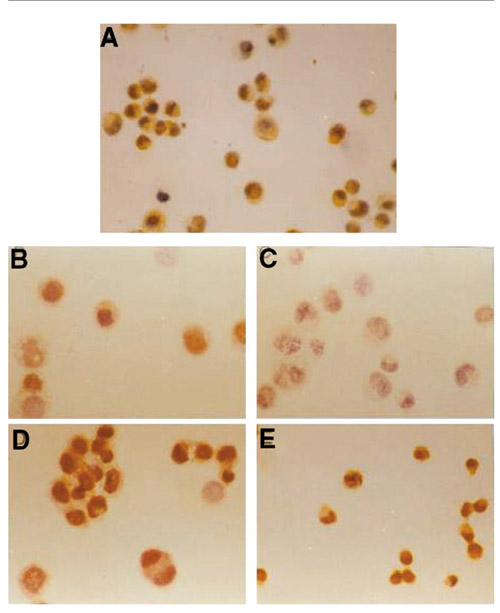
Expression of glutathione S-transferase Pi in NOM-pretreated MCF-7/ADR cells, detected with monoclonal antibody. The cell line was treated with NOM at the start (A) (control) and after 2 days (B), 3 days (C), 5 days (D) and 10 days (E). Magnification ×200.
ADR accumulation and cell cycle changes in NOM-treated cells
The accumulation of ADR in MCF7/ADR cells was much less than in MCF7/WT cells. Each cell line was treated with 20, 10 and 5 μM NOM. Marked increases in intracellular ADR accumulation were found in MCF7/ADR cells, the increases being 2.7-fold, 2.3-fold and 1.5-fold, respectively. However, NOM did not increase ADR accumulation in MCF7/WT cells (Fig. 6). FCM analysis was performed in MCF7/ADR cells treated for 48 h with ADR alone or in combination with 20 μM NOM. The results showed that when the ADR concentration was increased from 5 μM to 40 μM, only a small increase in arrested cells at the G2M phase was found in MCF7/ADR cells treated with ADR alone. However, when ADR was used in combination with 20 μM NOM, a marked arrest was seen. NOM alone failed to induce any change in the cell cycle (Fig. 7 and Table 4).
Figure 6.
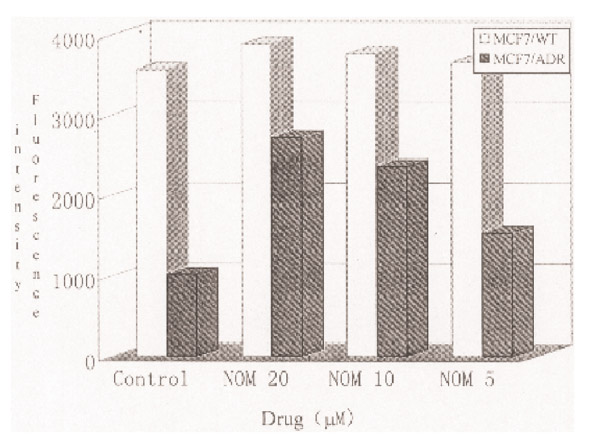
Accumulation of adriamycin in NOM-pretreated MCF-7/ADR and MCF-7/WT cells. The fluorescence intensity, measured by FACScan flow cytometry, is the mean fluorescence of 104 cells.
Figure 7.
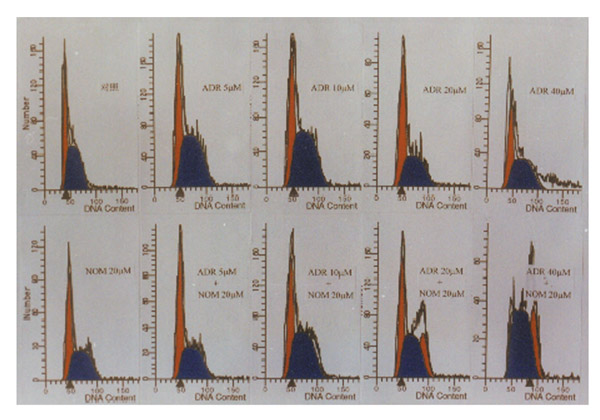
Cell cycle analysis of MCF-7/ADR cells treated with ADR either alone or in combination with nomegestrol (20 μM). The percentages of cells in the G0G1, S and G2/M phases are detailed in Table 2. Data refer to one experiment.
Table 4.
Effects of nomegestrol alone or in combination with adriamycin on cell cycle in MCF7/ADR cells
| ADR alone | ADR+ NOM (20 μM) | |||||
| ADR concentration (μM) | G0G1 | S | G2/M | G0G1 | S | G2/M |
| 0 (control) | 40 | 60 | 0 | 47 | 52.8 | 0.2 |
| 5 | 40 | 60 | 0 | 52 | 47.8 | 0.2 |
| 10 | 39 | 60 | 1 | 38 | 55.6 | 6.4 |
| 20 | 52 | 46 | 2 | 30 | 46 | 24 |
| 40 | 50 | 45 | 5 | 3 | 66.6 | 30.4 |
Values are percentages of cells at the indicated cell cycle stages. ADR, adriamycin; NOM, nomegestrol.
Discussion
The overexpression of P-gp in tumour cell membrane is considered to be the major mechanism of MDR. P-gp is able to pump various anticancer drugs out of cells, thus resulting in a low intracellular drug concentration that is insufficient to kill tumour cells [18]. To use compounds with low toxicity, or with none at all, to bind P-gp and block its transport function is the most common method of reversing MDR [19]. Progesterone and MG belong to the progestogen group. Although progesterone is not a substrate for P-gp, it can directly bind to P-gp and block the transport of drug efflux [20]. MG is a strong reversal agent: its capacity to increase intracellular accumulation of vincristine is 2-3-fold that of progesterone [6]. Previous results have shown that a daily dose of 800 mg MG resulted in a plasma concentration of 2 μM [21]; however, to reverse MDR, 5 μM is necessary in theory. Because high doses of MG induce vomiting, oedema, dizziness and androgen-like side-effects, the ideal effective concentration is difficult to obtain in vivo. Therefore, the search for new drugs with low toxicity is in progress to meet an urgent need for clinical applications.
NOM is a progestogen compound used in family planning. In recent years, biological activities other than contraception have been found [22]. Animal experiments have demonstrated that NOM has almost no toxicity. Its full chemical name is 3,20-diketo-6-methyl-17-α-hydroxy-19-norpregna-4,6-diene [9].
The present study has demonstrated that NOM significantly sensitizes the MCF7/ADR cell line to ADR in a concentration-dependent manner, but has no similar effect on MCF7/WT cells. The reversal effects of 20, 10 and 5 μM NOM were 21-fold, 12-fold and 8-fold, respectively. The enhancement of chemosensitivity to ADR by NOM was stronger than that by MG, a precursor of NOM, at corresponding concentrations (P <0.05). The reversal effect of NOM was similar to that of VRP, which is a classical reversal agent with a high reversal activity; NOM therefore also has a strong reversal activity.
Both TAM and DRO are anti-oestrogen agents. TAM is a first-line drug used in endocrinotherapy for breast cancer. Anti-oestrogens can inhibit P-gp function by binding it [23]. In our study, the effect of NOM was significantly greater than that of DRO.
The development of MCF7/ADR cells was induced by treating MCF7/WT with ADR. Its MDR phenotype is not altered after 3 months of serial passaging in drug-free medium [24, 25]. MCF7/ADR cells have been demonstrated to overexpress MDR1/P-gp, as well as the GSTπ and MRP genes. They are therefore good cell models for studying the effect of NOM on breast cancer MDR.
Semiquantitative RT-PCR analysis is a sensitive and specific method [26, 27]. A variety of methods using either quantitative or semequantitative RT-PCR have been used to determine relative initial target mRNA levels in samples. However, in all of these methods undefined variations in amplification complicated the interpretation of results. Most investigators use internal amplification standards in an attempt to correct for variations between tubes. In the present study, as in many other studies, we chose to use endogenous standards. The reference mRNA and target mRNA are usually processed together throughout the experiments, from RNA extraction until PCR amplification. This tends to minimize differences in RNA yields between samples.
It has been considered that the overexpression of MDR1/P-gp is a major mechanism of MDR in tumour cells [28]. A few studies have reported that some reversal agents can inhibit MDR1/P-gp expression. Stein et al [29] have found that cytokines such as interleukin-2, interferon-γ and tumour necrosis factor-α were capable of decreasing the expression of MDR1 mRNA in the colon carcinoma cell lines HCT15 and HCT16. Liu et al [30] treated MCF7/ADR cells with the Chinese herb Fructus psoraleae for 48 h and found that P-gp expression became undetectable. However, no results on whether or not progestogen can modulate the expression of MDR1/P-gp have been reported. The present study has demonstrated that NOM significantly inhibited MDR1/P-gp expression on the third day after treatment. The MDR mRNA expression level was very low, and that of P-gp almost ceased.
It is worthy to note that on the fifth day after NOM treatment, the expression of MDR1/P-gp began to increase again. This phenomenon was also observed when treating colon carcinoma cell line with cytokines [29]. Some studies have also found that P-gp antagonists such as VRP, cyclosporin A and reserpine could induce MDR1/P-gp expression in the colon carcinoma cell line LS180-Ad50 [31, 32]. A few papers have reported MDR1 expression after treatment with progesterone. Lee et al [33] showed that progesterone interacted with P-gp, inducing its expression in the granulose cell in preovulatory follicles to modulate steroid efflux. Our results show that NOM modulated the expression of MDR1 in a time-dependent manner. Over 5 days of treatment with NOM, MDR1 expression became elevated. From these findings it seems that NOM and other progestogen compounds show an effect of upregulated MDR1 expression in long-term treatment. It is unclear how NOM effects this time-dependent modulation of the expression of MDR1/P-gp. If a similar modulation of the MDR phenotype occurs in vivo, then the duration of treatment with NOM becomes important. It will be of prime importance to verify the time course for modulation by reversal agents and to design appropriate clinical trials.
In investigating the P-gp-induced MDR phenotype, the overexpression of some non-P-gp MDR-related proteins have been demonstrated. These proteins are important in drug resistance in some tumour cell lines [34, 35]. We also observed the expression of the non-P-gp MDR-related genes GSTπ, TopoIIα and MRP. We found that NOM markedly decreased the level of expression of GSTπ mRNA and protein with a time course similar to that of MDR1/P-gp. The results suggest that NOM possibly acts as a reversal agent of GSTπ. MRP belongs to the superfamily of ATP-binding cassette protein transporters. Like P-gp, MRP is located in the plasma membrane of resistant tumour cells; however, the mechanism of MRP might be fundamentally different from that of P-gp. The extrusion of several drugs by MRP requires glutathione. MRP might be identical to the GS-X pump [36]. MRP-related MDR was not reversed by classic reversal agents of P-gp such as VRP and cyclosporin A [37, 38, 39]. Our results suggest that NOM cannot modulate MRP gene expression either.
TopoII, as a kind of nuclease, is important in the processes of DNA metabolism such as transcription, replication and chromosome partitioning during cell division. TopoIIα is present mainly in the S phase [40]. TopoIIα in tumour cells is a target enzyme of some anticancer agents. If TopoIIα activity decreases or a TopoIIα gene mutation develops, TopoIIα-related MDR results [41, 42].
Our results showed that TopoIIα expression in MCF7/ADR cells was weaker than in MCF7/WT cells. Because TopoIIα is a target enzyme of ADR, MDR to ADR in MCF7/ADR cells might to some extent be related to TopoIIα. However, no change in TopoIIα gene expression level was detected in NOM-treated MCF7/ADR cells, indicating that the reversal effect of NOM on MDR in MCF7/ADR cells cannot result from modulating the gene expression of TopoIIα.
P-gp belongs to the ATP-dependent transporters. It acts as a pump or hydrophobic vacuum cleaner, effectively increasing drug efflux and decreasing drug influx [43]. Many studies have demonstrated that progestogen compounds directly bind P-gp and block the function of the pump efflux [10, 44]. We studied the effect of NOM on intracellular ADR accumulation in MCF7/ADR cells by using FCM assays. ADR can emit fluorescence; its intensity represents its accumulation [25]. After 2 h of treatment with NOM, the intracellular fluorescence intensity of ADR was markedly enhanced, suggesting that NOM directly inhibits the P-gp pump efflux function in the same way as other progestogen compounds do.
It has been reported that ADR induces dose-dependent G2M arrest and that cells in this phase are particularly sensitive to chemical drugs [45]. Cell-cycle DNA content assays by FCM suggest that NOM enhances the blocking activity of ADR on the cell cycle. This enhancement of blocking activity might be partly responsible for the reversal effect of NOM.
In brief, our results indicate that in MCF7/ADR cells NOM can significantly sensitize their chemosensitivity to ADR, down-regulate MDR1/P-gp and GSTπ expression levels in time-dependent manner, markedly increase intracellular ADR accumulation and enhance the cell cycle blocking activity of ADR. At present, although a variety of agents inhibit the function of P-gp in vitro, their clinical use is also limited by the toxicity associated with the doses required to reverse MDR. Because NOM can be safely administered at high doses, it might be a good candidate for an MDR reversal agent in clinics.
Abbreviations
ADR = adriamycin; DRO = droloxifen; GSTπ = glutathione S-transferase Pi; MDR = multidrug resistance; MG = megestrol; MRP = MDR-related protein; MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; NOM = nomegestrol; PBS = phosphate-buffered saline; P-gp = P-glycoprotein; RT-PCR = reverse transcriptase polymerase chain reaction; TAM = tamoxifen; TopoIIα = topoisomerase IIα; VRP = verapamil.
Acknowledgments
Acknowledgement
We thank Professor Lian-Fang He for a critical reading of the manuscript.
References
- Early Breast Cancer Trialists' Collaborative Group Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy: 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women. Lancet. 1992;339:71–84. [PubMed] [Google Scholar]
- Gottesman MM. How cancer cells evade chemotherapy. Cancer Res. 1993;53:747–754. [PubMed] [Google Scholar]
- Ferry DR, Traunecker H, Kerr DJ. Clinical trials of P-glycoprotein reversal in solid tumours. Eur J Cancer. 1996;32A:1070–1081. doi: 10.1016/0959-8049(96)00091-3. [DOI] [PubMed] [Google Scholar]
- Panasci L, Jean-Claude BJ, Vasilescu D, Mustafa A, Damian S, Damian Z, Georges E, Liu Z, Batist G, Leyland-Jones B. Sensitization to doxorubicin resistance in breast cancer cell lines by tamoxifen and megestrol acetate. Biochem Pharmacol. 1996;52:1097–1102. doi: 10.1016/0006-2952(96)00456-x. [DOI] [PubMed] [Google Scholar]
- Claudio JA, Emerman JT. The effects of cyclosporin A, tamoxifen, and medroxyprogesterone acetate on the enhancement of adriamycin cytotoxicity in primary cultures of human breast epethelial cells. Breast Cancer Res Treat. 1996;41:111–122. doi: 10.1007/BF01807156. [DOI] [PubMed] [Google Scholar]
- Fleming GF, Amato JM, Agresti M, Safa AR. Megestrol acetate reverses multidrug resistance and interacts with P-glycoprotein. Cancer Chemother Pharmacol. 1992;29:445–449. doi: 10.1007/BF00684845. [DOI] [PubMed] [Google Scholar]
- Aisner J, Tchekmedyian NS, Tait N, Parnes H, Novak M. Studies of high-dose megestrol acetate: potential application in cachexia. Semin Oncol. 1988;15S:68–75. [PubMed] [Google Scholar]
- Grulol DJ, Bourgeois S. Chemosensitizing steroids: glucocorticoid receptor agonists capable of inhibiting P-glycoprotein function. Cancer Res. 1997;54:720–727. [PubMed] [Google Scholar]
- Gasteaud JM. 3,20-Diketo, 6-methyl, 17-α-hydroxy 19-norpregna 4,6-diene, its esters and the uses thereof. US Patent 4544555. 1985.
- Yang C-PH, DePinho SH, Greenberger LM, Arceci RJ, Horwitz SB. Progesterone interacts with P-glycoprotein in multidrug-resistant cells and in the endometrium of gravid uterus. J Biol Chem. 1989;264:782–785. [PubMed] [Google Scholar]
- Wang L, Yang CP, Horwitz SB, Trail PA, Casazza AM. Reversal of the human multidrug-resistance phenotype with megestrol acetate. Cancer Chemother Pharmacol. 1994;34:96–102. doi: 10.1007/BF00685925. [DOI] [PubMed] [Google Scholar]
- Bojar H, Stuschke M, Staib W. Effects of high-dose medrox-yprogesterone acetate on plasma membrane lipid mobility. Prog Cancer Res Ther. 1984;31:115–119. [Google Scholar]
- Vickers PJ, Dickson RB, Shoemaker R, Cowan KH. A multidrug-resistant MCF7 human cancer cell line which exhibits cross-resistance to anti-estrogens and hormone-independent tumor growth. Mol Endocrinol. 1988;2:886–892. doi: 10.1210/mend-2-10-886. [DOI] [PubMed] [Google Scholar]
- Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrzolium based semiautomated colormetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936. [PubMed] [Google Scholar]
- O'Driscoll L, Kennedy S, McDermott E, Kelehan P, Clynes M. Multiple drug resistance-related messenger RNA expression in archival formalin-fixed paraffin-embedded human breast tumour tissue. Eur J Cancer. 1996;32A:128–133. doi: 10.1016/0959-8049(95)00552-8. [DOI] [PubMed] [Google Scholar]
- Zhou G. Routine histoimmunochemistry methods. In Practical Methodology of Oncopathology [in Chinese] Edited by Xu L Shanghai: Shanghai Medical University Press, Changshu Printing Factory; 1997. pp. 173–174.
- Xu L, Yang W. Standards of determining histoimmunochemistry staining results [in Chinese]. China Oncol. 1996;6:229–231. [Google Scholar]
- Ling V. P-glycoprotein and resistance to anticancer drugs. Cancer. 1992;69:2693–2609. doi: 10.1002/1097-0142(19920515)69:10<2603::aid-cncr2820691034>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Ford JM. Experimental reversal of P-glycoprotein-mediated multidrug resistance by pharmacological chemosensitisers. Eur J Cancer. 1996;32A:991–1001. doi: 10.1016/0959-8049(96)00047-0. [DOI] [PubMed] [Google Scholar]
- Yang CP, Cohen D, Greenberger LM, Hsu SI, Horwitz SB. Differential transport properties of two mdr gene products are distinguished by progesterone. J Biol Chem. 1990;265:10282–10288. [PubMed] [Google Scholar]
- Wang L, Yang C-PH, Trial P, Horwitz SB, Casazza AM. Reversal of the multidrug resistance (MDR) phenotype with megesterol acetate. (MA). Proc Am Assoc Cancer Res. 1991;32:377. [Google Scholar]
- Pasqualini JR, Paris J, Sitruk-Ware R, Chetrite G, Botella J. Progestins and breast cancer. J Steroid Biochem Mol Biol. 1998;65:225–235. doi: 10.1016/s0960-0760(98)00028-4. [DOI] [PubMed] [Google Scholar]
- Rao US, Fine RL, Scarborough GA. Antiestrogens and steroid hormones: substrates of the human P-glycoprotein. Biochem Pharmacol. 1994;48:287–292. doi: 10.1016/0006-2952(94)90099-x. [DOI] [PubMed] [Google Scholar]
- Batist G, Tulpule A, Sinha BK, Katki AG, Myers CE, Cowan KH. Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells. J Biol Chem. 1986;261:15544–15549. [PubMed] [Google Scholar]
- Ford JM, Brufferman EP. Cellular and biochemical characterization of thioxanthenes for reversal of multidrug resistance in human and murine cell lines. Cancer Res. 1990;50:1748–1756. [PubMed] [Google Scholar]
- Wunder JS, Andrulis IL, Gazdar AF, Willman CL, Griffith B, Von Hoff DD. Quantitative analysis of MDR1 (multidrug resistance) gene expression in human tumors by polymerase chain reaction. Proc Natl Acad Sci USA. 1990;87:7160–7164. doi: 10.1073/pnas.87.18.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog CE, Trepel JB, Mickley LA, Bates SE, Fojo AT. Various methods of analysis of mdr1/P-glycoprotein in human colon cancer cell lines. J Natl Cancer Inst. 1992;84:711–716. doi: 10.1093/jnci/84.9.711. [DOI] [PubMed] [Google Scholar]
- Lehnert M. Clinical multidrug resistance in cancer: a multifactorial problem. Eur J Cancer. 1996;32A:912–920. doi: 10.1016/0959-8049(96)00069-x. [DOI] [PubMed] [Google Scholar]
- Stein U, Walther W, Shoemaker RH. Modulation of mdr1 expression by cytokines in human colon carcinoma cells: an approach for reversal of multidrug resistance. Br J Cancer. 1996;74:1384–1391. doi: 10.1038/bjc.1996.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Meng S, Yang J, Ping W. Reversal effect of R3 (extract from a Chinese herb Bu-Gu-Zhi) on MDR of MCF7/ADR cell line [in Chinese]. Chinese J Clin Oncol. 1997;24:325–330. [Google Scholar]
- Herzog CE, Tsokos M, Bates SE, Fojo AT. Increased mdr-1/P-glycoprotein expression after treatment of human colon carcinoma cells with P-glycoprotein antagonists. J Biol Chem. 1993;268:2946–2952. [PubMed] [Google Scholar]
- Bhat UG, Winter MA, Pearce HL, Beck WT. A structure-function relationship among reserpine/yohimbine analogs in their ability to increase expression of mdr1 and P-glycoprotein in a colon carcinoma cell line. Mol Pharmacol. 1995;48:682–689. [PubMed] [Google Scholar]
- Lee GY, Croop JM, Anderson E. Multidrug resistance gene expression correlates with progesterone production in dehydroepiandrosterone-induced polycystic and equine chorionic gonadotropin-stimulated ovaries of prepubertal rats. Biol Reprod. 1998;58:330–337. doi: 10.1095/biolreprod58.2.330. [DOI] [PubMed] [Google Scholar]
- Danks MK, Schmidt CA, Cirtain MC, Suttle DP, Beck WT. Altered catalytic activity of and DNA cleavage topoisomeraseII from human leukemic cells selected for resistance to VM-26. Biochemistry. 1988;27:8861–8869. doi: 10.1021/bi00424a026. [DOI] [PubMed] [Google Scholar]
- Danks MK, Yalowich JC, Beck WT. Atypical multiple drug resistance in a human leukemic cell line selected for resistance to teniposide (VM-26). Cancer Res. 1987;47:1297–1301. [PubMed] [Google Scholar]
- Loe DW, Deeley RG, Cole SP. Biology of the multidrug resistance-associated protein, MRP. Eur J Cancer. 1996;32A:945–957. doi: 10.1016/0959-8049(96)00046-9. [DOI] [PubMed] [Google Scholar]
- Loe DW, Deeley RG, Cole SP. Chemosensitisation and drug accumulation effects of cyclosporin A, PSC833 and verapamil in human MDR large cell lung cancer cells expressing a 190k membrane protein distinct from P-glycoprotein. Eur J Cancer. 1993;29A:408–415. doi: 10.1016/0959-8049(93)90397-x. [DOI] [PubMed] [Google Scholar]
- Leier I, Jedlitschky G, Buchholz U, Cole SP, Deeley RG, Keppler D. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J Biol Chem. 1994;269:27807–27810. [PubMed] [Google Scholar]
- Zaman GJ, Flens MJ, van Leusden MR, de Haas M, Mulder HS, Lankelma J, Pinedo HM, Scheper RJ, Baas F, Broxterman HJ. The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc Natl Acad Sci USA. 1994;91:8822–8826. doi: 10.1073/pnas.91.19.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Hwong CL. Cellular regulation of mammalian DNA topoisomerases. Adv Pharmacol. 1994;29A:167–189. doi: 10.1016/s1054-3589(08)60545-1. [DOI] [PubMed] [Google Scholar]
- de Jong S, Zijlstra JG, de Vries EG, Mulder NH. Reduced DNA topoisomerase and drug-induced DNA cleavage activity in an adriamycin-resistant human small cell lung carcinoma cell line. Cancer Res. 1990;50:304–309. [PubMed] [Google Scholar]
- Bugg BY, Danks MK, Beck WT, Suttle DP. Expression of a mutant DNA topoisomerase etioposide in CCRF-CEM human leukemic cells selected for resistance to teniposide. Proc Natl Acad Sci USA. 1991;88:7654–7658. doi: 10.1073/pnas.88.17.7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germann UA. P-glycoprotein-a mediator of multidrug resistance in tumour cells. Eur J Cancer. 1996;32A:927–944. doi: 10.1016/0959-8049(96)00057-3. [DOI] [PubMed] [Google Scholar]
- Claudo JA, Emerman JT. The effects of cyclosporin A, tamoxifen, and medroxyprogesterone acetate on the enhancement of adriamycin cytotoxicity in primary cultures of human breast epithelial cells. Breast Cancer Res Treat. 1996;41:111–122. doi: 10.1007/BF01807156. [DOI] [PubMed] [Google Scholar]
- Wadler S, Green MD, Basch R, Muggia FM. Lethal and sublethal effects of the combination of doxorubicin and the bis-dioxopiperazine(+)-1,2-bis(3,5-diozopeperazinyl-l-yl) propane (ICRF 187) on murine sarcoma S180 in vitro. Biochem Pharmacol. 1987;9:1495–1501. doi: 10.1016/0006-2952(87)90116-x. [DOI] [PubMed] [Google Scholar]